
Abstract
Demyelinating diseases with inflammation affect both the central and peripheral nervous systems. The histopathological hallmark of these devastating, and sometimes life-threatening, diseases is segmental demyelination of long axonal tracts and varying degrees of inflammation, with important long-term consequences for the function and survival of axons and neurons. Mitochondria are essential for the function of all cells, but maybe more so for neurons, in both physiological and pathological conditions. Healthy neurons utilise a sensitive, responsive and dynamic mitochondrial population for their varied basal metabolism. It is not surprising, then, that mitochondria also play a fundamental role in adaptive mechanisms which enable neurons to survive pathological events such as loss of myelin sheath and exposure to inflammatory mediators. Indeed, it is believed that the survival of axons in such an environment, and perhaps recovery to full functionality, critically depends on the adaptive capacity of its mitochondrial population. This chapter will present and interpret currently available, published knowledge relating to the role of neuronal mitochondria in neuroprotection and neurodegeneration during demyelinating inflammatory diseases of the central and peripheral nervous systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Multiple sclerosis
- Guillain-Barré syndrome
- Mitochondria
- Mitochondrial dynamics
- Demyelination
- Inflammation
1 Multiple Sclerosis
Multiple sclerosis (MS) is the most common demyelinating inflammatory disease of the central nervous system (CNS) and the leading cause of non-traumatic disability in young adults in the developed world [1, 2]. As a distinct neurological entity, MS was based on clinical criteria established by the French physician Jean-Martin Charcot more than 150 years ago [3]. The disease typically follows a relapsing-remitting course, whereby periods of loss of neurological function called relapses or attacks are followed by periods of complete or incomplete recovery (remissions). Over time, complete remissions become rare and neurological dysfunction gradually accumulates, leading to permanent, and often severe, disability.
Over 100 years ago, Otto Marburg described the pathology of what we now recognise as a partially remyelinated MS ‘plaque’. Since then, a large number of descriptive histopathological studies of post-mortem tissue have provided a detailed picture of MS lesions. These and, in more recent years, MRI-based imaging studies in patients together revealed unique, and to date unexplained, characteristics of MS – frequently appearing and disappearing inflammatory-demyelinating lesions that are well demarcated from the normal-appearing surrounding tissue and scattered throughout the CNS in a seemingly random fashion. Widespread neurodegeneration is a more recently recognised major feature of MS and often suspected as the likely cause of permanent disability.
The ubiquitous presence of infiltrating immune cells in the examined lesions has led to the often stated but as yet unproven hypothesis that multiple sclerosis is an autoimmune disease. Great efforts have been dedicated to identifying a common, specific autoimmune target antigen in MS, with particular focus on components of the myelin sheath. However, to date, all attempts to identify an autoantigen have failed [4].
The main pathological characteristics of MS lesions are (1) presence of scattered inflammatory foci in the brain and spinal cord parenchyma, mainly consisting of macrophages and lymphocytes and often centred on a blood vessel, (2) segmental demyelination, (3) neuronal and axonal degeneration and (4) focal gliosis [5]. Although a major feature, inflammation is not pathognomonic of MS. Other neurological diseases with progressive loss of neurons, such as Alzheimer’s [6] and Parkinson’s disease [7], also show a degree of inflammation [8, 9]. Although a varying degree of white matter loss has been reported in both of these neurodegenerative conditions [10–12], the feature that distinguishes MS from all other CNS diseases is the type of demyelination. Namely, MS is the only CNS disease characterised by the so-called segmental (i.e. spreading from one node to the next one) demyelination. Indeed, recent histopathological evidence from exceptionally early MS lesions suggests that primary demyelination is the earliest event in the lesion formation [13–15]. The loss of the myelin sheath along a section of an axon has considerable consequences for its function, most obviously for impulse conduction. This early event sets off a series of adaptive mechanisms aimed at preserving the axon structure, restoring its primary function in impulse conduction and restoring myelin. Mitochondria play a central role in a number of these adaptive, survival mechanisms. This chapter will review currently available, published knowledge on the role of mitochondria in axonal and neuronal protection against degeneration during inflammatory-demyelinating disease.
1.1 The Role of Myelin in Axonal Energy Homeostasis
The main role of myelin is generally believed to be in enabling saltatory impulse conduction in myelinated axons. This is separate from any metabolic support for the axon which may be provided by the myelinating glial cell. As in all cells, the neuron maintains ionic disequilibrium across its plasma membrane via the activity of ATP utilising pumps. The maintained ionic concentration gradient is measurable as a resting potential. A feature more specific to neurons however is the presence in the membrane of voltage-dependent Na+ and K+ gates. These gates open transiently in response to a critical level of depolarisation, particularly the Na+ gates, allowing ions to flow along the concentration gradient and producing a transient reversal of potential – the action potential – before closing and allowing restoration of the resting potential. The opening of the channels, and hence the action potential, is an all-or-nothing phenomenon. The depolarisation and accompanying ionic flow last long enough to open adjacent gates. However, gates once opened and closed require a finite time to achieve the initial configuration and cannot be immediately reopened; consequently, the action potential progresses along the axon in one direction, giving impulse conduction. The immediate free energy required for the action potential is derived from the local change in entropy. The resting potential relies on the continuous activity of the Na+/K+ ATPase, which is distributed evenly along the axolemma (axonal membrane) in both myelinated and nonmyelinated axons. This would establish a requirement for an even distribution of mitochondria.
In contrast to unmyelinated axons, the distribution of sodium channels in myelinated axons (Fig. 8.1) is restricted to narrow gaps between the segments of the myelin sheath called nodes of Ranvier (~1 μm) where there may be 1,000–2,000 channels per μm2. Neighbouring nodes, for example, in human sural nerve, a sensory nerve which descends down the posterior lateral side of the calf and lateral side of foot dorsum, may be separated by up to 1.4 mm of continuous myelin, called internodal segments [16]. Such clustering of channels at regular, but distant, intervals means that the flux of sodium and potassium ‘jumps’ from node to node skipping long internodal stretches, thus vastly increasing the speed of conduction. Furthermore, myelin acts as an electrical insulator, reducing the overall capacitance of the axolemma and thereby maximising the effect of potential change brought about by the inward sodium current at the node [17–19]. In other words, in the absence of myelin, the depolarising current quickly dissipates as it flows along the axons rather than staying ‘focused’ to the narrow nodal gap, resulting in insufficient current available to depolarise the next node and saltatory conduction fails. Having such an effect on preventing the loss of current, it follows that myelin reduces the net amount of ions required for successful membrane depolarisation across the axolemma. In this way, myelin also minimises axonal energy consumption, as far fewer sodium and potassium ions are required to be actively transported by Na+/K+ATPase in order to re-establish resting potential than in unmyelinated axons.

Impulse conduction in unmyelinated and myelinated axons. In unmyelinated axons (top panel), sodium channels are distributed along the whole surface of axonal membrane. During impulse conduction, sodium ions enter intra-axonal space from extracellular compartment along its entire length, sequentially changing membrane potential and thus creating small, continuous depolarising currents which spread slowly from one end of the axon to another. In myelinated axons (bottom panel), channel distribution is highly organised within specific subregions. Sodium channels are restricted to narrow gaps between the segments of the myelin sheath, called nodes of Ranvier. Myelin is attached to axolemma at paranodal regions by a number of anchoring proteins such as Caspr. Voltage-gated potassium channels and sodium/potassium pump are located in the juxtaparanodal regions. During impulse conduction, ion fluxes are limited to these narrow regions, thus greatly increasing speed of conduction
As mitochondrial oxidative phosphorylation is the main source of ATP in neurons, both neuronal cell bodies, dendrites and axons are highly dependent on correct functioning of these organelles for reliable and constant ATP supply. Furthermore, neurons are morphologically the most complex cells in the body, many having thousands of dendrites and axons of considerable length; thus, their function is critically dependent not only on the correct function but also on the precise and correct location of mitochondria. For example, neurons exhibit great variations in impulse activity over time, thus the relative energy demands of different neuronal sub-compartments are often rapidly changing. Although mitochondrial DNA synthesis has been reported to occur within some vertebrate peripheral axons [20], given that the majority of mitochondrial proteins are encoded in the nuclear genome [21], the bulk of mitochondrial biogenesis is thought to occur in the vicinity of the cell body. Therefore, any increase in metabolic demand at a distance from the soma would require increased mitochondrial transport via the long-distance, axonal transport system. This complex transport machinery provides optimal mitochondrial supply to all neuronal areas simultaneously, including sites remote from the cell body. Indeed, axonal impulse activity and mitochondrial axonal transport rate are positively correlated in both myelinated and unmyelinated peripheral nerve fibres [22]. Clearly, delivering mitochondria to areas of high metabolic demand (the right place) at the right time is essential. One such area appears to be the unmyelinated or demyelinated segment. This difference in axonal energy requirements depending on the presence of myelin is shown clearly by the example of optic nerve fibres crossing lamina cribrosa, a mesh-like structure at the posterior part of the sclera. Namely, the axons of the optic nerve are in many mammals unmyelinated on the intraocular side, but the same axons are myelinated on the extraocular side of lamina cribrosa. Interestingly, both the total amount and the activity of mitochondrial complex IV (cytochrome c oxidase) have been found to be significantly greater in the unmyelinated part of these axons than in their myelinated segments [23, 24]. As complex IV is the major consumer of cellular oxygen, this increase in complex IV activity suggests that the demand for the oxygen, and by inference the ATP, is higher in the unmyelinated than in the myelinated segment of the axon. Furthermore, given that the diameter of the optic nerve axons on either side of the lamina cribrosa is the same [25], the increase in the level of the complex IV subunit II [23] suggests a greater density of mitochondria in the unmyelinated segment of optic nerve axons. A similar high relative density of mitochondria in unmyelinated, compared with myelinated, axons has been observed in peripheral nerve axons (Fig. 8.2). This inverse correlation between myelination and mitochondrial density is entirely consistent with the energy-saving effect of the myelin sheath on axonal metabolic demand arising directly from impulse conduction, i.e. if conduction increases axonal transport, then this is an additional demand for energy consequential to conduction.

In vivo confocal image of mitochondria in myelinated and unmyelinated fibres in Thy1-CFP-S positive (mito-S) mouse peripheral nerve. Exposed saphenous nerve axons are labelled in situ with Alexa Fluor 488 conjugated Griffonia isolectin IB4 (green) and tetramethylrhodamine methyl ester (TMRM, red). Isolectin IB4 is a glycoprotein isolated from the seeds of the African legume, Griffonia simplicifolia, known to specifically bind to a subgroup of unmyelinated fibres, hence named IB4+ C fibres. TMRM is a potentiometric dye which preferentially accumulates within polarised (healthy) mitochondria. This combination of unmyelinated fibre labelling, functional mitochondrial labelling and constitutively expressed mitochondrial CFP enables visualisation of polarised mitochondria (c, d) within unmyelinated fibres (a, d). In CFP-S mice, the cyan fluorescent protein is constitutively expressed only in mitochondria within 40–60 % of myelinated axons (not expressed in unmyelinated fibres), thus enabling distinction between mitochondria in myelinated fibres and mitochondria in Schwann (and other) cells (Note that the density of mitochondria within unmyelinated fibres (green arrows in a) is noticeably greater (red arrows in c and in merged image in d) than in larger diameter, myelinated fibres (white arrows). Scale bar = 10 μm)
1.2 Energy Metabolism of Demyelinated Axon
Having in mind the above, it is not surprising that the loss of myelin sheath (i.e. demyelination during multiple sclerosis) renders axons vulnerable to a range of insults, including relative energy insufficiency. One of the earliest, and the most obvious, consequences of myelin loss is the impairment of impulse conduction propagation across the demyelinated segment. The effect of demyelination on impulse conduction has been studied in a range of focal, experimentally induced demyelinating lesions, typically using Schwann cell and oligodendrocyte toxins which inhibit protein synthesis such as diphtheria toxin and ethidium bromide or agents that cause destruction of myelin without affecting the myelin-forming cells, such as lysolecithin [26]. In such lesions, demyelination begins at the paranodal regions and spreads in both directions along the juxtaparanode and internode, as myelin loses its compact structure and detaches from axons [18]. This structural change affects the electrical properties of the axolemma in such a way that the overall membrane capacitance increases [17]. Owing to this increase in capacitance of a demyelinating axon, greater inward current is necessary in order to depolarise the next node, which means that more sodium moves into the axon and it takes a longer time to reach the excitation threshold than in myelinated axons. It has been shown in experimentally induced demyelinated lesions that internodal conduction may be 30 times slower than in normal, myelinated internodes [17]. Furthermore, repetitive impulse activity along such demyelinated segments leads to a gradual build-up of intracellular sodium, eventually overwhelming the sodium pump, and subsequently completely blocking the conduction [17]. Nonetheless, as the loss of myelin progresses and axons become completely denuded, impulse conduction is established again, albeit with different characteristics. Instead of being saltatory conduction, over the 2 or 3 weeks of persistent demyelination, the conduction re-establishes as slow and continuous, reminiscent of conduction in unmyelinated axons [27]. Furthermore, the demyelinated axons may exhibit spontaneous, ectopic impulse activity [28].
These elegant studies of axonal conduction along demyelinated segments, performed over 30 years ago, predicted that profound changes in distribution of ion channels along the axonal membrane must underlie the re-establishment, and the change in the nature, of impulse conduction. Indeed, it was later reported in animal models of multiple sclerosis [29, 30] and in MS lesions [31, 32] that the pattern of distribution of voltage-gated sodium channels changes dramatically following demyelination. Specifically, a subgroup of sodium channels called persistent current NaV1.6 channels, and other Na channels such as Nav1.2, become diffusely distributed along the demyelinated axolemma, instead of highly concentrated in a narrow area of the node [31]. Similarly, it has been shown that the distribution of potassium channels changes from their strict paranodal location in myelinated axons to diffuse localisation along demyelinated or dysmyelinated internodes [33–36]. As in healthy unmyelinated fibres, the widespread distribution of these changes allow for greater amounts of ion movements across the axolemma during impulse conduction, thereby necessitating greater activity of Na+/K+ ATPase, which accordingly increases ATP demand. It has been shown in models of ischemia [37, 38] and neurotrauma [39] that when such increased energy demand cannot be met due to insufficient ATP, the ensuing energy imbalance leads to accumulation of sodium ions in the axoplasm. Although harder to prove, high impulse conduction load is presumed to have a similar effect on intra-axonal sodium concentration, as the repetitive firing may lead to fast depletion of ATP. The high intra-axonal concentration of sodium, in turn, activates another pump called the sodium/calcium exchanger (Na+/Ca2+ exchanger) to work in reverse [39], importing calcium ions from the extracellular space in exchange for intracellular sodium, whereas it normally exports calcium from the axoplasm. These changes lead to pathologically high intracellular concentrations of calcium within demyelinated axons [38, 39] and activation of dangerous Ca-mediated cascades which, in experimental models of inflammatory-demyelination, result in axonal degeneration [30, 32].
From all the above, it is clear that two critical factors that determine the future of demyelinated axons are (1) the availability of ATP for the increased demand in the demyelinated segment and (2) the capacity of that segment to buffer excess calcium load. Both of these functions are performed by mitochondria; hence, unsurprisingly, mitochondria play a major role in determining the long-term outcome of demyelination. Importantly, however, following demyelination, the increased demand for mitochondria arises within an area that may be a long way away from the cell body. Therefore, other aspects of axonal function, such as those regulating mitochondrial transport from cell body along the axon, are essential for delivering mitochondria to the vulnerable site and thus at least equally important for long-term axonal survival.
1.3 The Role of Mitochondria in Survival of Demyelinated Axon
How long demyelinated axons survive in vivo is unclear. Answering this question is particularly difficult due to extremely limited availability of human tissue from demyelinated lesions. Even so, it has been apparent from the earliest descriptions of MS lesions, such as those described by Otto Marburg in 1906 [40], as well as from the more recent ones [41, 42] that some demyelinated axons can persist within the lesions long enough to become remyelinated. Autopsy material from patients who died early in the course of MS revealed crucial information about natural evolution of demyelinated axons in human brains during early stages of the disease. These studies showed that demyelinated axons that had newly arisen within previously unaffected white matter often become remyelinated, forming so-called shadow plaques [43, 44]. Furthermore, in one study of 98 lesions, approximately 15 % of old shadow plaques showed evidence that axons within this area had been recently demyelinated for the second time [42]. Similar focal recurrence of new and chronic lesions has been reported in longitudinal MRI examinations [45–47].
Experimental models of demyelination and dysmyelination further elucidated the role of myelin in axonal survival. A number of significant differences between these, typically genetically modified, rodent models of myelin loss and multiple sclerosis, a disease that is yet to be faithfully reproduced in experimental animals, preclude us from drawing parallels between human and rodent demyelinated axons. Nonetheless, studies in which one myelin component has been replaced by another provided an important insight into the role of myelin as trophic support for axons. For example, a study in which the CNS myelin protein called proteolipid protein has been replaced by a peripheral nervous system (PNS) myelin protein called P0 in the CNS of mice showed that despite appropriate myelination and apparently intact compact myelin, these mice show increased rate of axonal degeneration and subsequent neurological deficit in comparison with wild-type mice [48]. This and other studies [49, 50] have emphasised the role of myelin-forming cells in axonal health and survival independently of the role of myelin itself.
Given that myelin-forming cells provide trophic support [51] and the role of myelin in energy saving, successful remyelination of demyelinated axons, enables better and more long-lasting functionality and overall protection to axons than would be the case for the chronically demyelinated axons, as experimentally confirmed in mice [52]. Indeed, axonal degeneration represents a major feature of multiple sclerosis, as reported originally by Charcot [3]. Furthermore, axonal loss increases progressively over the duration of the disease [53, 54] and has been identified as a major determinant in the irreversible loss of neurological function [55]. Changes in mitochondrial activity and in organisation of the mitochondrial network help to promote axonal survival and thus play an important role in determining the overall disease progression rate.
1.3.1 Changes in Mitochondrial Distribution Associated with Demyelination
As mitochondria are the main source of ATP in neurons, the increased demand for ATP brought about by myelin loss and subsequent changes in the nature of impulse conduction and the expression pattern of ion channels are closely accompanied by changes in the pattern and distribution of axonal mitochondria. More specifically, axonal mitochondria aggregate within demyelinated regions. For example, in experimentally induced segmental demyelination of cat optic nerve, the number of mitochondria per unit area, as assessed by the number of mitochondrial profiles on the cross-section of electron micrograph, was found to be up to 2.5 times greater in demyelinated than in control optic nerve axons [56]. Interestingly, the greatest mitochondrial density within these demyelinated segments coincided with periods of impulse conduction re-establishment (i.e. the transition from conduction block to slow, continuous conduction) [57] when the energy demand is predicted to be the highest (as long as the frequency of conduction is not greatly reduced) and then decreased in parallel with advancing remyelination and thus the decrease in axonal energy demand. Furthermore, the peak density of mitochondria in the demyelinated segments of the same optic nerve axons was nearly identical to the density of mitochondria in their unmyelinated, intraocular, counterparts which were not affected by this experimental demyelination [57]. Such closely comparable mitochondrial densities suggest that the levels of energy demand in demyelinated and unmyelinated axons of similar calibres are similar.
In vitro, the effect of demyelination on mitochondrial distribution within axons has been examined in organotypic cerebellar slice cultures and myelinating dorsal root ganglion (DRG) cultures [58]. Although being examined at postnatal day 8 or 9, axons in organotypic cerebellar slice cultures do not possess all the characteristics of adult myelinated axons. Nonetheless, like in the adult cat axons described above, their exposure to demyelinating agent lysolecithin resulted in significant increase in mitochondrial content in comparison with axons from control cultures, as assessed by electron microscopy [59]. A similar effect has been reported in cultures of rat embryonic peripheral nerve axons. Namely, mixed cultures of DRG and Schwann cells, isolated from embryonic day 16, can be maintained in culture for up to 9 weeks during which time Schwann cells can be induced to myelinate the growing DRG axons by addition of ascorbic acid to the culture medium [60]. When lysolecithin is added to these cultures, axons become demyelinated. Electron microscopy assessment has shown that following exposure to lysolecithin, the size of stationary mitochondrial sites within demyelinated axons significantly increases compared with myelinated axons [58]. Also, larger mitochondria become significantly more abundant in demyelinated than in myelinated axons. However, in this model system unlike in adult mice, despite this increase in size, the number of stationary mitochondrial sites per unit of axonal length remained identical in myelinated and demyelinated axons, suggesting that some smaller, newly aggregated, mitochondria may have fused in order to form large mitochondria. This difference between axons demyelinated in culture and demyelinated in situ likely reflects the fact that axonal growth is continuing in vitro and other differences in the axon maturity and type of myelination between these model systems. In another words, developing axons and adult axons may employ different mechanisms to increase mitochondrial mass within demyelinated region.
A significant increase in axonal mitochondrial content has also been observed in the corpus callosum of mice fed cuprizone [59], a toxin which causes demyelination by selectively killing oligodendrocytes [61], in comparison with control fed mice. In this elegant study, authors used a serial block scanning electron microscope to precisely reconstruct individual mitochondria within both demyelinated and myelinated axons and quantify mitochondrial number, length and volume. Using this technique, the study found that the number, length and volume of individual mitochondria were significantly greater in demyelinated than in myelinated axons. Overall mitochondrial volume per axonal volume of demyelinated axons was found to be double that of myelinated axons [59]. Another experimental model of demyelination also showed the increase in mitochondrial content in demyelinated axons. Targeted demyelination induced by stereotactic injection of ethidium bromide (EB) to the caudal cerebellar peduncle was also associated with increased axonal mitochondrial content, reaching the peak levels during demyelination and early remyelination, but staying high even in fully remyelinated axons [62]. The difference between remyelinated and myelinated axons was attributed to a significant increase in mitochondrial number rather than their size [62]. It is worth noting that the glial trophic support to the axons is removed in the above models, thus the increased myelin content may represent a response to the combination of increased ATP and trophic demand. In vivo, real-time confocal imaging data from our laboratory showed that in demyelination of the peripheral nerve induced with lysolecithin, the increase in mitochondrial density in the demyelinated segment faithfully coincides with the period of re-establishment of continuous conduction and thus the greatest energy demand, in such axons.
Finally, the change in mitochondrial density has also been reported in axons in multiple sclerosis lesions. Immunohistochemical labelling for a mitochondrial marker called endoplasmic reticulum-associated binding protein (ERAB) showed a significant increase in the level of this protein in axons within active demyelinating lesions in comparison with normal-appearing white matter [63]. Similarly, a significant increase in mitochondrial density judged by the level of voltage-gated anion channel (VDAC), a protein specific for the outer membrane of all mitochondria [64], has been found in axons within chronically demyelinated lesions compared with control and normal-appearing white matter [65]. The increase in density of mitochondria in some human lesions seems to persist beyond the duration of demyelination, remaining significantly greater in remyelinated than in normally myelinated axons [62]. Although the stage and the length of human lesions are difficult to assign and immunohistochemical studies are less powerful in determining the precise number and volume of mitochondria per axon area than serial electron microscopy, these studies nonetheless suggest that an increase in mitochondrial mass is an important mechanism for matching increased energy and metabolic demand in human, as well as in experimental, axonal demyelination.
1.4 Mechanisms Underlying Mitochondrial Redistribution Along Demyelinated Axons
Mitochondria are remarkably dynamic organelles. In excised, intercostal nerve axons, about 15 % of all axonal mitochondria appear mobile [66]. In electrically active PNS axons conducting within physiological range, the number, the size and the velocity of mobile mitochondria increase proportionally to increase in conduction frequency. Remarkably, this increased mobilisation of mitochondria within conducting axons preferentially affects anterogradely moving mitochondria, thus resulting in accumulation of mitochondria in peripheral nerve terminals [22]. Thus, expectedly, the mechanism underlying the redistribution of mitochondria to energetically demanding regions of axons following demyelination is closely coupled with axonal transport of these organelles.
1.4.1 Axonal Transport of Mitochondria
Mitochondrial axonal transport is complex and highly regulated process which involves specialised molecular machinery (Fig. 8.3). The two major components of this machinery are (1) microtubules, which serve as tracks, and (2) molecular motors, which carry mitochondria along the tracks.

Mitochondrial axonal transport machinery. (a) The canonical structure of kinesin 1, the main anterograde motor. Kinesin 1 consists of motor domains, heavy and light chains. The motor domains contain ATPase-hydrolysing moiety and bind to microtubules (shown in part c). Adaptor proteins Milton and Miro1, which also contain Ca2+-binding domain, bind kinesin to mitochondria (shown in part c) and play a part in regulation of mitochondrial arrest at sites of high Ca2+ concentration. Using energy from ATP hydrolysis, kinesin makes step-like movements along microtubules towards its positive end (c), carrying mitochondria along the microtubule track. (b) Dynein is a multi-protein complex which acts as the major retrograde motor. Dynein also uses ATP for movement along the microtubules. For detailed structure of dynein see reference [76]. (c) Mitochondrial trafficking. Mitochondria are transported along microtubules by molecular motors kinesin 1 and dynein. Kinesin 1 moves towards the ‘plus end’ of the microtubule, which is typically furthest from the cell body. In contrast, dynein predominantly moves towards the ‘minus end’ of the microtubule, typically nearest the cell body. Stationary mitochondria appear to be bound to cytoskeletal proteins neurofilament medium (NF-M) and neurofilament heavy (NF-H) chain side arm bound to neurofilament light (NF-L) chain
Microtubules are long, cylindrical structures spanning the length of axon, formed by polymerisation of α- and β-tubulin heterodimers. The heterodimers are assembled in such a way that α-tubulin is always exposed at one end of the microtubule and β-tubulin is exposed on the opposite end, thus giving intrinsic polarity to microtubules. By convention, the cell body-anchored end is called the ‘minus end’, and the growing end (with exposed α-tubulin) is called the ‘plus end’. Such orientation whereby the ‘plus end’ points towards the axon terminal is nearly uniform in all axons [67] and plays an important part in axonal transport as molecular motors move preferentially in one direction. The major molecular motors for axonal transport are kinesin 1 and dynein [68]. These motors comprise of a number of specialised domains assembled into large, multiunit complexes (see Fig. 8.3). Two of the domains associate with microtubules and are thus called microtubule-binding domains. They are linked by a hinge region, resembling two feet (microtubule-binding domains), legs (light chains) and the hip (hinge region). The microtubule-binding domains exert ATP-hydrolysing activity [69]. Using the energy released by ATP hydrolysis, the ‘legs’ alternate in attaching and detaching from microtubules in a stepwise motion, thus pulling along the cargo [70, 71]. One ATP molecule is hydrolysed for every 8 nm step of kinesin [72]. The kinesin family of molecular motors predominantly moves towards the ‘plus end’ of microtubules, thus enabling anterograde (from cell body towards the terminal) transport of cargoes [74, 75].
Other domains of molecular motors, often called adaptor domains, have functions such as binding cargoes and sensing changes in the local environment. In the kinesin family, major adaptor proteins are cargo-binding Milton and Miro1. Other adaptor proteins connecting kinesin and mitochondria include syntabulin, fasciculation and elongation protein zeta 1 (FEZ1) and RAN binding protein 2 (RANBP2). Miro1 contains a Ca2+-sensing and Ca2+-binding domain which plays a critical part in mitochondrial arrest at sites of high Ca2+ concentration. Namely, the linkage between Miro1 and mitochondria is disrupted by Ca2+ binding to Miro1, e.g. at sites of high (micromolar) intra-axonal concentrations of Ca2+ [73]. In this way, the Ca2+-dependent binding of Miro1 to mitochondria regulates mitochondrial retention at sites of high Ca2+ concentrations.
Dynein is a particularly complex multi-protein motor (for detailed structure, see review by Hirokawa et al. [76]), which also uses ATP for movement along the microtubules. The dynein family, and a related large complex called dynactin, can move in both directions, but favour movement towards the ‘minus end’ of microtubules, enabling retrograde (towards the cell body) transport [77–79].
A number of studies have (reviewed in [34]) proposed that the purpose of mobilisation of axonal mitochondria by means of axonal transport is a physiological response which enables delivery of sufficient number of mitochondria to sites of high-energy demand or high Ca2+ concentration, such as peripheral nerve terminals, active growth cones and synapses. Data from our lab has shown that mitochondria are preferentially mobilised from the areas proximal to the segment depleted of ATP (i.e. from the side of the cell body or through the anterograde axonal transport). Also, it has been shown in cultured rat cortical and cerebellar neurons that mitochondrial motility affects other aspects of mitochondrial dynamics, such as fission and fusion [80]. For example, it has been shown that the probability that a mobile mitochondrion will get engaged in fusion with a stationary mitochondrion increases proportionally to the velocity of the moving partner, whereby the higher the speed at impact, the higher the chance that the colliding mitochondria will fuse [80].
Studies of mitochondrial transport in myelinating DRG cultures showed that the velocity of mitochondrial transported along the axons increases by almost 50 % following demyelination, returning to the same level as prior to demyelination once axons become remyelinated. On the other hand, data from our lab shows that, in contrast to myelinating DRG cultures, axonal mitochondrial transport within demyelinated peripheral nerve axons in vivo becomes significantly reduced just prior to demyelination. It is unclear what accounts for this difference, as the same agent, lysolecithin, is used in both systems. It seems likely that in cultured DRGs, which are unlikely to be conducting, and which are still growing while being examined, mitochondria need to be transported towards the growing cone [81] to populate the growing axons, whereas in adult axons imaged in vivo mitochondria had already been positioned along axons prior to demyelination [22].
Once recruited from other parts of the axon and transported towards the demyelinated segments, a specific mechanism for mitochondrial anchoring is activated to ensure that mitochondria remain within the demyelinated area. The protein which tethers mitochondria to microtubules and thus immobilises them is called syntaphilin [82]. Immunoreactivity for this protein has been shown to significantly increase in experimentally demyelinated corpus callosum in mice, when compared with controls [59]. Indeed, deletion of syntaphilin prevented mitochondrial accumulation within demyelinated axons in mice [59]. Furthermore, demyelinated axons in these syntaphilin-deficient mice show significantly higher susceptibility to degeneration than those in wild-type controls [59]. Also, the study showed that the degeneration of demyelinated axons in syntaphilin-deficient mice could be minimised when mice were treated with sodium channel blocker flecainide, thus supporting the hypothesis that impulse load-induced sodium/calcium cascade initiates axonal degeneration in such axons. In human axons obtained from post-mortem brains from patients with multiple sclerosis, increased immunohistological labelling for syntaphilin associated with mitochondrial labelling has also been reported [65]. Therefore, focal mitochondrial immobilisation mediated by the anchoring protein syntaphilin appears to represent a common and important adaptation mechanism for providing energy, and/or a local calcium buffer to areas where ion fluxes of sodium and Ca2+ into denuded axon segments may become augmented.
1.4.2 Changes in Mitochondrial Function Associated with Demyelination
Apart from the mitochondrial mass, changes in mitochondrial function have also been reported within demyelinated axons. In experimental demyelination, induced in mice fed a cuprizone diet, mitochondrial complex IV activity was significantly increased in comparison with the control diet-fed mice [83]. However, in this study, the increase in complex IV activity was found only in the early stages of the treatment, i.e. prior to immunohistochemically detectible demyelination, the level of this complex activity being similar between the groups during the overt demyelination [83]. In another study, using ethidium bromide injection to induce focal demyelination, the activity of mitochondrial complex IV has been found to be significantly increased in remyelinated axons in comparison with myelinated axons [62]. However, the study does not report the complex IV activity in demyelinated axons.
In human axons, an increase in activity of complex IV has been reported within MS lesions negative for proteolipid protein (PLP), one of the major components of myelin, using immunohistochemistry, histochemistry and biochemical assays [84]. Also, the increase in activity of complex IV was found in a subset of axons within inactive lesions [65]. Importantly, the axons showing increased complex IV activity appeared structurally intact and had no pathological accumulation of amyloid precursor protein or dephosphorylated neurofilament. It is possible, therefore, that the greater mitochondrial activity in these axons reflects the successful adaptation to conditions in such lesions, in the form of high local ATP production to enable survival. However, only direct measurements of produced ATP, a methodology that is currently not available for human tissue, could directly address this hypothesis. Furthermore, studies that have examined lesions at different stages have described the opposite effect, i.e. the decrease of activity of mitochondrial complexes, as discussed below.
1.5 Effects of Inflammation on Mitochondrial Function and Transport in Inflammatory-Demyelinating Lesions
Inflammation is a major characteristic of MS. Importantly, a haematogenous inflammatory reaction develops within the CNS rapidly following initiation of lesions [85, 86]. The relative abundance of inflammatory cells has led to one of the most widely used classifications of MS lesions into active or inactive lesions. Typically, active MS lesions are characterised by the abundant presence of infiltrating inflammatory cells, mainly phagocytic macrophages filled with the initial degradation products of myelin, interspersed between segmentally demyelinated, relatively spared axons [87]. On the other hand, inactive (or chronic) lesions are typically characterised by fewer inflammatory cells scattered between demyelinated, thinly myelinated and damaged axons and intense glial scar [87]. Importantly, the extent of inflammation strongly and positively correlates with the magnitude of axonal degeneration [88, 89]. The mechanism underlying this high rate of axonal degeneration is partly attributed to inflammatory damage to mitochondria. Inflammatory cells produce a wide range of mediators [90–94], of which reactive nitrogen [95] and oxygen [96] species have been shown to be particularly toxic to mitochondria [97–102]. Indeed, while the increase in the activity of complex IV has been found in a subset of chronic, inactive white matter lesions [65, 84], at the active edge of these lesions where the inflammation is prominent, the complex IV activity has been found to be significantly decreased [65]. This pattern of complex IV activity is consistent with findings that reactive nitrogen and oxygen species produced by activated microglia and macrophages are potent inhibitors of complexes I and IV [103]. In the motor cortex of patients with MS, activity of complexes I, III and V (ATP synthase) has been found to be significantly decreased in comparison with non-neurological controls, even in the areas that had been classified as ‘not lesioned’ by conventional ultra- and immunohistochemical methods [48]. Interestingly, the activity of complex IV in patient tissue was comparable to that of controls, raising the possibility that the increase in activity of complex IV is a compensatory mechanism to maintain mitochondrial membrane potential. Nonetheless, the decrease in activity of complex V in this study suggests that neurons in the cortex of MS patients were deprived of ATP even before they become demyelinated. The decrease in the activity of the complexes in this study may be explained by the low expression of nuclear genes encoding for mitochondrial respiratory chain proteins [48]. However, it is unclear what caused the low expression pattern of these genes. Another study that reported a decrease in activity of complex I in chronic active lesions suggested that the cause may be the oxidative damage to mitochondrial DNA [104]. Experimental studies in acute, lipopolysaccharide (LPS)-induced model of systemic inflammation (sepsis) in rats showed that, compared with controls, systemic inflammation reduces activity of all mitochondrial complexes and oxygen consumption and significantly lowers production of ATP in mitochondria isolated from the liver [105] or heart muscle [106] and that this effect is prevented with antioxidant therapy. Also, injection of LPS in mouse brain has been shown to increase lipid peroxidation and decrease glutathione content at the site of injection, causing loss of mitochondrial membrane potential and mitochondrial integrity [107].
Experimental studies have further elucidated mechanisms by which mitochondria participate in axonal damage within active MS lesions. For example, in vivo confocal imaging of spinal cord axons of mice with experimental autoimmune encephalomyelitis (EAE), a widely used model of MS, demonstrated changes in the shape of intra-axonal mitochondria consistent with mitochondrial fragmentation [108]. Similar morphological changes in axonal mitochondria could be reproduced by external addition of reactive oxygen species, implicating this mediator in mitochondrial damage in EAE [108]. Functional status of such fragmented mitochondria is difficult to assess, although the fragmentation occurred preferentially within the axonal ovoids, the formations often preceding axonal transection. Studies from our lab suggest that fragmentation of axonal mitochondria occurs at the onset of neurological deficit in EAE and that it is associated with loss of mitochondrial membrane potential. Further in vivo confocal imaging studies showed that axonal transport of mitochondria and other cargoes is significantly reduced in EAE in comparison with axons from control mice, leading to loss of mitochondria in distal segments of affected axons [109]. Similarly to mitochondrial fragmentation, this axonal transport deficit preceded structural deficits and could be rescued by redox scavengers [109].
Inducible nitric oxide synthase that synthesises nitric oxide (NO) production in macrophages, another potent mitochondrial toxin, is often found in inflammatory foci in MS [95, 110]. Apart from possibly inhibiting mitochondrial respiration and thus ATP depletion, NO has another detrimental effect. Specifically, NO [111], as well as ROS [112], has been shown to play a role in the induction of the so-called mitochondrial permeability transition pore (mPTP), an inner mitochondrial membrane channel permeable for small molecules and solutes [113]. Abrupt opening of mPTP leads to loss of mitochondrial membrane potential, uncoupling of oxidative phosphorylation and rapid mitochondrial swelling. Perhaps most dangerously for the axon, the opening of mPTP releases intra-mitochondrial Ca2+ into the axoplasm, raising its concentration to pathologically high levels. Bearing in mind the capacity of mitochondria to store large amount of Ca2+ [114], the opening of mPTP is expected to be particularly detrimental within demyelinated segments of axons which already exhibit high mitochondrial density. Furthermore, mitochondria in such segments are likely to contain high Ca2+ concentration, given the altered ionic balance of demyelinated axon, including the described reversal of Na+/Ca2+ exchanger.
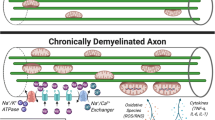
Taken together, the strong association between inflammation and impairment of mitochondrial function demonstrated in studies above suggests that inflammatory environment may be the deciding factor with regard to mitochondrial involvement in axonal survival vs. axonal degeneration (summarised in Fig. 8.4). More specifically, in the absence of inflammation, intrinsic capacity of axonal mitochondrial network to match the increase of the local demand for energy production and Ca2+ handling appears sufficient to enable long-term axonal survival in adverse conditions such as demyelination. However, the presence of focal inflammation may disturb this balance by damaging mitochondria. This effect of inflammation on mitochondria may be particularly harmful in areas of axons in which mitochondrial mass had previously increased in order to buffer excess Ca2+.

Schematic representation of main axonal and mitochondrial changes following demyelination. Top panel: In myelinated axon, sodium channels are concentrated within narrow nodes of Ranvier. Sodium/potassium ATPase pumps sodium ions out of the axons following action potential propagation, thus re-establishing resting membrane potential. Mitochondria are distributed along the internode to provide ATP for the action of sodium pump. Middle panel: Following demyelination, sodium channels become distributed along the whole denuded surface of axolemma, thus facilitating continuous conduction of action potentials. The increased axoplasmic concentration of sodium ions brought about by continuous conduction necessitates greater activity of sodium pump. In addition, Na+/Ca2+ exchanger operates in reverse, i.e. exports Na+ in exchange for Ca2+. Increased ATP demand and Ca2+ concentration induce increase in the number and size of mitochondria within the demyelinated segment, thus enabling its function and survival. Bottom panel: Inflammation disturbs the balance between the energy demand and supply established within demyelinated axons. Inflammatory mediators inhibit mitochondrial respiration, thus depleting ATP, and open mitochondrial permeability transition pore, releasing small solutes from mitochondrial matrix, including Ca2+, which triggers a number of pathological processes leading to rapid axonal degeneration
2 Guillain-Barré Syndrome and Inflammatory Neuropathies
Guillain-Barré syndrome (GBS) is a life-threatening, postinfectious, immune-mediated disease of the peripheral nervous system characterised by ascending muscle weakness. It is believed that the disease arises when the subject’s immune response generates pathogen-specific antibodies that are cross-reactive with components of the peripheral nerve myelin or axolemma, resulting in myelin and/or axonal damage. Most patients recover; however, about a half retain residual neurological deficits [115]; and a proportion develop a chronic form of the disease known as chronic inflammatory-demyelinating polyradiculoneuropathy (CIDP) [116]. Although the triggers of GBS and CIDP have not been identified, and may be different, both of these diseases are characterised by inflammatory infiltration of the peripheral nerves, predominantly by lymphocytes and activated macrophages [117–121]. It has been shown that these cells produce the usual pro-inflammatory mediators, such as nitric oxide, ROS, proteases and pro-inflammatory cytokines [122]. However, little is known about the effect(s) of inflammation on mitochondria within the affected nerves. Also, it is not known whether mitochondrial dysfunction, either pre-existing (perhaps hidden by compensatory mechanisms active in noninflamed nerves) or secondary (caused by the inflammation), contributes to the pathogenesis of immune-mediated peripheral neuropathies. As mentioned above, some of these pro-inflammatory factors interfere with mitochondrial function and dynamics. For example, nitric oxide, produced by inducible nitric oxide synthase (iNOS) in activated macrophages, has been shown to reversibly inhibit mitochondrial respiration [97, 98]. It is likely that such NO-induced damage to mitochondria occurs in inflamed peripheral nerve axons, as the presence of nitrotyrosine, a marker of nitric oxide-mediated peroxynitrite formation, has been identified in nerve biopsies from patients with CIDP [122]. In neuron and Schwann cell cocultures, neurons seem intrinsically more susceptible than Schwann cells to detrimental effects of pathological concentrations of NO [122]. Furthermore, nitric oxide has been shown to lead to excessive mitochondrial fission in cultured cortical neurons via S-nitrosylation of dynamin-related protein 1 [123, 124]. It is possible that NO produced by iNOS-positive inflammatory cells may result in excessive mitochondrial fission in peripheral nerve axons during the course of inflammatory neuropathies. However, whether and how inflammation alters mitochondrial function and transport in axons in vivo, it is entirely unknown. It could be speculated that, as in the CNS [109] and other cells [125], certain inflammatory signals may impair mitochondrial transport in peripheral nerve axons. For example, in cultured fibroblasts, tumour necrosis factor alpha (TNFα) has been shown to inhibit mitochondrial transport by function of hyperphosphorylation of motor protein kinesin 1 [125]. Disruption of microtubule networks, as shown to occur during experimental autoimmune encephalomyelitis in mice [126], may further contribute to impairment of mitochondrial transport in peripheral nerve axons.
3 Conclusion
Impulse conduction, myelination status and effects of inflammatory insult to the axons are closely interconnected and interdependent. Highly dynamic axonal mitochondria represent a major adaptive mechanism for maintaining functionality and structural integrity of axons under pathological conditions. To this effect, mitochondria act as a network rather than as individual organelles. Utilising upregulation in mitochondrial complex activity, changes in fusion/fission balance and redistribution of mitochondrial mass, mitochondrial network can be effectively reorganised to achieve optimal Ca2+ storage and supply of ATP to vulnerable regions of long axonal tracts. Such reorganisation and functional adaptation enables both short- and long-term protection against neurodegeneration. These adaptive modifications may, however, become exhausted under sustained or aggressive inflammatory attack. Furthermore, toxic effects of inflammatory mediators on Ca2+-laden axonal mitochondria may result in destructive rise in intra-axonal Ca2+ levels and subsequent rapid axonal degeneration. Further evidence is hence needed to fully establish the role of mitochondria in the pathogenesis and/or progression of the inflammatory diseases of central and peripheral nervous systems.
References
Pugliatti M, Rosati G, Carton H, Riise T, Drulovic J, Vécsei L, et al. The epidemiology of multiple sclerosis in Europe. Eur J Neurol. 2006;7:700–22.
Tullman MJ. Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am J Manag Care [Internet]. 2013;19(2 Suppl):S15–20.
Charcot JM. Histologie de la sclerose en plaques. Gaz Hop. 1868;41:554–66.
Trapp BD. Pathogenesis of multiple sclerosis: the eyes only see what the mind is prepared to comprehend. Ann Neurol. 2004;55(4):455–7.
Compston A, McDonald I, Noseworthy J, Lassmann H, Miller D, Smith K, Wekerle H, Confavreux C, editors. McAlpine’s multiple sclerosis. 4th ed. Philadelphia: Elsevier; 2006.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–72.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–97.
Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–69.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–34.
Bohnen NI, Albin RL. White matter lesions in Parkinson disease. Nat Rev Neurol. 2011;7(4):229–36.
Agosta F, Dalla Libera D, Spinelli EG, Finardi A, Canu E, Bergami A, et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and Alzheimer disease. Ann Neurol. 2014;76(6):813–25.
Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128(Pt 11):2705–12.
Gay FW, Drye TJ, Dick GWA, Esiri MM. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis: identification and characterization of the primary demyelinating lesion. Brain. 1997;120(8):1461–83.
Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55(4):458–68.
Henderson APD, Barnett MH, Parratt JDE, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol. 2009;66(6):739–53.
Jacobs JM. On internodal length. J Anat. 1988;157:153–62.
Rasminsky M, Sears TA. Internodal conduction in undissected demyelinated nerve fibres. J Physiol. 1972;227(2):323–50.
Bostock H, Sears TA. The internodal axon membrane: electrical excitability and continuous conduction in segmental demyelination. J Physiol. 1978;280:273–301.
Smith KJ, McDonald WI. The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Philos Trans R Soc Lond B Biol Sci. 1999;354(1390):1649–73.
Amiri M, Hollenbeck PJ. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol [Internet]. 2008;68(11):1348–61.
Cotter D, Guda P, Fahy E, Subramaniam S. MitoProteome: mitochondrial protein sequence database and annotation system. Nucleic Acids Res. 2004;32(Database issue):D463–7.
Sajic M, Mastrolia V, Lee CY, Trigo D, Sadeghian M, Mosley AJ, et al. Impulse conduction increases mitochondrial transport in adult mammalian peripheral nerves in vivo. PLoS Biol. 2013;11(12):e1001754.
Bristow EA. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol. 2002;120(6):791.
Barron MJ, Griffiths P, Turnbull DM, Bates D, Nichols P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br J Ophthalmol. 2004;88(2):286–90.
Minckler DS, McLean IW, Tso MO. Distribution of axonal and glial elements in the rhesus optic nerve head studied by electron microscopy. Am J Ophthalmol. 1976;82(2):179–87.
Hall SM, Gregson NA. The in vivo and ultrastructural effects of injection of lysophosphatidylcholine into myelinated peripheral nerve fibres of the adult mouse. J Cell Sci. 1971;9(3):769–89.
Smith KJ, Bostock H, Hall SM. Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J Neurol Sci. 1982;54(1):13–31.
Smith KJ, McDonald WI. Spontaneous and evoked electrical discharges from a central demyelinating lesion. J Neurol Sci. 1982;55(1):39–47.
Noebels JL, Marcom PK, Jalilian-Tehrani MH. Sodium channel density in hypomyelinated brain increased by myelin basic protein gene deletion. Nature. 1991;352(6334):431–4.
Craner MJ, Lo AC, Black JA, Waxman SG. Abnormal sodium channel distribution in optic nerve axons in a model of inflammatory demyelination. Brain. 2003;126(Pt 7):1552–61.
Moll C, Mourre C, Lazdunski M, Ulrich J. Increase of sodium channels in demyelinated lesions of multiple sclerosis. Brain Res. 1991;556(2):311–6.
Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101(21):8168–73.
Dupree JL, Girault JA, Popko B. Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol. 1999;147(6):1145–52.
Mathis C, Denisenko-Nehrbass N, Girault J-A, Borrelli E. Essential role of oligodendrocytes in the formation and maintenance of central nervous system nodal regions. Development. 2001;128(23):4881–90.
Boyle MET, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30(2):385–97.
Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, et al. Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci. 2001;21(19):7568–75.
Stys PK, Waxman SG, Ransom BR. Na(+)-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol. 1991;30(3):375–80.
Stys P, Waxman S, Ransom B. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci. 1992;12(2):430–9.
Li S, Jiang Q, Stys PK. Important role of reverse Na+-Ca2+ exchange in spinal cord white matter injury at physiological temperature. J Neurophysiol. 2000;84(2):1116–9.
Die Sogenannte OM. Akute multiple sklerose. Jahrb Psychiatr. 1906;27:211–312.
Prineas JW, Connell F. Remyelination in multiple sclerosis. Ann Neurol. 1979;5(1):22–31.
Prineas JW, Barnard RO, Revesz T, Kwon EE, Sharer L, Cho E-S. Multiple sclerosis. Brain. 1993;116(3):681–93.
Lassmann H. Comparative neuropathology of chronic experimental allergic encephalomyelitis and multiple sclerosis. Schriftenr Neurol. 1983;25(1):135.
Moore GR, Neumann PE, Suzuki K, Lijtmaer HN, Traugott U, Raine CS. Balo’s concentric sclerosis: new observations on lesion development. Ann Neurol. 1985;17(6):604–11.
Willoughby EW, Grochowski E, Li DK, Oger J, Kastrukoff LF, Paty DW. Serial magnetic resonance scanning in multiple sclerosis: a second prospective study in relapsing patients. Ann Neurol. 1989;25(1):43–9.
Koopmans RA, Li DK, Oger JJ, Kastrukoff LF, Jardine C, Costley L, et al. Chronic progressive multiple sclerosis: serial magnetic resonance brain imaging over six months. Ann Neurol. 1989;26(2):248–56.
Thompson AJ, Kermode AG, Wicks D, MacManus DG, Kendall BE, Kingsley DP, et al. Major differences in the dynamics of primary and secondary progressive multiple sclerosis. Ann Neurol. 1991;29(1):53–62.
Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59(3):478–89.
Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33(3):366–74.
Li C, Tropak MB, Gerlai R, Clapoff S, Abramow-Newerly W, Trapp B, et al. Myelination in the absence of myelin-associated glycoprotein. Nature. 1994;369(6483):747–50.
Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485(7399):517–21.
Irvine KA, Blakemore WF. Remyelination protects axons from demyelination-associated axon degeneration. Brain. 2008;131(Pt 6):1464–77.
Lovas G, Szilagyi N, Majtenyi K, Palkovits M, Komoly S. Axonal changes in chronic demyelinated cervical spinal cord plaques. Brain. 2000;123(2):308–17.
Bjartmar C, Kidd G, Mörk S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48(6):893–901.
De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS, et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121(Pt 8):1469–77.
Mutsaers SE, Carroll WM. Focal accumulation of intra-axonal mitochondria in demyelination of the cat optic nerve. Acta Neuropathol. 1998;96(2):139–43.
Carroll WM, Jennings AR, Mastaglia FL. Galactocerebroside antiserum causes demyelination of cat optic nerve. Brain Res. 1985;330(2):378–81.
Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci. 2010;30(19):6658–66.
Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, Ransohoff RM, et al. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci U S A. 2014;111(27):9953–8.
Fex Svenningsen A, Shan W-S, Colman DR, Pedraza L. Rapid method for culturing embryonic neuron-glial cell cocultures. J Neurosci Res. 2003;72(5):565–73.
Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11(1):107–16.
Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, Ziabreva I, et al. Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain. 2011;134(Pt 7):1901–13.
Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med. 2014;20(3):179–87.
Colombini M. Voltage gating in the mitochondrial channel, VDAC. J Membr Biol. 1989;111(2):103–11.
Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, et al. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132(Pt 5):1161–74.
Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4(7):559–61.
Dombeck DA, Kasischke KA, Vishwasrao HD, Ingelsson M, Hyman BT, Webb WW. Uniform polarity microtubule assemblies imaged in native brain tissue by second-harmonic generation microscopy. Proc Natl Acad Sci U S A. 2003;100(12):7081–6.
Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in drosophila motor axons. Mol Biol Cell. 2006;17(2):2057–68.
Hua W, Young EC, Fleming ML, Gelles J. Coupling of kinesin steps to ATP hydrolysis. Nature. 1997;388(6640):390–3. [Internet]. Available from: file:///Users/mbzcf/Documents/Papers/1997/Hua/Nature 1997 Hua.pdf\npapers://37ea81e1-bf1a-4112-927e-b768b470c325/Paper/p778.
Pfister KK, Shah PR, Hummerich H, Russ A, Cotton J, Annuar AA. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2(1):e1.
Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6(3):201–14.
Schnitzer MJ, Block SM. Kinesin hydrolyses one ATP per 8-nm step. Nature. 1997;388(6640):386–90.
MacAskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541–55.
Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST. Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol. 1991;114(2):295–302.
Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42(1):39–50.
Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68(4):610–38.
Ross JL, Wallace K, Shuman H, Goldman YE, Holzbaur ELF. Processive bidirectional motion of dynein-dynactin complexes in vitro. Nat Cell Biol. 2006;8(6):562–70.
Mallik R, Petrov D, Lex SA, King SJ, Gross SP. Building complexity: an in vitro study of cytoplasmic dynein with in vivo implications. Curr Biol. 2005;15(23):2075–85.
Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125(Pt 9):2095–104.
Cagalinec M, Safiulina D, Liiv M, Liiv J, Choubey V, Wareski P, et al. Principles of the mitochondrial fusion and fission cycle in neurons. J Cell Sci. 2013;126(pt10):2187–97.
Ruthel G, Hollenbeck PJ. Response of mitochondrial traffic to axon determination and differential branch growth. J Neurosci. 2003;23(24):8618–24.
Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132(1):137–48. Synaptic Function Section, The Porter Neuroscience Research Center, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Building 35, Room 3B203, 35 Convent Drive, Bethesda, MD 20892, USA.
Acs P, Selak MA, Komoly S, Kalman B. Distribution of oligodendrocyte loss and mitochondrial toxicity in the cuprizone-induced experimental demyelination model. J Neuroimmunol. 2013;262(1–2):128–31.
Witte ME, Bø L, Rodenburg RJ, Belien JA, Musters R, Hazes T, et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol. 2009;219(2):193–204.
Traugott U, Reinherz EL, Raine CS. Multiple sclerosis. Distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J Neuroimmunol. 1983;4(3):201–21.
Esiri MM, Reading MC. Macrophage populations associated with multiple sclerosis plaques. Neuropathol Appl Neurobiol. 1987;13(6):451–65.
Brück W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol. 1995;38(5):788–96.
Ferguson B. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(3):393–9. [Internet].
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338(5):278–85.
Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann Neurol. 1995;37(4):424–35.
Fabriek BO, Van Haastert ES, Galea I, Polfliet MMJ, Döpp ED, Van Den Heuvel MM, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia. 2005;51(4):297–305.
Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130(Pt 11):2800–15.
Cuzner ML, Opdenakker G. Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol. 1999;94(1–2):1–14.
Lindberg RLP. The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain. 2001;124(9):1743–53.
Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol. 2001;158(6):2057–66.
Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Brück W, et al. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62(1):25–33.
Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345(1):50–4.
Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95(13):7631–6.
Granger DL, Lehninger AL. Sites of inhibition of mitochondrial electron transport in macrophage-injured neoplastic cells. J Cell Biol. 1982;95(2 Pt 1):527–35.
Bolaños JP, Peuchen S, Heales SJR, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem. 2002;63(3):910–6.
Geng Y, Hansson GK, Holme E. Interferon-gamma and tumor necrosis factor synergize to induce nitric oxide production and inhibit mitochondrial respiration in vascular smooth muscle cells. Circ Res. 1992;71(5):1268–76.
McCord JM, Keele BB, Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci. 1971;68(5):1024–7.
Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Mitochondrial protein nitration primes neurodegeneration in experimental autoimmune encephalomyelitis. J Biol Chem. 2006;281(42):31950–62.
Lu F, Selak M, O’Connor J, Croul S, Lorenzana C, Butunoi C, et al. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J Neurol Sci. 2000;177(2):95–103.
Lowes DA, Webster NR, Murphy MP, Galley HF. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth. 2013;110(3):472–80.
Vanasco V, Magnani ND, Cimolai MC, Valdez LB, Evelson P, Boveris A, et al. Endotoxemia impairs heart mitochondrial function by decreasing electron transfer, ATP synthesis and ATP content without affecting membrane potential. J Bioenerg Biomembr. 2012;44(2):243–52.
Noble F, Rubira E, Boulanouar M, Palmier B, Plotkine M, Warnet J-M, et al. Acute systemic inflammation induces central mitochondrial damage and mnesic deficit in adult Swiss mice. Neurosci Lett. 2007;424(2):106–10.
Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17(4):495–9. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.
Sorbara CD, Wagner NE, Ladwig A, Nikić I, Merkler D, Kleele T, et al. Pervasive axonal transport deficits in multiple sclerosis models. Neuron. 2014;84(6):1183–90.
Bö L, Dawson TM, Wesselingh S, Mörk S, Choi S, Kong PA, et al. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol. 1994;36:778–86.
Martin LJ, Adams NA, Pan Y, Price A, Wong M. The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J Neurosci. 2011;31(1):359–70.
Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria. Cell. 2008;134(2):279–90.
Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. Arch Biochem Biophys. 1979;195(2):460–7.
Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358(6384):325–7. Department of Biomedical Sciences, University of Padova Italy.
Dornonville de la Cour C, Jakobsen J. Residual neuropathy in long-term population-based follow-up of Guillain-Barré syndrome. Neurology. 2005;64(2):246–53.
Hughes R, Sanders E, Hall S, Atkinson P, Colchester A, Payan P. Subacute idiopathic demyelinating polyradiculoneuropathy. Arch Neurol. 1992;49:612–6.
Miyakawa T, Murayama E, Sumiyoshi S, Deshimaru M, Kamano A. A biopsy case of Landry-Guillain-Barré syndrome. Acta Neuropathol. 1971;17(3):181–7.
Wiśniewski H. Landry-Guillain-Barré syndrome. Arch Neurol. 1969;21(3):269.
Illes Z. Accumulation of V 7.2-J 33 invariant T cells in human autoimmune inflammatory lesions in the nervous system. Int Immunol. 2004;16(2):223–30.
Winer J, Hughes S, Cooper J, Ben-Smith A, Savage C. Gamma delta T cells infiltrating sensory nerve biopsies from patients with inflammatory neuropathy. J Neurol. 2002;249(5):616–21.
Wanschitz J, Maier H, Lassmann H, Budka H, Berger T. Distinct time pattern of complement activation and cytotoxic T cell response in Guillain-Barré syndrome. Brain. 2003;126(Pt 9):2034–42.
Lehmann HC, Köhne A, Meyer zu Hörste G, Dehmel T, Kiehl O, Hartung H-P, et al. Role of nitric oxide as mediator of nerve injury in inflammatory neuropathies. J Neuropathol Exp Neurol. 2007;66(4):305–12.
Cho D-H, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–5.
Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25(16):3900–11.
De Vos K, Severin F, Van Herreweghe F, Vancompernolle K, Goossens V, Hyman A, et al. Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J Cell Biol. 2000;149(6):1207–14.
Shriver LP, Dittel BN. T-cell-mediated disruption of the neuronal microtubule network: correlation with early reversible axonal dysfunction in acute experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169(3):999–1011.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing
About this chapter
Cite this chapter
Sajic, M. (2016). Mitochondrial Dysfunction and Transport in Demyelinating Disease with Inflammation. In: Reeve, A., Simcox, E., Duchen, M., Turnbull, D. (eds) Mitochondrial Dysfunction in Neurodegenerative Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-28637-2_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-28637-2_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28635-8
Online ISBN: 978-3-319-28637-2
eBook Packages: MedicineMedicine (R0)