
Abstract
Invasion of various tissues and vessels depends on distinct cellular structures involved in cell adhesion, initiation of tissue invasion, and degradation of the extracellular matrix (ECM). These structures are generally summarized under the term invadosomes, which in turn comprises the two major types of these specialized structures, i.e., podosomes and invadopodia. Podosomes are matrix-degrading adhesive structures formed by certain normal cells, such as macrophages and endothelial cells, and by invading cancer cells. The generation of podosomes strongly depends on protein kinase C-related signaling pathways. Podosomes can degrade ECM components through the shedding of wide array of proteases, specifically matrix metalloproteinases (MMPs). A special type of podosomes is invadopodia, which mediates basement membrane and tissue invasion of cancer cells. These protrusions are actin-based dynamic structures or “organelles” of transformed cells that are critically required for tumor cell invasion and extravasation. Tumor cells can extend invadopodia through the endothelial lining of vessels to the extravascular space prior to extravasation and metastatic growth.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Matrix Metalloproteinases
- Gallbladder Carcinoma
- Biliary Tract Cancer
- Thrombospondin Motif
- Podosome Formation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Tissue Invasion: Invadosomes as Pacemakers of Invasion
Recent findings led to the identification of specialized cellular structures involved in adhesion, initiation of tissue invasion, and matrix degradation. These structures are generally summarized under the term invadosomes, which in turn contains the two major types of these structures, i.e., podosomes and invadopodia (Block et al. 2008). Podosomes and in particular invadosomes are characterized by the production of a wide array of enzymes that serve as histolytic factors that degrade tissues and specifically the extracellular matrix of tumors.
Podosomes
General Aspects
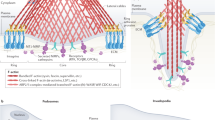
Podosomes (“feet bodies/particles”) are matrix-degrading adhesive structures formed by invading normal cells, specifically macrophages, osteoclasts, and dendritic cell (Linder and Aepfelbacher 2003; Linder and Kopp 2005; Stylli et al. 2008; Symons 2008; Gawden-Bone et al. 2010; Murphy and Courtneidge 2011; Starnes et al. 2011; Schachtner et al. 2013). Podosomes or podosome rosettes also play a significant role in endothelial cell biology where they are involved in sprouty angiogenesis (Schlaepfer et al. 2004). Podosomes are induced by factors activating protein kinase C (Tatin et al. 2006), such as diacylglycerols and phorbol esters, and can, following their complete formation and activation, degrade EMC components through the shedding of proteases (Linder 2007). Podosomes are cylindrical cell protrusions located to the outer surface of the cell membrane, measuring up to 2 μm in diameter. Both podosomes and the similar invadopodia are actin-based structures that develop from the ventral cell membrane of cells in contact with a substratum. Podosomes carry the lipid membrane of the cell surface region from which they emerge (Yamaguchi and Oikawa 2010). In locomoting cells, podosomes are situated at the front border (leading edge) between lamellipodia and the lamellum. Podosomes have a central role in migration and invasion of matrix mediated by matrix degradation and therefore are important structures in inflammation and cancer. In contrast to invadopodia, which are small structures, podosomes grow to relatively large structures. Podosomes are usually visualized by phalloidin staining, this toxic cyclopeptide binding to filamentous actin/F-actin. In phalloidin or antibody staining, podosomes appear as isolated punctate structures on the ventral surface of cells. They are often localized behind the leading edge in case of podosomes, while invadopodia are more often clustered under the nucleus. Podosomes consist of a central core structure containing F-actin and actin regulators and a peripheral ring structure (the podosome ring) showing integrins, vinculin, paxillin, and other proteins (Wernimont et al. 2008). In podosome rings, plectin, alpha-II spectrin, talin, and focal adhesion kinase are present, while these proteins are absent in invadopodia (Takkunen et al. 2010). In the course of the formation of a podosome ring, plectin accumulates at the early ring domain of the cell surface, whereby this deposition requires myosin contractility (Gad et al. 2008). In part of cells, including cancer cells, podosomes are clustered in the form of ringlike arrangements, the podosome rosettes. Rosette formation is strongly stimulated by Src kinases, protein kinase C, Rho family GTPases, ezrin, and certain integrins (review: Murphy and Courtneidge 2011). Formation of podosome rosettes is stimulated by ezrin interacting with cortactin and associated with adhesion of cells to fibronectin (Kocher et al. 2009). Furthermore, phosphorylation of moesin by Jun N-terminal kinase is important for podosome rosette formation (Pan et al. 2013a). Podosome rosette formation is stimulated by focal adhesion kinase (Pan et al. 2011) and by hepatoma-derived growth factor through the activation of the phosphatidylinositol 3-kinase/Akt pathway (Kung et al. 2012). Podosome rosette formation is suppressed by protein tyrosine phosphatase SHP2 (Pan et al. 2013b), and Src-induced rosette formation is inhibited by p53 via the upregulation of caldesmon (Mukhopadhyay et al. 2009).
Biogenesis and Morphogenesis of Podosomes
Podosomes contain a complex set of proteins making part of the actin-based engine, including actin, myosin IIA, the Arp2/3 complex, N-WASP, cortactin, gelsolin, and cofilin. For the generation of podosomes, actin cytoskeleton remodeling through Arp2/3-mediated actin polymerization and dynamic microtubules are required (Linder et al. 2000; Snyder et al. 2011; van den Dries et al. 2013). Rapid and spatially restricted remodeling of the actin cytoskeleton is a crucial step in the initiation of podosome formation. The site of first appearance is a surface microdomain where actin-containing stress fibers and focal adhesions intersect (Kaverina et al. 2003). Certain actin-associated proteins are involved in this process. A central role is played by the Arp2/3 pathway, whereby the Arp2/3 complex promotes actin assembly and competes with caldesmon in this function (Morita et al. 2007). Other actin-associated protein comprises Src-cortactin (Luxenburg et al. 2006; Clark et al. 2007), WASP (Tsuboi 2007), caldesmon, and paxillin (Badowski et al. 2008). Caldesmon has an important role in podosome biogenesis and function. Ectopic expression of caldesmon reduces the number of both podosomes and invadopodia (Yoshio et al. 2007). Furthermore, the actin-mediated morphogenesis of podosomes is modulated by integrins located at the podosome site. Integrin-linked kinase/ILK, located in an area surrounding the actin-rich podosome core, regulates podosome maturation (Griera et al. 2014). WASP (Wiskott-Aldrich syndrome protein) is an adaptor protein critical for podosome formation (Dovas et al. 2009) and operates in conjunction with the WASP-interacting protein/WIP. WIP is responsible for the stability and localization of WASP to sites of actin assembly in podosomes (Chou et al. 2006; Garcia et al. 2012) and is involved in ruffle formation (Banon-Rodriguez et al. 2013). WASP also interacts with the F-BAR domain protein, FBP17, a protein that promotes actin nucleation (Tsujita et al. 2013). Integrins can prevent the Src-induced cell rounding, but cannot impair the formation of podosomes (Huveneers et al. 2008).
It is suggested that an actin-microtubule cross talk in the cytoskeleton is critical for the morphogenesis of podosomes. There is evidence that protrusion of the podosome depends on the activity and function of the microtubule-associated kinesin motor component, KIF1C (Kopp et al. 2006). In early sites of podosome formation, vimentin deposition colocalizes at the stress fiber-focal adhesion interface, and plectin colocalizes with vimentin. Plectin is an important protein in cytoskeleton organization (Wiche 1998). Apart from well-known members of the actin assembly engine, podosomes also contain other proteins, such as the cytoplasmic scaffold protein Tks5 (synonym: Fish), which accumulates in podosomes under stimulation by Src. The presence of Trk5 in podosomes is required for cell invasion (Courtneidge et al. 2005). In the process of podosome formation, Tks5 recruits AFAP-110, p190RhoGAP, and cortactin, essential for podosome morphogenesis (Crimaldi et al. 2009). The early morphogenesis of podosomes is preceded by the formation of cell surface ruffles at the dorsal surface of cells. Ruffle formation is associated with morphogenesis of podosomes and depends on an interaction between palladin and the receptor tyrosine kinase, Eps8, this interaction promoting assembly of the actin cytoskeleton (Goicoechea et al. 2006). Palladin is a protein that promotes podosome formation and assembly (Goicoechea et al. 2009), but it also regulates cell and ECM interactions via maintenance of a normal actin cytoskeleton architecture (Liu et al. 2007). Podosome size and number are regulated in macrophages by the Rho GTPase effector PAK4, a member of the p21-associated kinase family, and its regulator alphaPIX (PAK-interacting exchange factor), two factors which induce highly localized changes in actin dynamics (Gringel et al. 2006).
Podosome Functions
Podosomes are dynamic mechanosensors that sense torsional tractions underneath the podosome rings, whereby interactions between myosin tension and actin dynamics are essential for podosome regulation (Collin et al. 2008). Podosomes play an important role in the degradation of extracellular proteins. Podosomes express sets of proteases that hydrolyze several proteins and glycoproteins of the ECM. In dendritic cells, protrusive podosomes employ MMP-14 for matrix degradation and endocytosis (Gawden-Bone et al. 2010). Fully developed podosomes operate within a network of stimulatory and inhibiting factors. Podosome-mediated degradation of basement membrane collagen is induced by TGF-beta (Rottiers et al. 2009). Other proteins known to modulate podosome function are the Cdc42 regulators. Cdc42 GTPase-activating protein/CDC42GAP regulates podosome-associated cell motility through mediation of extracellular signal-related kinase/ERK activity (Szczur et al. 2006).
Invadopodia
General Aspects
There are several structures at the cell surface that are specialized in creating distinct intercellular and cell-matrix adhesions. A long-known example is the focal adhesion, which contains clusters of transmembrane integrin receptors which are tethered at one end to the ECM and at the other end to stress fibers of the actin network. A second adhesion structure which is critically involved in cancer invasion is the invadopodia.
Selected References
(Gimona and Buccione 2006; Weaver 2006; Yamaguchi et al. 2006; Gimona et al. 2008; Stylli et al. 2008; Albiges-Rizo et al. 2009; Buccione et al. 2009; Saltel et al. 2011; Bravo-Cordero et al. 2012; Klemke 2012; Sibony-Benyamini and Gil-Henn 2012; Yamaguchi 2012; Génot and Gligorijevic 2014; Paz et al. 2014).
Invadopodia mediate basement membrane and tissue invasion of cancer cells and are characterized by the integration of several cytoskeletal processes that involve modulation of contractile forces and mechanotransduction (Lohmer et al. 2014; Tokui et al. 2014). Invadopodia are actin-based protrusions or “organelles” of transformed cells and tumor cells that form distinct cell-matrix adhesion sites. Invadosomes are critically required for cancer cell extravasation. In the course of intravascular spread of tumor cells, these cells can, after homing to distant endothelial surfaces, extend invadopodia through endothelium into the extravascular space prior to their extravasation and metastatic growth (Leong et al. 2014).
Invadopodia share several features with podosomes, but are morphologically different from podosomes, as they have a different structure, occur in larger numbers, and are more active in their capability to degrade matrix (Tolde et al. 2010; Artym et al. 2011). Adhesion rings surround invadopodia, a feature shared with podosomes (Branch et al. 2012). Typical invadopodia are smaller than podosomes and occur in larger numbers per cell. They measure from 0.1 to 0.8 μm in diameter and can reach a length of 2 μm or more. Invadopodia can form clusters around membrane invaginations close to the site of the Golgi complex.
Molecular Composition and Biogenesis of Invadopodia
Similar to podosomes, invadopodia possess a complex microfilament machinery regulating F/G-actin switching mediated by a now impressive number of actin-associated proteins, including N-WASP, cortactin, tensin, formins, Src kinases, the Arp2/3 complex, paxillin, cofilin, and gelsolin (Gimona 2008; Albiges-Rizo et al. 2009). Invadopodia are constructed of an N-WASP-dependent branched actin network and a Rho GTPase Cdc42-based pathway involved in invadopodial-membrane protrusion (Albiges-Rizo et al. 2009). The actin network of invadopodia, which contains cortactin, is regulated by a multimolecular complex containing Src kinase, the formin mDia1, actin, and Spire1, a protein which serves as the connection to Rab3A GTPase (Weaver 2008; Lagal et al. 2014). As an important component of the invadopodial machine, myosin 1e is located to the invadopodial core where it may act as a scaffold, linking the plasma membrane to the actin cytoskeleton (Oudekirk and Krendel 2014). In their interaction with the extracellular matrix, invadopodia are potently induced by dense fibrillar collagen via a kindlin2 serine phosphorylation signaling pathway (Artym et al. 2015). Furthermore, invadopodia are equipped with a complex set of regulatory and signaling proteins (Table 1). In particular, pathways leading to invadopodia formation are linked with a signaling cascade that also critically affects cell proliferation and differentiation, Wnt/beta-catenin signaling. Specifically, the Wnt5a-Ror2 axis is involved in enhanced formation of invadopodia (Endo et al. 2015). Biogenesis of invadopodia is strongly induced by Cdc42, while the adaptor protein is required for the degradative activity of invadosomes (Di Martino et al. 2014). A further protein which markedly promotes formation of invadopodia is the cytoskeletal protein WAVE3, which induces cancer cell invasion and metastasis via induction of MMP-9 and other MMPs (Davuluri et al. 2014). Invadopodia biogenesis depends on diaphanous-related formins (Lizarraga et al. 2009). Paxillin, which plays a role in several steps of invadopodia biogenesis and function, is a focal adhesion-associated, phosphotyrosine-containing protein that possesses several domains for protein-protein interactions. These domains are the necessary docking site for several cytoskeletal proteins, including vinculin and actopaxin, tyrosine kinases and serine/threonine kinases, and GTPase-activating proteins and numerous adaptor proteins (Turner 2000; Schaller 2001; Pignatelli et al. 2012b). Paxillin is associated with the paxillin kinase linker/PKL, a protein regulating directed cell migration (Yu et al. 2009). At sites of ECM degradation, an invasion-related complex constructed of paxillin, cortactin, and protein kinase Cmu associated with invadopodia (Bowden et al. 1999). For the function of the actin-based network, the atypical GTP-binding protein dynamin is required, dynamin being a central modulator of cellular protrusive events (McNiven et al. 2004). In the protruding edge of invadopodia and similar structures, actin-associated proteins are accumulated. The interaction of these proteins is coordinated by a member of the F-BAR family of proteins, CIP4/Cdc42-interacting protein 4, in a Rac1/WAVE1-dependent manner (Saengsawang et al. 2013).
Induction and Regulation of Invadopodia in Cancer Cells
In cancer cells, numerous factors can elicit the biogenesis of invadopodia. A major role is played by a complex interaction of cancer cells with the extracellular matrix (ECM) located to tumor stroma. Invadopodia are strongly induced upon contact of tumor cells with distinct domains of the ECM (review: Hoshino et al. 2013a). This contact promotes the activity of several signaling pathways that induce actin cytoskeleton rearrangement, the recruiting of actin-associated proteins, and the generation of cell protrusions (Destaing et al. 2011; Linder et al. 2011). Similar to lamellipodia and related cell protrusions, formation of invadopodia depends on interactions between focal adhesions and the actin cytoskeleton. Via focal adhesions and integrin receptors, invadopodia interact with proteins in the extracellular matrix/ECM. Among ECM proteoglycans and proteins, hyaluronan interacting proteins RHAMM play a role in invadopodia induction (Gurksy et al. 2012). Generally, adhesion signaling has a central role in invadopodia formation, with the critical involvement of protein kinase C and Src kinases (Destaing et al. 2011). Src signaling to induced invadopodia is a regulated protein involved in cell migration and invasion, TM4SF5/transmembrane 4 L six family member 5 (Jung et al. 2013). Src kinase associates with diaphanous formin-1, actin, and Spire-1 in a complex that contributes to invadopodia biogenesis (Lagal et al. 2014). Invadopodia formation is stimulated by proteins that induce epithelial-mesenchymal transition/EMT, e.g., Twist1 (Eckert et al. 2011) and the focal adhesion protein Hic-5, a paxillin homologue (Pignatelli et al. 2012a), linking EMT with mechanisms that determine tissue invasion. Invadopodia are induced by Src-mediated phosphorylation of ASAP1 (Bharti et al. 2007) and by the activation of Cdc42, e.g., by the stromal cell-derived protein palladin (Goicoechea et al. 2014). Src kinases orchestrate several steps of invadopodia biogenesis in a distinct spatiotemporal pattern (Boateng and Huttenlocher 2012). The latter mechanism illustrates the close interaction between tumor cell and matrix for the induction of invadosomes. Invadopodia are induced by a key regulator of the F/G-actin switch mechanism, Abl interactor 1/Abi 1, which affects invadopodia formation in an Src-dependent manner (Sun et al. 2009). Formation of invadopodia depends on the activity of GTPases, e.g., RhoGTPases, which provide signals to the cytoskeleton through small G proteins of the Rho family (Spuul et al. 2014). Invadopodia contain the Arf GTPase-activating protein ASAP1, whereas ASAP3 is not expressed (Ha et al. 2008). The contact between cancer cells and ECM also involves a complex mode of pH sensing. Several pH regulators such as V-ATPases and Na(+)/H(+) exchangers are expressed in invadosomes, and these sensors/regulators control the acid microenvironment to provide a milieu for appropriate activation of MMPs and other degradases (Brisson et al. 2012).
A second major pathway that induces invadopodia in cancer cells is epithelial-mesenchymal transition (EMT). Formation of invadopodia is stimulated by EMT taking place upon contact of tumor cells with the extracellular matrix. Apart from its central role in EMT induction, the transcription factor Twist1 is capable to induce invadopodia formation, probably via its transcriptional target, PDGFRalpha (Eckert et al. 2011). Also the EMT-inducing factor TGF-beta induces invadopodia formation via ectopic expression of the focal adhesion protein, Hic-5 (Pignatelli et al. 2012a). In the setting of EMT, podosome-like structures occurring in noninvasive carcinoma cells switch to actin comet-embedded invadopodia containing MMP-1. In the course of this transition, the podosomes become smaller and achieve the shape of numerous invadopodia, structures in which talin is replaced by tensin. Therefore, EMT can induce the production of the potent degrading engine required for cancer invasion (Takkunen et al. 2010). Invadopodia are also induced by a factor involved in the regulation of EMT, CD147/basigin (Grass et al. 2012).
Invadopodia maintenance and function are regulated by integrins, Src tyrosine kinase signaling, cortactin, AMAP/ASAP1, the adaptor protein Tks5/Fish, receptor tyrosine kinases, Rho family GTPases, ARF6, and dynamin 2. Beta1 integrins are required for the formation of competent invadopodia, whereby beta1 integrin interacts with the tyrosine kinase Arg and stimulates Arg-dependent phosphorylation of cortactin (Beaty et al. 2013). Beta1A integrin is a master regulator of podosome and invadopodia organization and function (review: Destaing et al. 2010). ASAP1 is an Arf GTPase-activating protein/GAP containing a BAR domain and is a substrate for Src kinase.
Tissue and Matrix Destruction (Histolysis) as a Major Mechanism of Cancer Invasion
Introduction
Both invadopodia and lamellipodia engage in contacts with the underlying extracellular matrix (ECM) and are then stimulated to secrete matrix metalloproteinases (MMPs; see below; Coussens and Werb 1996; Stamenkovic 2000; Linder 2007; Linder et al. 2011; van Horssen et al. 2013). The contact between invadopodia and ECM is a complex process that involves signals from matrix rigidity and myosin II-FAK/Cas pathways (Alexander et al. 2008). Typical MMPs produced by invadopodia are MMP-2 and MMP-9 and in part of neoplasms also MT1-MMP (Watanabe et al. 2013). Expression and secretion of these enzymes causes pericellular proteolysis in cancer (Sevenich and Joyce 2014). Invadopodia express latent MMP-2 and membrane type 1 MPP at the cell surface, where they are activated (Chen and Wang 1999). ECM degradation by invadopodia is regulated by the action of the phosphoinositide-binding protein, ZF21, a protein that promotes cancer cell migration and invasion (Hoshino et al. 2013b). The function of invadopodia also requires several types of ADAMs, which interact with integrins in invadopodia and cooperate with certain invadopodial adaptor proteins, such as Tks5/Fish. Physiological type I collagen can induce a special type of invadosomes, the linear invadosomes. Linear invadosomes, cortactin- and N-WASP-containing protrusions that can degrade matrix, result through replacement of podosomes or invadopodia upon contact with type I collagen present in collagen-rich ECMs (Juin et al. 2012).
Matrix Metalloproteinases (MMPs): Types and Classification
Matrix metalloproteinases (MMPs) are enzymes that are implicated in remodeling of the extracellular matrix (EMC), including the basement membranes (Deryugina and Quigley 2006; Friedl and Wolf 2009; Shiomi et al. 2010). MMPs are classified as two main types of proteases, i.e., those that are anchored to the cell surface membrane and a second group that is secreted into the extracellular space (Table 2).
General Roles and Regulation of MMPs in Liver Cancer
Generally, MMPs play pleiotropic roles in cancer, exceeding aspects of mere “degradomics” but also including effects on cell adhesion, growth signaling, and other pathways (Kleiner and Stetler-Stevenson 1999; Overall and Dean 2006). The activity of MMPs is regulated and controlled at several levels, such as enzyme activation or inhibition at the cell surface, complex formation, and compartmentalization in various subcellular compartments. Within normal and cancer cells, MMPs can be localized at the cell surface, in the cytosol, and in organelles and the nucleus. The role of intracellular MMPs has not yet been clarified in detail (review: Mannello and Medda 2012). Downregulation of the Notch signaling pathway impairs HCC cell migration (Zhou et al. 2013b) and inhibits HCC cell invasion by inactivating MMP-2 and MMP-9 expression via the extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling pathways (Zhou et al. 2012), while high levels of Notch1 augment MMP-2 and MMP-9 and promote a highly invasive HCC phenotype. Conversely, sonic hedgehog, a ligand of hedgehog, is frequently expressed in HCC cells and decreases the expression of MMP-9 via the ERK pathway (Lu et al. 2012). High Notch1 expression in HCCs was correlated with tumor size, tumor grade, venous invasion, and the metastatic state (Zhou et al. 2013a). In its inhibitory action on migration and invasion, Notch1 operates by regulating CD44v6, E-cadherin, MMPs, and uPA via the COX-2 and ERK1/2 pathways (Zhou et al. 2013). Downregulation of Notch1 inhibits HCC cell invasion by inactivating the COX2/Snail/E-cadherin pathway involved in EMT (Zhou et al. 2013c). Secretion of multiple MMPs is promoted by HGF via the transcription factor Ets-1 which activates MMP transcription (Ozaki et al. 2003). Secretion of MMPs by stromal cells is stimulated by CD147 (EMMPRIN; basigin), a cell surface glycoprotein that belongs to the immunoglobulin superfamily and is strongly expressed on the surface of many tumor cell types. Basigin exists in several isoforms with differential functions. Basigin stimulates the secretion of VEGF and hyaluronan, promoting angiogenesis and anchorage-independent growth (Nabeshima et al. 2006). These effects are abolished by depletion of CD147, mainly downregulating MMP-11 and VEGF (Jia et al. 2007). CD147 exerts its pro-metastatic effects also in HCCs (Xu et al. 2007; Zhang et al. 2007c; Jia et al. 2008). In HCC cells, downregulation of CD147 inhibits gelatinase production, interferes with tumor cell adhesion to type IV collagen, and alters cytoskeletal structure (Qian et al. 2008). One basigin isoform, basigin-3, inhibited proliferation of HCC cells and MMP induction (Liao et al. 2011). CD147 interacts with integrin alpha6beta1, stimulating invasion and MMP secretion in HCC cells (Dai et al. 2009). A further protein with which CD147 interacts is annexin, a Ca(2+)- and phospholipid-binding protein subject to phosphorylation by tyrosine kinases and protein kinase C. This interaction promotes migration and MMP-mediated invasion of HCC cells. The pro-invasive and pro-metastatic effect of basigin/CD147 is counteracted by the basigin isoform, basigin-3, in HCC (Liao et al. 2011).
Expression of the Diverse Metalloproteinases in Liver Cancer
Several members of the large MMP family are expressed or overexpressed in HCCs and cholangiocarcinomas and affect the development of a metastatic phenotype (Yamamoto et al. 1999; Bodey et al. 2000; Giannelli et al. 2002; McKenna et al. 2002; Ishii et al. 2003; Ozaki et al. 2003; Matsunaga et al. 2004; Okamoto et al. 2005; Gao et al. 2006; Altadill et al. 2009). Generally, expression of MMPs in cancer cells is augmented by EMT. The EMT factor, Twist1, activates MMPs and by this induces tumor cell invasion (Zhao et al. 2011). Snail, an inducer of EMT, accelerates cancer invasion by an EMT-associated upregulation of MMPs (Miyoshi et al. 2004, 2005). Expression of MMPs increases as a function of increasing stage (Gao et al. 2006). Secretion of MMPs by stromal myofibroblasts is suppressed by TGF-beta1, while the hormone relaxin binding to its cognate receptor, relaxin family peptide receptor 1, upregulates MMPs (Chow et al. 2012).
MMP-1
MMP-1 is involved in both tumor cell invasion and the generation of metastases, also in HCCs (Ogasawara et al. 2005). MMP-1 proteolytically activates protease-activated receptor-1 (PAR-1). In HCCs, the MMP-1/PAR-1 signaling axis is strongly activated and associated with tumor invasion and progression (Liao et al. 2011, 2012). Increased expression of MMP-1 and MMP-2 in HCCs is correlated with tumor differentiation (Ogata et al. 1999). However, other studies arrived at different results. For example, high levels of MMP-1 transcripts were detected in well-differentiated cancer cells of early HCCs, but not in moderately to poorly differentiated HCCs, suggesting an MMP-1-mediated role in early invasive processes, such as destruction of portal tracts (Okazaki et al. 1997).
MMP-2
Expression of MMP-2 is involved in HCC invasion, and its activity is influenced by several genetic variants that modulate the enzyme’s effects in the invasive pathway (Wu et al. 2008). Expression of MMP-2 in HCCs is associated with dedifferentiation in HCC (Ogata et al. 1999) and was correlated with intra- and extrahepatic metastases (Liu et al. 2003). Expression of MMP-2 predicted lymph node metastasis in HCCs (Xiang et al. 2011). The secretion of MMP-2 is stimulated by the transcription factor, Snail, which is expressed in HCC cells and mediates EMT. On the other hand, downregulation of Snail in HCC cells is associated with increased expression of the adhesion molecule, E-cadherin, and downregulation of MMP-2 (Chen et al. 2012a). MMP-2 is also upregulated by osteopontin (Chen et al. 2011a,b), through the SDF-1/CXC4 axis (Zhang et al. 2011), via Notch signaling (Zhou et al. 2012), by secreted clusterin (Chen et al. 2012a), Rock2 (Huang et al. 2014), and by sonic hedgehog signaling through focal adhesion kinase/AKT signaling (Chen et al. 2013a). Osteopontin itself is downregulated by microRNA-181a (Bhattacharyya et al. 2010). The activity of MMP-2 in HCC is strongly controlled by TIMP-2 (Musso et al. 1997; Giannelli et al. 2002). MMP-2 expression is downregulated by microRNA-29b, a microRNA that suppresses angiogenesis, invasion, and metastasis (Fang et al. 2011). As with other MMPs, MMP-2 is regulated by tissue inhibitor of metalloproteinase-2/TIMP-2. Strong expression of TIMP-2 in hilar cholangiocarcinomas is associated with inhibition of cancer invasion and metastasis (Xiao et al. 2004). MMP-2 is also expressed in subsets of gallbladder carcinoma (Karadag et al. 2008).
MMP-3
Expression of MMP-3 is associated with the prognosis of HCV-related HCCs (Okamoto et al. 2010). In HGF-induced invasion of HCC cells, MMP-3 is involved, in that HGF stimulates the secretion of pro-MMP-3 (Monvoisin et al. 2002). Upregulation of MMP-3 in hepatoma cells is associated with a downstream mediation of autocrine motility factor (Yu et al. 2004). MMP-3 is also induced by HBV X protein, which is a known promoter of cell migration (Yu et al. 2005). The pro-metastatic activity of MMP-3 depends on its genetic polymorphism, in that the MMP-3 5A allele was the most prominent MMP-3 enzyme to promote invasion (Okamoto et al. 2010). Expression of MMP-3 in HCC cells is reduced by microRNA-30a-3p (Wang et al. 2014).
MMP-7
Similar to other MMPs, MMP-7 has recently been demonstrated to act in the invasion cascade, also in metastatic HCCs (Gao et al. 2006). In particular, MMP-7 expression is a prognostic factor in cholangiocarcinoma, whereby cholangiocarcinoma cells show strong immunoreactivity for this enzyme (Miwa et al. 2002). Expression of MMP-7 is associated with poor prognosis in patients with intrahepatic cholangiocarcinoma (Hirashita et al. 2012) and is an unfavorable postoperative prognostic factor in perihilar, hilar, and extrahepatic cholangiocarcinomas (Itatsu et al. 2008), and serum MMP-7 seems to be a valuable diagnostic marker in discrimination of cholangiocarcinomas from reactive biliary pathologies (Leelawat et al. 2009). One non-synonymous variant of MMP-7 was found to confer risk of liver cirrhosis in patients with HCC (Hung et al. 2009). MMP-7 engages in a signaling pathway together with beta-catenin. Upregulation of MMP-7 expression by beta-catenin is promoted by DKK1, which is overexpressed in HCCs (Chen et al. 2013b). Similar to sclerostin, DKK1 (Dickkopf-related protein 1) is an antagonist of the Wnt/beta-catenin signaling cascade. DKK1 promotes invasion and metastasis in HCCs, associated with increased RhoA and JNK levels (Tao et al. 2013). In cholangiocarcinomas, DKK1 is related to lymphatic metastasis and an aggressive course (Shi et al. 2013). This phenomenon may be caused by the induction of an invasive phenotype by DKK1. MMP-7 expression is downregulated in HCCs by the tumor suppressor, fibulin-5 (Tu et al. 2014).
MMP-9
MMP-9 has an important role in HCC invasion (Arii et al. 1996; Sun et al. 2005; Nart et al. 2010; Tao et al. 2010; Yeh et al. 2010; Thieringer et al. 2012) and is also involved in invasion of hilar cholangiocarcinoma (Li et al. 2005). Upregulation of MMP-9 is found in a majority of HCCs and is subject to MMP-9 gene polymorphism (El-Samanoudy et al. 2014). Overexpression of MMP-9 mRNA may be associated with the progression of small HCCs with a diameter of <=2 cm (Sakamoto et al. 2000). MMP-9 expression is a significant predictor of recurrence after liver transplantation in HCC patients (Zhang et al. 2006). The MMP family members, MMP-2 and MMP-9, are critical for the invasive potential of numerous malignancies, including HCC. Overexpression of MMP-9 in HCCs is correlated with growth and invasion in small tumors already (Sakamoto et al. 2000) and is associated with macrovessel invasion in larger tumors (Nart et al. 2010). High expression of MMP-9 was associated with both time to recurrence and overall survival, while high expression of MMP-2 was only correlated with time to recurrence (Chen et al. 2012b). MMP-9 cleaves osteopontin to produce a fragment that is essential for osteopontin-induced invasion of HCC cells (Takafuji et al. 2007). MMP-9 is also expressed in tumor-associated macrophages (TAMs) located at the invasive front of murine HCC (Roderfeld et al. 2010). MMP-9 cooperates with focal adhesion kinase (FAK), whereby this mechanism that is active in HCC cell migration is stimulated by activated hepatic stellate cells (Han et al. 2014). In addition to its role as mediators of histolysis, MMPs may cause other biologic effects, e.g., angiogenesis in cancers (Bergers et al. 2000). In mice, hepatocyte-specific expression of MMP-9 promoted liver tumor development (Thieringer et al. 2012).
The induction and localization of MMP-9 are regulated by several factors. MMP-9 signaling in HCC is promoted by activated hepatic stellate cells (Han et al. 2014), which occur in tumor stroma. MMP-9 activity for mediating invasion is regulated by the calcium-binding protein S100A4 (Zhang et al. 2013) and is strongly regulated by protein kinase C-dependent NF-kappaB activation in HCC cells (Hah and Lee 2003). Trafficking of MMP-9 in invadopodia is regulated by Rab GTPases, specifically Rab40b (Jacob et al. 2013). Induction of MMP-9 expression in HCC is stimulated by interleukin 23 through NF-kappaB induction (Li et al. 2012). Expression of MMP-9 is promoted by the PRL (phosphatase of regenerating liver) phosphatase, a group of distinct protein tyrosine phosphatases. Activation of MMPs is mediated by Twist-induced EMT, whereby specifically MMP-2 and MMP-9 are secreted (Zhao et al. 2011). The activity of MMP-9 and MMP-2 is attenuated by the protein, RECK (reversion-inducing cysteine-rich protein with Kazal motifs), an important regulated metalloproteinase. Downregulating of RECK in cholangiocarcinomas is associated with enhanced MMP-2/MMP-9 activity and a metastatic phenotype (Namwat et al. 2011). HBV viral X protein induces MMP-9 gene expression via activation of ERK and PI-3K/AKT pathway activation (Chung et al. 2004), whereas hepatitis C virus NS3 protein enhances cancer cell invasion by activating MMP-9 activity (Lu et al. 2015). MicroRNA-133a inhibits HCC cell migration and invasion by targeting MMP-9 (Chen et al. 2015). The expression of MMP-9 is inhibited by microRNA-491, which also blocks EMT in HCC (Zhou et al. 2013d). MMP-9 is upregulated in other hepatic and biliary tract cancers, including cholangiocarcinoma. In cholangiocarcinomas, MMP-9 expression is enhanced by downregulation of microRNA-138 (Wang et al. 2013b). MMP-9 expression is enhanced in gallbladder carcinoma (Karadag et al. 2008).
MMP-10
Strong immunoreactivity for MMP-10 was found in HCCs, especially in the extracellular matrix adjacent to blood vessels (Bodey et al. 2000). MMP-10 can be activated by C-terminal-truncated HBV X protein, a process stimulating HCC invasion (Sze et al. 2013).
MMP-11
MMP-11 is strongly expressed in both tumor cells and stromal cells surrounding cancer. There is evidence that MMP-11 is a tumor lymphatic metastasis-associated MMP (Jia et al. 2007). MMP-11 is downregulated by CD147 (basigin), affecting the lymphatic metastasis pattern of HCC in a mouse model (Jia et al. 2007). Expression and secretion of MMP-11, together with VEGF, is inhibited in HCCs by ectopic expression of microRNA-125a (Bi et al. 2012).
MMP-12
Overexpression of MMP-12 was found in 58 % of human HCCs, and its expression was significantly correlated with an invasive phenotype (in particular venous invasion) and poor prognosis in HCC (Ng et al. 2011).
MMP-14
Membrane type 1 MPP (MT1-MMP, MMP-14) promotes invasiveness in HCC cell lines (Murakami et al. 1999), is overexpressed in highly invasive HCCs (Harada et al. 1998), and induces metastases in HCC, but MMP-14 gene polymorphisms also contribute to HCC susceptibility (Chen et al. 2011c). Increased MMP-14 mRNA expression by tumor cells in HCCs may have prognostic significance (Määtä et al. 2000). An aggressive phenotype is observed in HCCs that show atypical localization of MT1-MMP in the tumor cell nuclei (Ip et al. 2007). By its enzymatic activity, it not only degrades extracellular matrix but it also induces a signaling pathway stimulating cell adhesion and proliferation (Ip et al. 2005). MT1-MMP confers a proteolytic activity to invadopodia (see above), particularly upon tumor cell contact with ECM. This activity is regulated by the v-SNARE TI-VAMP/VAMP7 (Steffen et al. 2008). The cell surface level and endocytosis of MMP-14 are also regulated by the planar cell polarity-associated protein, VANGL2, a protein that also induced invadopodium formation (Williams et al. 2012). In tumor cells, endocytosis of MT1-MPP by clathrin- and caveolae-dependent pathways is counteracted by mechanisms stabilizing the enzyme at the cell surface (Poincloux et al. 2009). MT1-MPP is targeted by microRNA-150-5p which inhibits HCC migration and invasion. In metastasizing liver cancers, expression of this MiR is reduced (Li et al. 2014). MT1-MMP can be upregulated by HBV X protein (Ou et al. 2007). Similar to MT1-MMP, also MT3-MMP expression was associated with an invasive phenotype in HCCs (Arai et al. 2007).
Tissue Inhibitors of Metalloproteinases (TIMPs) in Liver Cancer
Tissue inhibitors of metalloproteinases (TIMPs; four members, TIMP-1, TIMP-2, TIMP-3, and TIMP-4) are important regulator proteins that interfere with the activity of certain MMPs. TIMP-1 inhibits MMP-9, while TIMP-2 inhibits MMP-2. Both TIMPs were detected in HCC tumor cells, stromal cells, and endothelial cells, whereby expression signals were stronger for TIMP-1 than for TIMP-2 (Joo et al. 2000; Matsumoto et al. 2004), which is of special interest in the light of the MMP expression patterns in HCCs. In one study, TIMP was preferentially expressed in the capsule of HCCs (Fukuda et al. 1991). Metastatic HCCs showed lower levels of TIMPs (Gao et al. 2006), and HCC tissue expression levels of TIMP-2 were higher in patients without metastases (Giannelli et al. 2002). However, TIMP-1 can create a premetastatic niche in the murine liver through stromal cell-derived factor 1/CXCR4-dependent neutrophil recruitment (Seubert et al. 2015). Expression of TIMPs is heterogeneous and varies considerably among HCC nodules, but there was no clear correlation between the TIMP expression levels, differentiation grade, and invasion, although TIMP expression was also detectable in HCC metastases (Nakatsukasa et al. 1996). In cholangiocarcinoma, expression of TIMP-1 was strong and was associated with the extent of invasion (Nakatsukasa et al. 1996). In cultured cells, expression of TIMP-1 was negatively regulated by the EMT effector, Twist1 (Okamura et al. 2009). In addition to TIMP-1 and TIMP-2, TIMP-3 is involved in the regulation of cancer invasion. Transfection of TIMP-3 in HCC cell lines suppresses invasive capacity (Zhang et al. 2007b). In HBV-associated HCC, TIMP3 can be epigenetically silenced (Lai and Lo 2005).
There are protease inhibitors different from TIMPs. The serine protease inhibitor, clade B, member 1/SERPINB1, is a member of the SERPINB family involved in suppression of migration and invasion in various cancers. SERPINB1 expression in HCCs is correlated with tumor invasion and an aggressive course (Cui et al. 2014).
Metzincin Superfamily Members: ADAMs
A disintegrin and metalloproteinases (ADAMs) or MDCs (metalloprotease-like, disintegrin-like, cysteine-rich proteins) are membrane-bound metzincins and the adamalysin subfamily. Twenty-two members of ADAM family are known (reviews: Porter et al. 2005; Edwards et al. 2008). ADAMs possess a domain structure similar to that of MMPs, but in contrast to the latter, ADAMs display a cysteine-rich domain, an epidermal growth factor-like repeat domain, and the disintegrin domain, but lack the hemopexin-like domain of MMPs (review: Klein and Bischoff 2011). The main functional activity of ADAM metalloproteases is ectodomain shedding of various receptors, growth factors, Notch, cytokines, and other signaling substances involved in cellular homeostasis (“ADAM sheddases”). For example, ADAM17 is critically involved in post-shedding activation of pro-TNF-alpha and is therefore also termed tumor necrosis factor-alpha convertase or TACE. For several cell membrane signaling proteins, cleave by ADAM sheddases prepares what is called regulated intramembrane proteolysis or RIP, resulting in intracellular domains that can be translocated to the nucleus for the regulation of gene transcription (Edwards et al. 2008).
Various malignant neoplasms express several species of ADAMs, particularly ADAM8, ADAM9, ADAM10, ADAM12, ADAM15, ADAM17, ADAM19, and ADAM28 (review: Mochizuki and Okada 2007; Duffy et al. 2009). In cancer invasion and spread, the critical function of ADAMs is the metalloproteinase activity of these enzymes (Rocks et al. 2008). Similar to MMPs, the metalloprotease domain is shielded by a pro-domain that involves the cysteine switch mechanism. For activation of the protease, this protecting shield is removed in the trans-Golgi network by the action of proprotein convertases, such as furin (Klein and Bischoff 2011). ADAM8 is overexpressed in HCCs in comparison with normal liver. Expression levels were positively correlated with tumor size, grade, recurrence, and metastasis (Zhang et al. 2013). Downregulation of ADAM10 expression inhibits invasiveness and metastasis of human HCC cells, associated with increased E-cadherin protein levels and a related change in cell migration (Yue et al. 2013). ADAM12 mediates the shedding of the epidermal growth factor receptor (EGFR) ligand, heparin-binding EGF-like growth factor/HB-EGF, in a Notch-dependent manner. Released EB-EGF induces the formation of invadopodia under hypoxic conditions (Diaz et al. 2013). ADAM17 is involved in the cleavage of the ectodomain of numerous transmembrane proteins. ADAM17 is overexpressed in HCC under hypoxic conditions and enhances the phosphorylation of the EGFR (Wang et al. 2013a). ADAM17 is targeted by microRNA-145 which suppresses invasion of HCC cells (Yang et al. 2014). In HCCs, ADAM17 is also regulated by microRNA-122, a tumor suppressor microRNA (Tsai et al. 2009). ADAM28 is implicated in tumor growth and progression in several human cancers (Mochizuki and Okada 2009). It operates through cleavage of the proapoptotic von Willebrand factor. Tumor cells with low expression levels of ADAM28 display a higher rate of apoptosis (Mochizuki et al. 2012).
ADAMTS and ADAMTS-Like Proteins
ADAMTS (a disintegrin and metalloprotease with thrombospondin motifs) are a family of extracellular proteases that is distinguished from ADAM metalloproteases by the presence of multiple copies of thrombospondin 1-like repeats. ADAMTS bind to and process various components of ECM proteins and proteoglycans, including heparin; procollagens I, II, and II; aggrecan; brevican; versican; decorin; fibronectin; and von Willebrand factor. ADAMTS are involved in the regulation of critically important processes such as morphogenesis, angiogenesis, and carcinogenesis. For example, of the at least 20 members in this family, ADAMTS2 is a procollagen N-proteinase; ADAMTS4 and ADAMTS5 are aggrecanases active in the hydrolysis of cartilage aggrecan (review: Tang 2001). Overexpression of ADAMTS1 results in shedding of semaphorin 3C from the ECM, a step that stimulates cell migration (Esselens et al. 2010). The seven ADAMTS-like proteins are proteins that lack proteolytic activity but possess ADAMTS ancillary domains. The proteins have important regulatory roles in the ECM (review: Apte 2009).
Other Proteases Involved in Cancer Invasion and Progression
In invadopodia, few other proteolytic enzymes are expressed, including serine proteinase/seprase, aspartate proteases, threonine proteases, dipeptidyl dipeptidase, and cysteine cathepsins (Jevnikar et al. 2012; Rakashanda et al. 2012). A group of calcium-dependent cysteine proteases is the calpains, enzymes involved in several homeostatic mechanisms. Among the various calpain members, m- and m-calpain are involved in regulating cell migration and invasion. In comparison with normal hepatocytes, these two calpains are elevated in HCC cells and regulate HCC cell migration and invasion (Chen et al. 2013c).
Several types of enzymes active on matrix proteins are produced and secreted by leukocytes infiltrating the tumor stroma. Macrophages express a distinct metalloelastase, a member of the MMP family. Expression of metalloelastase by tumor-associated macrophages in HCC is correlated with angiostatin generation and survival, in that tumors having no metalloelastase expression and hence a reduced angiostatin showed poorer survival (Gorrin Rivas et al. 1998). Human neutrophils produce an elastase that can degrade TIMP-1, a process accelerated by heparin (Nunes et al. 2011).
HCCs can express and secrete various ectopeptidases, including neprilysin (CD10), aminopeptidase N (CD13), and angiotensin-I-converting enzyme (CD143), whereby this expression differs considerably between normal liver and HCCs. In HCCs, neprilysin immunoreactivity is found on canalicular domains, a feature with diagnostic significance (Borscheri et al. 2001). In comparison with normal parenchyma, CD10 is low in HCCs, while CD13 and CD143 were mildly increased. CD13 may be involved in the regulation of cell polarity, whereas CD143 may influence angiogenesis (Rocken et al. 2004).
Thrombin can induce cell migration in HCCs. HCCs express several proteinase-activated receptors (PAR) , mainly the thrombin receptors PAR(1), PAR(3), and PAR(4). PAR form a novel G-protein-coupled receptor subfamily. Stimulation of HCC cells in vitro with thrombin increased migration across a collagen barrier (Kaufmann et al. 2007). The urokinase system involves three key components, urokinase-type plasminogen activator (uPA), uPA receptor (uPAR), and plasminogen activator inhibitor type 1 (PAI-1). This system is activated in several tumor types and potently affects tumor invasion and metastasis. Urokinase-type plasminogen activator (uPA) is a key protein in the plasminogen activation system and is critically involved in proteolytic steps in tumor invasion. In human HCCs, uPA and c-met overexpression are coordinated in a complex fashion. Following its binding to its c-met receptor, HGF can upregulate uPA in a dose-dependent manner and by this mechanism augments HCC invasion (Lee et al. 2008). High levels of uPA in HCCs were correlated with macrovessel invasion and metastasis, and in tumors expressing uPA, uPAR, and PAI-1, a more prominent invasion phenotype was observed (Zheng et al. 2000). Overexpression of microRNA-23b in HCC cells leads to uPA and c-met downregulation and to decreased migration (Salvi et al. 2009). Plasmin- and trypsin-mediated activation of MMPs is inhibited by TFPI-2 (tissue factor pathway inhibitor), an extracellular matrix-associated Kunitz-type serine proteinase inhibitor. TFPI-2 inhibits the invasion of HCC cells (Xu et al. 2011).
Non-protease Proteins Involved in ECM Degradation
In the setting of the invasion process, heparan sulfate chains of heparan sulfate proteoglycans located in the ECM are cleaved by the endoglycosidase heparanase. Heparanase is upregulated in HCCs with an invasive-metastatic phenotype (El-Assal et al. 2001; Xiao et al. 2003; Edovitsky et al. 2004; Zhang et al. 2007a). In these neoplasms, heparanase also promotes angiogenesis (Ilan et al. 2006). HCCs also synthesize heparin-degrading sulfatases, sulfatase 1 (SULF1) and sulfatase 2 (SULF2). These two enzymes desulfatize heparan sulfate proteoglycans localized to cell surface and ECM. SULF1 and SULF2 are differentially expressed in HCCs, in that SULF1 is downregulated in up to a third of HCCs, while SULF2 is upregulated in up to 60 % of primary HCCs, and SULF2 activates MAP kinase and AKT pathways, promoting HCC growth and invasion (Lai et al. 2008).
In the invasion process, several nonstructural proteins of the extracellular matrix play a significant role. These proteins are called matricellular proteins and belong to various protein families. Well-known members include CCN family member cysteine-rich angiogenic inducer 61 (Cyr61/CCN1), CCN6, osteopontin, secreted protein acidic and rich in cysteine (SPARC), angiopoietin-like protein 4 (ANGPTL4), and thrombospondin-1 and thrombospondin-2 (review: Chong et al. 2012).
References
Albiges-Rizo C, Destaing O, Fourcade B, Planus E, Block MR (2009) Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J Cell Sci 122:3037–3049
Alexander NR, Branch KM, Parelk A, Clark ES, Iwueke IC, Guelcher SA, Weaver AM (2008) Extracellular matrix rigidity promotes invadopodia activity. Curr Biol 18:1295–1299
Altadill A, Rodriguez M, Gonzalez LO, Junquera S, Corte MD, Gonzalez-Dieguez ML et al (2009) Liver expression of matrix metalloproteases and their inhibitors in hepatocellular carcinoma. Dig Liver Dis 41:740–748
Apte SS (2009) A disintegrin-like and metalloproteinase (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem 284:31493–31497
Arai I, Nagano H, Kondo M, Yamamoto H, Hiraoka N, Sugita Y, Ota H, Yoshioka S et al (2007) Overexpression of MT3-MMP in hepatocellular carcinoma correlates with capsular invasion. Hepatogastroenterology 54:167–171
Arii S, Mise M, Harada T, Furutani M, Ishigami S, Niwano M, Mizumoto M, Fukumoto M et al (1996) Overexpression of matrix metalloproteinase 9 gene in hepatocellular carcinoma with invasive potential. Hepatology 24:316–322
Artym VV, Matsumoto K, Mueller SC, Yamada KM (2011) Dynamic membrane remodeling at invadopodia differentiates invadopodia from podosomes. Eur J Cell Biol 90:172–180
Artym VV, Swatkoski S, Matsumoto K, Campbell CB, Petrie RJ, Dimitriadis EK, Li X, Mueller SC, Bugge TH, Gucek M et al (2015) Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J Cell Biol 208:331–350
Badowski C, Pawlak G, Grichine A, Chabadel A, Oddou C, Jurdic P, Pfaff M, Albigès-Rizo C et al (2008) Paxillin phosphorylation controls invadopodia /podosomes spatiotemporal organization. Mol Biol Cell 19:633–645
Banon-Rodriguez I, Saez de Guinoa J, Bernardini A, Ragazzini C, Fernandez E, Carrasco YR et al (2013) WIP regulates persistence of cell migration and ruffle formation in both mesenchymal and amoeboid modes of motility. PLoS One 8:e70364
Beaty BT, Sharma VP, Bravo-Cordero JJ, Simpson MA, Eddy RJ, Koleske AJ, Condeelis J (2013) b1 integrin regulates Arg to promote invadopodial maturation and matrix degradation. Mol Biol Cell 24:1661–1675
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2:737–744
Bharti S, Inoue H, Hirsch DS, Nie Z, Yoon HY, Artym V, Yamada KM, Mueller SC et al (2007) Src-dependent phosphorylation of ASAP1 regulates podosomes. Mol Cell Biol 27:8271–8283
Bhattacharyya SD, Garrison J, Guo H, Mi Z, Markovic J, Kim VM, Kuo PC (2010) Micro-RNA-181a regulates osteopontin-dependent metastatic function in hepatocellular cancer cell lines. Surgery 148:291–297
Bi Q, Tang S, Xia L, Du R, Fan R, Gao L, Jin J, Liang S, Chen Z, Xu G, Nie Y, Wu K et al (2012) Ectopic expression of MiR-125a inhibits the proliferation and metastasis of hepatocellular carcinoma by targeting MMP11 and VEGF. PLoS One 7:e40169
Block MR, Badowski C, Millon-Fremillon A, Bouvard D, Bouin AP, Faurobert E, Gerber-Scokaert D et al (2008) Podosome-type adhesions and focal adhesions, so alike yet so different. Eur J Cell Biol 87:491–506
Boateng LR, Huttenlocher A (2012) Spatiotemporal regulation of Src and its substrates at invadosomes. Eur J Cell Biol 91:878–888
Bodey B, Bodey B, Siegel SE, Kaiser HE (2000) Immunocytochemical detection of MMP-3 and -10 expression in hepatocellular carcinomas. Anticancer Res 20:4585–4590
Borscheri N, Roessner A, Röcken C (2001) Canalicular immunostaining of neprilysin (CD10) as a diagnostic marker for hepatocellular carcinomas. Am J Surg Pathol 25:1297–1303
Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC (1999) An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 18:4440–4449
Branch KM, Hoshino D, Weaver AM (2012) Adhesion rings surround invadopodia and promote maturation. Biol Open 1:711–722
Bravo-Cordero JJ, Hodgson L, Condeelis J (2012) Directed cell invasion and migration during metastasis. Curr Opin Cell Biol 24:277–283
Brisson L, Reshkin SJ, Goré J, Roger S (2012) pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur J Cell Biol 91:847–860
Buccione R, Caldieri G, Ayala I (2009) Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev 28:137–149
Chen WT, Wang JY (1999) Specialized surface protrusions of invasive cells, invadopodia and lamellipodia, have differential MT1-MMP, MMP-2, and TIMP-2 localization. Ann NY Acad Sci 878:361–371
Chen RX, Xia YH, Xue TC, Zhang H, Ye SL (2011a) Down-regulation of osteopontin inhibits metastasis of hepatocellular carcinoma cells via a mechanism involving MMP-2 and uPA. Oncol Rep 25:803–808
Chen RX, Xia YH, Xue TC, Ye SL (2011b) Osteopontin promotes hepatocellular carcinoma invasion by up-regulating MMP-2 and uPA expression. Mol Biol Rep 38:3671–3677
Chen TY, Li YC, Liu YF, Tsai CM, Hsieh YH, Lin CW, Yang SF, Weng CJ (2011c) Role of MMP14 gene polymorphisms in susceptibility and pathological development to hepatocellular carcinoma. Ann Surg Oncol 18:2348–2356
Chen D, Wang Y, Zhang K, Jiao X, Yan B, Liang J (2012a) Antisense oligonucleotide against clusterin regulates human hepatocellular carcinoma invasion through transcriptional regulation of matrix metalloproteinase-2 and e-cadherin. Int J Mol Sci 13:10594–10607
Chen R, Cui J, Xu C, Xue T, Guo T, Gao D, Liu Y, Ye S, Ren Z (2012b) The significance of MMP-9 over MMP-2 in HCC invasiveness and recurrence of hepatocellular carcinoma after curative resection. Ann Surg Oncol 19(Suppl 3):S375–S384
Chen JS, Huang XH, Wang Q, Huang JQ, Zhang LJ, Chen XL, Lei J, Cheng ZX (2013a) Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis 34:10–19
Chen L, Li M, Li Q, Wang CJ, Xie SQ (2013b) DKK1 promotes hepatocellular carcinoma cell migration and invasion through beta-catenin/MMP7 signaling pathway. Mol Cancer 12:157
Chen B, Tang J, Guo YS, Li Y, Chen ZN, Jiang JL (2013c) Calpains are required for invasive and metastatic potentials of human HCC cells. Cell Biol Int 37:643–652
Chen X, Bo L, Zhao X, Chen Q (2015) MicroRNA-133a inhibits cell proliferation, colony formation ability, migration and invasion by targeting matrix metalloproteinase 9 in hepatocellular carcinoma. Mol Med Rep. doi:10.3892/mmr.2015.3232
Chong HC, Tan CK, Huang RL, Tan NS (2012) Matricellular proteins: a sticky affair with cancers. J Oncol 2012:351089
Chou HC, Anton IM, Holt MR, Curcio C, Lanzardo S, Worth A, Burns S, Thrasher AJ et al (2006) WIP regulates the stability of localization of WASP to podosomes in migrating dendritic cells. Curr Biol 16:2337–2344
Chow BS, Chew EG, Zhao C, Bathgate RA, Hewitson TD, Samuel CS (2012) Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS One 7:e42714
Chung TW, Lee YC, Kim CH (2004) Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERK and Pl-3K/AKT pathways: involvement of invasive potential. FASEB J 18:1123–1125
Clark ES, Whigham AS, Yarbrough WG, Weaver AM (2007) Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res 67:4227–4235
Collin O, Na S, Chowdhury F, Hong M, Shin ME, Wang F, Wang N (2008) Self-organized podosomes are dynamic mechanosensors. Curr Biol 18:1288–1294
Courtneidge et al. (2005) http://www.ncbi.nlm.nih.gov/pubmed/16869750
Coussens LM, Werb Z (1996) Matrix metalloproteinases and the development of cancer. Chem Biol 3:895–904
Crimaldi L, Courtneidge SA, Gimona M (2009) Tks5 recruits AFAP-110, p190RhoGAP, and cortactin for podosome formation. Exp Cell Res 315:2581–2592
Cui X, Liu Y, Wan C, Lu C, Cai J, He S, Ni T, Zhu J, Wei L, Zhang Y, Qian H (2014) Decreased expression of SERPINB1 correlates with tumor invasion and poor prognosis in hepatocellular carcinoma. J Mol Histol 45:59–68
Dai JY, Dou KF, Wang CH, Zhao P, Lau WB, Tao L, Wu YM, Tang J, Jiang JL et al (2009) The interaction of HAb18G/CD147 with integrin alpha6beta1 and its implications for the invasion potential of human hepatoma cells. BMC Cancer 9:337
Davuluri G, Augoff K, Schiemann WP, Plow EF, Sossey-Alaoui K (2014) WAVE3-NFkB interplay is essential for the survival and invasion of cancer cells. PLoS One 9:e10627
Deryugina EI, Quigley JP (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 25:9–34
Destaing O, Planus E, Bouvard D, Oddou C, Badowski C, Bossy V, Raducanu A, Fourcade B et al (2010) b1A integrin is a master regulator of invadosome organization and function. Mol Biol Cell 21:4108–4119
Destaing O, Block MR, Planus E, Albiges-Rizo C (2011) Invadosome regulation by adhesion signaling. Curr Opin Cell Biol 23:597–606
Di Martino J, Paysan L, Gest C, Lagrée V, Juin A, Saltel F, Moreau V (2014) Cdc42 and Tks5: a minimal and universal molecular signature for functional invadosomes. Cell Adh Migr 8:280–292
Diaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA (2013) Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol 201:279–292
Dovas A, Gevrey JC, Grossi A, Park H, Abou-Kheir W, Cox D (2009) Regulation of podosome dynamics by WASP phosphorylation: implication in matrix degradation and chemotaxis in macrophages. J Cell Sci 122:3873–3882
Duffy MJ, McKiernan E, O’Donovan N, McGowan PM (2009) Role of ADAMs in cancer formation and progression. Clin Cancer Res 15:1140–1144
Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J (2011) Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19:372–386
Edovitsky E, Elkin M, Zcharia E, Peretz T, Vlodavsky I (2004) Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. J Natl Cancer Inst 96:1219–1230
Edwards DR, Handsley MM, Pennington CJ (2008) The ADAM metalloproteinases. Mol Aspects Med 29:258–289
El-Assal ON, Yamanoi A, Ono T, Kohno H, Nagasue N (2001) The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin Cancer Res 7:1299–1305
El-Samanoudy A, Monir R, Badawy A, Ibrahim L, Farag K, El Baz S, Alenizi D, Alenezy A (2014) Matrix metalloproteinase-9 gene polymorphism in hepatocellular carcinoma patients with hepatitis B and C viruses. Genet Mol Res 13:8025–8034
Endo M, Nishita M, Fujii M, Minami Y (2015) Insight into the role of wnt5a-induced signaling in normal and cancer cells. Int Rev Cell Mol Biol 314:117–148
Esselens C, Malapeira J, Colomé N, Casal C, Rodriguez-Manzaneque JC, Canals F et al (2010) The cleavage of semaphorin 3C induced by ADAMTS1 promotes cell migration. J Biol Chem 285:2463–2473
Fang JH, Zhou HC, Zeng C, Yang J, Liu Y, Huang X, Zhang JP, Guan XY, Zhuang SM (2011) MicroRNA-29b suppresses tumor angiogenesis, invasion and metastases by regulating MMP-2 expression. Hepatology 54:1729–1740
Friedl P, Wolf K (2009) Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev 28:129–135
Fukuda Y, Imoto M, Koyama Y, Miyazawa Y, Nakano I, Hattori M, Urano F, Kodama S et al (1991) Immunohistochemical study on tissue inhibitors of metalloproteinases in normal and pathological human livers. Gastroenterol Jpn 26:37–41
Gad A, Lach S, Crimaldi L, Gimona M (2008) Plectin deposition at podosome rings requires myosin contractility. Cell Motil Cytoskeleton 65:614–625
Gao ZH, Tretiakova MS, Liu WH, Gong C, Farris PD, Hart J (2006) Association of E-cadherin, matrix metalloproteinases, and tissue inhibitors of metalloproteinases with the progression and metastasis of hepatocellular carcinoma. Mod Pathol 19:533–540
Garcia E, Jones GE, Machesky LM, Anton IM (2012) WIP: wasp-interacting proteins at invadopodia and podosomes. Eur J Cell Biol 91:869–877
Gawden-Bone C, Zhou Z, King E, Prescott A, Watts C, Lucocq J (2010) Dendritic cell podosomes are protrusive and invade the extracellular matrix using metalloproteinase MMP-14. J Cell Sci 123:1427–1437
Génot E, Gligorijevic B (2014) Invadosomes in their natural habitat. Eur J Cell Biol 93:367–379
Gianelli G, Bergamini C, Marinosci F, Fransvea E, Quaranta M, Lupo L, Schiraldi O et al (2002) Clinical role of MMP-2/TIMP-2 imbalance in hepatocellular carcinoma. Int J Cancer 97:425–431
Giannelli G, Astigiano S, Antonaci S, Morini M, Barbieri O, Noonan DM, Albini A (2002) Role of the alpha3beta1 and alpha6beta4 integrins in tumor invasion. Clin Exp Metastasis 19:217–223
Gimona M (2008) The microfilament system in the formation of invasive adhesions. Semin Cancer Biol 18:23–34
Gimona M, Buccione R (2006) Adhesions that mediate invasion. Int J Biochem Cell Biol 38:1875–1892
Gimona M, Buccione R, Courtneidge SA, Linder S (2008) Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol 20:235–241
Goicoechea S, Arneman D, Disanza A, Garcia-Mata R, Scita G, Otey CA (2006) Palladin binds to Eps8 and enhances the formation of dorsal ruffles and podosomes in vascular smooth muscle cells. J Cell Sci 199:3316–3324
Goicoechea SM, Bednarski B, Garcia-Mata R, Prentice-Dunn H, Kim HJ, Otey CA (2009) Palladin contributes to invasive motility in human breast cancer cells. Oncogene 28:587–598
Goicoechea SM, Garcia-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, Hwang RF et al (2014) Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene 33:1265–1273
Gorrin Rivas MJ, Arii S, Furutani M, Harada T, Mizumoto M, Nishiyama H, Fujita J, Imamura M (1998) Expression of human macrophage metalloelastase gene in hepatocellular carcinoma: correlation with angiostatin generation and its clinical significance. Hepatology 28:986–993
Grass GD, Bratoeva M, Toole BP (2012) Regulation of invadopodia formation and activity by CD147. J Cell Sci 125:777–788
Griera M, Martin-Villar E, Banon-Rodriguez I, Blundell MP, Jones GE, Anton IM, Thrasher AJ et al (2014) Integrin linked kinase (ILK) regulates podosome maturation and stability in dendritic cells. Int J Biochem Cell Biol 50:47–54
Gringel A, Walz D, Rosenberger G, Minden A, Kutsche K, Kopp P, Linder S (2006) PAK4 and alphaPIX determine podosome size and number in macrophages through localized actin regulation. J Cell Physiol 209:568–579
Gurksy LA, Xu X, Labrada LN, Nguyen NT, Xiao L, van Golen KL, Jia X, Farach-Carson MC (2012) Hyaluronan (HA) interacting proteins RHAMM and hyaluronidase impact prostate cancer cell behavior and invadopodia formation in 3D HA-based hydrogels. PLoS One 7:e50075
Ha VL, Bharti S, Inoue H, Vass WC, Campa F, Nie Z, de Gramont A, Ward Y, Randazzo PA (2008) ASAP3 is a focal adhesion-associated Arf GAP that functions in cell migration and invasion. J Biol Chem 283:14915–14926
Hah N, Lee ST (2003) An absolute role of the PKC-dependent NF-kappaB activation for induction of MMP-9 in hepatocellular carcinoma cells. Biochem Biophys Res Commun 305:428–433
Han S, Han L, Yao Y, Sun H, Zan X, Liu Q (2014) Activated hepatic stellate cells promote hepatocellular carcinoma cell migration and invasion via the activation of FAK-MMP9 signaling. Oncol Rep 31:641–648
Harada T, Arii S, Mise M, Imamura T, Higashitsuji H, Furutani M, Niwano M, Ishigami S et al (1998) Membrane-type matrix metalloproteinase-1 (MT1-MTP) gene is overexpressed in highly invasive hepatocellular carcinomas. J Hepatol 28:231–239
Hirashita T, Iwashita Y, Ohta M, Komori Y, Eguchi H, Yada K, Kitano S (2012) Expression of matrix metalloproteinase-7 is an unfavorable prognostic factor in intrahepatic cholangiocarcinoma. J Gastrointest Surg 16:842–848
Hoshino D, Branch KM, Weaver AM (2013a) Signaling inputs to invadopodia and podosomes. J Cell Sci 126:2979–2989
Hoshino D, Nagano M, Saitoh A, Koshikawa N, Suzuki T, Seiki M (2013b) The phosphoinositide-binding protein ZF21 regulates ECM degradation by invadopodia. PLoS One 8:e50825
Huang D, Du X, Yuan R, Chen L, Liu T, Wen C, Huang M, Li M, Hao L, Shao J (2014) Rock2 promotes the invasion and metastasis of hepatocellular carcinoma by modifying MMP2 ubiquitination and degradation. Biochem Biophys Res Commun 453:49–56
Hung TM, Chang SC, Yu WH, Wang YW, Huang C, Lu SC, Lee PH, Chang MF (2009) A novel nonsynonymous variant of matrix metalloproteinase-7 confers risk of liver cirrhosis. Hepatology 50:1184–1193
Huveneers S, Arslan S, van de Water B, Sonnenberg A, Danen EH (2008) Integrins uncouple Src-induced morphological and oncogenic transformation. J Biol Chem 283:13243–13251
Ilan N, Elkin M, Vlodavsky I (2006) Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol 38:2018–2039
Ip YC, Cheung ST, Leing KL, Fan ST (2005) Mechanism of metastasis by membrane type 1-matrix metalloproteinase in hepatocellular carcinoma. World J Gastroenterol 11:6269–6276
Ip YC, Cheung ST, Fan ST (2007) Atypical localization of membrane type 1-matrix metalloproteinase in the nucleus is associated with aggressive features of hepatocellular carcinoma. Mol Carcinog 46:225–230
Ishii Y, Nakasato Y, Kobayashi S, Yamazaki Y, Aoki T (2003) A study on angiogenesis-related matrix metalloproteinase networks in primary hepatocellular carcinoma. J Exp Clin Cancer Res 22:461–470
Itatsu K, Zen Y, Yamaguchi J, Ohira S, Ishikawa A, Ikeda H, Sato Y, Harada K, Sasaki M et al (2008) Expression of matrix metalloproteinase 7 is an unfavorable postoperative prognostic factor in cholangiocarcinoma of the perihilar, hilar, and extrahepatic bile ducts. Hum Pathol 39:710–719
Jacob A, Jing J, Lee J, Schedin P, Gilbert SM, Peden AA, Junutula JR, Prekeris R (2013) Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. J Cell Sci 126:4647–4658
Jevnikar Z, Mirkovic B, Fonovic UP, Zidar N, Svajger U, Kos J (2012) Three-dimensional invasion of macrophages is mediated by cysteine cathepsins in protrusive podosomes. Eur J Immunol 42:3429–3441
Jia L, Cao J, Wei W, Wang S, Zuo Y, Zhang J (2007) CD147 depletion down-regulates matrix metalloproteinase-11, vascular endothelial growth factor-A expression and the lymphatic metastasis potential of murine hepatocarcinoma Hca-F cells. Int J Biochem Cell Biol 39:2135–2142
Jia L, Xu H, Zhao Y, Jiang L, Yu J, Zhang J (2008) Expression of CD147 mediates tumor cells invasion and multidrug resistance in hepatocellular carcinoma. Cancer Invest 26:977–983
Joo YE, Seo YH, Lee WS, Kim HS, Choi SK, Rew JS, Park CS, Kim SJ (2000) Expression of tissue inhibitors of metalloproteinases (TIMPs) in hepatocellular carcinoma. Korean J Intern Med 15:171–178
Juin A, Billottet C, Moreau V, Destaing O, Albiges-Rizo C, Rosenbaum J, Génot E et al (2012) Physiological type I collagen organization induces the formation of a novel class of linear invadosomes. Mol Biol Cell 23:297–309
Jung O, Choi YJ, Kwak TK, Kang M, Lee MS, Ryu J, Kim HJ, Lee JW (2013) The COOH-terminus of TM4SF5 in hepatoma cell lines regulates c-Src to form invasive protrusions via EGFR Tyr845 phosphorylation. Biochim Biophys Acta 1833:629–642
Karadag N, Kirimlioglu H, Isik B, Yilmaz S, Kirimlioglu V (2008) Expression of matrix metalloproteinase in gallbladder carcinoma and their significance in carcinogenesis. Appl Immunohistochem Mol Morphol 16:148–152
Kaufmann R, Rahn S, Pollrich K, Hertel J, Dittmar Y, Hommann M, Henklein P, Biskup C et al (2007) Thrombin-mediated hepatocellular carcinoma cell migration: cooperative action via proteinase-activated receptors 1 and 4. J Cell Physiol 211:699–707
Kaverina I, Stradal TE, Gimona M (2003) Podosome formation in cultured A7r5 vascular muscle cells requires Arp2/3-dependent de-novo actin polymerization at discrete microdomains. J Cell Sci 116:4915–4924
Klein T, Bischoff R (2011) Active metalloproteases of the A disintegrin and metalloprotease (ADAM) family: biological function and structure. J Proteome Res 10:1–33
Kleiner DE, Stetler-Stevenson WG (1999) Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol 43(Suppl):S42–S51
Klemke RL (2012) Trespassing cancer cells: ‘fingerprinting’ invasive protrusions reveals metastatic culprits. Curr Opin Cell Biol 24:662–669
Kocher HM, Sandle J, Mirza TA, Li NF, Hart IR (2009) Ezrin interacts with cortactin to form podosomal rosettes in pancreatic cancer cells. Gut 58:271–284
Kopp P, Lamers R, Aepfelbacher M, Woehlke G, Rudel T, Machuy N, Steffen W, Linder S (2006) The kinesin KIF1C and microtubule plus ends regulate podosome dynamics in macrophages. Mol Biol Cell 17:2811–2823
Kung ML, Tsai HE, Hu TH, Kuo HM, Liu LF, Chen SC, Lin PR, Ma YL, Wang EM et al (2012) Hepatoma-derived growth factor stimulates podosome rosettes formation in NIH/3T3 cells through the activation of phosphatidylinositol 3-kinase/Akt pathway. Biochem Biophys Res Commun 425:169–176
Lagal V, Abrivard M, Gonzalez V, Perazzi A, Popli S, Verzeroli E, Tardieux I (2014) Spire-1 contributes to the invadosome and its associated invasive properties. J Cell Sci 127:328–340
Lai HJ, Lo SJ (2005) Epigenetic methylation of TIMP-3 may play a role in HBV-associated hepatocellular carcinoma. Chang Gung Med J 28:453–455
Lai JP, Thompson JR, Sandhu DS, Roberts LR (2008) Heparin-degrading sulfatases in hepatocellular carcinoma: roles in pathogenesis and therapy targets. Future Oncol 4:803–814
Lee et al. (2008) http://www.ncbi.nlm.nih.gov/pubmed/17992475
Leelawat K, Sakchinabut S, Narong S, Wannaprasert J (2009) Detection of serum MMP-7 and MMP-9 in cholangiocarcinoma patients: evaluation of diagnostic accuracy. BMC Gastroenterol 9:30
Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K et al (2014) Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep 8:1558–1570
Li Y, Zhang Y, Zheng Q (2005) Expression of RECK gene and MMP-9 in hilar cholangiocarcinoma and its clinical significance. J Huazhong Univ Sci Technolog Med Sci 25:552–554
Li J, Lau G, Chen L, Yuan YF, Huang J, Luk JM, Xie D, Guan XY (2012) Interleukin 23 promotes hepatocellular carcinoma metastasis via NF-kappa B induced matrix metalloproteinase 9 expression. PLoS One 7:e46264
Li T, Xie J, Shen C, Cheng D, Shi Y, Wu Z, Zhan Q, Deng X, Chen H, Shen B, Peng C, Li H, Zhu Z (2014) miR-150-5p inhibits hepatoma cell migration and invasion by targeting MMP14. PLoS One 9:e15577
Liao CG, Kong LM, Song F, Xing JL, Wang LX, Sun ZJ, Tang H, Yao H, Zhang Y et al (2011) Characterization of basigin isoforms and the inhibitory function of basigin-3 in human hepatocellular carcinoma proliferation and invasion. Mol Cell Biol 31:2591–2604
Liao M, Tong P, Zhao J, Zhang Y, Li Z, Wang J, Feng X, Hu M, Pan Y (2012) Prognostic value of matrix metalloproteinase-1/proteinase-activated receptor-1 signaling axis in hepatocellular carcinoma. Pathol Oncol Res 18:397–403
Linder S (2007) The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol 17:107–117
Linder S, Aepfelbacher M (2003) Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol 13:376–385
Linder S, Kopp P (2005) Podosomes at a glance. J Cell Sci 118:2079–2082
Linder S, Hüfner K, Wintergerst U, Aepfelbacher M (2000) Microtubule-dependent formation of podosomal adhesion structures in primary human macrophages. J Cell Sci 23:4165–4176
Linder S, Wiesner C, Himmel M (2011) Degrading devices: invadosomes in proteolytic cell invasion. Annu Rev Cell Dev Biol 27:185–211
Liu Z, Yan L, Xiang T, Jiang L, Yang B (2003) Expression of vascular endothelial growth factor and matrix metalloproteinase-2 correlates with the invasion and metastasis of hepatocellular carcinoma. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 20:249–250
Liu XS, Luo HJ, Yang H, Wang L, Kong H, Jin YE, Wang F, Gu MM, Chen Z, Lu ZY et al (2007) Palladin regulates cell and extracellular matrix interaction through maintaining normal actin cytoskeleton architecture and stabilizing beta1-integrin. J Cell Biochem 100:1288–1300
Lizarraga F, Poincloux R, Romao M, Montagnac G, Le Dez G, Bonne I, Rigaill G, Raposo G et al (2009) Diaphanous-related formins are required for invadopodia formation and invasion of breast tumor cells. Cancer Res 69:2792–2800
Lohmer LL, Kelley LC, Hagedorn EJ, Sherwood DR (2014) Invadopodia and basement membrane invasion in vivo. Cell Adh Migr 8:246–255
Lu JT, Zhao WD, He W, Wei W (2012) Hedgehog signaling pathway mediates invasion and metastasis of hepatocellular carcinoma via ERK pathway. Acta Pharmacol Sin 33:691–700
Lu L, Zhang Q, Wu K, Chen X, Zheng Y, Zhu C, Wu J (2015) Hepatitis C virus NS3 protein enhances cancer cell invasion by activating matrix metalloproteinase-9 and cyclooxygenase-2 through ERK/p38/NK-kB signal cascade. Cancer Lett 356:470–478
Luxenburg C, Parsons JT, Addadi L, Geiger B (2006) Involvement of the Src-cortactin pathway in podosome formation and turnover during polarization of cultures osteoclasts. J Cell Sci 119:4878–4888
Määtä M, Soini Y, Liakka A, Autio-Harmainen H (2000) Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: implications for tumor progression and clinical prognosis. Clin Cancer Res 6:2726–2734
Mannello F, Medda V (2012) Nuclear localization of matrix metalloproteinases. Prog Histochem Cytochem 47:27–58
Matsumoto E, Nakatsukasa H, Nouso K, Kobayashi Y, Nakamura S, Suzuki M, Takuma Y et al (2004) Increased levels of tissue inhibitor of metalloproteinase-1 in human hepatocellular carcinoma. Liver Int 24:379–383
Matsunaga Y, Koda M, Murawaki Y (2004) Expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) in hepatocellular carcinoma tissue, compared with the surrounding non-tumor tissue. Res Commun Mol Pathol Pharmacol 115–116:143–150
McKenna GJ, Chen Y, Smith RM, Meneghetti A, Ong C, McMaster R, Scudamore CH et al (2002) A role of matrix metalloproteinases and tumor host interaction in hepatocellular carcinomas. Am J Surg 183:588–594
McNiven MA, Baldassarre M, Buccione R (2004) The role of dynamin in the assembly and function of podosomes and invadopodia. Front Biosci 9:1944–1953
Miwa S, Miyagawa S, Soeda J, Kawasaki S (2002) Matrix metalloproteinase-7 expression and biologic aggressiveness of cholangiocellular carcinoma. Cancer 94:428–434
Miyoshi A, Kitajima Y, Sumi K, Sato K, Hagiwara A, Koga Y, Miyazaki K (2004) Snail and SIP1 increase cancer invasion by upregulation MMP family in hepatocellular carcinoma cells. Br J Cancer 90:1265–1273
Miyoshi A, Kitajima Y, Kido S, Shimonishi T, Matsuyama S, Kitahara K, Miyazaki K (2005) Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer 92:252–258
Mochizuki S, Okada Y (2007) ADAMs in cancer cell proliferation and progression. Cancer Sci 98:621–628
Mochizuki S, Okada Y (2009) ADAM28 as a target for human cancers. Curr Pharm Des 15:2349–2358
Mochizuki S, Soeijima K, Shimoda M, Abe H, Sasaki A, Okano HJ, Okano H, Okada Y (2012) Effect of ADAM28 on carcinoma cell metastasis by cleavage of von Willebrand factor. J Natl Cancer Inst 104:906–922
Monvoisin et al. (2002) http://www.ncbi.nlm.nih.gov/pubmed/11774258
Morita T, Mayanagi T, Yoshio T, Sobue K (2007) Changes in the balance between caldesmon regulated by p21-activated kinases and the Arp2/3 complex govern podosome formation. J Biol Chem 282:8454–8463
Mukhopadhyay UK, Eves R, Jia L, Mooney P, Mak AS (2009) p53 suppresses Src-induced podosome and rosette formation and cellular invasiveness through the upregulation of caldesmon. Mol Cell Biol 29:3088–3098
Murakami K, Sakukawa R, Ikeda T, Matsuura T, Hasumura S, Nagamori S, Yamada Y et al (1999) Invasiveness of hepatocellular carcinoma cell lines: contribution of membrane-type 1 matrix metalloproteinase. Neoplasia 1:424–430
Murphy DA, Courtneidge SA (2011) The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Cell Biol 12:413–416
Musso O, Théret N, Campion JP, Turlin B, Milani S, Grappone C, Clément B (1997) In situ detection of matrix metalloproteinase-2 (MMP2) and the metalloproteinase inhibitor TIMP2 transcripts in human primary hepatocellular carcinoma and in liver metastasis. J Hepatol 26:593–605
Nabeshima K, Iwasaki H, Koga K, Hojo H, Suzumiya J, Kikuchi M (2006) Emmprin (basigin/CD147): matrix metalloproteinase modulator and multifunctional cell recognition molecule that plays a critical role in cancer progression. Pathol Int 56:359–367
Nakatsukasa H, Ashida K, Higashi T, Ohguchi S, Tsuboi S, Hino N, Nouso K, Urabe Y et al (1996) Cellular distribution of transcripts for tissue inhibitor of metalloproteinases 1 and 2 in human hepatocellular carcinomas. Hepatology 24:82–88
Namwat N, Puetkasichonpasutha J, Loilome W, Yongvanit P, Techasen A, Puapairoj A et al (2011) Downregulation of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) is associated with enhanced expression of matrix metalloproteinases and cholangiocarcinoma metastases. J Gastroenterol 46:664–675
Nart D, Yaman B, Yilmaz F, Zeytunlu M, Karasu Z, Kiliç M (2010) Expression of matrix metalloproteinase-9 in predicting prognosis of hepatocellular carcinoma after liver transplantation. Liver Transpl 16:621–630
Ng KT, Qi X, Kong KL, Cheung BY, Lo CM, Poon RT, Fan ST, Man K (2011) Overexpression of matrix metalloproteinase-12 (MMP-12) correlates with poor prognosis of hepatocellular carcinoma. Eur J Cancer 47:2299–2305
Nunes GL, Simoes A, Dyszy FH, Shida CS, Juliano MA, Juliano L, Gesteira TF et al (2011) Mechanism of heparin acceleration of tissue inhibitor of metalloproteases-1 (TIMP-1) degradation by the human neutrophil elastase. PLoS One 6:e21525
Ogasawara S, Yano H, Momosaki S, Nishida N, Takemoto Y, Kojiro S, Kojiro M (2005) Expression of matrix metalloproteinases (MMPs) in cultured hepatocellular carcinoma (HCC) cells and surgically resected HCC tissues. Oncol Rep 13:1043–1048
Ogata R, Tirmura T, Kin M, Ueno T, Tateishi Y, Kuromatsu R, Shimauchi Y, Sakamoto M et al (1999) Increased expression of membrane type 1 matrix metalloproteinase and matrix metalloproteinase-2 with tumor dedifferentiation in hepatocellular carcinomas. Hum Pathol 30:443–450
Okamoto K, Mandai M, Mimura K, Murakawi Y, Yuasa I (2005) The association of MMP-1, -3 and -9 genotypes with the prognosis of HCV-related hepatocellular carcinoma patients. Res Commun Mol Pathol Pharmacol 117–118:77–89
Okamoto K, Ishida C, Ikebuchi Y, Mandai M, Mimura K, Murakawi Y, Yuasa I (2010) The genotypes of IL-1 beta and MMP-3 are associated with prognosis of HCV-related hepatocellular carcinoma. Intern Med 49:887–895
Okamura H, Yoshida K, Haneji T (2009) Negative regulation of TIMP1 is mediated by transcription factor TWIST1. Int J Oncol 35:181–186
Okazaki I, Wada N, Nakano M, Saito A, Takasaki K, Doi M, Kameyama K, Otani Y et al (1997) Difference in gene expression for matrix metalloproteinase-1 between early and advanced hepatocellular carcinomas. Hepatology 25:580–584
Ou DP, Tao YM, Tang FQ, Yang LY (2007) The hepatitis B virus X promotes hepatocellular carcinoma metastasis by upregulation of matrix metalloproteinases. Int J Cancer 120:1208–1214
Oudekirk JL, Krendel M (2014) Myosin 1e is a component of the invadosome core that contributes to regulation of invadosome dynamics. Exp Cell Res 322:265–276
Overall CM, Dean RA (2006) Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev 25:69–75
Ozaki I, Mizuta T, Zhao G, Zhang H, Yoshimura T, Kawazoe S, Eguchi Y, Yasutake T et al (2003) Induction of multiple matrix metalloproteinase genes in human hepatocellular carcinoma by hepatocyte growth factor via a transcription factor Ets-1. Hepatol Res 27:289–301
Pan YR, Tseng WS, Chang PW, Chen HC (2013a) Phosphorylation of moesin by Jun N-terminal kinase is important for podosome rosette formation in Src-transformed fibroblasts. J Cell Sci 126:5670–5680
Pan YR, Cho KH, Lee HH, Chang ZF, Chen HC (2013b) Protein tyrosine phosphatase SHP2 suppresses podosome rosette formation in Src-transformed fibroblasts. J Cell Sci 126:657–666
Paz H, Pathak N, Yang J (2014) Invading one step at a time : the role of invadopodia in tumor metastasis. Oncogene 33:4193–4202
Pignatelli J, Tumbarello DA, Schmidt RP, Turner CE (2012a) Hic-5 promotes invadopodia formation and invasion during TGF-β-induced epithelial-mesenchymal transition. J Cell Biol 197:421–437
Pignatelli J, LaLonde SE, LaLonde DP, Clarke D, Tunrer CE (2012b) Actopaxin (a-parvin) phosphorylation is required for matrix degradation and cancer cell invasion. J Biol Chem 287:37309–37320
Poincloux R, Lizarraga F, Chavrier P (2009) Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci 122:3015–3024
Porter S, Clark IM, Kevorkian L, Edwards DR (2005) The ADAMTS metalloproteinases. Biochem J 386:15–27
Qian AR, Zhang W, Cao JP, Yang PF, Gao X, Wang Z, Xu HY, Weng YY, Shang P (2008) Downregulation of CD147 expression alters cytoskeleton architecture and inhibits gelatinase production and SAPK pathway in human hepatocellular carcinoma cells. J Exp Clin Cancer Res 27:50
Rakashanda S, Rana F, Rafiq S, Masood A, Amin S (2012) Role of proteases in cancer: a review. Biotechnol Mol Biol Rev 7:90–101
Rocken C, Carl-McGrath S, Grantzdorffer I, Mantke R, Roessner A, Lendeckel U (2004) Ectopeptidases are differentially expressed in hepatocellular carcinomas. Int J Oncol 24:487–495
Rocks N, Paulissen G, El Hour M, Quesada F, Crahay C, Gueders M, Foidart JM et al (2008) Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie 90:369–379
Roderfeld M, Rath T, Lammert F, Dierkes C, Graf J, Roeb E (2010) Innovative immunohistochemistry identifies MMP-9 expressing macrophages at the invasive front of murine HCC. World J Hepatol 2:175–179
Rottiers P, Saltel F, Daubon T, Chaigne-Delalande B, Tridon V, Billottet C, Reuzeau E, Génot E (2009) TGFbeta-induced endothelial podosomes mediate basement membrane collagen degradation in arterial vessels. J Cell Sci 122:4311–4318
Saengsawang W, Taylor KL, Lumbard DC, Mitok K, Price A, Pietila L, Gomez TM, Dent EW (2013) CIP4 coordinates with phospholipids and actin-associated proteins to localize to the protruding edge and produce actin ribs and veils. J Cell Sci 126:2411–2423
Sakamoto Y, Mafune K, Mori M, Shiraishi T, Imamura H, Mori M, Takayama T, Makuuchi M (2000) Overexpression of MMP-9 correlates with growth of small hepatocellular carcinoma. Int J Oncol 17:237–243
Saltel F, Daubon T, Juin A, Ganuza IE, Veillat V, Génot E (2011) Invadosomes: intriguing structures with promise. Eur J Cell Biol 90:100–107
Salvi A, Sabelli C, Moncini S, Venturin M, Arici B, Riva P, Portolani N, Giulini SM et al (2009) Micro-RNA-23b mediates urokinase and c-met downmodulation and a decreased migration of human hepatocellular carcinoma cells. FEBS J 276:2966–2982
Schachtner H, Calaminus SD, Thomas SG, Machesky LM (2013) Podosomes in adhesion, migration, mechanosensing and matrix remodeling. Cytoskeleton (Hoboken) 70:572–589
Schaller MD (2001) Paxillin: a focal adhesion-associated adaptor protein. Oncogene 20:6459–6472
Schlaepfer DD, Mitra SK, Ilic D (2004) Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta 1692:77–102
Seubert B, Grünwald B, Kobuch J, Cui H, Schelter F, Schaten S, Siveke JT, Lim NH, Nagase H, Simonavicius N et al (2015) Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology 61:238–248
Sevenich L, Joyce JA (2014) Pericellular proteolysis in cancer. Genes Dev 28:2331–2347
Shi RY, Yang XR, Shen QJ, Yang LX, Xu Y, Qiu SJ, Sun YF, Zhang X, Wang Z et al (2013) High expression of Dickkopf-related protein 1 is related to lymphatic metastasis and indicates poor prognosis in intrahepatic cholangiocarcinoma patients after surgery. Cancer 199:993–1003
Shiomi T, Lemaître V, D’Armiento J, Okada Y (2010) Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic disease. Pathol Int 60:477–496
Sibony-Benyamini H, Gil-Henn H (2012) Invadopodia: the leading force. Eur J Cell Biol 91:896–901
Snyder BN, Cho Y, Qian Y, Coad JE, Flynn DC, Cunnick JM (2011) AFAP1L1 is a novel adaptor protein of the AFAP family that interacts with cortactin and localizes to invadosomes. Eur J Cell Biol 90:376–389
Spuul P, Ciufici P, Veillat V, Leclercq A, Daubon T, Kramer I, Génot E (2014) Importance of RhoGTPases in formation, characteristics, and functions of invadosomes. Small GTPases 5:e28195
Stamenkovic I (2000) Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol 10:415–433
Starnes TW, Cortesio CL, Huttenlocher A (2011) Imaging podosome dynamics and matrix degradation. Methods Mol Biol 769:111–136
Steffen A, Le Dez G, Poincloux R, Recchi C, Nassoy P, Rottner K, Galli T, Chavrier P (2008) MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr Biol 18:926–931
Stylli SS, Kaye AH, Lock P (2008) Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci 15:725–737
Sun MH, Han XC, Jia MK, Jiang WD, Wang M, Zhang H, Han G, Jiang Y (2005) Expressions of inducible nitric oxide synthase and matrix metalloproteinase-9 and their effects on angiogenesis and progression of hepatocellular carcinoma. World J Gastroenterol 11:5931–5977
Sun X, Li C, Zhuang C, Gilmore WC, Cobos E, Tao Y, Dai Z (2009) Abl interactor 1 regulates Src-Id1-matrix metalloproteinase 9 axis and is required for invadopodia formation, extracellular matrix degradation and tumor growth of human breast cancer cells. Carcinogenesis 30:2109–2116
Symons M (2008) Cell biology: watching the first steps of podosome formation. Curr Biol 18:R925–R927
Szczur K, Xu H, Atkinson S, Zheng Y, Filippi MD (2006) Rho GTPase CDC42 regulates directionality and random movement via distinct MAPK pathways in neutrophils. Blood 108:4205–4213
Sze KM, Chu GK, Lee JM, Ng IO (2013) C-terminal truncated HBx is associated with metastasis and enhances invasiveness via C-Jun/MMP10 activation in hepatocellular carcinoma. Hepatology 57:131–139
Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW (2007) An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 26:6361–6371
Takkunen M, Hukkanen M, Liljeström M, Grenman R, Virtanen I (2010) Podosome-like structures of non-invasive carcinoma cells are replaced in epithelial-mesenchymal transition by actin comet-embedded invadopodia. J Cell Mol Med 14:1569–1593
Tang BL (2001) ADAMTS: a novel family of extracellular matrix proteases. Int J Biochem Cell Biol 33:33–44
Tao K, Qian N, Tang Y, Ti Z, Song W, Cao D, Dou K (2010) Increased expression of a disintegrin and metalloproteinase-9 in hepatocellular carcinoma: implications for tumor progression and prognosis. Jpn J Clin Oncol 40:645–651
Tao YM, Liu Z, Liu HL (2013) Dickkopf-1 (DKK1) promotes invasion and metastasis of hepatocellular carcinoma. Dig Liver Dis 45:251–257
Tatin F, Varon C, Génot E, Moreau V (2006) A signalling cascade involving PKC, Src and Cdc42 regulates podosome assembly in cultured endothelial cells in response to phorbol ester. J Cell Sci 119:769–781
Thieringer FR, Maass T, Anthon B, Meyer E, Schirmacher P, Longerich T, Galle PR et al (2012) Liver-specific overexpression of matrix metalloproteinase 9 (MMP-9) in transgenic mice accelerates development of hepatocellular carcinoma. Mol Carcinog 51:439–448
Tokui N, Yoneyama MS, Hatakeyama S, Yamamoto H, Koie T, Saitoh H, Yamaya K, Funyu T, Nakamura T, Ohyama C et al (2014) Extravasation during bladder cancer metastasis requires cortactin-mediated invadopodia formation. Mol Med Rep 9:1142–1146
Tolde O, Rösel D, Vesely P, Folk P, Brabek J (2010) The structure of invadopodia in a complex 3D environment. Eur J Cell Biol 89:674–680
Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, Lin CD, Liao YL, Wang JL et al (2009) MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology 49:1571–1582
Tsuboi S (2007) Requirement for a complex of Wiskott-Aldrich syndrome protein (WASP) with WASP interacting protein in podosome formation in macrophages. J Immunol 178:2987–2995
Tsujita K, Kondo A, Kurisu S, Hasegawa J, Itoh T, Takenawa T (2013) Antagonistic regulation of F-BAR protein assemblies controls actin polymerization during podosome formation. J Cell Sci 126:2267–2278
Tu K, Dou C, Zheng X, Li C, Yang W, Yao Y, Liu Q (2014) Fibulin-5 inhibits hepatocellular carcinoma cell migration and invasion by down-regulating matrix metalloproteinase-7 expression. BMC Cancer 14:938
Turner CE (2000) Paxillin interactions. J Cell Sci 23:4139–4140
van den Dries K, Medders MB, de Keijzer S, Shekhar S, Subramaniam V, Figdor CG et al (2013) Interplay between myosin IIA-mediated contractility and actin network integrity orchestrates podosome composition and oscillations. Nat Commun 4:1412
Van Horssen R, Buccione R, Willemse M, Cingir S, Wieringa B, Attansio F (2013) Cancer cell metabolism regulates extracellular matrix degradation by invadopodia. Eur J Cell Biol 92:113–121
Wang XJ, Feng CW, Li M (2013a) ADAM17 mediates hypoxia-induced drug resistance in hepatocellular carcinoma cells through activation of EGFR/PI3K/Akt pathway. Mol Cell Biochem 380:57–66
Wang Q, Tang H, Yin S, Dong C (2013b) Downregulation of microRNA-138 enhances the proliferation, migration and invasion of cholangiocarcinoma cells through the upregulation of RhoC/p-ERK/MMP-2/MMP-9. Oncol Rep 29:2046–2052
Wang W, Lin H, Zhou L, Zhu Q, Gao S, Xie H, Liu Z, Xu Z, Wie J, Huang X, Zheng S (2014) MicroRNA-30a-3p inhibits tumor proliferation, invasiveness and metastasis and is downregulated in hepatocellular carcinoma. Eur J Surg Oncol 40:1586–1594
Watanabe A, Hoshino D, Koshikawa N, Seiki M, Suzuki T, Ichikawa K (2013) Critical role of transient activity of MT1-MMP for ECM degradation in invadopodia. PLoS Comput Biol 9:e1003086
Weaver AM (2006) Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis 23:97–105
Weaver AM (2008) Cortactin in tumor invasiveness. Cancer Lett 265:157–166
Wernimont SA, Cortesio CL, Simonson WT, Huttenlocher A (2008) Adhesion ring: a structural comparison between podosomes and the immune synapse. Eur J Cell Biol 87:507–515
Wiche G (1998) Role of plectin in cytoskeleton organization and dynamics. J Cell Sci 111:2477–2486
Williams BB, Mundell N, Dunlap J, Jessen J (2012) The planar cell polarity protein VANGl2 coordinates remodeling of the extracellular matrix. Commun Integr Biol 5:325–328
Wu LM, Zhang F, Xie HY, Xu X, Chen QX, Yin SY, Liu XC, Zhou L, Xu XB, Sun YL et al (2008) MMP2 promoter polymorphism (C-1306T) and risk of recurrence in patients with hepatocellular carcinoma after transplantation. Clin Genet 73:273–278
Xiang ZL, Zeng ZC, Tang ZY, Fan J, Sun HC, Tan YS (2011) Expression of cytokeratin 19 and matrix metalloproteinase 2 predicts lymph node metastasis in hepatocellular carcinoma. Mol Biol Rep 38:3531–3539
Xiao Y, Kleeff J, Shi X, Büchler MW, Friess H (2003) Heparanase expression in hepatocellular carcinoma and the cirrhotic liver. Hepatol Res 26:192–198
Xiao M, Zhou NX, Huang ZQ, Lu YL, Chen LH, Wang DJ, Chang WL (2004) The ratio of MMP-2 and TIMP-2 in hilar cholangiocarcinoma: a semi-quantitative study. Hepatobiliary Pancreat Dis Int 3:599–602
Xu J, Xu HY, Zhang Q, Song F, Jiang JL, Yang XM, Mi L, Wen N, Tian R, Wang L et al (2007) HAb18G/CD147 functions in invasion and metastasis of hepatocellular carcinoma. Mol Cancer Res 5:605–614
Xu Y, Qin X, Zhou J, Tu Z, Bi X, Li W, Fan X, Zhang Y (2011) Tissue factor pathway inhibitor-2 inhibits the growth and invasion of hepatocellular carcinoma cells and is inactivated in human hepatocellular carcinoma. Oncol Lett 2:779–783
Yamaguchi H (2012) Pathological roles of invadopodia in cancer invasion and metastasis. Eur J Cell Biol 91:902–907
Yamaguchi H, Oikawa T (2010) Membrane lipids in invadopodia and podosomes: key structures for cancer invasion and metastasis. Oncotarget 1:320–328
Yamaguchi H, Pixley F, Condeelis J (2006) Invadopodia and podosomes in tumor invasion. Eur J Cell Biol 85:213–218
Yamamoto H, Itoh F, Dachi Y, Fukushima H, Itoh H, Sasaki S, Hinoda Y, Imai K (1999) Messenger RNA expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human hepatocellular carcinoma. Jpn J Clin Oncol 29:58–62
Yang XW, Zhang LJ, Huang XH, Chen LZ, Su Q, Zeng WT, Li W, Wang Q (2014) miR-145 suppresses cell invasion in hepatocellular carcinoma cells: miR-145 targets ADAM17. Hepatol Res 44:551–559
Yeh HC, Lin SM, Chen MF, Pan TL, Wang PW, Yeh CT (2010) Evaluation of serum matrix metalloproteinase (MMP)-9 to MMP-2 ratio as a biomarker in hepatocellular carcinoma. Hepatogastroenterology 57:98–102
Yoshio T, Morita T, Kimura Y, Tsujii M, Hayashi N, Sobue K (2007) Caldesmon suppresses cancer cell invasion by regulating podosome/invadopodium formation. FEBS Lett 581:3777–3782
Yu FL, Liao MH, Lee JW, Shih WL (2004) Induction of hepatoma cells migration by phosphoglucose isomerase/autocrine motility factor through the upregulation of matrix metalloproteinase-3. Biochem Biophys Res Commun 314:76–82
Yu FL, Liu HJ, Lee JW, Liao MH, Shih WL (2005) Hepatitis B virus X protein promotes cell migration by inducing matrix metalloproteinase-3. J Hepatol 42:520–527
Yu JA, Deakin NO, Turner CE (2009) Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Mol Biol Cell 20:4706–4719
Yue Y, Shao Y, Luo Q, Shi L, Wang Z (2013) Downregulation of ADAM10 HepG2 cells. Biomed Res Int 2013:434561
Zhang Q, Chen X, Zhou J, Zhang L, Zhao Q, Chen G, Xu J, Qian F, Chen Z (2006) CD147, MMP-2, MMP-9 and MVD-CD34 are significant predictors of recurrence after liver transplantation in hepatocellular carcinoma patients. Cancer Biol Ther 5:808–814
Zhang Y, Li L, Wang Y, Zhang J, Wei G, Sun Y, Shen F (2007a) Downregulating the expression of heparanase inhibits the invasion, angiogenesis and metastasis of human hepatocellular carcinoma. Biochem Biophys Res Commun 358:124–129
Zhang H, Wang YS, Han G, Shi Y (2007b) TIMP-3 gene transfection suppresses invasive and metastatic capacity of human hepatocarcinoma cell line HCC-7721. Hepatobiliary Pancreat Dis Int 6:487–491
Zhang Q, Zhou J, Ku XM, Chen XG, Zhang L, Xu J, Chen GS, Li Q, Qian F, Tian R et al (2007c) Expression of CD147 as a significantly unfavorable prognostic factor in hepatocellular carcinoma. Eur J Cancer Prev 16:196–202
Zhang R, Pan X, Huang Z, Weber GF, Zhang G (2011) Osteopontin enhances the expression and activity of MMP-2 via the SDF-1/CXCR4 axis in hepatocellular carcinoma cell lines. PLoS One 6:e23831
Zhang J, Zhang DL, Jiao XL, Dong Q (2013a) S100A4 regulates migration and invasion in hepatocellular carcinoma HepG2 cells via NF-kB-dependent MMP-9 signal. Eur Rev Med Pharmacol Sci 17:2372–2382
Zhang Y, Tan YF, Jiang C, Zhang K, Zha TZ, Zhang M (2013b) High ADAM8 expression is associated with poor prognosis in patients with hepatocellular carcinoma. Pathol Oncol Res 19:79–88
Zhao XL, Sun T, Chen N, Sun D, Zhao N, Dong XY, Gu Q, Yao Z, Sun BC (2011) Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition regulator Twist1. J Cell Mol Med 15:691–700
Zheng Q, Tang ZY, Xue Q, Shi DR, Song HY, Tang HB (2000) Invasion and metastasis of hepatocellular carcinoma in relation to urokinase-type plasminogen activator, its receptor and inhibitor. J Cancer Res Clin Oncol 126:641–646
Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF (2012) Downregulation of the Notch signaling pathway inhibits hepatocellular carcinoma cell invasion by inactivation of matrix metalloproteinase-2 and -9 and vascular endothelial growth factor. Oncol Rep 28:874–882
Zhou L, Zhang N, Li QJ, Sun W, Zhang Y, Wang DS, Dou KF (2013a) Associations between high levels of Notch1 expression and high invasion and poor overall survival in hepatocellular carcinoma. Tumour Biol 34:543–553
Zhou L, Zhang N, Song W, You N, Li Q, Sun W, Zhang Y, Wang D, Dou K (2013b) The significance of Notch1 compared with Notch3 in high metastasis and poor overall survival in hepatocellular carcinoma. PLoS One 8:e57382
Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF (2013c) The down-regulation of Notch1 inhibits the invasion and migration of hepatocellular carcinoma cells by inactivating the cyclooxygenase-2/snail/E-cadherin pathway in viro. Dig Dis Sci 58:1016–1025
Zhou Y, Li Y, Ye J, Jiang R, Yan H, Yang X, Liu Q, Zhang J (2013d) MicroRNA-491 is involved in metastasis of hepatocellular carcinoma by inhibition of matrix metalloproteinase and epithelial to mesenchymal transition. Liver Int 33:1271–1280
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this entry
Cite this entry
Zimmermann, A. (2017). Mechanisms of Invasion and Metastasis: Tissue Invasion. In: Tumors and Tumor-Like Lesions of the Hepatobiliary Tract. Springer, Cham. https://doi.org/10.1007/978-3-319-26956-6_182
Download citation
DOI: https://doi.org/10.1007/978-3-319-26956-6_182
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26954-2
Online ISBN: 978-3-319-26956-6
eBook Packages: MedicineReference Module Medicine