
Abstract
Patients undergoing surgery require anesthesia that involves using a variety of medications that promote sedation, pain mitigation, and abate any response to stimulation. Early agents used for sedation induction were thiopental and etomidate. Although ketamine is commonly used in veterinary medicine, this agent is often employed in combination with a benzodiazepine to induce analgesia and sedation. Ketamine is a racemic mixture where the S(+) isomer is two to four times more potent. Midazolam is a water-soluble benzodiazepine where at pH>4, the molecule’s ring structure closes, and it becomes a highly lipophilic agent. Both ketamine and midazolam pharmacokinetics fit into a two-compartment open model and primarily metabolized by CYP3A4. The muscle relaxant agents succinyolcholine, d-tubocurarine, roncuronium, and vencuronium induce muscle paralysis used for anesthesia. Succinylcholine pharamacokinetics has been described as a one-compartment open model whereas the other agents a two- or three-compartment open model. Their pharmacodynamics effects are closely linked with their pharmacokinetic profiles. The short-acting opioids fentanyl, sufentanil, and alfentanil are used in anesthesia for pain management and maintain cardiovascular stability. The pharmacokinetics of these agents are expressed as either a two- or three-compartment open model and mainly metabolized by CYP3A4. Propofol and thiopental display a three-compartment open model. Various factors can alter anesthetic drug disposition and their pharmacodynamic actions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Etomidate
- Ketamine
- Midazolam
- Succinylcholine
- d-Tubocurarine
- Roncuronium
- Vecuronium
- Muscle relaxants
- Opioids
- Fentanyl
- Sufenantil
- Alfentanil
- Propofol
- Thiopental
1 Introduction
Anesthesia induction of patients undergoing surgery requires various combinations of medications to promote sleep or loss of consciousness, alleviate pain, and diminish response to any stimulation. Anesthesia is commonly achieved with a minimum of two different types of pharmacologic agents such as a hypnotic and an opioid analgesic [1]. In addition to the pharmacologic agents for hypnosis and analgesia, inhalation anesthetics and muscle relaxants are often employed prior to their usage. Physical signs have served as distinct pharmacodynamic (PD) biomarkers for the anesthesia that include respiratory patterns, somatic muscle tone, ocular signs, hemodynamic parameters, and the minimum alveolar concentration (MAC) [2]. This chapter will focus only on the pharmacologic agents used in anesthesia. For a review of the inhalational anesthetic agents, these agents display a three-compartment model as shown in Fig. 15.1 and the reader is referred to these references [3, 4].

Three-compartment open pharmacokinetic model
A summary of the anesthetic agents covered in this chapter is presented in Table 15.1. Pharmacologic anesthetic agents given intravenously (IV) or orally were used since the 1930s with thiopental, but integrating their pharmacokinetic properties with the PD effects occurred 45 years later. Since then, sophisticated PK/PD modeling methods have been developed and continually to be revised that enhances the clinical utility of these agents and development of newer agents. Anesthetic agents PK and PD have increased the comprehension of other central nervous system (CNS) drugs that have been employed to treat neurological and psychiatric medical conditions. This chapter will include the muscle relaxants used in anesthesia as distinct PK/PD models that are used in clinical practice. Some of the anesthetic agents have an extensive array of PK and PD studies (e.g., midazolam, propofol) and only selected key articles were selected for inclusion in this chapter. Although at least two different anesthetic drugs are used in clinical practice, the PK/PD of these agents is reviewed individually and the reader is referred to the drug-drug interactions with the anesthetic drugs in Chap. 24.
2 Etomidate
Etomidate (ETD) is a carboxylated imidazole with hypnotic properties and the FDA approved the drug as an IV anesthetic induction agent [5]. Preclinical studies reported that ETD may possess a wider margin of safety compared to thiopental [6]. CNS depressant actions were related to the stimulation of the gamma-aminobutyric acid (GABA) receptors. However, pain upon injection and myoclonia were reported to be ETD’s most undesirable adverse side effects. Nausea and vomiting occurring more frequently (as high as 50 %) have been reported in various studies with multiple ETD dosing [7].
ETD Pharmacokinetics and Pharmacodynamics
ETD has been described similar to thiopental with an open three-compartment pharmacokinetic (PK) model shown in Fig. 15.1. Table 15.1 describes the general ETD PK parameters. ETD 0.3 mg/kg was administered IV to eight patients who underwent eye or ear surgery with 14 blood samples obtained over the following 10 h [8]. The following PK properties (mean ± s.d.) were found with ETD that included volume of distribution (Vd) of 4.5 ± 2.2 L/kg, CL of 860 ± 230 mL/min, and elimination half-life of 4.6 ± 2.6 h. The free fraction of ETD was about 7 % and the hepatic extraction ratio of 0.5 was determined. A later study reported that ETD with a rapid distribution half-life of 2.81 ± 1.64 min and a protein binding of 77 % almost totally to albumin was metabolized by hydrolyzation in the plasma and in the liver at the ETD ester forming carboxylic acid [7].
A PD dose-response relationship was found with ETD in patients undergoing electroconvulsive therapy (ECT) that received no other premedication treatment [9]. ETD was given at doses of 0.1, 0.2, and 0.4 mg/kg. The higher ETD doses had significantly greater effects on waking time and the late recovery time (p < 0.01). The ETD 0.1 mg/kg dose had a reported mean (± s.d.) waking time of 7.52 ± 1.07 min and a mean recovery time of 20.41 ± 1.17 min. The ETD doses of 0.2 mg/kg and 0.4 mg/kg reported mean waking times of 10.02 ± 0.78 min and 14.05 ± 1.46 min, respectively. The ETD 0.2 mg/kg and 0.4 mg/kg reported mean late recovery times of 27.60 ± 1.74 min and 38.05 ± 2.70 min, respectively. ETD (N = 10) 0.3 mg/kg was compared to thiopental (N = 5) 3.5 mg/kg in patients with various elective surgeries and PD actions assessed by the EEG [10]. The main differences between the two agents were the lack of beta activity and a longer duration of “deep stage” sleep with ETD. ETD had higher incidences of pain and myoclonic and tonic movements than thiopental but these effects were not associated with epileptiform discharges.
3 Ketamine
Ketamine (KTM) is an N-methyl-D-aspartate (NMDA) receptor antagonist that blocks glutamatergic functions with opioid receptor activity [11, 12]. KTM has been used in veterinary medicine and when combined with benzodiazepine anesthetics employed for analgesia and sedation in adult and pediatric populations [13]. KTM has efficacy in neuropathic and nociceptive pain. A variety of administration routes for KTM have been utilized including parenteral, oral, rectal, subcutaneous, transdermal, and intranasal [11]. Only the routes of administration when KTM is used for anesthesia will be presented in this chapter.
KTM Pharmacokinetics and Pharmacodynamics
KTM PK was investigated in five adult healthy male volunteers given a single dose of 0.125 and 0.250 μg/kg separated by 1 week [14]. Blood samples were obtained prior to drug administration and for 7 h afterward. Pain assessment was conducted by using sphygmomanometers known as “tourniquet time.” KTM plasma concentration time data were fitted by a two-compartment open model. The KTM metabolite nor-KTM was also characterized. The mean KTM PK parameters for both doses reported were clearance (CL) of 17 mL/min/kg, elimination half-life of 186 ± 8 min, and volume of distribution (Vd) of 3.1 L/kg. Nor-KTM peak plasma concentrations (mean ± S.E.M.) were reached at 75 min (40 ± 14 ng/mL) and 45 min (21 ± 3 ng/mL) for the 0.250 μg/kg and 0.125 μg/kg doses, respectively. Both KTM doses extended the period of pain-free time at KTM plasma concentrations greater than 100 ng/mL. KTM 0.5 mg/kg single dose was given intramuscularly (IM) and an oral solution to six healthy volunteers and the pain evaluation was conducted by the tourniquet test by ischemic exercise [15]. KTM bioavailability was found to be 93 % and 16 % for the IM and oral solution, respectively. The mean (± s.d.) peak plasma concentration occurred at 22 ± 4 min and 30 ± 5 min for the IM and oral solution, respectively. The mean elimination half-life for the IM and oral routes were 115 ± 12 min and 174 ± 50 min, respectively, with plasma concentrations associated with analgesia at 150–160 ng/mL [15, 16]. Nor-KTM plasma concentrations were 2- to 5-fold higher than KTM plasma concentrations noted from oral administration. Nor-KTM plasma concentrations were generally lower than the KTM plasma levels when given by the IM route [15].
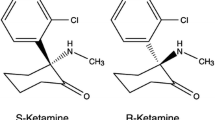
KTM is also a racemic mixture of two enantiomers S(+) KTM and R(−) KTM where the S isomer has been suggested to be two to four times more potent in pain alleviation and causes fewer adverse side effects than the racemic KTM [17]. The S(+) isomer was also reported to be twice as potent as the R(−) isomer on the NMDA receptors [18]. KTM is extensively metabolized by hepatic CYP3A4 (to nor-KTM) and to a lesser extent CYP2B6 and CYP2C9 [19]. Potential drug-drug interactions can occur with KTM via CYP enzymes (see Drug-Drug Interactions Anesthetic Agents Chapter).
The PK properties for racemic and S(+) KTM were evaluated in 50 adult patients undergoing minor surgery [20]. The patients were divided into two groups of 25 patients that received racemic KTM 2 mg/kg and S(+) KTM 1 mg/kg. Using the change of systolic arterial pressure, the sample size of 22 patients per group was calculated that achieved at 90 % power (alpha) at a 5 % level (beta). The PK parameters of S(+) KTM did not significantly differ from the racemic KTM. For example, the S(+) KTM mean (± s.d.) elimination half-life was 2.39 ± 1.26 h, CL 16.4 ± 5.7 mL/min/kg, and Vd 2.84 ± 1.59 L/kg. Systolic and diastolic arterial pressure significantly increased with both agents (p < 0.005). S(+) KTM had significantly (p < 0.005) higher systolic and diastolic arterial pressure than racemic KTM noted at 1, 3, and 15 min after drug administration. S(+) and R(−) KTM PK were examined in ten healthy male volunteers given two infusion cycles of S(+) KTM 0.1 and 0.2 μg/mL/min of R(−) KTM (11). KTM PK was estimated using a 2- and 3-compartment model. S(+) KTM showed a significantly (p < 0.05) higher mean (± s.d.) CL of 26.3 ± 3.5 mL/min/kg than R(−) KTM 13.8 ± 1.3 mL/min/kg. The authors suggested that R(−) KTM may inhibit S(+) KTM elimination although a mechanism was not proposed. Further studies would be needed to confirm the actions of R(−) KTM on S(+) KTM PK.
4 Midazolam
Midazolam (MDZ) is the first water-soluble benzodiazepine and used for sedation and sleep induction for anesthesia [21, 22]. Under the environmental pH = 4, the ring diazepine structure opens reversibly that produces a highly water-soluble molecule. At pH > 4, the ring closes resulting in a highly lipophilic molecule that under physiological pH rapidly enters the CNS after drug administration [21]. MDZ can be given IV, IM, and orally to induce sedation and anesthesia. Due to wealth of information with MDZ, the pharmacokinetic and pharmacodynamic sections were separated with selected information presented.
MDZ Pharmacokinetics
A summary of MDZ PK is presented in Table 15.1 and its elimination half-life was shown not to be significantly different when given either IV, IM, or oral administration [21]. MDZ PK will be briefly presented in this section as an open two-compartment model was described after IV administration [23]. MDZ PK was examined in six healthy volunteers given 5 mg IV, 10 mg oral solution, and 10 mg oral tablet [23]. The mean (± s.d.) MDZ bioavailability was reported to be low at 0.36 (±0.09) indicating a significant first-pass effect. The mean Vd and total MDZ CL after the IV dose were 1.14 ± 0.57 L/kg and 0.383 ± 0.094 L/kg/h, respectively. The mean terminal elimination half-life was 1.77 ± 0.83 h from the three different administration routes. After IV administration, sleep induction occurred within 1–2 min with continued sedation for an average of 1.33 h. Sleep induction took place later with the oral solution and tablet with an average of 0.38 h (range 0.25–0.55 h) and sedation was maintained for an average of 1.17 h (range 0.5 ± to 2.33 h). MDZ bioavailability was investigated with the oral doses 10 mg, 20 mg, and 40 mg versus an IV dose of 0.15 mg/kg in six healthy volunteers [24]. The mean MDZ bioavailability range for the 10 and 20 mg dose was identical (0.46 ± 0.11 and 0.48 ± 0.12, respectively). The MDZ bioavailability was greater with the 40 mg dose (range 0.63–0.72), but only three subjects were able to tolerate the high dose. The MDZ distribution of blood/plasma concentration coefficient (λ) was reported to be 0.53 indicating only a small extent of binding to red blood cell erythrocytes. MDZ is primarily metabolized by the CYP3A4 to its 1-OH and 4-OH MDZ metabolite. The 1-OH metabolite is converted to another metabolite 1,4 OH compound. All three metabolites are glucuronidated and then renally eliminated [21, 25].
Factors That Influence MDZ Pharmacokinetics
The factors reported to affect MDZ PK disposition were age and obesity [26]. It was found that in elderly males versus adult males, a decrease in drug CL took place. Differences in MDZ PK between elderly females and adult females were not found. Vd was slightly increased in the elderly females and males, but it was not significant. Based upon these findings, MDZ doses should be reduced by about 50 % in elderly men. MDZ disposition was significantly altered in morbidly obese persons reflected by the Vd with enhanced drug distribution into peripheral adipose tissues. This action produces a significant (p < 0.05) prolongation in MDZ elimination half-life. Therefore, MDZ doses should be increased proportionally to the patient’s total body weight. Significant differences in MDZ PK were not found between patients with chronic renal failure and normal controls and dosage adjustments are not recommended.
MDZ Pharmacodynamics
MDZ can induce and maintain anesthesia, used as a premedication agent prior to surgery and as an adjunct to local or regional anesthesia for sedation [26]. During anesthesia, MDZ affects respiration via CNS depression, but lacks significant cardiovascular effects, and produces only a slight decrease in cerebral perfusion and oxygenation [27]. IV MDZ 0.15 mg/kg was given to healthy unpremedicated volunteers (N = 20) where CNS and cardiovascular effects were monitored [28]. Significant adverse effects were not found and blood pressure only slightly decreased after 3 min postdrug administration. Anterograde amnesia and drowsiness were observed in all subjects,
The PD effects of oral MDZ 10 mg, 20 mg, and 40 mg or IV MDZ 0.15 mg/kg were investigated in six healthy volunteers [29]. The PD tests included the tracing test, reaction time, subject’s self-assessment (sedation, muscle relaxation, and concentration capacity), and investigator subjective assessment. After drug administration, MDZ PK parameters were determined and the plasma concentrations were linked with the PD effects using the sigmoid Hill Equation E max model: E = E max × Cp/EC50 × Cp where E = the intensity of action, E max = maximal effect, Cp = concentration linked to the effect, and EC 50 = plasma concentration at 50 % of E max. Peak effects from the oral route occurred at 30 min in reducing the PD effects. The duration of the PD effects were 2 h for the IV route and the 10 mg oral dose. The 20 and 40 mg doses had a significantly longer effect of 4 h (p < 0.05). The minimum effective MDZ plasma concentration to affect the subject’s reduced PD actions ranged from 30 to 100 ng/ml. The PD effects were correlated with the E max model. The respiratory and cardiovascular effects of MDZ and diazepam (DZ) were compared in eight healthy volunteers [30]. MDZ 0.05 mg/kg and DZ 0.15 mg/kg were given via the IV route using a randomized double-blind crossover method. Blood pressure, blood gases, and PaCO2 with plasma drug concentrations were obtained. The PD effects of blood pressure and PaCO2 followed a sigmoidal E max model where the MDZ EC50 was from 50 to 60 ng/ml. Correlation between respiratory effects and plasma drug concentrations was not found. In another study with IV MDZ 0.15 mg/kg and DZ 0.3 mg/kg given to eight healthy volunteers, both agents produced an equal effect in respiratory depression measured by ventilatory and mouth occlusion pressure response to CO2 [31]. Therefore, MDZ like DZ can cause significant respiratory depression. Physostigmine 2.0 mg given IV was reported in three case reports to reverse the MDZ-induced sedation [32]. Flumazenil, a benzodiazepine receptor antagonist, was reported to reverse the respiratory depression effects of MDZ alone and MDZ plus alfentanil in healthy volunteers (N = 20) using a starting dose of 1 mg IV followed by an infusion of 20 μg/min [33]. Flumazenil has become the standard “rescue” medication for reversing the actions of MDZ and other benzodiazepines. MDZ has been compared to other anesthetics and benzodiazepines for anesthesia induction with comparable effects [21].
5 Muscle Relaxants
Muscle relaxants are commonly used as adjunctive medications for anesthesia for endotracheal intubation and to reduce muscle tone during surgery [34]. These agents are given to patients undergoing general anesthesia and usually not to normal healthy volunteers. Patients with normal hepatic and renal function are considered as “normal” patients. Muscle relaxant PK and PD actions contributed toward the understanding of integrating PK to PD effects. A large number of muscle relaxants are available and beyond the scope of this chapter to cover. Muscle relaxants are neuromuscular-blocking agents with their main pharmacologic action to inhibit transmission of nerve impulses by acetylcholine at the skeletal neuromuscular junction [34]. Inactivation of neuromuscular blockers occurs via plasma cholinesterase by hydrolysis dependent on three factors: (1) intrinsic speed of reaction, (2) drug concentration, and (3) esterase concentration [35]. Based upon these pharmacologic mechanism, neuromuscular agents either depolarizing (non-competitive) or non-depolarizing (competitive) agents are deactivated [34]. Extensive review articles on the clinical pharmacokinetics of these agents have been published with various factors including pregnancy that can influence their disposition [34, 36–39]. This section will present only selected agents with succinylcholine as the depolarizing agent. Tubocurarine (d-isomer) is considered the prototypical non-depolarizing agent and the “newer” agents rocuronium and vecuronium are discussed in this section.
Succinylcholine (SCL) Pharmacokinetics and Pharmacodynamics
The typical adult dose of SCL is about 1.0 mg/kg that results in complete neuromuscular blockade with a 50 % recovery time in approximately 10 min [37]. SCL PK can be estimated by a one-compartment model and elimination: C o = C × e −kτ. The PD model can be determined as E = E o − k m × t d/2.30, where E o = the effect at time zero and t d = the time to whatever blockade percentage is selected at minute in a typical 70 kg patient with 3.5 l of plasma. It was reported that the in vivo rate of SCL hydrolysis was between 3 and 7 mg/L/min and that an infusion rate of 4 mg/min was needed to maintain a 90 % reduction in the human twitch movement [36]. About 1/2800 persons possess an atypically low amount of plasma cholinesterase which results in a slower rate of hydrolysis and in those persons, the SCL dose was suggested to be significantly reduced by tenfold or greater [40]. However, presently, patient identification is not yet possible and clinicians must carefully monitor every patient.
Tubocurarine (dTC) Pharmacokinetics and Pharmacodynamics
Early dTC PK reported studies have been limited to blood sampling collection times of up to 60 min postdrug administration. The use of dTC has been reported since the 1960s [41]. dTC PK studies with longer sample collection times up to 24 h reported a terminal half-life as long as 3.5 h, but a rapid recovery from muscle relaxant effects occurred after 15 min of drug cessation. The volume of distribution from the central compartment (V c) for dTC was estimated to be 72–97.7 mL/kg [37]. From the central compartment shown in Fig. 15.1, the k e leads to the sigmoidal E max PD model [37]. A significant linear correlation between serum dTC concentration and muscle twitch tension was found and recovery from the twitch tension was estimated to be 0.7 μg/mL [42].
dTC infusion was given to patients (N = 12) to maintain a “steady-state” concentration of 1.09 μg/mL and after the infusion cessation, twitch response returned in half of patients with a full recovery for all patients in 30 min [43]. Based upon these results, a dTC bolus dose of 540 μg/kg and infusion rate of 2.0 μg/kg should produce continuous paralysis in the average patient. PK of dTC was reported not to significantly differ between infants, children, and adults and patients with hypothermia [44, 45]. dTC was simultaneously modeled using the open three-compartment model and the sigmoidal E max model [46]. The results reported that the mean rate constant for dTC equilibrium for a paralysis effect from infusion was a serum concentration of 0.13 ± 0.04 μg/min and the mean steady-state serum concentration of 0.37 ± 0.05 μg/mL to achieve 50 % paralysis. These studies suggest that dTC has been well characterized for its PK and PD effects in a variety of patient populations [34, 46].
Rocuronium (RCM) Pharmacokinetics and Pharmacodynamics
RCM is a neuromuscular-blocking drug that has a similar PK profile as vecuronium (see vecuronium section) and time course of action except that it has a more rapid onset of action with an ED95 of 0.3 mg/kg [38]. RCM had an average CL and terminal elimination half-life was 0.27 L/h/kg and 83 min, respectively. Age, renal failure, smoking, and hypothermia were reported not be significant factors in RCM CL and Vd when comparing these parameters to the adult population [38, 47]. RCM protein binding was found to be at 25 % and the only metabolite detected was 17-desacetyl-RCM present in very low concentrations. The metabolite is only 1/20 as potent as RCM and likely clinically insignificant [38]. RCM PK and PD was evaluated in patients (N = 10) who were stabilized under either propofol or isoflurane anesthesia [48]. Differences in RCM PK were not found between the two groups. After a second RCM bolus dose of 0.5 mg/kg, the duration of neuromuscular blockade was significantly longer in the isoflurane group versus the propofol group (20 ± 6 min versus 39 ± 8 min, p < 0.05). Using the sigmoidal E max model, the EC50 was significantly higher under propofol anesthesia (1008 μg/L versus 592 μg/L, p < 0.05).
The effects of RCM PK in patients with mild to moderate cirrhosis (N = 17) was compared to healthy patients (N = 21) given an RCM bolus dose of 0.6 mg/kg [49]. Blood samples were obtained for the next 8 h after RCM administration with the twitch response assessed. Using a three-compartment open model, RCL CL was significantly reduced in the cirrhotic group (2.66 mL/kg/min versus 3.70 mL/kg/min, p < 0.005) and elimination half-life was also significantly prolonged in the cirrhotic group (28.3 ± 12.1 min versus 16.8 ± 4.6 min, p < 0.005; 143 ± 80 min versus 92 ± 40 min, p < 0.05, respectively). The time for clinical effect did not differ between the groups, but the mean time to recovery was significantly longer in the patients with cirrhosis (T50 % recovery 73.9 ± 33.9 min versus 52.6 ± 19.8 min, p < 0.05). The increased PD recovery time reflects the prolonged PK effects of RCM in patients with cirrhosis. A similar finding in RCM PK and PD (recovery time) was reported in patients with obstructive jaundice (OJ, N = 27) and control patients (N = 26) given RCM of 0.9 mg/kg [50]. RCM plasma concentrations were significantly higher (p < 0.05) from 30 to 120 min post RCM bolus injection with a corresponding longer recovery time (T25 % OJ 80.8 ± 16.9 min versus 62.8 ± 13.2 min, p < 0.01). From these studies, hepatic impairment but not renal impairment can significantly alter RCM disposition and prolong PD effects.
Vecuronium (VCM) Pharmacokinetics and Pharmacodynamics
VCM and its active 3-desacetyl VCM metabolite PK and PD were reported in 12 healthy volunteers [51]. Animal models reported that 3-desacetyl VCM had about 50–70 % neuromuscular-blocking activity as VCM [52]. The VCM and metabolite PK data were fit into a two- and three-compartment open model with the PD twitch model analysis using the sigmoidal E max model. The VCM CL was significantly greater than the metabolite (5.39 [range 5.04–7.19] mL/kg/min versus 3.51 [range 2.11–6.57] mL/kg/min, p < 0.05). The metabolite had significantly greater Vd and longer elimination half-life than VCM (254 [range 215–411] L/kg versus 152 [range 111–170] L/kg; 116 [44–672] minutes versus 34 [range 25–61] minutes, p < 0.05). The EC50 for the VCM and its metabolite was 123 [range 109–154] ng/mL versus 102 [range 71–123] ng/mL (p < 0.05), respectively. These findings show that the 3-desacetyl VCM is a potent active metabolite and can prolong VCM PD actions in patients [51].
The PK and PD of VCM were compared to pancuronium (PCM) in nine patients undergoing surgery [53]. VCM was shown to have a 50% shorter mean elimination half-life than PCM (71 ± 20 min versus 140 ± 25 min, p < 0.05) and corresponding increase in CL. The EC50 was similar for both agents. The PK and PD of VCM were compared to PCM in twelve children aged 3–6 years [54]. The elimination half-life of VCM did not significantly differ from PCM. However, the VCM Vd and CL were significantly greater than PCM with shorter VCM duration of action and recovery index (p < 0.05). The shorter action duration for VCM was suggested to be probably due to the larger Vd and higher CL. Elderly patients (age 70–84 years) were found not to have any significant differences in PK and PD actions compared to young adults (age 30–57 years) when given VCM and PCM [55].
VCM PK and PD effects were compared in normal patients (N = 7) and patients with renal failure (N = 12) given 0.1 mg/kg [56]. Mean VCM CL was significantly reduced in patients with renal failure (3.08 ± 0.83 mL/kg/min versus 5.29 ± 2.17 mL/kg/min, p < 0.05) and duration of action significantly longer (98.6 ± 37.7 min versus 54.1 ± 25.2 min, p < 0.05). A significant correlation between VCM CL and duration of action was found to be r 2 = 0.869. VCM PK was significantly altered in burn patients (N = 20) compared to normal patients (N = 20) given a single bolus of 0.12 mg/kg [57]. A three-compartment open model best described the VCM profile with an enhanced VCM CL in burn patients (0.12 L/min versus 0.095 L/min, p < 0.001) and shorter elimination half-life (5.5 h versus 6.6 h, p < 0.001). This shorter time period of VCM exposure in burn patients may explain the resistance to VCM in these patients.
6 Opioids
The role of opioids in anesthesia has evolved from use as a premedicant or adjunctive agent to the inhalants and postoperative pain management to a primary anesthetic drug due to their PD actions to maintain cardiovascular stability during surgery [58]. Only the short-acting opioid agents fentanyl (Fen), sufentanil (Suf), and alfentanil (Alf) will be presented in this section as these agents are the most commonly used opioids in anesthesia. Fen was introduced in the 1960s with Suf and Alf being the “newer” opioids. The main advantages of these opioids over morphine are a faster onset of analgesia and shorter elimination half-life shown in Table 15.2 and that allows for enhanced dosing flexibility in anesthesia management [58]. Additionally, these three drugs also have a lack of hyperglycemic response to surgery, decreased catecholamine levels, and increased lipid solubility [59].
Opioid Pharmacokinetics and Pharmacodynamics
Fen and Suf PK have been described as a three-compartment open model [58]. Suf and Alf PK were described as both a two- and three-compartment model [58, 60]. The Gepts Model has been utilized as a foundational PK approach for Suf studies [61]. Peak brain concentrations in patients (N = 19) were reached for Alf at 45 s, Suf at 5 min, and Fen at 6 min during the postacute stage of head injury with normal intracranial pressure [62].These opioid PK parameters and their comparison to morphine are presented in Table 15.2 [58, 62]. Suf Vd and elimination half-life were found to be between Fen and Alf. All three opioids are highly protein bound [63]. Fen and Alf are metabolized by hepatic CYP3A4 [64, 65]. Suf is also metabolized by N-dealkylation and O-demethylation, but a specific CYP enzyme was not reported [66]. Later, it was reported that CYP3A4 was responsible for Suf metabolism to the N-dealkylation metabolite [67]. Erythromycin is a known CYP3A4 inhibitor and was shown not to significantly affect Suf disposition [68]. An explanation for the lack of erythromycin effect on Suf PK may be due to its extraction ratio. Fen and Suf are agents with high hepatic extraction ratios of 0.8 and 1.0, respectively [69]. Compounds with a high extraction ratio could be less prone to metabolic inhibitors and more dependent on hepatic blood flow. Alf was found to have a low to moderate hepatic extraction ratio from 0.14 to 0.4 [58, 69, 70]. The three opioids were also reported not to be P-glycoprotein substrates but were shown to be inhibitors using the Caco-2 cell model [70]. The PD effects of Alf using the ED95 serum concentration-response curves to maintain hemodynamic stability in surgical patients (N = 64) were reported be 300 ng/ml and 400 ng/ml for superficial and intra-abdominal operations, respectively [71]. Concentration-response relationships for hemodynamic control under target-controlled infusion (TCI) with Suf and Fen were achiever with concentrations of 0.71 ± 0.13 ng/mL and 7.3 ± 1.3 ng/mL, respectively [72]. Higher Suf and Fen mean concentrations of ≥1.25 ng/mL and 13.5 ng/mL, respectively, did not improve hemodynamic control. Slightly lower Suf Fen mean concentrations were reported also to be effective for patients undergoing CABS with 0.59 ± 0.13 ng/mL and 5.8 ± 1.9 ng/mL, respectively [73]. Using the Gepts Model for Suf TCI, anesthesia was managed for patients (N = 34) with CABS with Suf concentrations as low as 0.4 ng/mL and equally effective as 0.8 ng/mL [74].
Factors That Can Alter Opioid Pharmacokinetics and Pharmacodynamics
Opioids are typically administered as a bolus injection and/or by continuous infusion. Other administration routes that can be used include epidural, intrathecal, transdermal, and intranasal in which each of these routes alters the PK of these three opioids [75]. Factors have been shown to influence opioid PK that include age, obesity, plasma protein content, acid-base status, hepatic, and surgical procedures such as cardiopulmonary bypass [75]. Renal impairment was reported not to significantly alter opioid disposition due to their PK profiles and hepatic extraction ratios. Age-related effects for opioid disposition result from changes in increased body fat, decreases in protein binding, hepatic blood flow, and enzyme capacity [75]. Children ages 9 months to 10 years had reported significantly higher Alf CL than adults (p < 0.05) and a much shorter elimination half-life (41.6 min versus 55 min, p < 0.05). These PK changes were likely due to the increased CYP3A4 hepatic enzyme activity found in the children [76]. A similar finding with Suf was reported with children ages 2–8 years with an average CL of 1.83 L/kg/h [77]. Using EEG assessments, it was found that a 50 % reduction in Alf or Fen dose in the elderly was needed to produce similar effects in EEG suppression compared to the adults although significant PK differences were not found between the elderly and the adults [78].
Obesity was found to be a significant factor in opioid PK as these agents are highly lipophilic due to the peripheral compartment (either two or three) that contains a high adipose content. This factor increases opioid Vd which prolongs the drug’s elimination half-life. All three agents are highly protein bound (see Table 15.2); however, only 50 % of Fen and Suf are bound to albumin. Alf binding to albumin is lower at 33 % [63, 75]. These agents are also bound to α1-acid glycoprotein (AAG) and changes in AAG levels can either increase or decrease free drug concentrations. Acid-base changes in pH influence protein binding as alkalosis leads to increased protein binding and acidosis results in decreased protein binding. The pH changes has greater effects for Fen > Suf > Alf [63]. As previously mentioned, hepatic blood flow is a major factor in opioid PK due to their extraction ratio. Hepatic impairment could affect opioid disposition but varying results have been reported and therefore, dosage adjustments may not be necessary except for patients with moderate to severe impairment [75]. Different surgical procedures including cardiovascular bypass have been reported to alter opioid PK properties [79–83]. Surgery such as in CABS produces factors such as hemodilution, relative hypotension, and hypothermia [75]. Hemodilution results in lower plasma protein binding and increases drug Vd. Hypotension reduces hepatic blood flow and hypothermia reduces enzyme metabolic capacity. Each factor alters opioid serum or plasma concentrations and those changes can lead to an enhanced or reduced PD effects by prolonging or diminishing the opioid pharmacologic actions.
7 Propofol
Propofol (Ppf) is an anesthetic agent introduced in the 1980s to induce anesthesia. Unfortunately, a high incidence of pain upon IV injection and anaphylactoid reactions resulted in the development of an emulsion formation [84]. Ppf can be given as a bolus injection and by controlled infusion to induce and maintain anesthesia. Selected articles are presented to describe Ppf disposition and its PD effects.
Ppf Pharmacokinetics and Pharmacodynamics
Ppf disposition was reported in 12 adult patients (six males and six females) given a single bolus IV injection of 2.5 mg/kg with blood samples collected for 8 h post-administration [85]. Ppf displayed a three-compartment open model as shown in Fig. 15.1; however, a subsequent second peak drug concentration occurred indicating a redistribution effect at 60 min. Ppf PK did not significantly differ between males and females and the mean (±SEM) CL was 1.80 L/kg for both groups. The elimination half-life for males was slightly greater than females (56.0 ± 4.0 min versus 44.9 ± 4.0 min, p = n.s.). Ppf PK was compared between the elderly (N = 12) aged 65–80 years and the adults (N = 12) aged 18–35 years [86]. Ppf doses were a single bolus of 2.0 mg/kg and 2.5 mg/kg for the elderly and adult groups, respectively. Ppf CL was significantly lower in the elderly group than the adult group (1.43 ± 0.09 L/min versus 1.78 ± 0.12 L/min, p = 0.03) and a smaller Vd (19.6 ± 5.2 L versus 26.3 ± 2.9 L, p = 0.046). Plasma protein binding did not differ between the groups.
Ppf PK covariates given by bolus injection and a 60 min infusion were evaluated in 24 patients with the PK data fitted to a three-compartment model [87]. Using a population PK approach, age was reported to be a significant covariate for volume of distribution and CL. Ppf CL was influenced by weight, lean body weight, and height. When taken together, these three variables significantly improved the model (p < 0.01). PK of Ppf in children (N = 20) aged 2–10 years was compared to adults under infusion to maintain a target Ppf plasma concentration of 15 μg/mL for anesthesia (10 μg/mL is used for the adults) [88]. The volume of distribution in the central compartment (Vc) was about 50 % greater than adults (343 mL/kg versus 228 mL/kg) and a higher CL (34.30 mL/kg/min versus 27.36 mL/kg/min). A larger Ppf bolus dose of 50 % and a higher maintenance infusion dose of 25 % were recommended for children. Lower Ppf Vd and CL were reported with the Asian population (Indian and Chinese) and dosage adjustments may be needed [89, 90].
Ppf was reported with in vitro models to be metabolized mainly by CYP2B6 (although this enzyme is about 3–6 % of the total hepatic enzyme content) and to a lesser extend CYP2C9 [91–93]. The CYP2B6 and UGT1A9 genotypes were reported to be significantly affected by Ppf plasma concentrations in patients (N = 51) aged 42–81 years [92]. The group had a mean age of 65 years with 29 subjects >65 years and statistical analysis indicated patients with advanced age of 65 years had a higher Ppf risk score when factored with the two genotypes that influence Ppf PK and PD.
A number of studies have indicated that Ppf PK can be scaled allometrically and directly proportionally to lean body mass (LBM) [94, 95]. Ppf PK and PD effects were reported in adult patients (N = 42) and the sigmoidal Emax model was used to determine effective concentration (EC) to achieve a Bispectral Index Score (BIS) between 40 and 60 [96]. The Ppf maintenance EC50 was found to be 2.23 mg/L (95 % C.I. 1.95–2.51). Ppf LBM was sued in dosing and sex was found not to influence the PK model. Using body weight as the key indicator for Ppf dosing, this concept was evaluated in adult morbidly obese patients (N = 66, BMI ≥ 40 kg/m2) that used a preset Ppf concentration of 2.5 μg/mL during infusion to maintain anesthesia [97]. The Bispectral Index < 60 score (BIS) was used as the biomarker for anesthesia that determined the effective concentration (ECe). A probit regression model was used to calculate the ECe50 and ECe95 for Ppf. Total body weight (TBW) was found to be the best factor in Ppf dosing and the ECe50 was 3.4 μg/mL and ECe95 was 4.2 μg/mL. A higher Ppf target concentration was needed in morbidly obese patients most likely due to the much larger Ppf Vd. Similar findings using TBW for Ppf dosing and BIS in morbidly obese children and adolescents were found [98, 99].
Although initial Ppf studies did not report sex as a significant factor, later studies that measured Ppf metabolites reported that females had significantly higher Ppf glucuronide (1.25 fold, p < 0.03), 4-OH Ppf-1-glucuronide (2.1 fold, p = 0.0009), and 4-OH Ppf-4-glucuronide (1.7 fold, p = 0.02) concentrations than males [100, 101]. Significant effects of CYP2B6 and UGT1A9 genotypes were not found to be factors. However, females tended to recover faster than males from Ppf anesthesia and that PD effect can be due to the increased Ppf metabolism.
Surgery can influence Ppf disposition. CABS effects on Ppf disposition was reported in patients (N = 19) given 4 mg/kg/h infusion [102]. Total Ppf concentrations remained unchanged, but unbound Ppf amounts increased by twofold during surgery and decreased back to baseline levels at surgery completion. Careful patient monitoring is recommended during surgery. Ppf CL appears to change during liver transplantation as described in ten patients [103]. The following mean (± s.d.) Ppf CL were reported in the dissection, anhepatic, and reperfusion phases as 1.89 ± 0.48 L/min, 1.08 ± 0.25 L/min, and 1.53 ± 0.51 L/min, respectively. The Ppf mean extraction ratio was found to be 0.24 ± 0.12 without changes in Ppf concentrations between radial and pulmonary arteries. Ppf CL decreased about 42 % during the anhepatic phase and after reperfusion, Ppf metabolism resumes to prior capacity. A population PK and PD model for Ppf in patients (N = 23) undergoing lung cancer surgery reported a lower EC50 of 1.4 mg/L during Ppf infusion 8 mg/kg/h [104]. However, the Population PK variables for CL was 2.38 L/min and volume of distribution was 189 L, which did not differ from previous Ppf studies. It was suggested that the use of Fen 3 μg/kg IV bolus dose may have influence the Ppf EC50 or cancer patients could be more sensitive due to chemotherapy and other therapeutic approaches to cancer. Other factors may influence Ppf PK and PD that remains to be elucidated. However, when making these assessments, Ppf modeling has generally utilized two specific approaches examining Ppf effect-site concentrations [105]. Computer simulation models have observed Ppf concentrations to range between 1.3 and 4.4 μg/mL with a desired effect noted at 2 min after the bolus dose. Continuous infusion dosing may require an EC50 from 1.5 to 4.0 μg/mL depending upon the patient’s body weight and other factors that influence Ppf PK and PD.
8 Thiopental
Thiopental (TPL) is a barbiturate agent which was introduced into clinical anesthetic practice in 1934 and a popular anesthetic agent for many years. Unfortunately, thiopental produces respiratory and myocardial depression; causes spontaneous tremor, muscle movements, and hiccoughs in some patients; and is contraindicated in patients with porphyria and demyelinating diseases [106].
TPL Pharmacokinetics and Pharmacodynamics
Thiopental’s very short PD actions were originally thought to be related to drug metabolism. It was not until the 1950s and 1960s that TPL PK was modeled to describe its redistribution from the brain and plasma to less perfused fat tissues in the body that accounted for its actions [106]. A low extraction ratio for thiopental of 0.1 was reported that suggests that hepatic metabolism may not account for the PD actions [107]. The basic PK parameters of thiopental are shown in Table 15.1. After a bolus intravenous (IV) injection of TPL, a basic PK model with a distribution and elimination phase was described [108]. However, early studies initially suggested that thiopental protein binding markedly decreases at plasma concentrations of 100 μg/mL or greater indicating increased availability of additional free thiopental concentrations [109]. Based upon these findings, studies reported the lack of fit in describing TPL using a one- or two-compartment model [108].
It was not until the 1980s that a three-compartment model was shown to accurately describe thiopental PK with one central compartment feeding into two separate peripheral compartments with rate constants (e.g., k 1,2 and k 2,1, k 1,3 and k 3,1) to and from each compartment [109]. Drug dosing proceeds directly into the central compartment with metabolism and elimination (k e ) from the central compartment. Using the following equations, the total cumulative thiopental lost from the central compartment from metabolism was metabolic loss = CL × ∫ Cpssdt from zero to time t and total drug loss = V × [Cpss (0) – Cpss (t)] where V = volume of distribution, CL = total body clearance, Cpss thiopental plasma concentration, 0 = time zero, and t = thiopental plasma concentration at time t. Using this mathematical approach, the following thiopental PK parameters were reported in 11 surgical patients (mean ± s.d.): CL = 3.4 ± 0.4 mL/min/kg; V (central compartment) = 0.53 ± 0.18 L/kg; Vd (steady state) = 2.34 ± 0.75 L/kg; and terminal elimination half-life = 719 ± 329 min. Further, protein binding was examined and found to be not different with TPL plasma concentrations >100 μg/mL and remained consistent at about 83 % [110]. The hepatic extraction ratio from the central compartment was reported to be 0.14. A TPL isomer (1-ethylpropyl) was identified but was present in only about 6–7 % in the TPL preparation. The isomer displayed similar PK properties as thiopental and anesthetic potency in mice [111]. TPL metabolism including its CYP profile has not yet been reported due to early development of the drug.
Factors Influencing Thiopental Pharmacokinetics and Pharmacodynamics
Various factors were evaluated that could affect TPL disposition. TPL PK was reported to not significantly differ between young women (N = 8) and young men (N = 8) with an age range from 20 to 40 years that used a three-compartment model as previously described [112]. TPL protein binding and PK were determined in a pediatric population (N = 24) age range from 5 months to 13 years and compared to adult patients (N = 11) where each patient received a single thiopental bolus IV injection [113]. Protein binding (87 %) and Vd at steady state of TPL were similar between the pediatric and adult groups. Total TPL drug CL was significantly greater in the pediatric patients than the adult patients (6.6 ± 2.2 mL/min/kg versus 3.1 ± 0.5 mL/min/kg, p < 0.001). Elimination half-life was also significantly longer in the pediatric group compared to the adult group (6.1 ± 3.3 h versus 12 ± 6 h, p < 0.005). The shorter elimination half-life in infants and children was solely due to the greater hepatic CL.
TPL was given to elderly women (N = 8) and elderly men (N = 8) age range from 60 to 79 years and was described by a three-compartment model [114]. The TPL PK parameters volume of distribution, elimination half-life, and CL did not significantly differ between the elderly women and men. When the elderly data was compared to previous studies of young men and young women [112], both elderly populations had significantly higher Vd than the young adult group (p < 0.05) and longer elimination half-lives (elderly women mean 990 min, range 616–2223; elderly men mean 791 min, range 440–1580). However, only CL was found to be significantly greater in the elderly women (mean 0.19 L/min, range 0.137–0.269; p < 0.05) when compared to young adult women (mean 0.131 L/min, range 0.047–0.209). Induction of sleep onset had lower TPL plasma concentrations in both elderly women and 3.9 % elderly men compared to young adult women and men but this finding was not statistically significant. The average induction dose for TPL dose was significantly lower for the elderly groups versus the young adult groups (p < 0.05). Therefore, the elderly can have a longer TPL elimination half-life due to the volume of distribution but need lower drug doses which induce sleep at an earlier time frame [115].
TPL PK parameters were reported to be similar between patients with chronic renal failure (N = 7) and with age-matched normal patients undergoing surgery [116]. Intrinsic TPL CL was reported to be significantly lower in patients undergoing renal transplantation (p < 0.05) and higher protein binding (83 % versus 89 %, p < 0.05); however, the PD cardiovascular effects and cardiac output were unchanged for both groups [116]. Thiopental PK in patients with cirrhosis (N = 8) was compared to patients with normal hepatic and renal function undergoing elective or orthopedic surgery [117]. The TPL Vd was significantly lower in patients with cirrhosis than normal patients (2.3 ± 0.5 L/kg versus 3.5 ± 1.9 L/kg, p < 0.05) although CL did not significantly differ. Mean TPL protein binding in patients with cirrhosis was almost twice that of the normal patients (25.2 ± 3.9 % versus 14.5 ± 3.4 %, p < 0.05) which can be explained by the lower serum albumin concentrations [117]. However, due to TPL’s low extraction ratio, dosing adjustments and PD effects are not clinically needed. Significant differences were not found between patients with chronic alcoholism (N = 10) and normal control patients (N = 9) when given TPL [118, 119].
9 Conclusions
Anesthetic agents have specific PD functions to induce sleep, reduce pain, and maintain anesthesia. A single anesthetic drug does not fulfill all these requirements. Yet, the understanding of their PK and PD individually contributes to the overall anesthesia management of patients undergoing surgery. Without anesthetic drugs, medical treatments are significantly impacted. Anesthetic agents have formed the basis of the two- and three-compartment PK models and integration with their PD effects has shaped the foundations for modeling central nervous system drugs.
References
Eilers H Niemann (2003) Clinically important drug interactions with intravenous anesthetic agents in older patients. Drug Aging 20:969–980
Stanski DR, Shafer SL (1995) Quantifying anesthetic drug interactions. Anesthesiology 83:1–5
Feingold A, Holaday DA (1997) The pharmacokinetics of metabolism of inhalation anaesthetics. Br J Anaesth 49:155–162
Tanner G (1982) Pharmacokinetics of inhalation anesthetics: a three-compartment linear model. Anesth Analg 61:587–594
Doenicke A (1975) Etomidate: a new intravenous hypnotic. Acta Anaesthesia Belg 25:5–8
Morgan N, Lumley J, Whitwam JA (1975) Etomidate, a new water insoluble nonbarbiturate intravenous induction agent. Lancet 1:955–956
Geise JL, Stanley TH (1983) Etomidate: a new intravenous anesthetic induction agent. Pharmacotherapy 3:251–258
Van Hamme MJ, Ghoneim MM, Ambre JJ (1978) Pharmacokinetics of etomidate, a new intravenous anesthetic. Anesthesiology 49:274–277
Kay B (1976) A dose-response relationship for etomidate, with some observations on cumulation. Br J Anaesth 48:213–216
Ghoneim MM, Yamada T (1977) Etomidate: a clinical and electroencephalographic comparison to thiopental. Anesthesia Analg 56:479–485
Austin KR (1976) Ketamine hydrochloride: a potent analgesic. Br Med J 2:943–945
Kronenberg RH (2002) Ketamine as an analgesic: parenteral, oral, rectal, subcutaneous, transdermal, and intranasal administration. J Pain Palliat Care Pharmacother 16:27–35
Hatch RC (1973) Ketamine – excellent anesthetic. J Am Vet Assoc 162:835
Clements JA, Nimmo WS (1981) Pharmacokinetics and analgesic effect of ketamine in man. Br J Anaesth 53:27–30
Grant IS, Nimmo WS, Clements JA (1981) Pharmacokinetics and analgesic effects of IM and oral ketamine. Br J Anaesth 53:805–810
Clements JA, Nimmo WS, Grant IS (1982) Bioavailability, pharmacokinetics, and analgesic activity of ketamine in humans. J Pharm Sci 71:539–541
Oye I, Paulsen O, Maurset A (1992) Effects of ketamine on sensory perception: evidence for a role of NMDA receptors. J Pharmacol Exp Ther 260:1209–1213
Zeilhofer HU, Swandulala D, Geisslinger G, Brune K (1992) Differential effects of ketamine enantiomers on NMDA receptor currents in cultured neurons. Eur J Pharmacol 213:155–158
Hijazi Y, Boulieu R (2002) Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos 30:853–858
Geisslinger G, Hering W, Thomann P et al (1993) Pharmacokinetics and pharmacodynamics of ketamine enantiomers in surgical patients using a stereoselective analytical method. Br J Anaesth 70:666–671
Ihmsen H, Geisslinger G, Schuttler J (2001) Stereoselective pharmacokinetics of ketamine: R(−) ketamine inhibits the elimination of S(+) ketamine. Clin Pharmacol Ther 70:431–438
Kanto JH (1985) Midazolam: the first water-soluble benzodiazepine. Pharmacotherapy 5:138–155
Kanto JH, Allonen H (1983) Pharmacokinetics and the sedative effect of midazolam. Int J Clin Pharmacol Ther Toxic 21:460–463
Smith MH, Eadie MJ, Brophy TO (1981) The pharmacokinetic of midazolam in man. Eur J Clin Pharmacol 19:271–278
Heizmann P, Eckert M, Ziegler WH (1983) Pharmacokinetics and bioavailability in man. Br J Clin Pharmacol 16:43S–49S
Kronbach T, Mathys D, Umeno M et al (1989) Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol 30:89–96
Reves JG, Fragen RJ, Vinik R, Greenblatt DJ (1985) Midazolam: pharmacology and uses. Anesthesiology 62:310–324
Crevoisier C, Ziegler WH, Eckert M, Heizmann P (1983) Relationship between plasma concentration and effect of midazolam after oral and intravenous administration. Br J Clin Pharmacol 16:51S–61S
Gemperle M, Kapp WK (1983) Midazolam and anaesthesia. Br J Clin Pharmacol 16:187S–190S
Forster A, Gardaz JP, Suter PM, Gemperle M (1980) IV midazolam as an induction agent for anaesthesia: a study in volunteers. Br J Anaesth 52:907–911
Sunzel M, Paalzow L, Berggren L, Eriksson I (1988) Respiratory and cardiovascular effects in relation to plasma levels of midazolam and diazepam. Br J Clin Pharmacol 25:561–569
Forster A, Gardaz JP, Suter PM, Gemperle M (1980) Respirator depression by midazolam and diazepam. Anesthesiology 53:494–497
Caldwell CB, Gross JB (1982) Physostigmine reversal of midazolam-induced sedation. Anesthesiology 57:125–127
Gross JB, Blouin RT, Zandsberg S et al (1996) Effect of flumazenil on ventilatory drive during sedation with midazolam and alfentanil. Anesthesiology 85:713–720
Ramzan MJ, Somogyi AA, Walker JS et al (1981) Clinical pharmacokinetics of the non-depolarising muscle relaxants. Clin Pharamcokinet 6:25–60
Kalow W (1956) The relation of plasma cholinesterases to response to clinical doses of succinylcholine. J Arch Can Anaesth 3:22–30
Wingard LB, Cook DR (1977) Clinical pharmacokinetics of muscle relaxants. Clin Pharmacokinet 2:330–343
Agoston S, Vandenbrom RH, Wierda JM (1992) Clinical pharmacokinetics of neuromuscular blocking drugs. Clin Pharmacokinet 22:94–115
Paul D, Atherton L, Huter JM (1999) Clinical pharmacokinetics of the newer neuromuscular blocking drugs. Clin Pharmacokinet 36:169–189
Guay J, Grenier Y, Varin F (1998) Clinical pharmacokinetics of neuromuscular relaxants in pregnancy. Clin Pharmacokinet 34:483–497
Kalow W, Gunn DR (1959) Some statistical data on atypical cholinesterase of human serum. Am J Hum Genet 23:239–250
Ryan AR (1964) Tubocurarine administration based upon its disappearance and accumulation curves in anaesthesized man. Br J Anaesth 38:287
Matteo RS, Spector S, Horowitz PE (1974) Relation of serum d-tubocurarine concentration to neuromuscular block in man. Anesthesiology 41:440–448
Ramzan MI, Shanks CA, Triggs EJ (1980) Pharmacokinetics of tubocurarine administered by combined IV bolus and infusion. Br J Anaesth 52:893–899
Fisher DM, O’Keefe C, Stanski DR et al (1982) Pharmacokinetics and pharmacodynamics of d-tubocurarine in infants, children, and adults. Anesthesiology 57:203–208
Ham J, Stanski DR, Newfield P, Miller RD (1981) Pharmacokinetics and dynamics of d-tubocurarine during hypothermia in humans. Anesthesiology 55:631–633
Sheiner LB, Stanski DR, Vozeh S et al (1979) Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin Pharmacol Ther 25:358–371
Ltorre F, deAlemeida MC, Stanek A, Kleeman PP (1997) The effect of smoking on neuromuscular transmission after rocuronium. Anaesthesist 46L:493–495
Drage A, Varin F, Plaud B, Donati F (2002) Rocuronium pharmacokinetics-pharmacodynamic relationship under stable propofol or isoflurane anesthesia. Can J Anesth 49:353–360
Van Miert MM, Estwood NB, Boyd AH et al (1997) The pharmacokinetic and pharmacodynamics of rocuronium in patients with hepatic cirrhosis. Br J Clin Pharmacol 44:139–144
Wang ZM, Zhang P, Lin MJ et al (2013) Influence of obstructive jaundice on pharmacodynamics of rocuronium. PLoS One 8:e78052
Caldwell JE, Szenohradszky J, Segredo V et al (1994) The pharmacodynamics and pharmacokinetics of the metabolite 3-desacetylvencuronium (ORG 7268) and its parent compound, vecuronium, in human volunteers. J Pharm Exp Ther 270:1216–1222
Marshall IG, Gibb AJ, Durant NN (1983) Neuromuscular and vagal blocking actions of pancuronium bromide, its metabolites, and vecuronium bromide (Org NC45) and its potential metabolites in the anesthetized cat. Br J Anaesth 55:703–714
Cronnelly R, Fisher DM, Miller RD et al (1983) Pharmacokinetics and pharmacodynamics of vecuronium (ORG NC 45) and pancuronium in anaesthesized patients. Anesthesiology 58:405–408
Meistelman C, Agoston S, Kersten UW et al (1996) Pharmacokinetics and pharmacodynamics of vencuronium and pancuronium in anesthetized children. Anesth Analg 65:1319–1322
Rupp SM, Castagnoili KP, Fisher DM, Miller RD (1987) Pancuronium and vencuronium pharmacokinetics and pharmacodynamics in younger and elderly adults. Anesthesiology 67:45–49
Lyman DP, Cronnelly R, Castagnoli KP et al (1988) The pharmacodynamics and pharmacokinetics of vecuronium in patients anesthetized with isoflurane with normal renal function or with renal failure. Anesthesiology 69:227–231
Vego-villa KR, Kaneda K, Yamashita S et al (2014) Vecuronium pharmacokinetics in patients with major burns. Br J Anaesthesia 112:304–310
Davis PJ, Cook DR (1986) Clinical pharmacokinetics of the newer intravenous anaesthetic agents. Clin Pharmacokinet 11:18–35
Willens JS, Myslinski NR (1993) Pharmacodynamics, pharmacokinetics, and clinical uses of fentanyl, sufentanil, and alfentanil. Heart Lung 22:239–251
Fung DL, Eisele JH (1980) Fentanyl pharmacokinetics in awake volunteers. J Clin Pharmacol 20:652–658
Gepts E, Shafer SL, Camu F et al (1998) Linearity of pharmacokinetics and model estimation of sufentanil. Anesthesiology 83:1194–1204
Metz C, Gobel L, Gruber M et al (2000) Pharmacokinetics of human cerebral opioid extraction: a comparative study on sufentanil, fentanyl, and alfentanil in a patient after severe head injury. Anesthesiology 92:1559–1567
Meuldermans WE, Hurkmans RM, Heykants JJ (1982) Plasma protein binding and distribution of fentanyl, sufentanil, alfentanil and lofentanil in blood. Arch Int Pharmacodyn Ther 257:4–19
Labroo RB, Paine FP, Thummel KE, Khararsch ED (1997) Fentanyl metabolism by human hepatic and intestinal cytochrome P450 3A4: implications for interindividual variability in disposition, efficacy, and drug interactions. Drug Metab Dispos 25:1072–1080
Yun CH, Wood M, Wood AJ (1992) Identification of the pharmacogenetic determination of alfentanil metabolism cytochrome P450 3A4. An explanation of the variable elimination clearance. Anesthesiology 77:467–474
Roscow CE (1984) Sufentanil citrate: a new opioid analgesic for use in anesthesia. Pharmacotherapy 4:11–19
Lundberg S, Roelofse JA (2011) Aspects of pharmacokinetics and pharmacodynamics of sufentanil in pediatric practice. Paediatr Anaesth 21:274–279
Bartkowski RR, Goldberg ME, Huffnagle S, Epstein RH (1993) Sufentanil disposition. Is it affected by erythromycin administration? Anesthesiology 78:260–265
Ibrahim AE, Feldman J, Karim A, Kharasch ED (2003) Simultaneous assessment of drug interactions with low and high extraction opioids. Anesthesiology 98:853–861
Wandel C, Kim R, Wood M, Wood A (2002) Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology 96:913–920
Shafer A, Sung ML, White PF (1986) Pharmacokinetic and pharmacodynamics of alfentanil infusions during general anesthesia. Anesth Analg 65:1021–1028
Thomson IR, Henderson BT, Singh K, Hudson RJ (1998) Concentration-response relationship for fentanyl and sufentanil in patients undergoing coronary artery bypass grafting. Anesthesiology 89:852–861
Thompson IR, Harding G, Hudson RJ (2000) A comparison of fentanyl and sufentanil in patients undergoing coronary artery bypass graft surgery. J Cardiothoracic Vas Anesth 14:652–656
Jeleazcov C, Saari TI, Ihmsen H et al (2012) Changes in total and unbound concentrations of sufentanil during target controlled infusion for cardiac surgery with cardiopulmonary bypass. Br J Anaesth 109:698–706
Scholz J, Steinfath M, Schulz M (1996) Clinical pharmacokinetics of alfentanil, fentanyl, and sufentanil. An update. Clin Pharmacokinet 31:275–292
Davis PJ, Killian A, Stiller RL et al (1988) Alfentanil pharmacokinetics in premature infants and older children. Anesthesiology 69:A758
Guay J, Gaudreault P, Tang A et al (1992) Pharmacokinetics of sufentanil in normal children. Can J Anaesth 39:14–20
Scott JC, Stanski DR (1987) Decreased fentanyl and alfentanil dose requirements with age: a simultaneous pharmacokinetic and pharmacodynamics evaluation. J Pharmacol Exp Ther 240:159–163
Bovill JG, Sebel PS, Blackburn CL, Oei-Lim V, Heykants JJ (1984) The pharmacokinetics of sufentanil in surgical patients. Anesthesiology 61:502–506
Hudson RJ, Bergstrom RG, Thomson IR et al (1989) Pharmacokinetics of sufentanil in patients undergoing abdominal aortic surgery. Anesthesiology 70:426–431
Hudson RJ, Thomson IR, Burgess PM, Rosenbloom M (1991) Alfentanil pharmacokinetics in patients undergoing abdominal aortic surgery. Can J Anaesth 38:61–67
Baylaen WA, Herregods LL, Mortier EP et al (1989) Cardiopulmonary bypass and the pharmacokinetics of drugs. An update. Clin Pharmacokinet 17:10–22
Hall R (1991) The pharmacokinetic behavior of opioids administered during cardiac surgery. Can J Anaesth 38:747–756
Cummings GC, Dixon J, Kay NH et al (1984) Dose requirements of ICI 35868 (propofol, Diprivan) in a new formulation for induction of anaesthesia. Anaesthesia 39:1168–1173
Kay NH, Sear JW, Uppington J et al (1986) Disposition of propofol in patients undergoing surgery. Br J Anaesth 58:1075–1079
Kirkpatirck T, Cockshott ID, Douglas EJ, Nimmo WS (1988) Pharmacokinetics of propofol (Diprivan) in elderly patients. Br J Anaesth 60:146–150
Schnider TW, Minto CF, Gambus PL et al (1998) The influence of method of administration and covariates on the pharmacokinetics of propofol in adult volunteers. Anesthesiology 88:1170–1182
Marsh B, White M, Morton N, Kenny GNC (1991) Pharmacokinetic model driven infusion of propofol in children. Br J Anaesth 67:41–48
Puri A, Medhi B, Panda NB et al (2012) Propofol pharmacokinetics in young healthy Indian subjects. Indian J Pharmacol 44:402–406
Ye HB, Li JH, Rui JZ et al (2012) Propofol pharmacokinetics in China: a multicentric study. Indian J Pharmacol 44:393–397
Court MH, Duan SX, Hesse LM et al (2001) Cytochrome P450 2B6 is responsible for interindividual variability of propofol hydroxylation by human liver microsomes. Anesthesiology 94:110–119
Guitton J, Buronfosse T, Desage M et al (1998) Possible involvement of multiple human cytochrome P450 isoforms in the liver metabolism of propofol. Br J Anaesth 80:788–795
Turpeinen M, Zanger UM (2012) Cytochrome P450 2B6: function, genetics, and clinical relevance. Drug Metab Drug Interact 27:185–197
Kansaku F, Kuma T, Sasaki K et al (2011) Individual differences in pharmacokinetics and pharmacodynamics of anesthetic agent propofol with regard to CYP2B6 and UGR1A9 genotype and patient age. Drug Metab Pharmacokinet 26:532–537
Coetzee JF (2012) Allometric or lean body mass scaling of propofol pharmacokinetics. Clin Pharmacokinet 51:137–145
Martin-Mateos I, Perez JAM, Reboso JA, Leon A (2013) Modeling propofol pharmacodynamics using BIS-guided anaesthesia. Anaesthesia 68:1132–1140
Echevarria CG, Elgueta MF, Donoso MT et al (2012) The effective effect-site propofol concentration for induction and intubation with two pharmacokinetic models in morbidly obese patients using total body weight. Anesth Analg 115:823–829
Diepstraten J, Chidambaran V, Sadhasivan S et al (2012) Propofol clearance in morbidly obese children and adolescents. Clin Pharmacokinet 51:543–551
Chidambaran V, Sadhasivan S, Diepstraten J et al (2013) Evaluation of propofol anesthesia in morbidly obese children and adolescents. BMC Anesthesiol 13:1–9
Loryan I, Lindqvist M, Johansson I et al (2012) Influence of sex on propofol metabolism, a pilot study: implications for propofol anesthesia. Eur J Clin Pharmacol 68:397–406
Choong E, Loryan I, Lindqvist M et al (2013) Sex difference in formation of propofol metabolites: a replication study. Basic Clin Pharmacol Toxic 113:126–131
Hiraoka H, Yamamoto K, Okana N et al (2004) Changes in drug plasma concentrations of an extensively bound and highly extracted drug propofol, in response to altered plasma binding. Clin Pharmacol Ther 75:324–330
Takizawa D, Sato E, Hiraoka H et al (2005) Changes in apparent systemic clearance of propofol during transplantation of living related donor liver. Br J Anaesth 95:643–647
Przybylowski K, Tyczka J, Szczesny D et al (2015) Pharmacokinetic and pharmacodynamics of propofol in cancer patients undergoing major kung surgery. J Pharmacokinet Pharmacodyn 42:111–122
Glen JB (2013) Propofol effect-site concentration: hunt the Keo. Anesth Anal 117:535–536
Guerra F (1980) Thiopental forever after. In: Aldrete JA, Stanley TH (eds) Trends in intravenous anesthesia. Yearbook, Chicago, pp 143–151
Bischoff KB, Dedrick RL (1968) Thiopental pharmacokinetics. J Pharm Sci 57:1346–1351
Morgan DJ, Blackman GL, Paull JD et al (1981) Pharmacokinetics and plasma binding of thiopental. I. Studies in surgical patients. Anesthesiology 54:468–473
Stanski DR (1981) Pharmacokinetic modelling of thiopental. Anesthesiology 54:446–448
Burch PG, Stanski DR (1983) The role of metabolism and protein binding in thiopental anesthesia. Anesthesiology 58:146–152
Stanski DR, Burch PG, Harapat S, Richards RK (1983) Pharmacokinetics and anesthetic potency of a thiopental isomer. J Pharm Sci 72:937–940
Christensen JH, Andreasen F, Jansen JA (1980) Pharmacokinetics of thiopentone in a group of young women and a group of young men. Br J Anesth 52:913–918
Sorbo S, Hudson RJ, Loomis JC (1984) The pharmacokinetics of thiopental in pediatric surgical patients. Anesthesiology 61:666–670
Christensen JH, Andreasen F, Jansen JA (1981) Influence of age and sex on the pharmacokinetics of thiopentone. Br J Anesth 53:1189–1195
Christensen JH, Andreasen F, Jansen JA (1982) Pharmacokinetics and pharmacodynamics of thiopentone, a comparison between young and elderly patients. Anaesthesia 37:398–404
Christensen JH, Andreasen F, Jansen JA (1983) Pharmacokinetics and pharmacodynamics of thiopentone in patients undergoing renal transplantation. Acta Anaesthesiol Scand 27:513–518
Pandele G, Chaux F, Salvadori C et al (1985) Thiopental pharmacokinetics in patients with cirrhosis. Anesthesiology 59:123–125
Couderc E, Ferrier C, Haberer JP et al (1984) Thiopentone pharmacokinetics in patients with chronic alcoholism. Br J Anaesth 56:1393–1397
Kharasch KD, Russell M, Mautz D et al (1997) The role of cytochrome P450 3A4 in alfentanil clearance. Implications for interindividual variability in disposition and perioperative drug interactions. Anesthesiology 87:36–50
Santamaria R, Pailleux F, Beaudry F (2013) In vitro ketamine CYP3A-mediated metabolism study using mammalian liver S9 fractions, cDNA expressed enzymes and liquid chromatography tandem mass spectrometry. Biomed Chromatogr. doi:10.1002/bmc3199
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Jann, M.W. (2016). Anesthetic Drugs Pharmacokinetics and Pharmacodynamics. In: Jann, M., Penzak, S., Cohen, L. (eds) Applied Clinical Pharmacokinetics and Pharmacodynamics of Psychopharmacological Agents. Adis, Cham. https://doi.org/10.1007/978-3-319-27883-4_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-27883-4_15
Published:
Publisher Name: Adis, Cham
Print ISBN: 978-3-319-27881-0
Online ISBN: 978-3-319-27883-4
eBook Packages: MedicineMedicine (R0)