
Abstract
Polyolefins are among the top 10 products in chemical industry. The annual production of polyethylene (PE) and polypropylene (PP) occupies 50 % of plastic production worldwide and will be 60 % upon including polystyrene. The annual production of PE and PP is estimated to exceed 130 million tons as they are the most widely used polymers in the world. They are commonly produced using Ziegler and chromium-based catalysts.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Coordination Polymerization
- Ethylene Polymerization
- Anionic Polymerization
- Polymerization Catalyst
- Olefin Polymerization
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
3.1 Introduction
Polyolefins are among the top 10 products in chemical industry. The annual production of polyethylene (PE) and polypropylene (PP) occupies 50 % of plastic production worldwide and will be 60 % upon including polystyrene. The annual production of PE and PP is estimated to exceed 130 million tons as they are the most widely used polymers in the world. They are commonly produced using Ziegler- and chromium-based catalysts. The continued increase in global production and consumption (about 7 % annual increase) of polymeric and petrochemical materials in the last two decades has resulted in widespread academic and industrial research activities, particularly in the field of polyolefins [ 1, 2 ]. During the last few decades, development of knowledge in olefinic polymers passed through several distinct periods. This chapter will be concerned with the comprehension of the developing chemistry of olefin polymerization in some details.
3.2 Principles of Polymerization
The basic implication for occurrence of polymerization is to have a monomer capable of linking by chemical reaction. Keeping this concept into account, many polymers comprising various properties have been prepared. Addition polymerization processes can take place through more than one type of mechanism based on the initiator. In the addition polymerization, the produced polymers have identical empirical formula to that of the monomers from which they are formed.
The more commonly used descriptive name “chain-growth polymerization” is given to these addition polymerization synthetic methods.
In this polymerization, chain will grow after an initiating step. As a result, a macromolecule with a reactive end-group that can be an anion, radical, or cation is formed. In the sections below, the detailed mechanism for each type of polymerization will be discussed.
3.2.1 Free Radical Mechanism
A free radical is a short-lived intermediate. It is a species possessing an unpaired electron due to deficiency of one electron and usually results from homolytic cleavage of a covalent bond or addition of radical to a multiple bond. A typical carbon radical is sp2 hybridized with the unpaired electron in the perpendicular unhybridized p-orbital.
Radical polymerization is the most widely practiced method at the present and in the old times [3]. It is commonly adopted in polymerization of olefins because C=C bond is most susceptible to be attacked by a radical during polymerization. This would result in a new radical active center (then the process will go on and on), as demonstrated in Scheme 3.1.

Free radical polymerization propagation step
With every addition of radical to an olefinic monomer, the radical active center is transferred to a newly formed chain end [3, 4].
As of 1990, this conventional free radical polymerization is developed to the newer reversible deactivation radical polymerization. More details of each will be discussed here.
3.2.1.1 Mechanism of Conventional Free Radical Polymerization
This method is divided into three distinct steps, namely initiation, propagation, and termination [5].
-
(A)
Initiation
It mainly comprises the formation/creation of the free radical active center by the addition of radical initiator to the monomer molecule. The following list of reactions shows the formation method of most commonly used radical initiators.
-
(i)
Homolysis (thermal homolytic cleavage)
This process takes place under the influence of heat (thermolysis) or ultraviolet radiation (photolysis). The most common examples are shown in Scheme 3.2.
Scheme 3.2 
Free radical initiators through thermal homolytic cleavage
-
(ii)
Photolysis (photochemical initiators)
Homolytic bond cleavage could be brought about through ultraviolet radiation rather than heating, and examples are shown in Scheme 3.3.
Scheme 3.3 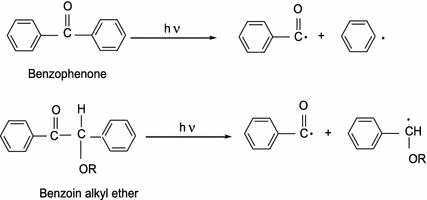
Radical initiators produced through photolysis
-
(iii)
Redox reactions (single-electron transfer)
This process is usually adopted when heating or photolysis is not appropriate. Free radical is produced through a redox reaction including an electron transfer process (see Scheme 3.4).
Scheme 3.4 
Radical initiators through single-electron transfer
Redox reactions are also observed when (Fe2+ or Co2+) is added to peroxides or hydroperoxides.
The generated radical initiator now attacks the olefinic monomer so that it adds to the least hindered carbon and produces the more stable radical. Steric and mesomeric (electronic) effects contribute to location of radical addition as demonstrated in the following diagram:

-
(i)
-
(B)
Propagation
This step involves a fast sequence of monomer additions leading to molecular growth and eventually to polymer. Several thousands of monomers can be added in few seconds to ethylene (a) or higher alkenes (b) as shown in Scheme 3.5.
Scheme 3.5 
a Propagation of addition reaction of olefin and b propagation of higher olefins (R=alkyl group) head to tail addition
-
(C)
Termination
Termination process can take place via either (a) coupling of two macroradicals or (b) disproportionation reaction, thus destroying the active center. Coupling of two growing chains would lead to a single linear polymer chain both with initiator fragment and with the other chain end, as shown in Scheme 3.6.
Scheme 3.6 
Chain termination reactions
Chain termination can also occur through chain transfer mechanisms by which the radical electron is transferred to other chain or molecule in the reaction medium.
Detailed strategies for performing industrial free radical polymerization are beyond the scope of this chapter.






3.2.2 Ionic Polymerization
In this type, olefinic polymerization proceeds via ionic active center. Ionic active center could be cationic or anionic depending on the stabilization affected by substituent. Stabilization can take place by inductive and/or mesomeric effects.
Cationic active center will be stabilized by electron-donating substituents that delocalize positive charge, while anionic active center is favored with electron-withdrawing or electronegative substituents which delocalize negative charge as shown in the following (a) carbocationic and (b) carbanionic structures:
Both cationic and anionic polymerization can be utilized when substituent on active center is capable of delocalizing both positive and negative charges (e.g., styrene and 1, 3-butadiene). The counterion in ionic polymerization has a significant effect on the stereochemistry of the resulting polymer.
Termination step of ionic polymerization takes place through a different path from that of radical polymerization. The following discussion will shed some light on ionic polymerization.
3.2.3 Cationic Polymerization Mechanism [6, 7]
-
(A)
Chain initiation
The cationic active center will be the product of adding an electrophile to the alkylated olefin (e.g., adding H+ from sulfuric or perchloric acid) as initiator. More commonly used initiators are Lewis acids such as AlCl3, BF3, or SnCl4 which are used with water or alkyl halide.

-
(B)
Chain propagation:
An attack on the olefinic monomer by the electrophile (H+ or R+) will generate the cationic active center (carbocation). The addition of the electrophile is oriented toward production of the more stable carbocation (Markovnikov orientation) as given in Scheme 3.7.
Scheme 3.7 
Chain propagation step in cationic polymerization
-
(C)
Chain termination step:
Chain reaction termination could be accomplished by two plausible mechanisms, namely
-
(a)
removal of vicinal hydrogen to give terminal C=C bond at the end of the polymer
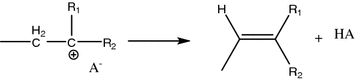
-
(b)
chain transfer of vicinal (H+) to a molecule of the monomer which results in the formation of monomer carbocation according to the following equation:

-
(a)




3.2.4 Anionic Polymerization
Bases were used as polymerization catalyst since early days of polymerization studies, but showed limited usefulness due to the encountered low extent of polymerization. Reaction conditions are adjusted to enhance living anionic polymerization as applied to styrene in liquid ammonia through initiation by K+NH2 −. Addition of the K+ NH2 − to olefinic part of the monomer generates the intermediate carbanion which propagates until termination is attained by abstraction of proton by the anion from the ammonia. If this type of termination occurred fast, then a limited polymerization process would be attained as shown in Scheme 3.8.

Anionic polymerization mechanism
In the absence of ammonia, the carbanionic polymer would stay alive and will grow again if more monomer is added. Butyl lithium was also used as initiator for anionic polymerization by adding the Bu− to the vinylic group of styrene (using THF as solvent would coordinate with Li+ ion).
In the termination step, the living carbanionic polymer can be deactivated by proton abstraction from protic solvent, by which process the end of the polymer is saturated.
Another way to deactivate carbanion is by reaction of the carbanion with carbon dioxide and dilute acid to give carboxylic terminal group or by reaction with alkene oxide (epoxide) to give a terminal alcohol.
3.3 Stereochemical Implications and Tacticity
Polymerization of olefins results in the variation of geometrical and configurational arrangements (named tacticity) [8]. Having control of these arrangements is an issue of particular importance because tacticity affects the physical properties of the polymer.
Atactic polymers are generally amorphous, soft, and flexible. Isotactic and syndiotactic polymers on the other hand are more crystalline (less flexible).
When the monomer comprises an olefinic carbon attached to two different groups (R and H), a chiral carbon will result upon polymerization. The groups R and H in the polymer can have either one of the two arrangements.
Upon polymerization, the resulting type of isomeric form (of the repeating monomer) will be important and might lead to any of the three modes of tacticity, namely
-
1.
Isotactic: In this mode, all repeated units show same configuration.

-
2.
Syndiotactic: Configuration of repeated units alternates between unit and the next in a synchronized manner.

-
3.
Atactic: In this mode, configuration of repeated units is placed randomly (irregularly) in an unpredictable manner.




In commercial application of polymers, isotactic is the most desired mode due to its crystalline nature and good mechanical properties (e.g., propylene). The atactic analogue with its irregular structure is found as amorphous material, waxlike (not very useful mechanical properties).
When the counterion is strongly coordinating with the active center of polymer’s terminal unit and the incoming monomer, isotactic polymer will be more favored. This task is hard to achieve when nonpolar monomers are incorporated. Polarity is attained when cationic or anionic polymerization mechanism is adopted. It is also useful to employ nonpolar solvent at low-temperature conditions. Polar solvents would disrupt coordination and consequently lose stereochemical control leading to syndiotactic or atactic polymer.
3.4 Stereo Chemistry of Conjugate Diene Polymerization
Conjugated dienes upon polymerization would result in either cis or trans isomeric polymer. One of the most important examples of this class is isoprene. Example of these isomeric polymers is shown in Scheme 3.9.

Cis and Trans isomeric polymers of isoprene
Natural polymerization of isoprene (natural rubber) produces the less symmetrical cis isomer as can be seen in the up- and down-orientation of the methyl group. This will result in the amorphous natural rubber. The synthetic more symmetrical trans isomer is a crystalline hard and rigid solid and will be obtained upon polymerization at low temperature. This transoid geometry is more stable than the cisoid due to the existence of the methyl group which facilitates the addition upon trans orientation.
Anionic isoprene polymerization using nonpolar solvent and Li+ counterion leads to predomination of the cis polymer. This is due to the formation of the coordinated intermediate shown below:
Coordination effect on stereochemistry of polymers inspired scientists to look for various catalysts that would result in a selected type of polymer. In the following section, the application of transition metal-based catalysts in polymerization of olefins will be demonstrated.
3.5 Coordination Polymerization
Coordination polymerization of olefins was first proposed in 1956 for the unusual, at that time, low-pressure polymerization of ethylene and polymerization of propylene with the transition metal catalysts discovered by Ziegler in 1953, and for the ferric chloride catalyzed ring-opening polymerization of propylene oxide to crystalline polymer reported by Pruitt et al. in a Dow patent. Polymerization carried out in the presence of a coordination catalyst is referred to as “coordination polymerization”. This term is used when each polymerization step involves the complexation of the monomer before its enchainment at the active site of the catalyst [9].
The majority of polyolefins is produced with titanium (Zeigler catalysts) and zirconium (metallocene catalysts) or by a free radical process (low-density polyethylene (LDPE)). Recently, late transition metals (LTMs), in particular nickel and palladium [10, 11], and iron and cobalt, are seeing a renewed interest as olefin polymerization catalysts [12, 13].
Interestingly, the discovery of methylaluminoxane (MAO) in 1980 played a crucial role in determining the course of action in polymer science [14]. This co-catalyst replaced the alkyl aluminum compounds because it acts not only as alkylating agent, but also as scavenging agent that led to a remarkable increase in the activity of the catalysts.
Unbridged metallocenes, in which the backbone of the catalyst is loosely bound, were the driving force for the development of the next generation of polymerization catalysts because of different microstructures produced due to the change in the symmetry of the active site of the metal center involved. Thus, a new horizon was born toward the synthesis of controlled microstructures.
In 1984, Ewen first reported the use of metallocene-based catalysts for the isospecific polymerization of propylene [15] and the polymerization of propylene at −45 °C using a Cp2TiPh2 (Fig. 3.1) and MAO. The catalyst system produced a partially isotactic polymer with a pentad content (mmmmm) of about 52 %, and a probability of finding meso dyads is P m = 0.85.

C 2v -Ewen’s precursor for the synthesis of partially isotactic polypropylene
Kaminsky et al. [16] reported the production of highly isotactic PP from the activated zirconium-based metallocene [ethylene bis(4,5,6,7-tetrahydro-1-indenyl)zirconium(IV) dichloride]; [(ether)ZrCl2], (Fig. 3.2) MAO serves as a co-catalyst. Their publication in Angewandte chemie can be considered as the starting point for a worldwide competition in the search for molecularly defined polymerization catalysts and for new materials [16].

Structure of a bridged C 2 -(ebthi) ZrCl2) precatalyst
MAO can form a complex with the metallocene even at −60 °C as proved by IR measurement, followed by a rapid alkylation of the transition metal center and dissociation of the complex into an ion pair (equations below). This pathway is considered as the initiation step in α-olefin polymerization with the aid of MAO [17].
According to this pathway, the Cl− or CH3 − can be abstracted by the bulky co-catalyst MAO (Me2AlO-(MeAIO)x-OAIMe2 with high molecular weight and 5 < x < 20) or borate under the formation of the bulky co-catalyst anion and a metallocene cation with a weak back-donation. The polymerization then happens by the coordination and subsequent insertion of the olefin into the metal–carbon bond of the catalytic active species, L2Zr-CH2R+. Most conventional MAO-activated metallocenes require a large excess of MAO with [AI]/[M] molar ratios >100 in order to shift the equilibrium toward the formation of active cationic metallocenes at the expense of inactive neutral complexes.
Many new developments on the so-called constrained geometry catalyst (CGC) (Fig. 3.3) system have been reported in the patent literature and were reviewed [18].

Structure of the constrained geometry catalyst: [{(tert-butylamido-N-dimethylsilyl}-(η 5-cyclopentadienyl) zirconium (IV) dichloride]
Worldwide research accomplishments on single-site metallocene-based catalysts have led to convincing improvements of poly(α-olefin) materials as well as to understanding of basic reaction mechanisms responsible for the growth of a stereoregular polymer chain at a metal center [10, 11, 16]. However, this catalyst generation allows only the use of a limited number of polar monomers bearing sterically hindered functionalities due to the sensitivity of early transition metal complexes to electron-donating functional groups [19]. Thus, copolymers of technically important polar monomers with ethylene are produced exclusively by radical polymerization routes in high-pressure processes. Therefore, there is still an unlimited interest to discover and develop new families of polymerization catalysts that can allow more control on the polymer material properties. Subsections below represent some of the most successfully applied transition metal-based polymerization catalysts.
3.6 Development of Polymerization Catalysts
3.6.1 α-Diimine-Based Catalysts
LTM-based catalysts are not as sensitive as Ziegler–Natta and metallocene catalysts to functional groups. Therefore, and because of the weak oxophilicity character, late transition metal complexes are the most attractive candidates because of their tolerance toward polar functionalities. However, until mid-1990s, only few reports were introduced utilizing these compounds as catalysts for the polymerization of α-olefins and ethylene [10, 11]. This could be due to the fact that these catalysts generally exhibit reduced activities for olefin insertion and β-hydride elimination, which steadily competes with chain growth resulting in the formation of oligomers.
In 1995, the quantum leap happened in the utilization of LTMs in the polymerization of ethylene, after the discovery of the α-diimines-based palladium(II) and nickel(II) catalysts (Scheme 3.10) which became the first polymerization catalysts to give high molar mass PE. This was attained by the application of sterically o-protected auxiliary ligands of the imino moiety, in such a way that the bulky substituents on the aryl groups of the ligands block an associative olefin exchange, thus effectively retarding chain transfer processes [20].

The highly active nickel (II) α-diimine-based catalysts reported by Johnson et al. for the polymerization of ethylene
Killian et al. [21] described the well-performing α-diimine-based catalysts after the development of a procedure for the living polymerization of α-olefins based on Ni α-diimine catalysts and application of this procedure to the synthesis of diblock and triblock poly(α-olefins) (Scheme 3.11). Moreover, they found that the ratio of 1,2—versus 2,1-insertion is sensitive to the nature of the α-diimine ligand [21].

Polymerization of α-olefins by α-diimine NiII-based catalyst
3.6.2 2,6-Bis(Imino)Pyridine-Based Catalysts
LTM complexes have attracted increasing attention [22, 23], especially after the reports of Brookhart and Gibson about new Fe (II)-based complexes containing 2,6-bis(imino) pyridyl ligands as efficient catalyst precursors for ethylene polymerization (Scheme 3.12). After MAO activation, the complexes show high activity and produce strictly linear PE [24]. The parent catalysts that were based on iron(II)- and cobalt(II)-based precursors showed that there is a clear relationship between the molar mass of the polymer produced and the bulky groups on the ortho position of the aromatic auxiliary ligands.

Brookhart/Gibson-type BIP polymerization catalyst
The presence of the two di-iso-propyl groups on the ortho position of the imino moiety produces strictly linear PE, thus blocking the chance of the β-hydride elimination step.
Following that year, many work groups studied the theoretical concepts concerning this type of catalytic system. Griffiths et al. [27] presented the first theoretical studies on this catalyst system, having assumed the generally accepted Cossee–Arlman polymerization mechanism [25–27] (Scheme 3.13).

Cossee–Arlman mechanism (Ziegler–Natta Ti-based catalyst), asterisk The other Ti atom was omitted for clarity
Abu-Surrah et al. [29] also reported on highly active 2,6-bis(arylimino) pyridine iron(II)- and cobalt(II)-based ethylene polymerization catalysts which lack the ortho alkyl substituents on the aryl groups. Modifications of the steric bulkiness of the aromatic groups in the tridentate ligands influenced not only the catalytic activity, but also the molecular weight, and for the first time the microstructure of the resulted material (Scheme 3.14) [28].

2,6-bis(arylimino)pyridineiron(II)- and cobalt(II)-based ethylene polymerization catalysts which lack the ortho alkyl substituents
McTavish et al. [30] reported another form of catalysts containing BIP-based ligands. The resultant iron dichloride complexes were highly active ethylene polymerization catalysts after the activation with methylaluminoxane (MAO). Activities in the range of 3000–18,000 g/mmol bar h were reached. The molecular weights (M n ) of the resultant PEs lie in the range of 6500–24,000 with broad molecular weight distributions (16.5–38.0). The nature of the imine carbon substituent has a marked effect on the polymer molecular weight, whereas the catalyst activity is largely unaffected by changes to this substituent [29].
The catalytically active species formed by the treatment of 2,6-bis(imino) pyridine iron(II) chloride complexes with MAO is generally proposed to be a highly reactive monomethylated iron(II) cation [LFe-Me]+ (L = 2,6-bis(imino)pyridine ligand) bearing a weakly coordinating counteranion [Me-MAO]−. Both monochloride and monoalkyl cationic species are expected to be present in the solution, their relative concentration depending on the MAO/Fe ratio [24].
At high loadings, the active species will be the dimethylated product, which does not polymerize ethylene alone, followed by reductive elimination (path B) with aluminum metal center to give the active species (Z; Scheme 3.15), which is presumably the same for metallocene-mediated MAO polymerization [23].

General mechanism for the activation processes, where open square represents the vacant coordination site
Scheme 3.16 represents different chain termination pathways, in which path (D) represents a propagation mechanism following a Cossee–Arlman mechanism involving migratory insertion of ethylene into a metal alkyl bond. The latter pathways are chain transfer pathways: β-H transfer reactions to the metal (two types of path E) or the monomer (path F) give one double bond per polymer chain (vinyl end groups), whereas chain transfer to aluminum (path G) gives saturated polymer chains. NMR and computational studies of cobalt complexes by Gibson and co-workers [30] have shown that β-H chain transfer proceeds via a stepped mechanism involving a cobalt hydride species (path E).

Different chain termination pathways
3.6.3 Salicyldimine-Based Complexes
Salen-type complexes are a fundamental class of compounds in coordination chemistry, known since 1933. They have been extensively studied, and more than 2500 have been synthesized. Interest in salen-type complexes intensified in 1990 when the groups of Jacobsen and Katsuki discovered the enantioselective epoxidation of unfunctionalized alkenes using chiral Mn(salen) complexes as catalysts. Since that time, an extremely wide variety of reactions catalyzed by salen complexes have been investigated.
3.6.4 Early Transition Metal-Based Phenoxy-Imines
In 1998, ligand-oriented design led to the discovery of phenoxy-imines that were utilized using group 4 metals initiated by Mitsui Chemicals led by T. Fujita and co-workers under the name “FI catalysts” which stands for the Japanese pronunciation of the ligand “Fenokishi-Imin Haiishi” and, at the same time, for “Fujita group Invented catalysts” [31, 32].
Phenoxy-imine ligands have the advantageous properties of diversity and tunability. Within the framework, there are three readily changeable substituents, which will sterically and electronically affect polymerization reactions (R1–R3, Fig. 3.4).

General structure of the FI-salicyldimine-based ligand
Those types of catalysts were based on continuous trial and error, and they found that group 4 transition metal complexes having phenoxy-imine ligands displayed very high activity for ethylene polymerization at 25 °C under atmospheric ethylene pressure. e.g. (complex 1, Fig. 3.5) bis[N-(3-tert-butylsalicylidene)anilinato]zirconium(IV) dichloride, exhibited 519 kgPE/mmolZr × h of activity, which is almost 20 times higher than the activities observed with Cp2ZrCl2/MAO (27 kgPE/mmolZr h) under the same polymerization conditions [33].

Structure of FI-zirconium (IV) and its Ti(IV)-phenoxy-imine correspondent
Fujita and co-workers reported on the catalytic behavior of fluorinated bis(phenoxy-imine) titanium complexes bearing a series of substituents ortho to the phenoxy oxygen for ethylene/higher α-olefin (i.e., 1-hexene, 1-octene, and 1-decene) (co)polymerization (Fig. 3.6) [27].

General formula of the fluorinated FI-Ti(IV)-based catalysts
Independent of the magnitude of steric bulk of the ortho substituent, also, all complexes that were investigated produced PEs and ethylene/higher α-olefin copolymers with very narrow molecular weight distributions [e.g., PEs, M w/M n = 1.05–1.16, M. 44,000–412,000; ethylene and 1-hexene copolymers, M w/M n = 1.07–1.19, M n 49,000–102,000, 1-hexene content 3.2–22.6 mol%], indicative of living polymerization. The incorporation ability for higher α-olefins is highly dependent on the nature of the ortho substituent, and Ti complexes with a sterically less encumbered ortho substituent incorporated a higher amount of higher α-olefins. A number of unique block copolymers consisting of linear PE and ethylene/1-hexene copolymer segments were prepared using one of the living catalysts with enhanced incorporating capability for higher α-olefins. These block copolymers exhibited lower peak melting temperatures (T m) relative to the corresponding homo-PE.
3.6.4.1 Late Transition Metal Salicyldimine Complexes
Complexes containing ligands of N,O-chelate are particularly interesting and challenging for catalysis by mixed-donor ligand complexes, such as the Ni-based systems were shown to be effective in ethylene polymerization, because of their ease of preparation and simple modification of both steric and electronic effects.
Several nickel(II) salicylaldiminato systems have been developed which are highly active for the polymerization of ethylene (Fig. 3.7). Furthermore, bulky substituents in the 3-position of the salicylaldiminate ring were found to enhance the activity of the catalyst and lower the number of branches in the resulting PE. An electron-withdrawing group in the 5-position of the salicylaldiminate ring also increases catalyst activity. With these systems, moderately high molecular weight polymer with about 10–50 branches per 1000 carbons can be obtained. As observed in other late metal systems, branching can be controlled by the variation of both temperature and pressure [34].

Structural representation of the most productive catalyst reported in the study, using (Ni(COD)2 as a co-catalyst)
Zhang et al. [36] reported that a series of neutral Ni (II) complexes derived from anilino-substituted enone ligands bearing electron-withdrawing trifluoromethyl and trifluoroacetyl groups have been synthesized (Fig. 3.8). When activated with either Ni(COD)2 or B(C6F5), these complexes were active for the polymerization of ethylene to branched PEs. Complex is especially active and long-lived, with a turnover frequency (TOF) of 5 × 105 at 60 °C and 200 psig of ethylene, a half-life exceeding 15 h at 35 °C, and a total turnover number exceeding 106 at 35 °C and 200 psig [35].

Synthesis of substituted enone SD-NiII-based complex
3.6.5 Quinaldimine-Based Complexes
Quinaldimines are attractive special type of aldimine family that are used as backbone for the preparation of catalytically active material for the polymerization of ethylene or those containing polar functionalities especially (meth) acrylates.
Abu-Surrah et al. [37] reported the synthesis of C 2 -symmetric iron (II) and cobalt(II) complexes bearing new tetradentate ligands based on 2-bromomethylquinoline. The isolated complexes were the first C 2 -symmetric octahedral complexes that resemble the structure of the tetrahedral, C 2 -symmetric metallocene-based catalysts (Scheme 3.17) [36].

Fe(II)-quinaldimine-based complex
Britovsek et al. [38] reported a bidentate imino quinolinyl nickel dibromide complex (Fig. 3.9) for the polymerization of ethylene, but it showed moderate activity toward the formation of oligomers rather than polymers [37].

Structural representation of the bidentate Ni(II)-based complex
Yliheikkila et al. [39] reported a series of manganese (II) dichloro complexes for ethylene polymerization using MAO as a co-catalyst (Fig. 3.10) [38]. From the series are two of the most active octahedral manganese (II) complexes and those bearing tetradentate nitrogen ligands with chiral backbone [39], (Abu-Surrah et al. [40]). The highest activity in ethylene polymerization \(\left( { 6 7.0\,{\text{kg}}_{\text{PE}} \,{\text{mol}}^{ - 1}_{\text{Mn}} \,{\text{h}}^{ - 1} } \right)\) was obtained with A 1/MAO at 80 °C under 5 bars of ethylene pressure.

Quadridentate octahedral Mn(II)-based complexes
3.6.6 Ruthenium-Based Polymerization Catalysts
Olefin metathesis (carbene) polymerization is also known as ring closure metathesis. It was introduced by Grubbs and his co-workers in the 1970s and became an important organic synthesis tool. It included the stable ruthenium-based catalyst [40]. Later, it was subjected to much advancement and became more versatile tool to make new polymers versatile [41].
In metathesis polymerization, the catalytically active species is a stable metal–carbene bond that is formed between the metal and the alkene. Upon reaction with cycloalkane, a living moiety capable of chain growth is formed. The olefin metathesis reaction mechanism is shown in Scheme 3.18.

Ring opening metathesis polymerization mechanism
Applications of Grubbs catalyst in ring closure metathesis and its second-generation catalyst for phase-transfer ring closure metathesis were the subject of more recent reports [42, 43].
3.7 Remarks and Outlook
Over the last 60 years, only few discoveries have had such a visible impact on the development of our modern society than Ziegler–Natta olefin polymerization catalysts. They have facilitated large-scale production of synthetic polyolefins and rubbers and subsequently the introduction of cheap commodity materials in our everyday life.
The discovery of a highly active family of catalysts based on iron, a metal that had no previous track record in this field, has highlighted the possibilities of further new catalyst discoveries. The search for new catalysts be restricted to metals that have a history of giving polymerization-active centers was no longer needed. The LTMs especially are likely to provide fertile ground for future development, and the greater functional group tolerance of the LTMs also offers the attractive prospect of polar co-monomer incorporation. A relatively small amount of functionality can dramatically transform the adhesion and wettability properties of polyolefins; more heavily functionalized products offer the prospect of materials with totally new properties and performance parameters. It is clear that, for olefin polymerization catalysis, the process of catalyst discovery and development is far from over.
Still, pollution caused by these nondegradable common plastics, oil stock declining, and concerns about greenhouse effect have directed research efforts toward sustainable alternatives to polyolefins and other fossil feedstock-derived polymers [44].
Ideally, sustainable materials should match or exceed the physical and mechanical properties of the replaced polymer, be available at a competitive price, be issued from renewable resources, and be environmentally friendly, i.e., entirely recyclable without the release of hazardous and persistent substances [45, 46].
The contemporary advancements in “olefin polymerization” impose several challenges to be faced by polymer industries.
The following is the list of the most urgent tasks:
-
1.
Reduce process variability (while retaining cost-effectiveness).
-
2.
Inefficiency and improvement of product.
-
3.
Produce prime product, while keeping up with customer demand.
-
4.
Polymer recycling and sustainability of material feedstock.
-
5.
Manufacturers are skeptical about using the latest technology.
-
6.
Gearing toward reducing pollution caused by industry and to produce less wastage.
-
7.
High demand for new and advanced material with the need to find solutions to growing market and upscaling production.
-
8.
Quality of material on demand; high-weight material with controllable properties through structural and functional capability.
3.8 Conclusion
Olefin polymerization is becoming one of the most significant concerns in research and development of chemical industries.
The history and early developments in the pioneering work of polymers up to the most recent advancements are covered. Olefin polymerization started about 100 years back without involvement of metals, following anionic or radical pathways. The contemporary olefin polymerization industry depends mainly on the use of metal complexes as catalysts. At the present time, metal-catalyzed polymerization represents the most successful, conceivable, and sustainable procedure toward the synthesis of polyolefins. Nowadays, the list of metals includes several transition metals in the generation of catalysts. In doing their catalytic role, they follow various mechanisms that lead to a wide range of polymeric products.
The 1950s witnessed the introduction of heterogeneous metal catalysts, leading to a broad range of applications. Two decades later, metallocene led to active homogeneous catalysts which allowed the rational of specific polymer structures.
The better understanding and evolution in the 1990s led to new industrial implementations of the new post or nonmetallocenes, which is the outcome of the recent advancements. They are easier to assemble and produce higher control and activity over the desired polymer structures. This would also be reflected in endless applications of these polymers in many aspects of our life.
In conclusion, it must be emphasized that coordination polymerization of olefins is now considered among the most important areas in polymer research. This area occupies the most prominent place in polymer science and technology that chemical industries intend to cope with.
References
G. Odian (ed.), Principles of Polymerization, 4th edn. (Wiley, Hoboken, 2004) 832 p, ISBN 978-0-471-27400-1
R.J. Young, P.A. Lovelle, Introduction to Polymers, 3rd edn. (CRC press, Boca Raton, 2011), ISBN 978-0-8493-3929-5
K. Matyjaszewski, T.P. Davis (eds.), Handbook of Radical Polymerization (Wiley, Hobken, 2002)
G. Moad, D.H. Soloman, The Chemistry of Radical Polymerization, 2nd edn. (Elsevier, Oxford, 2006)
A.A. Gridnev, S.D. Ittel, Catalytic chain transfer in free radical polymerization. Chem. Rev. 101, 3611 (2001)
K. Matyjaszewski, P. Sigwalt, Unified approach to living and non-living cationic polymerization of alkenes. Polym. Int. 35, 1 (1994)
M. Sawamoto, Modern cationic vinyl polymerization. Prog. Polym. Sci. 16, 111 (1991)
L.S. Baugh, J.M.M. Canich (eds.), Stereoselective Polymerization with Single Site Catalyst (CRC Press, Boca Raton, 2008)
W. Kuran, Principles of Coordination Polymerization (Wiley, Hoboken, 2001) ISBN 0-479-84141-9
A. Abu-Surrah, B. Rieger, Late transition metal complexes: catalysts for a new generation of polymers. Angew. Chem. Int. Ed. Engl. 35, 2475–2477 (1996)
L. Johnson, C. Killian, M. Brookhart, New Pd (I1)- and Ni(I1)-based catalysts for polymerization of ethylene and α-olefins. J. Am. Chem. Soc. 117(23), 6414–6415 (1995)
B. Small, M. Brookhart, A. Bennett, Highly active iron and cobalt catalysts for the polymerization of ethylene. J. Am. Chem. Soc. 120, 4049–4050 (1998)
G. Britovsek, V. Gibson, B. Kimberley, P. Maddox, S. McTavish, G. Solan, A. White, D. Williams, Novel olefin polymerization catalysts based on iron and cobalt. Chem. Commun. 7, 849–850 (1998)
W. Kaminsky, The discovery of metallocene catalysts and their present state of the art. J. Polym. Sci. A: Polym. Chem. 42, 3911–3921 (2004)
J. Ewen, Mechanisms of stereochemical control in propylene polymerizations with soluble group 4B metallocene/methylalumoxane catalysts. J. Am. Chem. Soc. 106(21), 6355–6364 (1984)
W. Kaminsky, K. Külper, H.H. Brintzinger, F. Wild, Polymerization of propene and butene with a chiral zirconocene and methylalumoxane co-catalyst. Angew. Chem. Int. Ed. Engl. 24(6), 507–508 (1985)
W. Kaminsky, A. Funk, H. Haehnsen, New application for metallocene catalysts in olefin polymerization. Dalton Trans. 41, 8803–8810 (2009)
A. McKnight, R. Waymouth, Group 4 ansa-cyclopentadienyl-amido catalysts for olefin polymerization. Chem. Rev. 98, 2587–2598 (1998)
M. Kesti, G. Coates, R. Waymouth, Homogeneous Ziegler-Natta polymerization of functionalized monomers catalyzed by cationic Group IV metallocenes. J. Am. Chem. Soc. 114, 9679–9680 (1992)
L. Johnson, C. Killian, M. Brookhart, New Pd(I1)- and Ni(I1)-based catalysts for polymerization of ethylene and α-olefins. J. Am. Chem. Soc. 117(23), 6414–6415 (1995)
C. Killian, D. Tempel, L. Johnson, M. Brookhart, Living polymerization of α-olefins using ΝιΙΙ α-diimine catalysts. Synthesis of new block polymers based on α-olefins, J. Am. Chem. Soc. 118(46), 11664–11665 (1996)
C. Pellecchia, M. Mazzeo, D. Pappalardo, Isotactic-specific polymerization of propene with an iron-based catalyst: polymer end groups and regiochemistry of propagation Macromol. Rapid Commun. 19(12), 651–655 (1998)
V. Gibson, C. Redshaw, A. White, D. Williams, Novel aluminum containing ring systems: an octanuclear structural analogue of calix [4]pyrrole. Angew. Chem. Int. Ed. Engl. 38(7), 961–964 (1999)
V. Gibson, S. Spitzmesser, Advances in non-metallocene olefin polymerization catalysis. Chem. Rev. 103(1), 283–315 (2003)
B. Small, M. Brookhart, A. Bennett, Highly active iron and cobalt catalysts for the polymerization of ethylene. J. Am. Chem. Soc. 120, 4049–4050 (1998)
G. Britovsek, V. Gibson, B. Kimberley, P. Maddox, S. McTavish, G. Solan, A. White, D. Williams, Novel olefin polymerization catalysts based on iron and cobalt. Chem. Commun. 7, 849–850 (1998)
E. Griffiths, G. Britovsek, V. Gibson, I. Gould, Highly active ethylene polymerization catalysts based on iron: an ab-initio study. Chem. Commun. 14, 1333–1334 (1999)
P. Cossee, Ziegler-Natta catalysis I. Mechanism of polymerization of □-olefins with Ziegler-Natta catalysts. J. Catal. 3, 80–88 (1964)
A. Abu-Surrah, K. Lappalainen, U. Piironen, P. Lehmus, T. Repo, M. Leskela, New bis(imino)pyridine Iron(II) and Cobalt(II)-based catalysts: synthesis, characterization and activity towards polymerization of ethylene. J. Organomet. Chem. 648, 55–61 (2002)
S. McTavish, G. Britovsek, T. Smit, V. Gibson, A. White, D. Williams, Iron-based ethylene polymerization catalysts supported by Bis(imino)pyridine ligands: derivatization via deprotonation/alkylation at the ketimine methyl position. J. Mol. Cat. A: Chem. 261, 293–300 (2007)
K. Tellmann, M. Humphries, H. Rzepa, V. Gibson, Experimental and computational study of β-H transfer between cobalt (I) and alkenes. Organometallics 23(23), 5503–5513 (2004)
V. Gibson, K. Tellmann, M. Humphries, D. Gould, Bis(imino)pyridine cobalt alkyl complexes and their reactivity towards ethylene: a model system for β-hydrogen chain transfer. Chem. Commun. 20, 2316-2317 (2002)
R. Furuyama, M. Mitani, J.-I. Mobri, R. Mori, H. Tanaka, T. Fujita, Ethylene/higher α olefin copolymerization behavior of fluorinated bis(phenoxy- imine) titanium complexes with methylalumoxane: synthesis of new polyethylene-based block copolymers. Macromolecules 38(5), 1546–1552 (2005)
H. Makio, N. Kashiwa, T. Fujita, FI catalysts: a new family of high performance catalysts for olefin polymerization. Adv. Synth. Catal. 344(5): 477–493 (2002)
C. Wang, S. Friedrich, T. Younkin, R. Li, R. Grubbs, D. Banseleben, M. Day, Neutral nickel(II)-based catalysts for ethylene polymerization. Organometallics 17(15), 3149–3151 (1998)
L. Zhang, M. Brookhart, P. White, Synthesis, characterization, and ethylene polymerization activities of neutral nickel(II) complexes derived from anilino-substituted enone ligands bearing trifluoromethyl and trifluoroacetyl substituents. Organometallics 25(8), 1868–1974 (2006)
A.S. Abu-Surrah, B. Rieger, R. Fawzi, M. Steiman, Synthesis of chiral and C2-symmetric iron(II) and cobalt(II) complexes bearing a new tetradentate amine ligand system. J. Organomet. Chem. 497, 73–79 (1995)
G. Britovsek, S. Baugh, O. Hoarau, V. Gibson, D. Wass, A. White, D. Williams, The role of bulky substituents in the polymerization of ethylene using late transition metal catalysts: a comparative study of nickel and iron catalyst systems. Inorg. Chimica Acta 345, 279–291 (2003)
K. Yliheikkila, K. Axenov, M. Raisanen, M. Klinga, M. Lankinen, M. Kettunen, M. Leskela, T. Repo, Manganese(II) complexes in ethene polymerization. Organometallics 26, 980–987 (2007)
A. Abu-Surrah, T. Laine, R. Fawzi, M. Steiman, B. Rieger, An enantiomerically pure Schiff base ligand. Acta Cryst. C53, 1458–1459 (1997)
R.H. Grubbs, Olefin Metathesis catalyst for the preparation of Molecules and Materials. Angew. Chem. Int. Ed. 45, 3760–3765 (2006)
G.C. Vougioukalakis, R.H. Grubbs, Ruthenium-based heterocyclic carbene- coordinated olefin metathesis catalysts. Chem. Rev. 110, 1746–1787 (2010)
E. de Brito Sa’, J.M.E. de Matos, Ring closing metathesis by Hoveyda-Grubbs catalysts: a theoretical approach of some aspects of the initiation mechanism and the influence of solvent. Inorg. Chim. Acta 426, 20–28 (2015)
Y. Kobayoshi, S. Inukai, T. Watanabe, Y. Sogiyama, H. Hamamoto, T. Shioiri, M. Matsugi, A medium fluorous Grubbs-Hoveyda 2nd generation catalyst for phase transfer catalysis of ring closing metathesis reactions. Tet. Lett. 56(11), 1363–1366 (2015)
S. Mecking, Nature or petrochemistry?-biologically degradable materials. Angew. Chem. Int. Ed. 43(9), 1078 (2004)
A.J. Ragauskas, C.K. Williams, B.H. Davison, G. Britovsek, J. Cairney, C.A. Eckert, W.J. Frederick Jr, J.P. Hallett, D.J. Leak, C.L. Liotta, J.R. Mielenz, R. Murphy, R. Templer, T. Tschaplinski, The path forward for biofuels and biomaterials. Science 311, 484 (2006)
A.K. Mohanty, M. Misra, L.T. Drzal, Sustainable bio-composites from renewable resources: opportunities and challenges in the green materials world. J. Polym. Environ. 10(1–2), 19–26 (2002)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tabba, H.D., Hijji, Y.M., Abu-Surrah, A.S. (2016). Olefin Polymerization. In: Al-Ali AlMa'adeed, M., Krupa, I. (eds) Polyolefin Compounds and Materials. Springer Series on Polymer and Composite Materials. Springer, Cham. https://doi.org/10.1007/978-3-319-25982-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-25982-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-25980-2
Online ISBN: 978-3-319-25982-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)