
Abstract
The astrocytic Na+,K+-ATPase is important because increasing evidence indicates that increased extracellular K+ in brain following neuronal excitation initially is accumulated into astrocytes. This is due to higher Na+,K+-ATPase activity in astrocytes than in neurons and because the extracellular K+-sensitive site of the astrocytic Na+,K+-ATPase, in contrast to that of the neuronal enzyme, has low enough affinity for K+ to be further activated by increased K+ concentrations. However, K+ must eventually be re-accumulated into neurons in order to prevent neuronal K+ depletion. Accumulated astrocytic K+ is released through Kir4.1 channels, but a presently unsolved problem is how renewed astrocytic uptake is prevented. Experiments in well-differentiated cultured astrocytes providing a solution of this problem are discussed. At the same time subunit composition of the astrocytic Na+,K+-ATPase and its influence on the enzyme’s kinetic parameters is reviewed together with stimulation of the enzyme by noradrenaline and its functional importance. So are details of Na+,K+-ATPase signaling in response to submicromolar concentrations of ouabain and/or low mM K+ concentrations without which the catalytic activity of the astrocytic enzyme is abolished. Two pathophysiological conditions are discussed, cerebral ischemia/reperfusion and hepatic encephalopathy. In the former ouabain signaling dependence on extracellular Ca2+ is crucial and provides therapeutic possibilities. In the latter the ability of NH4 + to mimic K+ in both catalytic and signaling effects of the Na+,K+-ATPase is essential. In both conditions it is important that operation of the Na+, K+, Cl− and water cotransporter NKCC1 is dependent upon ion gradients created by the Na+,K+-ATPase.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Astrocyte
- Brain ischemia
- Brain potassium
- Glutamate
- Glycogenolysis
- Na+,K+-ATPase subunits
- Neuron
- NKCC1
- Ouabain signaling
- Potassium undershoot
1 Catalytic Activity of the Astrocytic Na+, K+-ATPase
1.1 The Astrocytic Na+,K+-ATPase, but not the Neuronal Enzyme, Is Stimulated by Above-Normal Extracellular K+ Concentrations
Activation of the Na+,K+-ATPase requires simultaneous binding of Na+ to an intracellular site and of K+ to an extracellular site of the enzyme [1, 2]. In excitable cells, such as neurons, the activity of the Na+,K+-ATPase is generally regulated by the intracellular concentration of Na+ ([Na+]i). The simultaneous increase in extracellular K+ ([K+]o) during the excitation has normally no effect on neuronal Na+,K+-ATPase activity, because the affinity of this site in neurons is so high that it is saturated at normal [K+]o. This was convincingly shown by Grisar et al. [3], who determined kinetic properties of the Na+,K+-ATPase in mechanically isolated glial cells, neuronal perikarya, and synaptosomes from rabbit brain cortex as well as human cells and observed no increases in Na+,K+-ATPase activity in the neuronal preparations (Fig. 12.1). However, it has been shown by gel electrophoresis that the brain contains two distinct molecular forms of the Na+,K+-ATPase, which can be separated in their active form by gentle tissue fractionation procedures [4]. One is the only Na+,K+-ATPase of astrocytes, while the other is the only Na+,K+-ATPase of the axonal membrane. Figure 12.1 shows that in contrast to the neuronal enzyme isolated glial cells show a distinct increase in Na+,K+-ATPase activity in response to an increase in [K+]o, confirming previous results by Henn et al. [5]. This indicates that the K+ affinity of the K+-sensitive site is lower in astrocytes than in neurons. Subsequent kinetic analysis in homogenates of cultured mouse cerebral astrocytes and neurons [6] showed conventional Michaelis–Menten kinetics with a K m value for K+ of 1.9 mM in astrocytes and a K m value of 0.43 mM in neurons. V max in astrocytes was approximately twice of that in neurons. A similar affinity for Na+,K+-ATPase-mediated K+ uptake was shown in rat cultures by Larsen et al. [7], consistent with the observation that the rate of active K+ uptake is similar in rat and mouse cultures [8].

Effect of different extracellular K+ concentrations ([K+]o) on Na+,K+-ATPase activity in astrocytes and neuronal perikarya isolated from a single human brain. From where further methodological details are described. Reproduced with permission, where further methodological details are described
The reason for the difference in K+ affinity between astrocytes and neurons is the different subunit composition. In freshly isolated cell fractions of mouse astrocytes and neurons, mRNA of the α1 subunit has twice as high an expression in astrocytes as in neurons, whereas the α2 subunit is almost restricted to astrocytes and the α3 subunit to neurons (Fig. 12.2a) Nevertheless, the traces of α3 in astrocytes and of α2 in neurons are probably representative of the in vivo situation, since cross-contamination between fractions should also have led to neuronal β2 expression. The β1 subunit is more highly expressed in neurons than in astrocytes, but only astrocytes express both β1 and β2. The expression of the auxiliary protein FXYD7 is equal in the two cell types (Fig. 12.2b). Neuron-selective expression of mRNA for α3 has also been shown by Cahoy et al. [9], and in cultures somewhat different from ours, Cameron et al. [10] reported that cortical astrocytes display α2 and β2 subunits and cerebellar granule neurons α3 and β1 subunits. The greater α1 expression in astrocytes than in neurons shown in Fig. 12.1 is also consistent with immunochemical data by MacGrail et al. [11] and with conclusions based on low-affinity ouabain binding, reflecting content of α1 protein, in our own cultured neurons and astrocytes [12].

Subunit composition of Na+,K+-ATPase from freshly isolated astrocytes and neurons obtained from mice where either the astrocyte-specific marker GFP or a neuronal marker, YFPH, had been linked to fluorescent compound, allowing isolation of an astrocytic respective a neuronal cell fraction by fluorescence-activated cell sorting (FACS). In each cell fraction mRNA expression was quantitated by reverse transcription polymerase chain reaction (RT-PCR). (A1): Products of PCR for α and β subunits and of the house-keeping gene TATA-binding protein (TBP) from three astrocytic and three neuronal samples; (A2): ratios between scanned expression of each subunit and TBP allowing quantitative determination of the expression of each subunit. Since different primers are used for each subunit the figure allows no quantitative comparison between expression of different subunits, but it provides reliable quantitation of astrocytic vs neuronal expression. (B1) and (B2): Similar results for FXYD7, the brain-specific FXYD. From Li et al. [17], reproduced with permission
Subunit composition is important for the kinetic properties of the Na+.K+-ATPase. In a study by Crambert et al. [13] nine different human Na+,K+-ATPase isozymes, composed of α and β isoforms, were expressed in Xenopus oocytes and analyzed for their transport and pharmacological properties. All human isozymes were functional but differed in their turnover rates depending on the α isoform. Variations in K+ affinity and activation were a result of a cooperative interaction between α and β isoforms with α2-β2 complexes having the lowest apparent K+ affinity. α Isoforms also influence the apparent internal Na+ affinity in the order α1 > α2 > α3 [13, 14]. FXYD7 decreases the apparent K+ affinity of α1-β1 and α2-β1, but not of α3-β1 isozymes [15]. These observations are consistent with the low affinity for K+-induced stimulation of Na+,K+-ATPase activity in astrocytes and the ensuing ability of the astrocytic enzyme to be stimulated by above-normal [K+]o. As seen in Fig. 12.3, the low affinity of the α2-β2 complex was confirmed by Larsen et al. [7], who added the new information that depolarization increased the affinity of this complex. The α2-β2 complex is preferentially immunoprecipitated in mouse brain, whereas no α1-β2 or α2-β1 complexes were demonstrated [16]. However, under some conditions, e.g., chronic treatment with the anti-bipolar drug carbamazepine the α2 subunit is induced in neurons without concomitant induction of the β2 subunit [17]. A submicromolar affinity for ouabain of the rat α2 and α3 subunits transfected into NIH 3 T3 cells is much lower than that of the α1 receptor of ~50 μM [18]. Inhibition of K+ uptake by different concentrations of ouabain is consistent with the values obtained by the binding studies ([19]; L. Hertz and W. Wal;z, unpublished experiments). The latter experiments also suggested that about three quarters of the K+ uptake was mediated by the α1 isoform.

Various subunit isoform compositions of the rat Na+/K+-ATPase were expressed in X. laevis oocytes. Na+,K+-ATPase activity as a percentage of V max was determined as a function of [K+]o and the curves fitted according to Michaelis–Menten kinetics. The graph shows the obtained activity at each [K+]o. From Larsen et al. [7], reproduced with permission
Since the Na+,K+-ATPase and the gastric H+,K+-ATPase are the only P-type ATPases forming α-β complexes, it is likely that the obligatory β subunit plays a major role for K+ transport [20, 21]. The β subunits facilitate correct membrane integration and packing of the catalytic α subunit, which is necessary for their resistance to degradation, acquisition of functional properties, routing to the cell surface, and determination of intrinsic transport properties [20]. In neurological diseases like familiar hemiplegic migraine type 2 (FHM2), the α2 subunit shows mutations which are expressed in astrocytes; some of these mutations are found close to the interaction loci between α and β subunits and another mutation causes a reduced apparent K+ affinity [21].
1.2 Both the Astrocytic and the Neuronal Na+,K+-ATPase Are Stimulated by Noradrenaline, but Different Subtype-Specific Receptors Are Involved
Both the astrocytic and the neuronal Na+,K+-ATPase are also stimulated by noradrenaline. In brain homogenates noradrenaline stimulation of Na+, K+-ATPase [22, 23] is inhibited by both α- and β-adrenergic antagonists [24]. Different noradrenergic receptor subtypes are involved in astrocytes and neurons with the β-adrenergic drug isoproterenol stimulating astrocytic but not neuronal Na+, K+-ATPase [6]. The noradrenergic stimulation occurs only at close to normal [K+]o, so any additive effect by simultaneous exposure to elevated [K+]o and noradrenaline is minimal (Fig. 12.4a) in either cell type. Rather, at aberrant [K+]o noradrenaline has an inhibitory effect, especially in neurons. Similarly K+-stimulated K+ uptake into astrocytes is only marginally increased by 1 μM isoproterenol (Fig. 12.4b). The identical effects on Na+, K+-ATPase activity and K+ uptake are important as the former is measured in a homogenate and the latter in intact cells. β1-Adrenergic stimulation of the K+ analogue rubidium has also been shown in pig hearts [25].

Effects of noradrenaline or the β-adrenergic agonist isoproterenol and of [K+]o on Na+,K+-ATPase activity or K+ uptake in cultured astrocytes. (a) Stimulation or inhibition (negative stimulation) of Na+,K+-ATPase activity in homogenates of cultured astrocytes (open columns) or mouse cerebral cortical interneurons (filled columns) by 10 μM noradrenaline at different [K+]o. The activity in the same homogenates in the absence of noradrenaline is indicated as 0 %. (b) Increase in intracellular K+ concentration in similar but intact cultures of astrocytes measured in arbitrary units by fluorescence of a K+-sensitive drug under control conditions, after addition of 1 μM of the β-adrenergic drug isoproterenol, 5 mM KCl, or simultaneous addition of isoproterenol plus 5 mM KCl. (a) From Hajek et al. [6], reproduced with permission; (b) From Hertz et al. [58], reproduced with permission
It is unknown why high K+ and noradrenaline do not have a synergistic effect on astrocytic and neuronal Na+,K+-ATPases. However, in the proximal convoluted tubule of the kidney noradrenaline acting on α-adrenergic receptors is known to stimulate Na+,K+-ATPase activity via an increase in [Ca2+]i and activation of the Ca2+-dependent protein phosphatase 2B, calcineurin [26]. The α1 isoform of Na+,K+-ATPase is the only catalytic Na+,K+-ATPase isoform expressed at this location and its dephosphorylation is increased at high [Na+]i, whereas protein kinase C (PKC) causes phosphorylation [27], which decreases Na+,K+-ATPase activity [28]. Ibarra et al. [27] concluded that the phosphorylation of a large pool of the Na+,K+-ATPase at a low [Na+]i allows dephosphorylation (and thus activation) by α-adrenergic receptor activation. The pathway for α-adrenergic stimulation of pyramidal neurons from rat cerebral cortex includes PKC stimulation [29] and increase in [Ca2+]i [30], and reduction in [K+]o increases [Na+]i in cerebral cortical neurons [31]. Similar effects as in the proximal convoluted tubule might therefore explain the noradrenergic stimulation of neuronal Na+,K+-ATPase activity at control levels of [K+]o and the lack of effect or inhibition at least at decreased [K+]o. Since β1-adrenergic stimulation of cultured astrocytes [32] leads to a G s/G i shift and subsequent PKC activation and increase in [Ca2+]i (Fig. 12.5) and [Na+]i is increased at low [K+]o [31], the interaction between [K+]o and noradrenaline on the astrocytic Na+,K+-ATPase can be explained in a similar manner at low K+. Astrocytic [Na+]i is not increased at high [K+]o [31] and the astrocytic Na+,K+-ATPase is not inhibited by noradrenaline at 12 mM [K+]o (Fig. 12.4a). It is reassuring that K+/noradrenaline interactions in cultured astrocytes may be explained by effects determined in freshly obtained cells from the rat proximal tubule.

Schematic illustration of stimulation of ERK phosphorylation by β-adrenergic receptors in astrocytes. Isoproterenol (ISO), binds to these receptors. At high concentrations (≥1 μM), the activation of the receptors induces a β1-adrenergic (red arrows), PKA-dependent “Gs/Gi switch,” which induces an enhancement of intracellular Ca2+ concentration by Ca2+ release from intracellular stores. The latter activates Zn-dependent metalloproteinases (MMPs) and leads to shedding of growth factor(s), such as heparin-binding epidermal growth factor (HB-EGF). The released HB-EGF stimulates autophosphorylation of the EGF receptor in the same and adjacent cells. The downstream target of the EGF receptor extracellular regulated kinases 1 and 2 ERK1/2 (shown in blue) is phosphorylated via Ras/Raf/MEK pathway, contingent upon recruitment of β-arrestin 1. ERK phosphorylation by isoproterenol at a high concentration can be inhibited by H-89, an inhibitor of PKA, by PTX, an inhibitor of Gi protein, by BAPTA/AM, an intracellular Ca2+ chelator, by GM6001, an inhibitor of Zn-dependent metalloproteinase, by AG1478, an inhibitor of the EGF receptor, by siRNA against β-arrestin 1, and by U0126, an MEK inhibitor (all shown in yellow). In contrast, at a low isoproterenol concentration (≤100 nM) β2-adrenergic (green arrows) activation of the receptors activates Src via the function of β-arrestin 2. Src stimulates ERK phosphorylation and phosphorylates the EGF receptor without involvement of the receptor-tyrosine kinase. ERK1/2 phosphorylation is secondary to MEK activation, which probably is induced by direct activation of Raf by Src, whereas Src-mediated phosphorylation of the EGF receptor may not participate in the phosphorylation of ERK1/2, which does not require recruitment of β-arrestin 1. The ERK phosphorylation by isoproterenol at low concentration can be inhibited by siRNA against β-arrestin 2, by PP1, a Src inhibitor, and by U0126, an MEK inhibitor. From Du et al. [32], reproduced with permission
2 Signaling Activity of the Astrocytic Na+,K+-ATPase
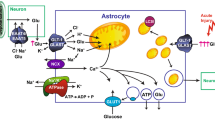
The Na+-K+-ATPase is also a signaling molecule reacting to endogenous ouabain-like compounds, which are present in brain [33, 34] including astrocytes [35], as well as to minor increases in [K+]o. Activation of the tyrosine kinase Src in intact cells by ouabain acting on the Na+,K+-ATPase was first shown by Haas et al. [36], who found rapid activation of Src when nontoxic concentrations of ouabain were added to cultured neonatal cardiac myocytes. Activation of Src stimulated a pathway leading to phosphorylation of the epidermal growth factor receptor (EGFR) via Ras and eventually to phosphorylation of extracellular regulated kinases 1 and 2 (ERK1/2). This pathway was confirmed by Zhang et al. [37], who also discovered an additional phospholipase C (PLC) and inositol trisphosphate (IP3) receptor pathway, which lead to an increase in [Ca2+]i. These pathways also operate in cultured astrocytes [38], as shown by inhibition of ERK1/2 phosphorylation induced by addition of 30 nM ouabain or 5 mM KCl by inhibitors of Src or EGF receptor phosphorylation (Fig. 12.6). An increase in [Ca2+]i by ultralow concentrations of ouabain had previously been shown in such cells by Forshammer et al. [39]. Xestospongin, an inhibitor of the IP3 receptor, inhibited a K+-induced K+ uptake (see Sect. 3). Thus, in contrast to the inhibition of the Na+,K+-ATPase by usually applied ouabain concentrations, very low concentrations, which replicate the effect of endogenous ouabains, enhance K+-mediated stimulation of the astrocytic Na+,K+-ATPase. Operation of a similar pathway in intact brain is shown by the demonstration that knock-out of the IP3 receptor, which is an intermediate in the pathway leading to the increase in [Ca2+]i, increases [K+]o, and abolishes the normal increase in [Ca2+]i in brain slices after high-frequency stimulation, and decreases the undershoot [40].

Signaling pathways in cultured astrocytes for ouabain and [K+]o increases ≤5 mM and ≥5 mM. The catalytic effects of the Na+,K+-ATPase and NKCC1 on ion fluxes are shown in the left side of the figure with Na+,K+-ATPase-mediated fluxes (+5 mM K+) in blue lettering at the bottom and NKCC1 fluxes (+10 mM K+) in red lettering at the top. All signaling pathways are shown in black lettering, with blue arrows for the signaling pathway of ouabain/Na+,K+-ATPase, activated by 30 nM ouabain or addition of 5 mM KCl (bottom) and red arrows for the signaling pathway leading to activation of NKCC1 (top). Transmembrane ion fluxes connected with signaling are shown by heavy red arrows. Increases in free cytosolic Ca2+ concentration ([Ca2+]i) are shown by black arrows. Key points were verified by abolishment of ERK phosphorylation or prevention of the normal increase in intracellular K+ content after addition of 5 mM KCl by the specific inhibitors or siRNA, shown in brown (the glycogenolysis inhibitor DAB) or yellow ovals. Note that the IP3 receptor participates in signaling after addition of 5 mM K+, but not after 10 mM K+, making its inhibitor xestospongin C an important tool for distinction between activation of the two pathways. In contrast phosphorylation of the EGF receptor (EGFR) and of extracellular regulated kinases 1 and 2 (ERK1/2), Src activation, increase in [Ca2+]i, and glycogenolysis occur in both pathways. Inhibition by amiloride of cellular increase in K+ after addition of 5 mM KCl suggests inhibition of the Na+ channel Na x . Inhibition of Ca2+ entry via the Ca2+/Na+ exchanger NCX, needed in the ouabain signaling pathway, was not tested in our experiments (but see, Fig. 12.9). However in Ca2+-free medium the K+ uptake normally induced by addition of 5 mM KCl was abolished. The pathway activated by addition of ≥10 mM KCl and leading to activation of NKCC1 shown in red in the upper part of the figure had previously been determined for inhibition of ERK phosphorylation (using specific inhibitors) and found to include depolarization-mediated L-channel opening and metalloproteinase-induced release of an agonist of EGFR causing its phosphorylation (pEGFR). Additional inhibitor experiments shown in the figure indicated its dependence on glycogenolysis (inhibition by DAB) and the metalloproteinase ADAM 17, which is not involved in the pathway activated by addition of 5 mM KCl. Signaling connections between ERK phosphorylation and activation of NKCC1 or between ERK phosphorylation or increase in [Ca2+]i and opening of Na x have not been investigated. Modified from Xu et al. [38], reproduced with permission
Even the slightest increase in [K+]o also increases glycogenolysis in brain (Fig. 12.7), and the effect increases in parallel with further augmentation of [K+]o [41]. The ouabain pathway opened by ouabain or 5 mM KCl also operates during K+-induced stimulation of glycogenolysis by small increases in [K+]o. Figure 12.8 shows that interference with the ouabain pathway (by the IP3 receptor antagonist xestospongin or a ouabain antagonist) inhibits stimulation of glycogenolysis by 5 mM KCl. In contrast nifedipine, an inhibitor of L-channel opening, does not impede glycogenolysis after addition of 5 mM K+, although it inhibits the effect of addition of 10 mM KCl. The latter finding will be discussed later in connection with increased activity of the cotransporter of Na+, K+ and 2 Cl− and water, NKCC1 [42, 43]. Here it suffices to mention that NKCC1 is a secondary active transporter, dependent on the ion gradients between extracellular and intracellular ion concentrations created by the Na+,K+-ATPase [44, 45] and that NKCC1 participates in astrocytic K+ uptake when [K+]o is increased by 10 mM or more [38]. It is also involved in the undershoot in [K+]o following intense neuronal stimulation, shown by a reduction of the undershoot by the NKCC1 inhibitor furosemide [46]. Under pathological conditions, it is of major importance for development of brain edema after ischemia/reperfusion [47] and for a NKCC1-mediated regulatory volume increase after cell shrinkage [48]. These effects will be discussed in detail in Sect. 3.

Effect of increases in [K+]o on mouse brain slices incubated in a physiological saline containing a total of 4 mM K+. Note absence of glycogenolysis at this [K+]o but increasing stimulation with increases in [K+]o. From Hof et al. [41], reproduced with permission, where further methodological details are described

Reduction of glycogenolysis, shown as decrease in glycogen content, caused by slightly elevated (+5 mM) [K+]o. The stimulation is inhibited by xestospongin and canrenone, an inhibitor of ouabain signaling, but not by nifedipine which inhibits L-channels for Ca2+ and stimulation of glycogenolysis by addition of ≥10 mM K+ [52]. These results are consistent with the pathways shown in Fig. 12.6. From Xu et al. [52], reproduced with permission
The Na+/Ca2+ exchanger NCX plays a major role in signaling by endogenous ouabains [49]. NCX is expressed in the plasma membrane, and most generally it extrudes one Ca2+ in exchange for 3 Na+. However the transporter can also mediate Ca2+ entry, and the transport direction depends on Na+ and Ca2+ gradients across the membrane and the membrane potential, which is influenced by [K+]o. Juxtaposition of plasma membrane and sarco(endo)plasmic reticulum membranes may permit NCX to regulate IP3 and ryanodine receptor-mediated Ca2+ signaling [50]. This is the case in arterial smooth muscle cells as shown in Fig. 12.9a, demonstrating that 100 nM ouabain causes an increase in [Ca2+]i, which is reduced by an NCX inhibitor and even more by removal of extracellular Ca2+. Along similar lines, Wang et al. [40] found that a PAR-1 agonist that increases [Ca2+]i in astrocytes, but apparently not in neurons [51], causes an elevation of intracellular K+ in cultured astrocytes, which is abolished by NCX inhibitors (Fig. 12.9b). The potency of ouabain as a K+ uptake inhibitor in our cultured astrocytes is greatly reduced in the absence of extracellular Ca2+ and concentrations as high as 0.1 and 0.3 μM ouabain may have a stimulatory effect (Fig. 12.9c). In these cultures uptake of K+, induced by a 5 mM increase in [K+]o (and thus dependent on nanomolar ouabain signaling), is abolished during incubation in Ca2+ free medium [47], whereas that evoked by the β1-adrenergic agonist dobutamine is maintained (Fig. 12.9d). Isoproterenol also stimulates astrocytic glycogenolysis, with no inhibitory effect by a β2-adrenergic inhibitor, but pronounced although perhaps not complete inhibition by a β1-adrenergic inhibitor [52].

Extracellular Ca2+ and its entry via NCX are required for ouabain-mediated increase in intracellular Ca2+ concentration ([Ca2+]i) and for K+ uptake stimulated by addition of 5 mM KCl, but not for that stimulated by a β1-adrenergic agonist. (a) In mouse mesenteric arteries [Ca2+]i is increased by ouabain, but the increase is abolished by the NCX inhibitor SEA0400, and [Ca2+]i further decreased in the absence of extracellular Ca2+. (b) In rat astrocytes the PAR1-selective agonist Thr-Phe-Leu-Leu-Arg-NH2 (TFLLR) which increases [Ca2+]i in astrocytes, but not in neurons, causes an increase in active uptake of the K+ analogue 86Rb, which is inhibited by two different inhibitors of NCX. (c) In the absence of extracellular Ca2+ the potency of ouabain on K+ uptake is drastically reduced, and (d) increase in intracellular K+ by addition of 5 mM KCl, measured by fluorescence of a K+-sensitive drug, is abolished during incubation in Ca2+-free incubation medium, whereas that by 10 μM of the β1-adrenergic agonist dobutamine is independent of Ca2+ depletion. (a) Modified from Blaustein et al. [50], reproduced with permission; (b) from Wang et al. [40], reproduced with permission; (c) from Song et al. [47], reproduced with permission; (d) from Song et al. [48], reproduced with permission
Studies of increase in intracellular K+ concentration in response to an increase in [K+]o have given some additional information about the ouabain signaling pathway as will be discussed in Sect. 3. They include the dependence of the K+ uptake upon glycogenolysis and Na+ channel activity, as illustrated in Fig. 12.6.
3 Na+, K+-ATPase and Physiological Brain K+ Homeostasis
3.1 Potassium Clearance
It is now well established that most clearance of increased [K+]o following neuronal excitation is active [46, 53]. However, at highly elevated [K+]o inwardly directed channel-mediated K+ may also play a role [54], and Larsen et al. [7] also found a minor channel-mediated uptake after focal iontophoretic administration of K+. A major reason why the astrocytic Na+,K+-ATPase is of interest is that it mediates the initial cellular re-uptake of K+ [7, 54–57]. Neuronal activity increases the extracellular K+ concentration [K+]o both due to stimulation of neuronal glutamatergic receptors and resulting K+ efflux [see 58] and due to action-potential-mediated cellular entry of Na+ followed by exit of K+ [55]. However, increases in [K+]o can also occur after intense stimulation of cortical neurons expressing GABAergic receptors [59]. In hippocampus this K+ release is dependent on bicarbonate-driven accumulation of Cl− and subsequent stimulation of outward flux via the K+, Cl− cotransporter KCC2 [60]. During normal neuronal activity the increases in [K+]o amount to ≤5 and often much less [61, 62]. Under these conditions cellular re-uptake is mediated exclusively by the Na+,K+-ATPase [38]. Released glutamate is predominantly [63, 64] and released GABA partly ([65], see however also [66]) taken up by astrocytes together with Na+.
Since the astrocytic Na+,K+-ATPase in contrast to the neuronal Na+,K+-ATPase has sufficiently low affinity for K+ to be stimulated by an increase in [K+]o above its normal concentration (Sect. 1), it plays a major role in the initial clearance of [K+]o. However, it is obviously also stimulated by normal [K+]o, but stimulation of the astrocytic Na+,K+-ATPase in the absence of elevated [K+]o may be prevented by its dependence on glycogenolysis. This dependence is indicated by the ability of the glycogenolysis inhibitor 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) to prevent K+-mediated uptake of K+ (Fig. 12.10). It should also be kept in mind that even a slight increase in [K+]o induces glycogenolysis in brain slices (Fig. 12.7). Such a complex regulation of the astrocytic Na+,K+-ATPase would allow neuronally released K+ to (1) initially be taken up mainly into astrocytes [7, 54–57]; (2) afterwards be re-released via Kir4.1 K+ channels [67], probably over a larger area due to connexin- and pannexin-mediated inter-astrocytic K+ fluxes [68], preventing that [K+]o is again increased; and (3) eventually be re-accumulated into neurons. The neuronal re-accumulation is a necessity in order to prevent depletion of neuronal K+, since K+ transport across the blood–brain barrier is very slow [69, 70]. This sequence does not explain how astrocytes re-accumulate lost K+ in the absence of local increases in [K+]o, but this may be a situation that only occurs under deep anesthesia.

The reason why astrocytic Na+,K+-ATPase depends on glycogenolysis is that glycogenolysis is required for its signaling function, which in turn is needed for K+-mediated stimulation of K+ uptake. The signaling pathway shown in Fig. 12.6 was further examined by showing that DAB had no inhibitory effect on K+ uptake into cultured astrocytes when extracellular Na+ was increased. This is shown in Fig. 12.11a and must be due to stimulation of Na+ uptake, since K+ uptake could be inhibited by amiloride (Fig. 12.11b), an inhibitor of Na+ channels, but at the concentration used probably not of NCX [71]. Consistency with the ouabain pathway shown in Fig. 12.6 is indicated by a similar ability of xestospongin, an inhibitor of the IP3 receptor to inhibit K+ uptake (Fig. 12.11c). Increase in [Na+]i is needed because the Na+,K+-ATPase’s intracellular site must be activated by Na+ concomitantly with the K+-mediated stimulation of the extracellular site. Since astrocytes are nonexcitable cells, the increased extracellular [K+]o after neuronal excitation is not accompanied by an increased intracellular [Na+]i in astrocytes. This complex regulation has up till now only been described in astrocyte cultures. However, during spreading depression large amounts of K+ is accumulated by astrocytes in intact brain tissue, and inhibition of glycogenolysis enhances the speed with which the depression spreads over brain cortex, indicating impaired cellular uptake of K+ [72]. An attempt to demonstrate that the rate of clearance of glutamate-induced increase in [K+]o in brain slices is increased when glycogenolysis is inhibited [58] gave a negative result, in all probability because glutamate also causes a pronounced increase in [Na+]i. Similar studies should be repeated with electrical stimulation of brain tissue or of optic nerve, where K+ release is secondary to action potential propagation. Another option might be to test DAB after iontophoretic application of K+ to brain slices.

(a) The effect of DAB on K+-mediated increase in intracellular K+ content is prevented by an increase in extracellular Na+ concentration (+10 mM). On the other hand addition to a normal medium of 200 μM amiloride (b) an inhibitor of Na+ channels or 500 μM xestospongine (c), an inhibitor of IP3 receptors have a similar inhibitory effect as DAB. The findings in (b) and (c) are consistent with the pathway shown in Fig. 12.6. From Xu et al. [38]
During spreading depression or seizures and in other situations with more highly elevated [K+]o such as after brain ischemia [61, 73], where [K+]o increases ≥10 mM occur, K+ is in addition re-accumulated into astrocytes by NKCC1. These K+ increases activate also the Na+,K+-ATPase, but not to any greater extent that K+ increases ≤5 mM, which saturate the K+-sensitive site of the Na+, K+-ATPase [38]. In the adult brain cortex NKCC1 is located both in glia cells, including astrocytes, where its activation by high [K+]o can cause life-threatening edema (reviewed by Hertz et al. [19]), and at GABAergic terminals located on the axon initial segment of cortical neurons where Cl− uptake via this transporter after intense stimulation is depolarizing and excitatory [74]. Cellular localization of NKCC1 is best determined by other than immunohistochemical techniques, since the immunological techniques can be deceptive [45, 75].
3.2 Post-stimulatory Undershoot in [K+]o
It is now well established that Na+,K+-ATPase-mediated K+ uptake plays the major role in cellular re-accumulation of increased [K+]o. However, the importance of NKCC1-mediated K+ uptake stimulated by the β-adrenergic agonist isoproterenol for the establishment of the post-stimulatory undershoot in [K+]o has only recently been suggested. In vivo evidence for such a mechanism includes that the undershoot is reduced by furosemide which inhibits NKCC1 [46] and its magnitude is increased by K+ channel inhibition [53]. Since [K+]o is not increased at this time, NKCC1 must be activated by a different stimulus. This is likely to be extracellular hypertonicity, known to occur after intense neuronal activity [76, 77] and possibly triggered by a 2:3 ratio between previous Na+,K+-ATPase-mediated cellular uptake of K+ and release of Na+ [78, 79], and causing cellular shrinkage. In cultured astrocytes bumetanide-inhibited NKCC1 activity is crucial for the subsequent regulatory volume increase (Fig. 12.12), and its rate is greatly enhanced by β1-adrenergic stimulation [47], which increases the ion gradients driving NKCC1 [48]. The cellular accumulation of Na+, K+, Cl− and water must lead to a corresponding decrease in extracellular ions, except for Na+, which is re-extruded by the Na+,K+-ATPase. Furosemide also inhibits another cotransporter KCC2, which is located in neurons, but KCC2 generally mediates outward transport [74], which would have the opposite effect on [K+]o, suggesting that this transporter is not involved. Extracellular hyperosmolarity also depresses population spikes and extracellular synaptic potentials [80], with neuronal gene expression changes blocked by the astrocyte-specific toxin fluoroacetate [81]. The transmitter-induced regulatory volume increase and concomitant reversal of extracellular hypertonicity may normalize neuronal activity and might play a role in inhibition of neuronal slow afterhyperpolarization, sAHP [58, 82]. As could be expected, the regulatory volume increase in cultured astrocytes is inhibited when glycogenolysis is prevented [47, 82]. Again, K+ accumulated into astrocytes may subsequently be released via Kir4.1 channels, as suggested by the increase in the magnitude of the undershoot when these channels are inhibited [53].

After an initial decrease of the volume in isotonic medium (V o) due to medium hypertonicity evoked by addition of 100 mM sucrose, a regulatory volume increase occurs. It is greatly accelerated by isoproterenol but this effect is inhibited by the NKCC1 inhibitor bumetanide. V 1: volume at any given time. From Song et al. [48], reproduced with permission
4 Na+, K+-ATPase and Glutamate Uptake
Like many other amino acids glutamate is accumulated into astrocytes in association with Na+ which provides the driving force and subsequently activates the intracellular Na+-sensitive site of the Na+,K+-ATPase. Glutamate is accumulated into astrocytes by the transporters GLT-1 and GLAST [63, 64] and GLT exists in a macromolecular complex that includes the Na+-K+-ATPase, most of the enzymes involved in glycolysis, and mitochondria [83].
It was previously mentioned that the affinity for Na+ is lower for α2 than for α1. It is even higher for α3 but that is of little relevance for glutamate uptake since most glutamate uptake occurs into astrocytes [63, 64]. Illarionova et al. [84] used very young astrocyte cultures expressing GLAST to study the importance of α1 and α2 on glutamate uptake. Selective inhibition of α2 resulted in a modest increase of [Na+]i together with large decrease in uptake of aspartate, a glutamate analogue that is less metabolizable than glutamate itself. Moreover exposure to 200 μM glutamate caused a larger increase in [Na+]i in α1 than in α2 overexpressing cells, and restoration of control levels of [Na+]i took longer time in α1 than in α2 overexpressing cells.
5 Na+,K+-ATPase and Pathophysiological Brain K+ Homeostasis
5.1 Brain Ischemia
During brain ischemia extracellular Ca2+ becomes greatly reduced (due to cellular uptake) whereas there is a large increase in [K+]o [73]. This leads to NKCC1-mediated brain edema, which only becomes significant after re-oxygenation (Table 12.1), reflecting its dependence on energy metabolism [47, 71]. There is abundant evidence that this edema occurs in astrocytes, but there must also be an effect on the blood–brain barrier bringing additional water into the brain (reviewed by Hertz et al. [85]). The specific NKCC1 inhibitor bumetanide [45] prevents the edema after ischemia/reperfusion (indicated by prevention of increase of water content in the tissue) and so do the same inhibitors (Table 12.2), which inhibit β1-adrenergic signaling in astrocytes (Fig. 12.5). Moreover, the edema is not significantly counteracted by the Ca2+-channel antagonist nimodipine [86], which prevents the Ca2+ uptake necessary for the development of NKCC1-mediated edema (Fig. 12.6). In the early experiments by the latter authors the edema developed already during the ischemic phase, possibly suggesting less complete arterial blockage than in the experiments shown in Tables 12.1 and 12.2. Both degrees of blockage may well be relevant for clinical stroke. The lack of effect by nimodipine points towards involvement of the other stimulus for NKCC1 activation, hypertonicity and cell shrinkage, and both of these were demonstrated by Matsuoka and Hossmann [86]. The inverse correlation between the magnitude of the increase in water space and the reduction of extracellular space demonstrated by these authors (Fig. 12.13) is consistent with swelling, and thus regulatory volume increase during the ischemic phase.

An inverse correlation between the size of the extracellular space and tissue hypertonicity (hyperosmolality—mosm/kg dry wt), i.e., a correlation between the largest reduction in extracellular space and highest degree of hypertonicity, is consistent with the concept of a correlation between reduction in extracellular space and NKCC1-mediated ion uptake as part of a regulatory volume increase. This is especially the case since the water transport by NKCC1 does no fully compensate osmotically for its ion uptake [43]. The authors of the original paper are not responsible for this interpretation, but did regard such correlations as reflections of the interrelationship between ischemia and the development of brain edema. From Matsuoka and Hossmann [86], reproduced with permission
The prevention of water increase shown in Table 12.2 by β1-adrenergic antagonists may seem peculiar because these inhibitors would normally not prevent the stimulation of the Na+,K+-ATPase driving NKCC1, which are mediated by the increased [K+]o. However, Fig. 12.9d showed that in the absence of extracellular Ca2+, increase in intracellular K+ mediated by an elevation of [K+]o is abolished in Ca2+-free media, whereas that mediated by the β1-adrenergic agonist dobutamine is unaltered, and in brain ischemia extracellular Ca2+ is greatly reduced due to cellular uptake [73].
Complete similarity between the inhibitors blocking the pathway for β1-adrenergic stimulation in cultured astrocytes (Fig. 12.5), those preventing regulatory volume increase in these cells [48], and inhibitors of brain edema in rats after ischemia and reperfusion (Table 12.2) supports the validity of the cultured astrocytes as models of astrocytes in situ. It is also of clinical significance. Goyagi et al. [87] have shown that administration of a β1-adrenergic agonist 30 min after the onset of a 2-h-long ischemic period drastically reduces infarct size and improves neurological deficit score in rats after 7 days. Administration of subtype-specific β1-adrenergic antagonists before experimental brain ischemia also provided neuroprotection against transient focal cerebral ischemia [88]. However, although the presence of β1-adrenergic antagonists beginning 30 min before the onset of ischemia and continued for 24 h provided long-term improvement of histological outcome, they had no effect on neurological outcome and spatial memory retention 14 days later [89]. Iwata et al. [90] also found that administration of antagonists specifically of the β1-adrenoceptor beginning 60 min after an 8-min bilateral carotid artery occlusion combined with hypotension reduced neuronal injury after forebrain ischemia, although motor activity was not improved. However, motor deficit index scores were significantly lower and neuronal survival better in rats treated with β1-adrenoceptor antagonists beginning 30 min before 10 min of spinal cord ischemia and continued for 24 h [91]. Perhaps it is important that the β1-adrenergic treatment, which also must have unwanted side effects on cognition and motor performance, is discontinued as soon as possible and not combined with other procedures that may enhance the side effects.
5.2 Hepatic Encephalopathy
It has been known for a long time that ammonia (NH4 +) can substitute for K+, but not for Na+ in the stimulation of both the Na+,K+-ATPase and active transport of Na+ and K+ [92]. In cultured astrocytes exposure to 5 mM NH4Cl activates NKCC after 24 h in a bumetanide-inhibited fashion [93]. A metabolic answer to NKCC1 activation, stimulation of oxygen consumption, is activated by even lower concentrations of ammonia than of K+ [94, 95]. A third similarity between K+ and NH4 + is that also NH4 + stimulates signaling by endogenous ouabains. In cultured astrocytes this is accompanied by an increased content of ouabain-like compounds [35]. Ouabain signaling activates production of reactive oxygen species (ROS) and nitrosactive agents which slowly sensitize NKCC1, explaining why cell swelling and brain edema normally take hours to develop after exposure to NH4 + ([96] and references therein). In cultured astrocytes, ammonia-induced cell swelling and ROS production (Fig. 12.14) can both be prevented by the main metabolite of spironolactone, canrenone, an aldosterone antagonist acting as a ouabain inhibitor [96, 97].

Ammonia-induced ROS production and cell swelling can be inhibited by canrenone, an inhibitor of ouabain. (a) Cells were incubated with 0 or 3 mM NH4Cl in the absence (control: no NH4Cl, no canrenone) or presence of 100 μM canrenone for 2 h. ROS was determined as fluorescence intensity of oxidized carboxy-H2DCFDA in individual cells in each of three cultures, averaged, and shown as mans ± SEM. (b) After incubation of the cells with 3 mM NH4Cl for 12 h, cell volume was determined as fluorescence intensity of calcein, again in individual cells from three coverslips and averaged and expressed as in (a). From Dai et al. [96], reproduced with permission
Since it is the α2 isoform of the Na+,K+-ATPase which is stimulated by ultralow concentrations of ouabain, it is also this isoform that shows upregulated gene expression during exposure to elevated ammonia concentrations both in cultured astrocytes and in the brain in vivo [98].
In a recent paper Hadjihambi et al. [99] have suggested that the demonstration that an inhibitory effect of the NKCC1 inhibitor, bumetanide, potently suppresses ammonia-induced neurological dysfunction [100] points to a potential new target for treatment of hepatic encephalopathy. The authors express concern that the expression of NKCC1 also on astrocytes and on endothelial cells may produce off-target actions. In this context it should be noted that Kelly et al. [101] showed that bumetanide prevented several ammonia-induced abnormalities in cultured astrocytes and that both Jayakumar et al. [93] and Song et al. [97] found that bumetanide inhibited ammonia-induced swelling in such cells. Moreover, Jayakumar et al. [102] based on experiments in brain cortical slices treated with ammonia concluded “that targeting NKCC may represent a useful therapeutic strategy in humans with acute liver failure.” Thus bumetanide treatment is not a new idea, and the effects on astrocytes and endothelial cells are therapeutic, probably for similar reasons as after ischemia and reperfusion.
Rangroo Thrane et al. [100] studied acute effects of very high plasma ammonia concentrations in intact, non-anesthetized mice and found evidence that the therapeutic effect of bumetanide was exerted on GABAergic neurons, where NKCC1 stimulation by NH4 + and an increased [K+]o over-activate NKCC1. In turn this compromises inhibitory neurotransmission. This is similar to the effect described in Sect. 3.1 as a response by cortical neurons to a stimulation-induced increase in [K+]o, where Cl− uptake via NKCC1 is depolarizing and excitatory [74]. The plasma ammonia concentrations obtained by Rangroo Thrane et al. [100] are at least one order of magnitude larger than those seen in hepatic encephalopathy [99]. This is especially important considering they were made in vivo in non-anesthetized animals, and hepatic disease leads to similar plasma concentration in rodents as in man [103]. Accordingly this study may be more directly relevant for the acute and deadly toxicity by very high concentrations of ammonia [104] a fact overlooked by Hadjihambi et al. [99]. Consistent with this concept virtually all animals died within 1 h and death was only postponed for ~10 min by bumetanide treatment [100]. This does not exclude that neuronal NKCC1 stimulation may contribute to the pathophysiology in hepatic encephalopathy provided the neuronal NKCC1 is also sensitized by oxidative and nitrosactive stress.
6 Conclusions
The present paper has attempted a comprehensive description of the mechanisms and roles of the astrocytic Na+,K+-ATPase. A considerable part of this is based upon experiments using mouse astrocytes in primary cultures and must ultimately be confirmed in intact brain tissue. However, several indications that they apply to astrocytes in situ are mentioned (similarity between K+ effects on cells isolated from brain and on our cultured cells; effects of glycogenolysis in spreading depression; confirmation of β1-adrenergic pathway determined in cultured astrocytes using specific inhibitors by the ability of the same transmitters to prevent edema after ischemia and re-oxygenation). Moreover, initial uptake of excess K+ in astrocytes is now well established and must be followed by return to neurons.
References
Skou JC (1957) The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 23:394–401
Skou JC (2004) The identification of the sodium pump. Biosci Rep 24:436–451
Grisar T, Franck G, Schoffeniels E (1980) Glial control of neuronal excitability in mammals: II. Enzymatic evidence: two molecular forms of the Na+, K+-ATPase in brain. Neurochem Int 2C:311–320
Sweadner KJ (1979) Two molecular forms of Na+ + K+-stimulated ATPase in brain. Separation, and difference in affinity for strophanthidin. J Biol Chem 254:6060–6067
Henn FA, Haljamäe H, Hamberger A (1972) Glial cell function: active control of extracellular K+ concentration. Brain Res 43:437–443
Hajek I, Subbarao KV, Hertz L (1996) Acute and chronic effects of potassium and noradrenaline on Na+, K+-ATPase activity in cultured mouse neurons and astrocytes. Neurochem Int 28:335–342
Larsen BR, Assentoft M, Cotrina ML et al (2014) Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62:608–622
Walz W, Kimelberg HK (1985) Differences in cation transport properties of primary astrocyte cultures from mouse and rat brain. Brain Res 340:333–340
Cahoy JD, Emery B, Kaushal A et al (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278
Cameron R, Klein L, Shyjan AW et al (1994) Neurons and astroglia express distinct subsets of Na, K-ATPase alpha and beta subunits. Mol Brain Res 21:333–343
McGrail KM, Phillips JM, Sweadner KJ (1991) Immunofluorescent localization of three Na, K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na, K-ATPase. J Neurosci 11:381–391
Peng L, Huang R, Zhang S, Hertz L (2010) Ouabain binding kinetics and FXYD7 expression in astrocytes and neurons in primary cultures: implications for cellular contributions to extracellular K+ homeostasis? Neuron Glia Biol 6:127–135
Crambert G, Hasler U, Beggah AT et al (2000) Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J Biol Chem 275:1976–1986
Zahler R, Zhang ZT, Manor M, Boron WF (1997) Sodium kinetics of Na, K-ATPase alpha isoforms in intact transfected HeLa cells. J Gen Physiol 110:201–213
Béguin P, Crambert G, Monnet-Tschudi F et al (2002) FXYD7 is a brain-specific regulator of Na, K-ATPase alpha 1-beta isozymes. EMBO J 21:3264–3273
Tokhtaeva E, Clifford RJ, Kaplan JH et al (2012) Subunit isoform selectivity in assembly of Na, K-ATPase α-β heterodimers. J Biol Chem 287:26115–26125
Li B, Hertz L, Peng L (2013) Cell-specific mRNA alterations in Na+, K+-ATPase α and β isoforms and FXYD in mice treated chronically with carbamazepine, an anti-bipolar drug. Neurochem Res 38:834–841
O’Brien WJ, Lingrel JB, Wallick ET (1994) Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys 310:32–39
Hertz L, Xu J, Chen Y et al (2014) Antagonists of the vasopressin V1 receptor and of the α1-adrenoceptor inhibit cytotoxic brain edema in stroke by effects on astrocytes - but the mechanisms differ. Curr Neuropharmacol 12:308–323
Geering K (2001) The functional role of beta subunits in oligomeric P-type ATPases. J Bioenerg Biomembr 33:425–438
Spiller S (2013) Struktur-Funktionsbeziehungen der Na+/K+-ATPase anhand elektrophysiologischer Untersuchungen C-terminaler und Migräne-assoziierter Mutationen. (Structure-function relationships of the Na+/K+-ATPase based on electrophysiological studies of C-terminal and migraine-associated mutations.) Doctoral Thesis, Technische Universität, Berlin
Yoshimura K (1973) Activation of Na-K activated ATPase in rat brain by catecholamine. J Biochem 74:389–391
Godfraind T, Koch MC, Verbeke N (1974) The action of EGTA on the catecholamines stimulation of rat brain Na-K-ATPase. Biochem Pharmacol 23:3505–3511
Wu PH, Phillis JW (1979) Receptor-mediated noradrenaline stimulation of Na+-K+ ATPase in rat brain cortical homogenates. Gen Pharmacol 10:189–192
Kupriyanov VV, Xiang B, Sun J, Jilkina O (2002) Effect of adrenergic stimulation on Rb+ uptake in normal and ischemic areas of isolated pig hearts: 87Rb MRI study. Magn Reson Med 48:15–20
Aperia A, Ibarra F, Svensson LB et al (1992) Calcineurin mediates alpha-adrenergic stimulation of Na+, K+-ATPase activity in renal tubule cells. Proc Natl Acad Sci U S A 89:7394–7397
Ibarra FR, Cheng SX, Agrén M et al (2002) Intracellular sodium modulates the state of protein kinase C phosphorylation of rat proximal tubule Na+, K+-ATPase. Acta Physiol Scand 175:165–171
de Lores Arnaiz GR, Ordieres MG (2014) Brain Na+, K+-ATPase activity in aging and disease. Int J Biomed Sci 10:85–102
Kobayashi M (2007) Differential regulation of synaptic transmission by adrenergic agonists via protein kinase A and protein kinase C in layer V pyramidal neurons of rat cerebral cortex. Neuroscience 146:1772–1784
Barth AM, Vizi ES, Lendvai B (2007) Noradrenergic enhancement of Ca2+ responses of basal dendrites in layer 5 pyramidal neurons of the prefrontal cortex. Neurochem Int 51:323–327
White HS, Chow SY, Yen-Chow YC, Woodbury DM (1992) Effect of elevated potassium on the ion content of mouse astrocytes and neurons. Can J Physiol Pharmacol 70(Suppl):S263–S268
Du T, Li B, Li H et al (2010) Signaling pathways of isoproterenol-induced ERK1/2 phosphorylation in primary cultures of astrocytes are concentration-dependent. J Neurochem 115:1007–1023
Fishman MC (1979) Endogenous digitalis-like activity in mammalian brain. Proc Natl Acad Sci U S A 76:4661–4663
Bagrov AY, Fedorova OV (2005) Cardenolide and bufadienolide ligands of the sodium pump. How they work together in NaCl sensitive hypertension. Front Biosci 10:2250–2256
Kala G, Kumarathasan R, Peng L, Leenen FH, Hertz L (2000) Stimulation of Na+, K+-ATPase activity, increase in potassium uptake, and enhanced production of ouabain-like compounds in ammonia-treated mouse astrocytes. Neurochem Int 36:203–211
Haas M, Askari A, Xie Z (2000) Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 275:27832–27837
Zhang L, Zhang Z, Guo H, Wang Y (2008) Na+/K+-ATPase-mediated signal transduction and Na+/K+-ATPase regulation. Fundam Clin Pharmacol 22:615–621
Xu J, Song D, Xue Z et al (2013) Requirement of glycogenolysis for uptake of increased extracellular K+ in astrocytes: potential implications for K+ homeostasis and glycogen usage in brain. Neurochem Res 38:472–485
Forshammar J, Block L, Lundborg C et al (2011) Naloxone and ouabain in ultralow concentrations restore Na+/K+-ATPase and cytoskeleton in lipopolysaccharide-treated astrocytes. J Biol Chem 286:31586–31597
Wang F, Smith NA, Xu Q, et al (2012) Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+. Sci Signal 5:ra26
Hof PR, Pascale E, Magistretti PJ (1988) K+ at concentrations reached in the extracellular space during neuronal activity promotes a Ca2+-dependent glycogen hydrolysis in mouse cerebral cortex. J Neurosci 8:1922–1928
Hamann S, Herrera-Perez JJ, Zeuthen T, Alvarez-Leefmans FJ (2010) Cotransport of water by the Na+-K+-2Cl- cotransporter NKCC1 in mammalian epithelial cells. J Physiol 588:4089–4101
Zeuthen T, Macaulay N (2012) Cotransport of water by Na+-K+-2Cl- cotransporters expressed in Xenopus oocytes: NKCC1 versus NKCC2. J Physiol 590:1139–1154
Pedersen SF, O’Donnell ME, Anderson SE, Cala PM (2006) Physiology and pathophysiology of Na+/H+ exchange and Na+-K+-2Cl- cotransport in the heart, brain, and blood. Am J Physiol Regul Integr Comp Physiol 291:R1–R25
Blaesse P, Airaksinen MS, Rivera C, Kaila K (2009) Cation-chloride cotransporters and neuronal function. Neuron 61:820–838
Xiong ZQ, Stringer JL (2000) Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol 83:1443–1451
Song D, Xu J, Du T et al (2014) Inhibition of brain swelling after ischemia-reperfusion by β-adrenergic antagonists – correlation with increased K+ and decreased Ca2+ concentrations in extracellular fluid. Biomed Res Int 2014:873590
Song D, Xu J, Hertz L, Peng L (2015) Regulatory volume increase in astrocytes exposed to hypertonic medium requires β1-adrenergic Na+/K+–ATPase stimulation and glycogenolysis. J Neurosci Res 93:130–13949
Song H, Thompson SM, Blaustein MP (2013) Nanomolar ouabain augments Ca2+ signaling in rat hippocampal neurones and glia. J Physiol 591:1671–1689
Blaustein MP, Zhang J, Chen L et al (2009) The pump, the exchanger, and endogenous ouabain: signaling mechanisms that link salt retention to hypertension. Hypertension 53:291–298
Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS (2008) Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci 28:6659–6663
Xu J, Song D, Bai Q et al (2014) Basic mechanism leading to stimulation of glycogenolysis by isoproterenol, EGF, elevated extracellular K+ concentrations, or GABA. Neurochem Res 39:661–667
D’Ambrosio R, Gordon DS, Winn HR (2002) Differential role of KIR channel and Na+/K+-pump in the regulation of extracellular K+ in rat hippocampus. J Neurophysiol 87:87–102
Somjen GG, Kager H, Wadman WJ (2008) Computer simulations of neuron-glia interactions mediated by ion flux. J Comput Neurosci 25:349–365
Ransom CB, Ransom BR, Sontheimer H (2000) Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol 522(Pt 3):427–442
Dufour S, Dufour P, Chever O et al (2011) In vivo simultaneous intra- and extracellular potassium recordings using a micro-optrode. J Neurosci Methods 194:206–217
Macaulay N, Zeuthen T (2012) Glial K+ clearance and cell swelling: key roles for cotransporters and pumps. Neurochem Res 37:2299–2309
Hertz L, Gerkau NJ, Xu J et al (2015) Roles of astrocytic Na+, K+-ATPase and glycogenolysis for K+ homeostasis in mammalian brain. J Neurosci Res 93(7):1019–1030
Louvel J, Papatheodoropoulos C, Siniscalchi A et al (2001) GABA-mediated synchronization in the human neocortex: elevations in extracellular potassium and presynaptic mechanisms. Neuroscience 105:803–813
Viitanen T, Ruusuvuori E, Kaila K, Voipio J (2010) The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol 588:1527–1540
Lothman E, Lamanna J, Cordingley G et al (1975) Responses of electrical potential, potassium levels, and oxidative metabolic activity of the cerebral neocortex of cats. Brain Res 88:15–36
Somjen GG (1980) Stimulus-evoked and seizure-related responses of extracellular calcium activity in spinal cord compared to those in cerebral cortex. J Neurophysiol 44:617–632
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Zhou Y, Danbolt NC (2013) GABA and glutamate transporters in brain. Front Endocrinol (Lausanne) 4:165
Schousboe A, Bak LK, Waagepetersen HS (2013) Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol (Lausanne) 4:102
Hertz, L, Rothman DL (2015) Glucose, lactate, β-hydroxybutyrate, acetate, GABA, and succinate as substrates for synthesis of glutamate and GABA. In Sonnewald U, Schousboe A (eds) The Glutamate/GABA – Glutamine Cycle: Amino Acid Neurotransmitter Homeostasis, Advances in Neurobiology vol 13 (Schousboe A (series ed). Springer, Heidelberg. In press.
Bay V, Butt AM (2012) Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia 60:651–660
Scemes E, Spray DC (2012) Extracellular K+ and astrocyte signaling via connexin and pannexin channels. Neurochem Res 37:2310–2316
Bradbury MW, Segal MB, Wilson J (1972) Transport of potassium at the blood-brain barrier. J Physiol 221:617–632
Kang EJ, Major S, Jorks D et al (2013) Blood-brain barrier opening to large molecules does not imply blood-brain barrier opening to small ions. Neurobiol Dis 52:204–218
Hertz L, Song D, Xu J, et al (2015) Role of the Astrocytic Na+, K+-ATPase in K+ homeostasis in brain: K+ uptake, signaling pathways and substrate utilization. Neurochem Res [Epub ahead of print]
Seidel JL, Shuttleworth CW (2011) Contribution of astrocyte glycogen stores to progression of spreading depression and related events in hippocampal slices. Neuroscience 192:295–303
Hansen AJ, Nedergaard M (1998) Brain ion homeostasis in cerebral ischemia. Neurochem Pathol 9:195–209
Khirug S, Yamada J, Afzalov R et al (2008) GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci 28:4635–4639
Peng L, Guo C, Wang T et al (2013) Methodological limitations in determining astrocytic gene expression. Front Endocrinol (Lausanne) 4:176
Dietzel I, Heinemann U, Hofmeier G, Lux HD (1982) Stimulus-induced changes in extracellular Na+ and Cl- concentration in relation to changes in the size of the extracellular space. Exp Brain Res 46:73–84
Dietzel I, Heinemann U, Lux HD (1989) Relations between slow extracellular potential changes, glial potassium buffering, and electrolyte and cellular volume changes during neuronal hyperactivity in cat brain. Glia 2:25–44
Thomas RC (1972) Electrogenic sodium pump in nerve and muscle cells. Physiol Rev 52:563–594
Clarke RJ, Apell HJ, Läuger P (1989) Pump current and Na+/K+ coupling ratio of Na+/K+-ATPase in reconstituted lipid vesicles. Biochim Biophys Acta 981:326–336
Huang R, Somjen GG (1995) The effect of graded hypertonia on interstitial volume, tissue resistance and synaptic transmission in rat hippocampal tissue slices. Brain Res 702:181–187
Yuan H, Gao B, Duan L et al (2010) Acute hyperosmotic stimulus-induced Fos expression in neurons depends on activation of astrocytes in the supraoptic nucleus of rats. J Neurosci Res 88:1364–1373
Hertz L, Xu J, Song D et al (2013) Astrocytic and neuronal accumulation of elevated extracellular K+ with a 2/3 K+/Na+ flux ratio-consequences for energy metabolism, osmolarity and higher brain function. Front Comput Neurosci 7:114
Genda EN, Jackson JG, Sheldon AL et al (2011) Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 31:18275–18288
Illarionava NB, Brismar H, Aperia A, Gunnarson E (2014) Role of Na, K-ATPase α1 and α2 isoforms in the support of astrocyte glutamate uptake. PLoS One 9:e98469
Hertz L, Peng L, Song D (2014) Ammonia, like K+, stimulates the Na+, K+, 2 Cl- cotransporter NKCC1 and the Na+,K+-ATPase and interacts with endogenous ouabain in astrocytes. Neurochem Res 40:241–257
Matsuoka Y, Hossmann KA (1982) Brain tissue osmolality after middle cerebral artery occlusion in cats. Exp Neurol 77:599–611
Goyagi T, Horiguchi T, Nishikawa T, Tobe Y (2010) Post-treatment with selective β1 adrenoceptor antagonists provides neuroprotection against transient focal ischemia in rats. Brain Res 1343:213–217
Goyagi T, Kimura T, Nishikawa T, Tobe Y et al (2006) Beta-adrenoreceptor antagonists attenuate brain injury after transient focal ischemia in rats. Anesth Analg 103:658–663
Goyagi T, Tobe Y, Nishikawa T (2012) Long-term and spatial memory effects of selective β1-antagonists after transient focal ischaemia in rats. Br J Anaesth 109:399–406
Iwata M, Inoue S, Kawaguchi M et al (2010) Posttreatment but not pretreatment with selective beta-adrenoreceptor 1 antagonists provides neuroprotection in the hippocampus in rats subjected to transient forebrain ischemia. Anesth Analg 110:1126–1132
Umehara S, Goyagi T, Nishikawa T et al (2010) Esmolol and landiolol, selective beta1-adrenoreceptor antagonists, provide neuroprotection against spinal cord ischemia and reperfusion in rats. Anesth Analg 110:1133–1137
Post RL, Merritt CR, Kinsolving CR, Albright CD (1960) Membrane adenosine triphosphatase as a participant in the active transport of sodium and potassium in the human erythrocyte. J Biol Chem 235:1796–1802
Jayakumar AR, Liu M, Moriyama M et al (2008) Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J Biol Chem 283:33874–33882
Hertz L, Schou M (1962) Univalent cations and the respiration of brain-cortex slices. Biochem J 85:93–104
Verkhratsky A, Nedergaard M, Hertz L (2014) Why are astrocytes important? Neurochem Res 40:389–401
Dai H, Song D, Xu J et al (2013) Ammonia-induced Na, K-ATPase/ouabain-mediated EGF receptor transactivation, MAPK/ERK and PI3K/AKT signaling and ROS formation cause astrocyte swelling. Neurochem Int 63:610–625
Song D, Du T (2014) Ammonium activates ouabain-activated signalling pathway in astrocytes: therapeutic potential of ouabain antagonist. Curr Neuropharmacol 12:334–341
Xue Z, Li B, Gu L et al (2010) Increased Na, K-ATPase α2 isoform gene expression by ammonia in astrocytes and in brain in vivo. Neurochem Int 57:395–403
Hadjihambi A, Rose CF, Jalan R (2014) Novel insights into ammonia-mediated neurotoxicity pointing to potential new therapeutic strategies. Hepatology 60:1101–1103
Rangroo Thrane V, Thrane AS, Wang F et al (2013) Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19:1643–1648
Kelly T, Kafitz KW, Roderigo C, Rose CR (2009) Ammonium-evoked alterations in intracellular sodium and pH reduce glial glutamate transport activity. Glia 57:921–934
Jayakumar AR, Valdes V, Norenberg MD (2011) The Na-K-Cl cotransporter in the brain edema of acute liver failure. J Hepatol 54:272–278
Jover-Cobos M, Noiret L, Lee K et al (2014) Ornithine phenylacetate targets alterations in the expression and activity of glutamine synthase and glutaminase to reduce ammonia levels in bile duct ligated rats. J Hepatol 60:545–553
Cauli O, López-Larrubia P, Rodrigues TB et al (2007) Magnetic resonance analysis of the effects of acute ammonia intoxication on rat brain. Role of NMDA receptors. J Neurochem 103:1334–1343
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hertz, L., Song, D., Peng, L. (2016). The Astrocytic Na+, K+-ATPase: Stimulation by Increased Extracellular K+, β-Adrenergic Activation, Ouabain-Mediated Signaling, and Interaction with the Transporter NKCC1. In: Chakraborti, S., Dhalla, N. (eds) Regulation of Membrane Na+-K+ ATPase. Advances in Biochemistry in Health and Disease, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-319-24750-2_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-24750-2_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24748-9
Online ISBN: 978-3-319-24750-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)