
Abstract
Rhabdomyosarcomas (RMS) are a group of aggressive malignancies derived from a skeletal muscle cell lineage. These neoplasms occur in several distinct body regions in children and adults. One subset of RMA develops in the hepatobiliary tract, mainly in the pediatric age group, associated with a distinct clinical and radiological syndrome. Hepatobiliary RMS is defined as a mucosal-type RMS arising from the wall of intrahepatic or extrahepatic bile ducts. This neoplasm is the most common biliary tract tumor in children, but is overall rare, accounting for only 0.8 % of all RMS and 1.3 % of malignant liver tumors in childhood. The neoplasm can form polypoid masses that protrude into the bile ducts. Histologically, most hepatobiliary RMS are embryonal RMA, whereas alveolar RMS is a very rare hepatic malignancy. Histologically, embryonal RMA of the liver shows a loose neoplastic tissue with desmin-positive stellate cells and cells resembling rhabdomyoblasts. Frequently, a cellular layer is found underneath the biliary tract epithelium, the so-called cambium. RMS of the hepatobiliary tract can rarely also occur in adults.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Bile Duct
- Choledochal Cyst
- Extrahepatic Bile Duct
- Desmoplastic Small Round Cell Tumor
- Alveolar Rhabdomyosarcoma
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Pediatric Hepatobiliary Rhabdomyosarcoma
ICD-O codes:
Embryonal rhabdomyosarcoma | 8910/3 |
Alveolar rhabdomyosarcoma | 8920/3 |
Pleomorphic rhabdomyosarcoma | 8901/3 |
Introduction
Rhabdomyosarcomas (RMS) are a group of aggressive mesenchymal malignancies well known to occur in several distinct body regions of children and histologically characterized by a growth of immature muscle cells with or without detectable cross-striation. Several types of RMS are now recognized, including embryonal RMS, alveolar RMS, pleomorphic RMS, spindle cell RMS, and anaplastic forms (Parham and Barr 2013).
Hepatobiliary rhabdomyosarcoma (HBRMS) is defined as a mucosal-type RMS arising from the wall of the intra- or extrahepatic bile ducts. This RMS is almost always an embryonal RMS. The lesion has first been described in 1875 (Wilks and Moxon 1875) and has later also been described as embryonic tumor with striated muscle (Sheehan 1930), a malignant mixed tumor of the choledochal duct (Goeters 1941), and a cystic liver tumor of the mixed type (Williams 1953). The case of Sheehan (1930), a 6-year-old girl, was described in great detail, and the author already pointed out that this type of tumor may present in the form of intrahepatic cysts representing dilated bile ducts obstructed by the intraluminally growing tumor. The lesion was further specified by Willis (1962).
Malignant muscle tumors have already been identified in the nineteenth century. The first case of RMS may have been described by the famous Viennese pathologist, Karl Freiherr von Rokitansky, who noted a tumor consisting of striated muscle cells in the tunica albuginea of the testis (von Rokitansky 1849). The first pediatric case might have been observed in a 3-year-old child by the Czech physician, Vilém Dusan Lambl, who called the tumor “muscle carcinoma” (Lambl 1860). Lambl is best known for his detection of an intestinal parasite which he called Cercomonas intestinalis (however, first observed by Anton van Leeuwenhoek) and which is now termed Giardia lamblia, an eponym based on Lambl and the French biologist, Alfred Mathieu Giard (1846–1908). Several classifications of RMS have been proposed (reviews: Parham 2001; Caillaud et al. 2006), and attempts for molecular classification have been undertaken (Davicioni et al. 2009).
Epidemiology
HBRMS is the most common biliary tract tumor in the pediatric age group and is, e.g., clearly more common as pediatric cholangiocarcinoma. But the tumor is, overall, rare, and it accounts for only 0.8 % of all rhabdomyosarcomas and 1.3 % of all malignant liver tumors in childhood. At the time of diagnosis, 75 % of the children are under 5 years of age, and the tumor most often occurs in children aged 3–4 years. Girls are generally more frequently involved (5:1). However, in a review of 26 patients reported between 1875 and 1981 (Lack et al. 1981), 13 were male, 12 were female, and in 1 patient, the sex was not specified. Rarely, HBRMS occurs in adolescents (Haider et al. 2013).
Clinical Features
Clinical signs and symptoms are rather nonspecific and include upper abdominal pain, abdominal distention, nausea with vomiting, and hepatomegaly The clinical presentation is closely linked to the distinct growth pattern of the tumor, which grows along and within the bile ducts, causing biliary obstruction and cholestatic jaundice. Jaundice with or without associated leukocytosis was found as the presenting sign in 60–80 % of cases and critically depends on the level of bile duct involvement, being common in RMS located to the porta hepatis and obligatory in RMS of the ampullary region. Jaundice is commonly associated with marked hepatomegaly, and this combination may be confused with forms of icteric and cholestatic hepatitis. Obstructive jaundice occurs relatively late in the course of the disease, i.e., when the tumors are adequately large and then obturating the lumina, because bile flow continues between the intraluminal tumor projections. As in other malignancies (Staalman and Umans 1993), HBRMS may be associated with the paraneoplastic syndrome, hypertrophic osteoarthropathy (Geary et al. 2004). Radiologically, HBRMS may mimic choledochal cyst (Zampieri et al. 2006; Rebollo Guelar et al. 2013; Margain-Deslandes et al. 2013).
Selected References
Goeters 1941; Gaubert 1965; Hays and Snyder 1965; Virenque et al. 1966; Soper and Dunphy 1968; Davis et al. 1969; Akers and Needham 1971; Corbineau et al. 1975; Babut et al. 1976; Taira et al. 1976; Nagaraj et al. 1977; Isaacson 1978; Cannon et al. 1979; Hashimoto et al. 1980; Coronado Perez and Angulo Hernandez 1981; Lack et al. 1981; Martinez et al. 1982; Mulet et al. 1982; Friedburg et al. 1984; Shamis et al. 1985; Arnaud et al. 1987; Horowitz et al. 1987; Shimada et al. 1987; Caty et al. 1990; Mann et al. 1990; Perisic et al. 1991; Gururangan et al. 1992; Babin-Boilletot et al. 1993; Lee et al. 1996; Pollono et al. 1998; Balkan et al. 1999; Hunt et al. 2002; Kebudi et al. 2003; Aggarwal et al. 2004; Zampieri et al. 2006; Huber et al. 2008; Zhao et al. 2011; Kumar et al. 2012; Diaconescu et al. 2013.
HBRMS of the Intrahepatic Bile Ducts
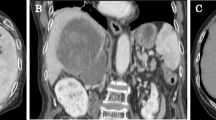
The intrahepatic biliary tree is the second most common manifestation of HBRMS, this tumor being more common in the extrahepatic duct system (see below). Sonography, conventional radiology, and CT display rather typical and, sometimes, pathognomonic changes. They comprise intrahepatic masses of heterogeneous structures with cystic areas and septations and bile duct stenosis with prestenotic duct dilatation (Witcombe 1979; Ruymann et al. 1985; Williams and Sheward 1986; Arnaud et al. 1987; Lee et al. 1996; Donnelly et al. 1998). MRI of the lesions show high signal intensity with heterogeneous enhancement in T2-weighted single-shot fast spin-echo axial MR images (Geary et al. 2004).
HBRMS of the Extrahepatic Bile Ducts
HBRMS involvement of the common bile duct is rare, but important pathology of the lower biliary tract in children, and is the most common site of HBRMS. Leriche (1934) reported the first case. The patient was a 4.5-year-old boy who presented with jaundice and a huge abdominal mass. At laparotomy, an enormous cystic dilatation of the common bile duct was seen. RMS of the large bile duct can mimic a choledochal cyst (Nemade et al. 2007; Ali et al. 2009). The dilated duct contained copious bloody, mucoid fluid and a tumor the size of a child’s head (Leriche 1934). Several other cases were reported since.
Selected References
Goeters 1941; Willis 1948; Werner 1951; Horn et al. 1955; Farinacci et al. 1956; Bernheim et al. 1962; Delany et al. 1966; Gout et al. 1974; Majmudar and Kumar 1976; Sarrazin et al. 1977; Taura et al. 1977; Phatak and Prabhu 1982; Friedburg et al. 1984; von der Oelsnitz et al. 1991; Verstandig et al. 1991; Sanz et al. 1997; Berghoff et al. 1998; Prasad et al. 2003; Aggarwal et al. 2004; Kirli et al. 2012.
Ultrasonography typically shows biliary dilatation (Friedburg et al. 1984; Arnaud et al. 1987; Geoffray et al. 1987) and a space-occupying process or intraductal mass within the common duct, usually surrounded by fluid which may reflect necrosis (Friedburg et al. 1984; Geoffray et al. 1987). The portal vein may be displaced by large lesions, but portal vein thrombosis has not been noted (Geoffray et al. 1987). Color Doppler imaging has revealed numerous abnormal tumor arteries with low resistive index (Roebuck et al. 1998). CT images disclose low-density irregularly shaped masses within the dilated duct, with hypodense and heterogeneous attenuation patterns (Geoffray et al. 1987). Low attenuation areas within the tumor have been reported (Miller and Greenspan 1985; Caty et al. 1990; Patil et al. 1992; Linstedt-Hilden and Brambs 1994). With contrast enhancement, the tumors show four patterns: strong heterogeneous, incomplete globular, mild, and none (Roebuck et al. 1998). The masses may be surrounded by fluid collections which represent bile fluid between the duct wall and the protruding tumor masses (Roebuck et al. 1998). This pattern is also seen in cholangiograms, showing irregular luminal defects (Friedburg et al. 1984), a large mass in the duct lumen (Sanz et al. 1997), or the replacement of the common duct by a multilocular cystic lesion associated with prestenotic marked bile duct dilatation (Kitagawa and Aida 2007). This lesion pattern has been described as early as 1979 by the use of percutaneous transhepatic cholangiography (Cannon et al. 1979). In the case of RMS located to the hepatic hilum, multilocular cystic lesions are seen in US and CT (Kitagawa and Aida 2007). The characteristic luminal alterations caused by RMS can also be demonstrated by use of endoscopic retrograde cholangiopancreatography (ERCP; Himes et al. 2008). In case of marked bile duct dilatation caused by stenosis, choledochal cyst was a frequent diagnosis before surgery (Ruymann et al. 1985; Caty et al. 1990; von der Oelsnitz et al. 1991; Tireli et al. 2005). It has been reported that coronal CT sections were particularly useful in demonstrating the complex relationship of common bile duct RMS to the porta hepatis, pancreas, and duodenum (Verstandig et al. 1991). In MR images, the tumors show predominantly low signal intensity on T1-weighted images with intense but inhomogeneous contrast enhancement. On T2-weighted images, the tumors are moderately to markedly hyperintense (Roebuck et al. 1998).
Pediatric Rhabdomyosarcoma of the Ampullary Region
Embryonal RMS located to the ampullary region is a very rare variant of hepatobiliary RMS causing biliary obstruction. This pathology was first described in 1968 based on autopsy findings on a 6-year-old child (Isaacson 1978). In another case, a 3-year-old boy, exploratory laparotomy performed for obstructive jaundice and a cystic ampullary mass at imaging showed a cystic tumor within the posterior aspect of the pancreatic head, contiguous with the distal common bile duct. The cystic mass was opened, and botryoid, gelatinous material was spontaneously extruded from the cavity. Histology displayed embryonal RMS. Pancreaticoduodenectomy followed by adjuvant chemotherapy and irradiation resulted in long-term survival (Caty et al. 1990). A third case related to a 20-month-old baby girl who presented with 1-month history of progressive jaundice. Contrast-enhanced CT showed a large heterogeneous mass which appeared to arise from the ampullary region causing biliary dilatation. Endoscopy revealed a large ulcerated lesion in the second part of the duodenum. In the resection specimen, an exophytically growing tumor of 11 cm diameter centered on the ampulla was found. Histology showed ampullary embryonal RMS with invasion of the distal common bile duct, duodenal wall, and pancreas (Perera et al. 2009).
HBRMS in Choledochal Cyst
Choledochal botryoid RMS was described in a 22-month-old boy who developed obstructive jaundice. Imaging first suggested cystic lymphangioma, but the cystic space was found at surgery to be a choledochal cyst harboring RMS (Sassi et al. 2008). A second reported patient was a 6-year-old girl who presented with recurrent jaundice and loss of weight. A large abdominal mass was palpable, and US and CT revealed a large subhepatic mass with multiple septations. The common bile duct opened directly into the mass, and the mass was continuous with the intrahepatic radicles. The resection specimen showed that the mass was choledochal cyst with secondary RMS (Patil et al. 1992).
Pediatric Rhabdomyosarcoma of the Liver Not Related to the Biliary Tract
Primary intrahepatic rhabdomyosarcoma not related to the biliary tract is extremely rare in children. Pack and Miller (1956) reported on a 14-year-old girl with a large tumor in the right liver lobe. Almost the entire lobe was replaced by a cystic thick-walled mass (up to 15.5 cm diameter) filled with amorphous necrotic brown-stained material and thin blood-tinged fluid. The histology showed embryonal RMS. Scheiden et al. (1988) described a 7-year-old boy who had presented with painful hepatomegaly and huge tumor in the left liver lobe. Surgical resection revealed an 18 cm-sized tumor in the left lobe, with extensive central regressive and cystic change. Histology showed embryonal RMS with immature-looking, desmin-positive tumor cells. The patient of Huang and coworkers (2003) was an 8-year-old boy presenting with abdominal pain, spiking fever, and a rapidly growing abdominal mass. Imaging revealed a large solid tumor in the right liver lobe, and the mass was removed by an extended right hepatectomy. Histology showed pleomorphic RMS. The patient died 2 months after resection due to tumor recurrence and massive internal hemorrhage. A second report described a 10-year-old boy who had a voluminous tumor at the inferior face of the right liver lobe, associated with thrombosis of the retrohepatic part of the inferior caval vein. The histology was embryonal rhabdomyosarcoma (Chat et al. 2007).
Rhabdomyosarcoma of the Hepatic Pedicle
In the pediatric age group, the hepatic pedicle or porta region represents an anatomical compartment where unusual tumors may arise, including liposarcoma and rhabdomyosarcoma. Embryonal RMS was observed in the mesenchyme of the liver pedicle in a 10-year-old child (Ferlicot et al. 1999).
Macroscopic Pathology
Most HBRMS exhibit a growth pattern centered on the intrahepatic bile ducts. Apart from this main pattern, RMS may also show a predominantly invasive growth pattern, with marked infiltration of the liver substance (Linstedt-Hilden and Brambs 1994). The tumor may present in the form of a mass filled with hemorrhagic and necrotic material (Kebudi et al. 2003). In resection specimens, one may note polypoid tumor masses that take their origin in the bile duct wall and protrude into the dilated bile duct lumen, sometimes resulting in the typical grape-like morphology that gave this RMS its name (botryoid meaning, grape-like). Resected polypoid, botryoid growths show a striking resemblance to nasal polyps. This botryoid pattern may result in voluminous papillary-polypoid growths (Leriche 1934; Davis et al. 1969). Smaller resection specimens may only consist of soft, polypoid, gelatinous, and/or transparent tissue fragments. In RMS mainly showing an intrahepatic growth pattern, the cut surface of resection specimens may show, within the mass, bulging tumor nodules representing the polypoid intraductal lesions (Lack et al. 1981).
Histopathology
The histopathology of HBRMS is, as such, the same for intra- and extrahepatic tumors and is therefore summarized for both paragraphs (Figs. 1, 2, and 3; Bässler and Voth 1962). Non-necrotic tumors have an intact surface toward the biliary lumen. Already Sheehan (1930), in his report of a large intrahepatic HBRMS, emphasized that “the mucous columnar epithelium of the included cysts and the bile ducts remained intact and healthy. Thus in the extension down the common bile-duct the large mass of tumour cells had spread underneath the epithelium which it had raised undamaged on its surface.” The intact biliary epithelium (with some degrees of epithelial disarray and/or atrophy) is expectedly positive for cytokeratins 7 and 19 and seems to play a significant role for the botryoid growth pattern. The subepithelial tumor generally shows a low cellularity, except a characteristic subepithelial zone of high cellularity, the so-called cambium layer well known from other mucosal sites of RMS. This layer contains most frequently embryonal rhabdomyoblasts, round cells with a myogenic cytoplasm, and racquet and strap cells. The latter possess large nuclei and dark, clumped chromatin and may show cross-striations. Within the cambium, foamy macrophages laden with lipids are not infrequently found (Davis et al. 1969). Interspersed among these neoplastic cells is a myxoid tissue component which becomes more prominent toward deeper layers of the tumor, where the neoplastic cells are more commonly stellate or spindled. The loose aspect of this tissue component may pose considerable differential diagnostic difficulties, because it may be misinterpreted as reactive edematous tissue in biopsies. This myxoid tissue may grow around septal and interlobular bile duct, producing a characteristic tissue sheath. Small bile ducts are usually surrounded by a hypercellular cambium which is in turn surrounded by a looser myxoid tissue (Davis et al. 1969). This tumor tissue induces stenotic changes with the formation of small bile duct cysts.

Hepatobiliary rhabdomyosarcoma, embryonal type. Immature tumor cells form a hypercellular subepithelial layer in a bile duct, the so-called cambium layer (hematoxylin and eosin stain)

Hepatobiliary rhabdomyosarcoma, embryonal type. Some of these neoplasms produce polypoid lesions with a loose hypocellular tumor tissue, protruding into bile ducts (botryoid rhabdomyosarcoma; hematoxylin and eosin stain)

Botryoid hepatobiliary rhabdomyosarcoma. The embryonal rhabdomyosarcoma consists of slender rhabdomyoblasts. The lesion is covered by damaged biliary epithelium. Botryoid rhabdomyosarcomas may lack a cambium layer (hematoxylin and eosin stain)
Immunohistochemistry
Immunohistochemistry has been considered to be the decisive diagnostic method in more than 20 % of RMS. RMS cells are consistently reactive for vimentin, and they express the skeletal muscle cell lineage markers, desmin, myogenin, and myogenic regulatory protein D1/MyoD1 (Fig. 4; Dias et al. 1990; Parham et al. 1991; Cessna et al. 2001; Sebire and Malone 2003). In a study of 956 cases (the majority of course being extrahepatic tumors), it was found that myogenin and MyoD1 were equally sensitive (positive for 97 % of RMS cases), with both also showing similar specificity (90 % vs. 91 % of cases), but expression was more consistent in alveolar RMS (ARMS) than embryonal RMS/ERMS (Morotti et al. 2006). In particular, nuclear myogenin expression is more pronounced in ARMS than in ERMS (Morgenstern et al. 2008). In pediatric liver tumors, polyclonal desmin and muscle-specific actin were variably immunoreactive in undifferentiated embryonal sarcoma (UES) and RMS; however, myogenin and MyoD1 were uniformly negative in UES and routinely positive in the majority of hepatobiliary RMS (Nicol et al. 2007). Embryonal RMS did not stain for the paired box transcription factor, PAX5, while 67 % of alveolar RMS were positive (Sullivan et al. 2009). It has been reported that diffuse myogenin expression in soft tissue pediatric RMS is an independent marker of poor survival (Heerema-McKenney et al. 2008). RMS with a component of round cells may be difficult to distinguish from other blue small cell tumors, e.g., neuroblastoma. Peripherin and alpha-internexin are specifically expressed in neuroblastoma but not in RMS. Microtubule-associated protein 1B (MAP1B) is strongly and diffusely expressed in all neuroblastomas but is also detectable in RMS. Nestin is diffusely expressed in RMA but also (multifocally) in a fraction of neuroblastomas (Willoughby et al. 2008). Part of RMS (more commonly ARMS than ERMS) may express anaplastic lymphoma kinase (ALK), most likely independent of the chromosomal fusion status (Corao et al. 2009). The intraluminal polypoid tumor masses in botryoid hepatobiliary RMS are lined by the preexisting cholangiocyte population (Fig. 5).

Hepatobiliary rhabdomyosarcoma, embryonal type. Tumor cells in the cambium layer and stellate cells deeper in the loose tissue are desmin positive (desmin immunostain)

Hepatobiliary rhabdomyosarcoma, embryonal type. A cytokeratin 19-positive cholangiocyte lining covers the bulging tumor. Note the vague cambium layer and the loose tumor tissue with stellate cells in the deeper part of the duct (CK19 immunostain)
Ultrastructure
At electron microscopy, cells remote from the biliary mucosal surface are usually separated by a greater amount of loose ground substance containing few scattered collagen fiber bundles. Small aggregates of tumor cells may be partially surrounded by basement membrane. Many tumor cells are so immature that a proper ultrastructural classification is not possible, but there are also rhabdomyoblasts with variable degrees of differentiation and sometimes well-defined, transversely oriented electron-dense areas and/or incomplete or deformed sarcomeres (Lack et al. 1981). In one electron microscopic study, the neoplasm was shown to consist of three types of cells: polygonal, elongated, and small cells. In contrast to the first two cell types, which revealed moderate to large amounts of myofibrils and occasional A, I, and Z bands, the small cell population was poor in myofibrils, but had dilated ER profiles (Taura et al. 1977).
Biology of Disease
HBRMS is an aggressive lesion that may infiltrate the liver substance and cause local and remote tumor spread, although this type of RMS is more prone to invasion of contiguous structures (Davis et al. 1969; Ruymann et al. 1985). In about 30 % of cases, metastases to the peritoneum, omentum, and locoregional lymph nodes occur. Distant metastases have been found in the lungs, pericardium, and bones of skull and extremities (Davis et al. 1969).
HBRMS remained incurable for a very long time period, until a child was cured by partial surgical resection, radiotherapy, and chemotherapy in 1971 (Akers and Needham 1971). In 1985, the Intergroup Rhabdomyosarcoma Study Group (IRSG) reported ten cases of HBRMS treated on the IRS I and IRS II protocols (Ruymann et al. 1985). Four of these ten patients survived, showing that, at that time, outcome was still poor. Aggressive surgery had been proposed (Martinez et al. 1982; Schweizer et al. 1994), but an analysis of 25 eligible patients with HBRMS enrolled in IRSG studies I through IV from 1972 to 1998 showed that gross total resection is rarely possible despite aggressive surgery and that outcome is good despite residual disease after surgery (Spunt et al. 2000). The signs and sequelae of obstructive jaundice/cholestasis may be relieved by external and internal-external biliary drainage (Roebuck and Stanley 2000). RMS located in soft tissues and classical sites will more and more be stratified for risk, e.g., via immunohistochemical marker expression or gene expression profiling (metagene patterns; Bortoluzzi et al. 2005). This has not yet been accomplished with hepatobiliary RMS, owing to the rarity of these lesions.
Hepatobiliary Alveolar Rhabdomyosarcoma
Hepatobiliary alveolar RMS is a very rare neoplasm (Fig. 6). Only in two reports there is evidence of an alveolar component. In a pediatric hepatic rhabdomyosarcoma with striated tumor cells and associated with mucin-producing cysts reported in 1930 (Sheehan 1930), one figure of the publication depicts what seem to be alveolar rhabdomyosarcoma. The other patient with an alveolar hepatic RMS was an adult male (68 years) with a tumor in the right liver lobe (Shibata et al. 1987).

Alveolar rhabdomyosarcoma of the liver (hematoxylin and eosin stain)
Primary Rhabdomyosarcoma of the Bile Ducts in Adults
Primary RMS of the large bile ducts in adults is extremely rare and has been described in the common bile duct (Aldabagh et al. 1986). The 40-year-old female patient presented with jaundice of short duration. A transhepatic percutaneous cholangiogram revealed an obstructive lesion involving the common bile duct with marked dilatation of the biliary system. At laparotomy, the tumor grossly involved the wall of the choledochus and extended into the cystic duct and common hepatic duct. Histologically, one noted an infiltration of the full thickness of the wall and the surrounding fatty tissue by a desmin- and myoglobin-reactive RMS with a distinct cambium layer. Elsewhere, the cells infiltrated in a single-file pattern with a desmoplastic reaction around normal structures (Aldabagh et al. 1986). Rhabdomyosarcoma may also develop in choledochal cysts. Pleomorphic RMS within a choledochal cysts has been described as a cause of obstructive jaundice (Tufail et al. 2006).
Primary Rhabdomyosarcoma of the Liver in Adults Not Clearly Related to the Biliary Tract
Adult-type rhabdomyosarcoma has been classically defined as a pleomorphic sarcoma with desmin expression occurring in the extremities and the trunk of adult patients. Classification of these lesions has been complex until a consensus was found (Palmer, SIOP, NCI, and International Classifications; review: Parham 2001). Histologically, these neoplasms occur in three categories, i.e., spindle cell RMS, pleomorphic RMS, and mixed forms (Stock et al. 2009). Spindle cell RMS seems to be a variant of embryonal RMS, and this tumor also occurs in the form of sclerosing spindle cell RMS (Gavino et al. 2010). Alveolar RMS is a distinct entity different in many aspects from classical adult-type RMS and shows up in a classical form and variants, including the clear cell variant. RMS occurring in the adult liver as a primary tumor seems to have complex features, in that also embryonal-type RMS and variants thereof have been reported.
Selected References
Miller and Pack 1956; Mori et al. 1979; Hatanaka et al. 1983; Watanabe et al. 1983; Morimoto et al 1986; McArdle et al. 1989; Cote and Urmacher 1990; Hayakawa et al. 1990; Zornig et al. 1992; Bürrig and Knauer 1994; Hiyama 1995; Tominaga et al. 1995; Meyer-Pannwitt et al. 1996; McRae and Lee 2005.
Two tumors were reported to be alveolar RMS (Shibata et al. 1987; Schoofs et al. 2011). In one of these neoplasms, FISH analysis revealed PAX3/FOXO1A fusion (Schoofs et al. 2011). The hepatic cell of origin for these sarcomas is not known. Hepatic RMS in the adult may coexist with synchronous hepatocellular carcinoma in the same liver (Hatanaka et al. 1983; Morimoto et al. 1986; Hayakawa et al. 1990).
Pathology
Macroscopically, the tumors were described to be mass lesion with a nodular border, whitish to yellowish, and either soft or firm. Sometimes, hemorrhage and necrosis occur (Watanabe et al. 1983). Embryonal RMS classically consists of small, undifferentiated round to spindled or stellate cells that may resemble embryonic skeletal muscle cells. These cells are embedded in a myxoid matrix, causing a typical “loose” aspect of the tissue, with hypocellular and hypercellular areas. Differentiation of the cell lineage presents as an increasing number of cells with eosinophilic cytoplasm (“tadpole cells” and “strap cells”). These cells contain myofibrils, may show cross-striation, and are rhabdomyoblasts. This phenotype is the classical variant of embryonal RMS. Part of the tumor cells may show distinct angulation of the muscle fibers – the so-called broken straw sign. The spindle cell variant has not been found in the liver so far. Immunohistochemically, more than 95 % of the tumor cells are reactive for desmin (Altmannsberger et al. 1985) and for HHF35, a monoclonal antibody directed against muscle actins (Schmidt et al. 1988). Nuclear positivity for myogenin is also diagnostically very useful (Cessna et al. 2001).
Rhabdomyosarcoma in/of the Inferior Vena Cava
Sarcoma of the inferior vena cava is usually leiomyosarcoma, with or without associated Budd-Chiari syndrome (see the respective chapter). More than 100 cases of this distinct entity have been reported. In contrast, other sarcomas are exceptional findings in this location. In particular, this refers to vascular RMS, which is mainly known from the pulmonary trunk and the pulmonary artery (Watanabe et al. 1985). A 63-year-old male patient had obstruction of the inferior vena cava caused by intraluminal pleomorphic RMS. This tumor was resected, but the patient died of local recurrence. Autopsy revealed RMS extending from the inferior vena cava just above the right renal vein to the right atrium and involving the caudate lobe of the liver. The tumor was associated with Budd-Chiari syndrome. It was difficult to decide whether the tumor took its origin in the vena cava or the liver (Fujita et al. 1993).
Intraperitoneal Involvement in Pediatric Rhabdomyosarcoma
Peritoneal manifestations of pediatric malignancy chiefly relate to desmoplastic small round cell tumors, lymphoma, germ cell tumors, neuroblastoma, Wilms tumor, and secondary seeding of intracranial tumors by ventriculoperitoneal shunts. Intraperitoneal neoplastic involvement in RMS is not common but may get into differential diagnostic considerations in case the lesions develop on the liver surface. It has been reported that approximately 10 % of children with abdominopelvic RMS may have intraperitoneal involvement either at the time of diagnosis or subsequently. The manifestations of intraperitoneal involvement comprise enhancing nodules or masses, a pseudomyxoma peritonei-like appearance, omental caking, and ascites (Chung et al. 1998). Intraperitoneal involvement has been observed in pelvic RMS, with enhanced nodular lesions in CT also located around the liver and in the lesser sac (Oto et al. 2001) .
Liver Metastasis in Pediatric and Adult Rhabdomyosarcoma
RMS may metastasize to almost all organs, but there are instances where patients with RMS present with a distinct mode of diffuse metastasis. These cases may be called the leukemic variant of RMS, because the children can have fever, generalized malaise, bone pain due to marrow involvement, and circulating RMS cells in the peripheral blood smear resembling leukemia (Cohen 1992). Hepatic metastases have also been described for sclerosing RMS (Kikuchi et al. 2013).
Hepatic Rhabdomyomatous Tumors
In the pediatric age group, rhabdomyomas typically occur in the heart and here mostly in the context of tuberous sclerosis. In 1862, Friedrich Daniel von Recklinghausen reported pigeon egg-sized myomas found in the heart of a newborn (von Recklinghausen 1862), and these lesions were termed rhabdomyoma by the German physician and pathologist, Friedrich Albert von Zenker (1864). In childhood, rhabdomyomatous tumors may arise in the liver as a late sequela of fetal rhabdomyomatous nephroblastoma. This lesion is considered to be a predominantly monophasic mesenchymal variant of Wilms tumor, sometimes bilateral, which has not been seen in patients older than 4 years. It exhibits a less aggressive biology than standard Wilms tumor despite its usually much larger size (Wigger 1976; Eble 1983; Joseph et al. 2003). Primary and metastatic lesions of this nephroblastoma variant can undergo maturation (Ishikawa et al. 2001). In a 14-year-old boy, a tumor with the features of a fetal rhabdomyoma arose in the liver 13 years after treatment for fetal rhabdomyomatous nephroblastoma (van der Kwast et al. 1992).
Other Liver Tumors with a Rhabdomyocytic/Rhabdomyoblastic Differentiation
In the pediatric age group, hepatoblastoma may develop rhabdomyoblastic components within the spectrum of mixed epithelial and mesenchymal hepatoblastoma with teratoid features (Shabanov et al. 1981). However, there are also pediatric liver tumors with a rhabdomyoid component that cannot easily be allocated to mixed hepatoblastomas. Williams (1953) described, in 2-year-old boy, a neoplasm which he termed liver tumor of mixed type. The tumor presented as a cystic mass located in the right liver lobe. At necropsy, the mass consisted of cysts with a diameter ranging from 1 to 3 cm and solid components. The largest cystic mass had a fibrous capsule and contained thin leaves of tissue separated by a glairy mucous substance. Microscopically, the cysts were lined by a mucus-secreting cuboid or flattened epithelium. The cysts were separated by a mesenchyme that showed embryonic features and hypercellular areas, containing round cells, spindle cells, or strap-like cells and focally immature muscle cells with cross-striation. A similar tumor had been described by Sheehan (1930). A 6-year-old girl showed, at necropsy, a liver tumor of 12 cm diameter. This tumor grossly consisted of whitish tissue and was fairly soft, but showed little necrosis and no hemorrhage. It had a well-defined but not encapsulated edge. An extension had spread down the bile duct, with formation of a bulging and bulbous intraluminal mass. Histology in part corresponded to rhabdomyosarcoma, with striated tumor cells. In addition, however, there were clusters of cysts associated with the tumor, measuring up to 2 cm in diameter. The cysts were lined by single layer of columnar epithelium which secreted mucin. There are primary hepatic tumors which co-express features of both, hepatocellular carcinoma and rhabdomyosarcoma (so-called rhabdomyosarco-hematoma; Goldman and Friedman 1969), or features of combined cholangiocarcinoma and rhabdomyosarcoma. These carcinosarcomas are discussed in a separate chapter. Gastrointestinal stromal tumors (GIST) can show, both in the primary tumor and liver metastasis, rhabdomyosarcomatous differentiation after tyrosine kinase inhibitor therapy (Liegl et al. 2009). Schmid and coworkers reported a liver tumor observed in a 36-year-old female patient who had developed intermittent fever and increasing asthenia (Schmid et al. 1979). In a right hepatectomy specimen, they found a spherical, well-circumscribed tumor of 8.5 cm diameter, composed of a whitish-gray, moderately firm tissue with small cystic areas. Histologically, the tumor did not invade the surrounding liver tissue, but rather caused perifocal tissue compression. The interface between the tumor and the liver was characterized by a dense inflammatory infiltration. The neoplasm itself consisted of elongated spindle cells and large and pleomorphic, often multinucleated cells with only rare mitotic figures. Some of the cells displayed a ribbon-shaped appearance like skeletal muscle fibers, however without cross-striation. Also the interior of the tumor showed an inflammatory reaction. These findings suggested a rhabdomyoblastic origin, but ultrastructurally, the tumor cells shared features with hepatocytes. Hence, this neoplasm may represent an example of epithelial-mesenchymal transition.
References
Aggarwal K, Pahuja S, Chadha R (2004) Botryoid rhabdomyosarcoma of common bile duct. Indian J Pediatr 71:363–364
Akers RD, Needham ME (1971) Sarcoma botryoides of the bile ducts with survival. J Pediatr Surg 6:474–479
Aldabagh SM, Shibata CS, Taxy JB (1986) Rhabdomyosarcoma of the common bile duct in an adult. Arch Pathol Lab Med 110:547–550
Ali S, Russo MA, Margraf L (2009) Biliary rhabdomyosarcoma mimicking choledochal cyst. J Gastrointestin Liver Dis 18:95–97
Altmannsberger et al (1985) http://www.ncbi.nlm.nih.gov/pubmed/3881039
Arnaud O, Boscq M, Asquier E, Michel J (1987) Embryonal rhabdomyosarcoma of the biliary tree in children: a case report. Pediatr Radiol 17:250–251
Babin-Boilletot A, Flamant F, Terrier-Lacombe MJ, Marsden B, van Unnik A, Deméocq F et al (1993) Primitive malignant nonepithelial hepatic tumors in children. Med Pediatr Oncol 21:634–639
Babut JM, Ferrand B, Bracq H, Feuillu J, Mention J, Lecornu M (1976) Rhabdomyosarcome hépatique chez l’enfant. Ann Chir 30:251–255
Balkan E, Kiristioglu I, Gurpinar A, Sinmaz K, Ozkan T, Dogruyol H (1999) Rhabdomyosarcoma of the biliary tree. Turk J Pediatr 41:245–248
Bässler R, Voth D (1962) Pathologie und submikroskopische Morphologie des sogenannten Sarcoma botryoides der grossen Gallengänge. Krebsforschung 65:44–55
Berghoff R, Drut R, Urrutia A, Cedola J (1998) Rhabdomyosarcoma of the extrahepatic biliary tree: initial treatment with chemotherapy and conservative surgery. Med Pediatr Oncol 30:290–293
Bernheim M, Feroldi J, Larbre F, Sterlin M (1962) Botryoid sarcoma of the common bile duct. Apropos of a personal case (in French ). Pediatrie 17:243–250
Bortoluzzi S, Bisognin A, Romualdi C, Danieli GA (2005) Novel genes, possible relevant for molecular diagnosis or therapy of human rhabdomyosarcoma, detected by genomic expression profiling. Gene 348:65–71
Bürrig KF, Knauer S (1994) Rhabdomyosarcoma of the liver in adulthood. Case report and review of the literature (in German). Pathologe 15:54–57
Caillaud JM, Gérard-Marchant R, Marsden HB, Van Unnik AJM, Rodary C, Rey A, Flamant F (2006) Histopathological classification of childhood rhabdomyosarcoma: a report from the international society of pediatric oncology pathology panel. Med Pediatr Oncol 17:391–400
Cannon PM, Legge DA, O’Donnell B (1979) The use of percutaneous transhepatic cholangiography in a case of embryonal rhabdomyosarcoma. Br J Radiol 52:326–327
Caty MG, Oldham KT, Prochownik EV (1990) Embryonal rhabdomyosarcoma of the ampulla of Vater with long-term survival following pancreaticoduodenectomy. J Pediatr Surg 25:1256–1258
Cessna MH, Zhou H, Perkins SL, Tripp SR, Layfield L, Daines C, Coffin CM (2001) Are myogenin and MyoD1 expression specific fo rhabdomyosarcoma? A study of 150 cases, with emphasis in spindle cell mimics. Am J Surg Pathol 25:1150–1157
Chat L, Mahi M, Chellaoui M, Khattab M, Benamour-Ammar H (2007) Embryonal rhabdomyosarcoma of the liver: an unusual site of origin (in French). J Radiol 88:72–74
Chung CJ, Fordham L:, Little S, Rayder S, Nimkin K, Kleinman PK, Watson C (1998) Intraperitoneal rhabdomyosarcoma involvement in children: incidence and imaging characteristics on CT. AJR Am J Roentgenol 170:1385–1387
Cohen MD (1992) Imaging of children with cancer, 1st edn. Mosby Year Book, St. Louis, pp 308–337
Corao DA, Biegel JA, Coffin CM, Barr FG, Wainwright LM, Ernst LM, Choi JK, Zhang PJ, Pawel BR (2009) ALK expression in rhabdomyosarcomas: correlation with histologic subtype and fusion status. Pediatr Dev Pathol 12:275–283
Corbineau D, Bertrand G, Simard C, Plane P, Delaitre R (1975) Sarcoma botryoide des voies biliares chez l’enfant. Ann Pediatr 22:735
Coronado Perez H, Angulo Hernandez O (1981) Liver neoplasms in children (in Spanish). Bol Med Hosp Infant Mex 38:723–740
Cote RJ, Urmacher C (1990) Rhabdomyosarcoma of the liver associated with long-term oral contraceptive use. Possible role of estrogens in the genesis of embryologically distinct liver tumors. Am J Surg Pathol 14:784–790
Davicioni et al (2009) http://www.ncbi.nlm.nih.gov/pubmed/19147825
Davis GL, Kissane JM, Ishak KG (1969) Embryonal rhabdomyosarcoma (sarcoma botryoides) of the biliary tree. Cancer 24:333–342
Delany HM, Driscoll PJ, Ainsworth H (1966) Sarcoma botryoides of the common bile duct. Report of a case and review of the literature. J Pediatr Surg 1:571–578
Diaconescu S, Burlea M, Miron I, Aprodu SG, Mihaila D, Olaru C, Miron L (2013) Childhood rhabdomyosarcoma. Anatomo-clinical and therapeutic study on 25 cases. Surgical implications. Rom J Morphol Embryol 54:531–537
Dias P, Parham DM, Shapiro DN, Webber BL, Houghton PJ (1990) Myogenic regulatory protein (MyoD1) expression in childhood solid tumors: diagnostic utility in rhabdomyosarcoma. Am J Pathol 137:1283–1291
Donnelly LF, Bisset GS, Frush DP (1998) Diagnosis please. Case 2: embryonal rhabdomyosarcoma of the biliary tree. Radiology 208:621–623
Eble JN (1983) Fetal rhabdomyomatous nephroblastoma. J Urol 130:541–543
Farinacci CJ, Fairchild JP, Sulak MH, Gilpatrick CW (1956) Sarcoma botryoides (a form of embryonal rhabdomyosarcoma) of the common bile duct: a report of two cases. Cancer 9:408–417
Ferlicot S, Quillard J, Chardot C, Caillou B, Oberlin O, Gauthier F, Fabre M (1999) Unusual site of an embryonal rhabdomyosarcoma of the mesenchymal hepatic pedicle ( in French ). Ann Pathol 19:521–524
Friedburg H, Kauffmann GW, Bohm N, Fiedler L, Jobke A (1984) Sonographic and computed tomographic features of embryonal rhabdomyosarcoma of the biliary tract. Pediatr Radiol 14:436–438
Fujita H, Kawata K, Sawada T, Mizutani T, Iwasaki Y, Shirono K, Kounosu H, Shirakata S (1993) Rhabdomyosarcoma in the inferior vena cava with secondary Budd-Chiari syndrome. Intern Med 32:67–71
Gaubert J (1965) Les tumeurs malignes du tube digestif et de ses annexes chez l’enfant. Pediatrie 20:545–554
Gavino AC, Spears MD, Peng Y (2010) Sclerosing spindle cell rhabdomyosarcoma in an adult: report of a new case and review of the literature. Int J Surg Pathol 18:394–397
Geary TR, Maclennan AC, Irwin GJ (2004) Hypertrophic osteoarthropathy in primary liver rhabdomyosarcoma. Pediatr Radiol 34:250–252
Geoffray A, Couanet D, Montagne JP, Leclère J, Flamant F (1987) Ultrasonography and computed tomography for diagnosis and follow-up of biliary duct rhabdomyosarcoma in children. Pediatr Radiol 17:127–131
Goeters W (1941) Bösartige Mischgeschwulst des Duktus choledochus bei einem Kleinkind. Arch Kinderheilk 122:217
Goldman RL, Friedman NB (1969) Rhabdomyosarcohepatoma in an adult and embryonal hepatoma in a child. Am J Clin Pathol 51:137–143
Gout JP, Pont J, Pasquier B, Sarrazin R, Rossignol AM, Roget J (1974) Botryoid sarcoma of the choledochus (in French). Pediatrie 29:689–702
Gururangan S, O’Meara A, MacMahon C, Guiney EJ, O’Donnell B, Fitzgerald RJ, Breatnach F (1992) Primary hepatic tumours in children: a 26-year review. J Surg Oncol 50:30–36
Haider N, Nadim MS, Piracha MN (2013) Primary embryonal rhabdomyosarcoma of the liver in a young male. J Coll Physicians Surg Pak 23:750–751
Hashimoto S, Tanike K, Eto T et al (1980) Botryoid sarcoma of the bile ducts. Rinsho Hashasen 25:507–510
Hatanaka S, Sawada S, Midorikawa O (1983) Autopsy case of hepatoma associated with rhabdomyosarcoma of a probable hepatic origin (in Japanese). Nippon Rinsho 41:895–900
Hayakawa K, Shibata T, Yamashita K, Hamanaka D, Tanigawa N, Uarata Y, Hosokawa Y et al (1990) Computed tomography and angiogram of primary hepatic rhabdomyosarcoma: report of two adult cases. Radiat Med 8:35–39
Hays DM, Snyder WH (1965) Botryoid sarcoma (rhabdomyosarcoma) of the bile ducts. Am J Dis Child 110:595–605
Heerema-McKenney A, Wijnaendts LC, Pulliam JF, Lopez-Terrada D, McKenney JK, Zhu S et al (2008) Diffuse myogenin expression by immunohistochemistry is an independent marker of poor survival in pediatric rhabdomyosarcoma: a tissue microarray study of 71 primary tumors including correlation with molecular phenotype. Am J Surg Pathol 32:1513–1522
Himes RW, Raijman I, Finegold MJ, Russell HV, Fishman DS (2008) Diagnostic and therapeutic role of endoscopic retrograde cholangiopancreatography in biliary rhabdomyosarcoma. World J Gastroenterol 14:4823–4825
Hiyama Y (1995) Primary hepatic leiomyosarcoma and rhabdomyosarcoma (in Japanese). Ryoikibetsu Shokogun Shirizu 7:463–466
Horn RC, Yakovac WC, Kaye R, Koop CE (1955) Rhabdomyosarcoma (sarcoma botryoides) of the common bile duct; report of a case. Cancer 8:468–477
Horowitz ME, Etcubanas E, Webber BL, Kun LE, Rao BN, Rao BN, Vogel RJ, Pratt CB (1987) Hepatic undifferentiated (embryonal) sarcoma and rhabdomyosarcoma in children. Results of therapy. Cancer 59:396–402
Huang FC, Eng HL, Chen CL, Ko SF (2003) Primary pleomorphic rhabdomyosarcoma of the liver: case report. Hepatogastroenterology 50:73–76
Huber J, Sovinz P, Freidl T, Jahnel J, Lackner H, Höllwarth M, Otte JB, Urban C (2008) Long term survival in two children with rhabdomyosarcoma of the biliary tract. Klin Padiatr 220:378–379
Hunt GC, Corless CL, Terry AB, Katon RM (2002) Embryonal rhabdomyosarcoma in a 3-year-old child diagnosed at ERCP: case report and review. Gastrointest Endosc 56:445–447
Isaacson C (1978) Embryonal rhabdomyosarcoma of the ampulla of Vater. Cancer 41:466–468
Ishikawa K, Toyoda Y, Fukuzato Y, Kato K, Ijiri R, Tanaka Y (2001) Maturation in the primary and metastatic lesions of fetal rhabdomyomatous nephroblastoma. Med Pediatr Oncol 37:62–63
Joseph JM, Suter OC, Nenadov-Beck M, Gudinchet F, Frey P, Meagher-Villemure K (2003) Repeated surgical excision for an unusual variant of nephroblastoma: case report and review of the literature. J Pediatr Surg 38:E13
Kebudi R, Görgun Ö, Ayan I, Cosar R, Bilgiç B (2003) Rhabdomyosarcoma of the biliary tree. Pediatr Int 45:469–471
Kirli AE, Parlak E, Oguz B, Talim B, Akçören Z, Karnak I (2012) Rhabdomyosarcoma of the common bile duct: an unusual cause of obstructive jaundice in a child. Turk J Pediatr 54:654–657
Kitagawa N, Aida N (2007) Biliary rhabdomyosarcoma. Pediatr Radiol 37:1059
Kumar V, Chaudhary S, Kumar M, Gangopadhyay AN (2012) Rhabdomyosarcoma of biliary tract – a diagnostic dilemma. Indian J Surg Oncol 3:314–316
Lack EE, Perez Atayde A, Schuster SR (1981) Botryoid rhabdomyosarcoma of the biliary tract. Am J Surg Pathol 5:643–652
Lambl VD (1860) Carcinoma tibiae mit Neubildung von quergestreiften Muskelnfassern. Aus dem Franz-Josef-Kinderhospitale in Prag. Theil I, p 191
Lee MJ, Chang ML, Huang PH, Lue WC (1996) Biliary tree rhabdomyosarcoma: report of one case. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 37:458–460
Leriche MR (1934) Volumineuse tumeur papillomateuse du choledoque chez un enfant. Soc Chir Lyon 31:598–602
Liegl B, Hornick JL, Antonescu CR, Corless CL, Fletcher CD (2009) Rhabdomyosarcomatous differentiation in gastrointestinal stromal tumors after tyrosinase inhibitor therapy: a novel form of tumor progression. Am J Surg Pathol 33:218–226
Linstedt-Hilden M, Brambs HJ (1994) Two different manifestations of botryoid sarcoma (embryonal rhabdomyosarcoma) of the biliary tree. Bildgebung 61:40–43
Majmudar B, Kumar VS (1976) Embryonal rhabdomyosarcoma (sarcoma botryoides) of the common bile duct: a case report. Hum Pathol 7:705–708
Mann JR, Kasthuri N, Raafat F, Pincott JR, Parkes SE, Muir KR, Ingram LC, Cameron AH (1990) Malignant hepatic tumours in children: incidence, clinical features and aetiology. Paediatr Perinat Epidemiol 4:276–289
Margain-Deslandes L, Gelas T, Bergeron C, Pracros JP, Collardeau-Frachon S, Lachaux A et al (2013) A botryoid rhabdomyosarcoma diagnosed as a choledochal cyst. Pediatr Blood Cancer 60:2089–2090
Martinez FLA, Haase GM, Koep LJ, Akers DR (1982) Rhabdomyosarcoma of the biliary tree: the case for aggressive surgery. J Pediatr Surg 17:508–511
McArdle JP, Hawley I, Sheyland J, Brain T (1989) Primary rhabdomyosarcoma of the adult liver. Am J Surg Pathol 13:961–965
McRae BL, Lee JR (2005) Pathologic quiz case: right upper quadrant abdominal mass in a young woman. Primary hepatorenal embryonal rhabdomyosarcoma with differentiation. Arch Pathol Lab Med 129:e51–e52
Meyer-Pannwitt U, Kummerfeldt K, Broelsch CE (1996) Primary pleomorphic rhabdomyosarcoma of the liver (in German). Langenbeck’s Arch Chir 381:75–81
Miller JH, Greenspan BS (1985) Integrated imaging of hepatic tumors in childhood. Part I: malignant lesions ( primary and metastatic ). Radiology 154:83–90
Miller TR, Pack GT (1956) Total right hepatic lobectomy for rhabdomyosarcoma. AMA Arch Surg 73:1060–1062
Morgenstern DA, Rees H, Sebire NJ, Shipley J, Anderson J (2008) Rhabdomyosarcoma subtyping by immunohistochemical assessment of myogenin: tissue array study and review of the literature. Pathol Oncol Res 14:233–238
Mori H, Matsubara N, Fujii M, Kawai T, Tanaka T, Takahashi M (1979) Alphafetoprotein producing rhabdomyosarcoma of the adult liver. Acta Pathol Jpn 29:485–491
Morimoto H, Takada Y, Akita T, Kato Y, Tanigawa N, Muraoka R, Urata Y (1986) A resected case of collision tumor of hepatocellular carcinoma and primary liver rhabdomyosarcoma (in Japanese). Nippon Geka Gakkai Zasshi 87:456–463
Morotti RA, Nicol KK, Parham DM, Teot LA, Moore J, Hayes J, Meyer W, Qualman SJ (2006) An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the children’s Oncology Group experience. Am J Surg Pathol 30:962–968
Mulet JF, Illa J, Mainou C, Ros J, Claret I (1982) Rhabdomyosarcoma of the bile ducts (in Spanish). An Esp Pediatr 17:65–70
Nagaraj HS, Kmetz DR, Leitner C (1977) Rhabdomyosarcoma of the bile ducts. J Pediatr Surg 12:1071–1074
Nemada B, Talapatra K, Shet T, Banavali S, Muckaden MA, Laskar S (2007) Embryonal rhabdomyosarcoma of the biliary tree mimicking a choledochal cyst. J Cancer Res Ther 3:40–42
Nicol K, Savell V, Moore J, Teot L, Spunt SL, Qualman S, Children’s Oncology Group, Soft Tissue Sarcoma Committee (2007) Distinguishing undifferentiated embryonal sarcoma of the liver from biliary tract rhabdomyosarcoma: a Children’s Oncology Group study. Pediatr Dev Pathol 10:89–97
Oto A, Basgün N, Kutluk T, Eryilmaz M, Oran M, Besim A (2001) Intraperitoneal involvement in rhabdomyosarcoma. CT findings in a child. Turk J Pediatr 43:342–344
Pack GT, Miller TR (1956) Total right lobectomy for rhabdomyosarcoma. AMA Arch Surg 73:1060–1062
Parham DM (2001) Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol 14:506–514
Parham DM, Barr FG (2013) Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol 20:387–397
Parham DM, Kelly DR, Donnelly WH, Douglass EC (1991) Immunohistochemical and ultrastructural spectrum of hepatic sarcomas of childhood: evidence for a common histogenesis. Mod Pathol 4:648–653
Patil KK, Omojola MF, Khurana P, Iyengar JK (1992) Embryonal rhabdomyosarcoma within a choledochal cyst. Can Assoc Radiol J 43:145–148
Perera MT, McKiernan PJ, Brundler MA, Hobin DA, Mayer DA, Mirza DF, Sharif K (2009) Embryonal rhabdomyosarcoma of the ampulla of Vater in early childhood: report of a case and review of literature. J Pediatr Surg 44:e9–e11
Perisic VN, Howard ER, Mihailovic T, Vujanic G, Milovanovic D, Ivanovsky P (1991) Cholestasis caused by biliary botryoid sarcoma. Eur J Pediatr Surg 1:242–243
Phatak AM, Prabhu SR (1982) Sarcoma botryoides of the common bile duct. A case report. Indian J Cancer 19:170–172
Pollono DG, Tomarchio S, Berghoff R, Drut R, Urrutia A, Cedola J (1998) Rhabdomyosarcoma of extrahepatic biliary tree: initial treatment with chemotherapy and conservative surgery. Med Pediatr Oncol 30:290–293
Prasad A, Chadha R, Choudhury SR (2003) Embryonal rhabdomyosarcoma of the common bile duct. Indian J Pediatr 70:91–92
Rebollo Guelar J, Lafont A, Garcia de Davila MT, Solemou V, Rose A, Bignon H (2013) Rhabdomyosarcoma of the biliary tree. Case report (in Spanish). Arch Argent Pediatr 111:e94–e96
Roebuck DJ, Stanley P (2000) External and internal-external biliary drainage in children with malignant obstructive jaundice. Pediatr Radiol 30:659–664
Roebuck DJ, Yang WT, Lam WW, Stanley P (1998) Hepatobiliary rhabdomyosarcoma in children: diagnostic radiology. Pediatr Radiol 28:101–108
Ruymann BF, Raney B, Crist W, Lawrence W, Lindberg R, Soule E, For the Intergroup Rhabdomyosarcoma Study Committee of CCSG and POG (1985) Rhabdomyosarcoma of the biliary tree in childhood. Cancer 56:575–581
Sanz N, de Mingo L, Florez F, Rollan V (1997) Rhabdomyosarcoma of the biliary tree. Pediatr Surg Int 12:200–201
Sarrazin R, Dyon JF, Brabant A, Pont J, Gout JP (1977) Botryoid sarcoma of the choledochus. Reflections apropos of a case and review of the literature (in French). Med Chir Dig 6:45–50
Sassi SH, Charfi L, Abes I, Mrad K, Dhouib R, Hamida NB, Oubiche F, Barsaoui S, Romdhane KB (2008) Cholestasis caused by a choledochal botryoid rhabdomyosarcoma in a 22-month-old boy (in French). Ann Pathol 28:45–48
Scheiden R, Kiesler J, Breitfellner G (1988) Primary embryonal rhabdomyosarcoma of the liver simulating malignant liver mesenchymoma (in German). Pathologe 9:37–41
Schmid C, Lombardi L, Giordano F, Delgrossi S (1979) Unusual morphologic features of a primary liver neoplasm. Tumori 65:649–655
Schmidt et al (1988) http://www.ncbi.nlm.nih.gov/pubmed/3354641
Schoofs G, Braeye L, Vanheste R, Verswijwel G, Debiec-Rychter M, Sciot R (2011) Hepatic rhabdomyosarcoma in an adult: a rare primary malignant liver tumor. Case report and literature review. Acta Gastroenterol Belg 74:576–581
Schweizer P, Schweizer M, Wehrmann M (1994) Major resection for embryonal rhabdomyosarcoma of the biliary tree. Pediatr Surg Int 9:268–273
Sebire NJ, Malone M (2003) Myogenin and MyoD1 expression in paediatric rhabdomyosarcomas. J Clin Pathol 56:412–416
Shabanov MA, Pokrovskaia NN, Iasonov AV (1981) Congenital hepatoblastoma with a rhabdomyoblastic component (in Russian). Arkh Patol 43:73–77
Shamis AI, Movchan VI, Lapina SP (1985) Rhabdomyoblastoma of the biliary tract in children (in Russian). Vestn Khir Im I I Grek 135:99–100
Sheehan HL (1930) An embryonic tumor of the liver containing striated muscle. J Pathol Bacteriol 33:251258
Shibata T, Hayakawa K, Yamashita K, Hamanaka D, Odori T, Ishii Y (1987) Two cases of primary hepatic rhabdomyosarcoma in adults (in Japanese). Rinsho Hoshasen 32:653–656
Shimada H, Newton WA, Soule EH, Beltangady MS, Maurer HM, For the Intergroup Rhabdomyosarcoma Study Committee (1987) Pathology of fatal rhabdomyosarcoma. Report from Intergroup Rhabdomyosarcoma Study (IRS-I and IRS-II). Cancer 59:459–465
Soper RT, Dunphy DL (1968) Sarcoma botryoides of the biliary tree. Surgery 63:1005–1011
Spunt SL, Lobe TE, Pappo AS, Parham DM, Wharam MD, Arndt C, Anderson JR, Crist WM et al (2000) Aggressive surgery is unwarranted for biliary tract rhabdomyosarcoma. J Pediatr Surg 35:309–316
Staalman CR, Umans U (1993) Hypertrophic osteoarthropathy in childhood malignancy. Med Pediatr Oncol 21:676–679
Stock N, Chibon F, Binh MB, Terrier P, Michels JJ, Valo I, Robin YM, Guillou L et al (2009) Adult-type rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am J Surg Pathol 33:1850–1859
Sullivan LM, Atkins KA, LeGallo RD (2009) PAX immunoreactivity identifies alveolar rhabdomyosarcoma. Am J Surg Pathol 33:775–780
Taira Y, Nakayama I, Moriuchi A, Takahara O, Ito T, Tsuchiya R et al (1976) Sarcoma botryoides arising from the bile duct of children. Acta Pathol Jpn 26:709–718
Taura S, Taura M, Tanaka N, Ito T, Tsuchiya R (1977) Ultrastructure of botryoid sarcoma of the common bile duct. Gastroenterol Jpn 12:305–310
Tireli GA, Sander S, Dervisoglu S, Demiralo O, Unal M (2005) Embryonal rhabdomyosarcoma of the common bile duct mimicking choledochal cyst. J Hepatobiliary Pancreat Surg 12:263–265
Tominaga J, Kawase S, Ochi T, Yoshida M, Izawa M (1995) A case of primary rhabdomyosarcoma of liver at autopsy – literature review of 7 cases reported in Japan (in Japanese). Nippon Shokakibyo Gakkai Zasshi 92:255–258
Tufail M, Rizwan MM, Majid HJ (2006) Pleomorphic rhabdomyosarcoma within a choledochal cyst; a rare cause of obstructive jaundice. J Pak Med Assoc 56:379–381
Van der Kwast TH, ten Kate FJ, Vuzevski VD, Madern GC, Terpstra OT (1992) Hepatic rhabdomyomatous tumor: late sequel of a fetal rhabdomyomatous nephroblastoma. Pediatr Pathol 12:449–456
Verstandig A, Bar-Ziv J, Abu-Dalu KI, Granot E, Schiller M (1991) Sarcoma botryoides of the common bile duct: preoperative diagnosis by coronal CT and PTC. Pediatr Radiol 21:152–153
Virenque J, Gaubert J, Bouissou H, Fabre MT (1966) A case of botryoid sarcoma of the bile ducts (in French). Ann Chir Infant 7:25–38
Von der Oelsnitz G, Spaar HJ, Lieber T, Münchow B, Booss D (1991) Embryonal rhabdomyosarcoma of the common bile duct. Eur J Pediatr Surg 1:161–165
Von Recklinghausen FD (1862) Verhandlung der Gesellschaft für Geburtshülfe in Berlin; Sitzung vom 25. März, 1862. Monatsschr. Geburtskunde 20, 1
Von Rokitansky K ( 1849) Ein aus quergestreiften Muskelfasern constituirtes Aftergewebe. Zschr Gesellsch Wiener Aerzte 331
Von Zenker FA (1864) Ueber die Veränderungen der willkürlichen Muskeln im Typhus abdominalis. Leipzig
Watanabe A, Motoi M, Mizobuchi K, Hara I, Nishimura K, Nagashima H (1983) An adult case with rhabdomyosarcoma of the liver. Jpn J Med Sci Biol 22:240–244
Watanabe K, Yasuda M, Inoue T et al (1985) A case of primary rhabdomyosarcoma of the pulmonary trunk (in Japanese). Nichikyoushitsukaishi 23:739
Werner E (1951) Primary sarcoma of the common bile duct as cause of obstructive jaundice (in German). Kinderarztl Prax 19:122–124
Wigger HJ (1976) Fetal rhabdomyomatous nephroblastoma – a variant of Wilms’ tumor. Hum Pathol 7:613–623
Wilks S, Moxon W (1875) Lectures on pathologic anatomy, 2nd edn. Brown Green, Longmans Roberts, London, pp 465–466
Williams AW (1953) Liver tumour of the mixed type. Br J Surg 41:13–15
Williams AG, Sheward SE (1986) Ultrasound appearance of biliary rhabdomyosarcoma. J Clin Ultrasound 14:63–65
Willis RA (1948) Pathology of tumours. Butterworth, London
Willis RA (1962) The pathology of tumors of children. Charles C. Thomas, Springfield, pp 67–75
Willoughby V, Sonowala A, Werland-Perurena A, Donner LR (2008) A comparative immunohistochemical analysis of small round cell tumors of childhood: utility of peripherin and alpha-internexin as markers for neuroblastomas. Appl Immunohistochem Mol Morphol 16:344–348
Witcombe JB (1979) Biliary rhabdomyosarcoma of childhood. Br J Radiol 52:1005–1006
Zampieri N, Camoglio F, Corroppolo M, Cecchetto M, Ornis S, Ottolenghi A (2006) Botryoid rhabdomyosarcoma of the biliary tract in children: a unique case report. Eur J Cancer Care (Engl) 15:463–466
Zhao M, Feng C, Wang JW, Liu Y, Tang SQ (2011) Childhood rhabdomyosarcoma: a retrospective review of 23 cases ( in Chinese ). Zhongguo Dang Dai Er Ke Za Zhi 13:657–660
Zornig C, Kremer B, Henne-Bruns D, Weh HJ, Schröder S, Brölsch CE (1992) Primary sarcoma of the liver in the adult. Report of five surgically treated patients. Hepatogastroenterology 39:319–321
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this entry
Cite this entry
Zimmermann, A. (2017). Tumors of the Striated Muscle Cell Lineage: Hepatobiliary Rhabdomyosarcoma and Rhabdomyoma. In: Tumors and Tumor-Like Lesions of the Hepatobiliary Tract. Springer, Cham. https://doi.org/10.1007/978-3-319-26956-6_64
Download citation
DOI: https://doi.org/10.1007/978-3-319-26956-6_64
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26954-2
Online ISBN: 978-3-319-26956-6
eBook Packages: MedicineReference Module Medicine