
Abstract
Alpha-1 antitrypsin deficiency (AATD) is a hereditary condition in which less than 10 % of the individuals are identified. This under-recognition is a barrier to lifestyle modifications, genetic counseling, and specific treatment. Targeted screening allows for high detection rates and lower costs than population screening. Little is known about the prevalence and demographic and clinical characteristics of individuals with the main AATD genotypes. We retrospectively analyzed data on adult subjects who underwent targeted screening for AATD from December 2003 to July 2009. AATD testing was ordered according to physician discretion, primarily in patients with COPD and unexplained chronic liver disease. We used an algorithm that begins with dried blood spot (DBS) genotyping and uses DBS alpha-1.
We included a total of 37,708 individuals of whom 1 % had the ZZ genotype, the most common genotype associated with severe alpha-1 antitrypsin deficiency in North America. This percentage corresponds to a 48-fold increase in the detection rate when compared to its estimated prevalence in the general population. The age at the time of testing was 57.3 ± 15 years in the overall population and 51.9 ± 13 in PI ZZ individuals (p < 0.001). Although individuals from different races were tested, PI ZZ subjects were of White (87 %), Hispanic (3.2 %), and mixed (0.3 %) race. In PI ZZ subjects, 11 % were current smokers. These individuals were diagnosed 9.2 (95 % CI: 4.2–14.3) years before never-smokers.
These data support targeted genotyping for the detection of alpha-1 antitrypsin deficiency in adults.
Authors’ Contributions
Adriano Tonelli participated in the study design IRB application, statistical analysis, interpretation of data, and writing and revision of the manuscript.
Farshid Rouhani participated in the study design data collection, interpretation of data, and writing and revision of the manuscript.
Pam Schreck participated in the IRB application data collection and writing and revision of the manuscript.
Mark Brantly participated in the study design data collection, interpretation of data, and writing and revision of the manuscript.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Alpha-1-antitrypsin deficiency (AATD) is an under-recognized hereditary condition characterized by low serum levels of alpha-1 antitrypsin (AAT) and an increased risk for the development of respiratory and hepatic disease [1–3]. Fewer than 10 % of the individuals with severe AATD are recognized clinically, typically after long diagnostic delays [4–6]. Its early detection is crucial as it may prompt specific interventions such as testing of family members, genetic counseling for patients and families, lifestyle changes (e.g., smoking cessation and occupation change), screening for other manifestations of the disease, and consideration of augmentation therapy [7].
An early diagnosis of AATD can be accomplished by screening the general population [8, 9] or by targeting high-risk groups. Population-based screening has the advantage of detecting clinically unaffected individuals at a younger age, but this modality is expensive and challenging to perform on a large scale [10]. Targeted detection is associated with higher recognition rates and lower costs. The diagnostic yield and costs of this approach depend largely on the characteristic of the individuals tested and the protocol used for processing the samples [11]. The ATS/ERS statement describes the conditions that should prompt testing for AATD, serving as the basis for targeted testing [1].
The gene that codes for AAT protein is located on the long arm of chromosome 14 (14q31–32.3). More than 120 variants of this gene exist. At least 32 of these variants are associated with altered, reduced, or absent serum AAT; however, the vast majority (~95 %) of AATD individuals with lung disease are homozygous for the Z allele [12, 13]. In addition, subjects heterozygous for the Z allele (PI MZ and PI SZ) have a modest increase in the risk of developing lung disease [1, 14–16].
Detection of AATD
Different algorithms for the detection of AATD that combine measurement of AAT protein in plasma, genotyping, and isoelectric focusing (IEF) have been proposed. The algorithm used in this study begins with genotyping for the two most common deficient alleles involved in the disease, namely PI*Z (Glu 342 GAG → Lys AAG) and PI*S (Glu 264GAA → Val GTA). Genotyping is less technically demanding than IEF and it allows us to identify the prevalence of the main genotypic variants in the population studied, a relevant analysis, since one of the main barriers for not testing for AATD individuals is the perceived low yield of the test [17].
Little is known about the demographic, clinical, and genotype characteristics of individuals who underwent target screening for AATD in the USA. We sought to determine the prevalence of the main AATD genotypic variants and describe the demographic and clinical characteristic of the patients who underwent testing in one of the largest targeted detection programs in the USA. In addition, we explored the relationship between age and genotype, gender, race, smoking history, year of testing, region of origin of the sample in the USA, and reason for testing.
National Detection Program
Data from subjects who underwent AATD testing as part of the National Detection Program for alpha-1-antitrypsin deficiency from December 2003 to July 2009 were retrospectively analyzed. Kits for alpha-1 antitrypsin detection, containing a healthcare provider’s guide to alpha-1 antitrypsin deficiency, the ATS/ERS statement on standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency [1], and several blood collection cards, were distributed to respiratory physicians throughout the country. A total of 5195 US physicians ordered at least one AATD test. Each blood collection card was accompanied by a questionnaire asking demographic and clinical information, including age, gender, race, geographic data, smoking history, diagnosis of respiratory or hepatic diseases, and family history of respiratory diseases. Family history of respiratory diseases referred to the presence of AATD, COPD, or emphysema in parents or siblings. Although a more recent version of the questionnaire discriminated among these conditions, for consistency these respiratory diseases were grouped together.
We recorded the US State where the sample was originated and grouped them in Regions (Northeast, Midwest, South, and West) as defined by the U.S. Census Bureau [18]. We excluded from the analysis those samples in which the origin was not recorded or they originated from Canada or Puerto Rico (n = 328).
Although we suggested testing potential AATD individuals following the recommendations proposed by the ATS/ERS statement [1], in practice, individuals were tested according to the discretion of physicians who received the kits. To simplify the analysis, we divided the reasons for screening into five main categories: (1) presence of respiratory diseases such as COPD, asthma, and bronchiectasis, (2) presence of hepatic diseases (hepatitis or cirrhosis) or abnormal liver function tests, (3) combination of respiratory and hepatic diseases, (4) presence of family history of COPD or AATD, and (5) family history of respiratory diseases plus respiratory and/or hepatic diseases.
Fresh whole blood was collected by fingertip puncture on a 903 filter paper. Blood was allowed to air dry before mailing. Once the dried blood spot (DBS) sample was received, we processed it following the algorithm shown in Fig. 5.1. PI*Z and PI*S genotyping was performed with End-point allelic discrimination on an ABI Prism 7500 Fast System (Applied Biosystems) using 3.2 mm DBS punches. Assays were run in a 96-well format according to the manufacturer’s instructions. Nephelometry determination was performed using methods described previously [19, 20]. For reflex testing, we performed single nucleotide polymorphisms (SNP) analysis and DNA sequencing using similar methodology to Ferrarotti et al. [21].

University of Florida detection Lab Testing Algorithm. Heterozygous and homozygous individuals for PiZ undergo reflex testing for confirmatory purposes. In the case of PiMZ and PiMS individuals with low DBS AAT levels, reflex testing is performed to rule out the combination of a Z or S allele with a Null or Rare allele. *Blood sample is requested for PI typing by single nucleotide polymorphism (SNP) and DNA sequencing
Dried blood spot collection methodology is minimally invasive, requires a small amount of sample, and is easier to preserve and ship [22]. In addition, we have demonstrated an excellent correlation between AAT protein concentration measured in plasma and in DBS (n = 347, r = 0.95) (Fig. 5.2).

Correlation between AAT protein concentration measured in plasma and DBS (panel a), and AAT protein concentration in DBS according to genotype (panel b) (n = 347)
Although this model confirmed the presence of S and Z alleles, and detected the majority of deficient individuals, it did not directly test for the common “wild-type” M variant. The presence of PI*M was assumed when genotyping for S and Z was negative and the DBS AAT protein level was concordant with this genotype. We used PI*M instead of PI non S-non Z (which is the correct term) for simplicity.
Results of the National Detection Program
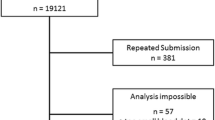
A total of 40,049 unique subjects underwent target screening for the presence of AATD from December 2003 to June 2009. For this analysis, we included adult patients (≥16 years) with an available genotype. Data from 37,708 individuals were used for further analysis (Fig. 5.3).

Flow diagram of the study population and results of genotyping
The genotypes were MM in 84 % (n = 31,670), MS in 7.8 % (n = 2957), SS in 0.3 % (n = 115), MZ in 6.2 % (n = 2344), SZ in 0.6 % (n = 244), and ZZ in 1 % (n = 378) (Fig. 5.2). One in 6.25 individuals had a genotype other than MM. At least one Z allele was present in 7.9 % of the samples (n = 2966). The S and Z allelic frequency was 4.55 % and 4.43 %, respectively.
Compared with data obtained from control cohorts of genetic epidemiological studies in the USA (n = 27,809) [23], we found an increase in the detection rate of genotypes other than PI MM in our program. The detection rate markedly increased for the genotype ZZ by a factor of approximately 48 times (Table 5.1).
Age
The age at the time of genetic testing for the entire cohort was 57.3 ± 15 years (Table 5.2). Individuals heterozygous for PI*Z were genetically diagnosed 3.8 (95 % CI: 0.3) years before subjects without any Z allele (p < 0.001). Individuals homozygous for PIZ were genetically diagnosed 1.96 (95 % CI: 0.7) years before subjects heterozygous for PI*Z (p = 0.007). PI*Z homozygous were genetically diagnosed 5.7 (95 % CI: 0.7) years earlier than subjects without any Z allele (p < 0.001).
The difference in age at the time of the testing between PI*MM and PI ZZ individuals was 5.73 years (95 % CI 4.2–7.2) (Table 5.3 and Fig. 5.4). PI ZZ individuals were genetically diagnosed at the age of 50 years or above and 70 years or above in 55 % and 9 % of the cases, respectively. The oldest individual diagnosed with the ZZ genotype was 87 years old.

Mean age (±2 Standard Error) according to genotype
Gender
Of all the subjects, 58 % were women. A lower percentage of women were observed in individuals heterozygous (55.2 %) and homozygous (49.2 %) for PiZ (p < 0.001) (Table 5.2). This female gender predominance was maintained for all genotypes, except for PI ZZ (Table 5.3).
In PI ZZ individuals, the age at the time of genetic testing did not differ between genders [men: 52.1 ± 11 years versus women: 51.7 ± 14 years (p = 0.75)].
Race
The race distribution of the entire cohort, individuals heterozygous and homozygous for PI*Z, divided by genotype, is shown in Tables 5.2 and 5.3. The PI*Z allele was present mainly in Caucasians and rarely in individuals of African-American, Asian-Pacific, or American-Indian descent. The PI*ZZ allele combination was not seen in the latter three groups (Table 5.2).
Smoking History
The smoking history for the entire cohort, individuals heterozygous and homozygous for PI*Z and divided by genotype, is shown in Tables 5.2 and 5.3. The percentage of never-smokers increased and the proportion of current smokers decreased when comparing subjects with no Z allele to subjects with one and two PI*Z alleles (p < 0.001) (Table 5.2). Interestingly, PI ZZ women included more never-smokers individuals than PI ZZ men (42.9 vs. 25.9 %, p < 0.001). In contrast, PI ZZ men included more ex-smokers subjects than PI ZZ women (49.7 vs. 28.8 %, p < 0.001).
Year of Testing
We tested 3142 samples in 2004, 3766 in 2005, 5257 in 2006, 7283 in 2007, 11,006 in 2008, and 7116 during the first 6 months of 2009. The corresponding mean ages at genetic testing were 54.4 ± 15 years, 58.2 ± 16 years, 57.3 ± 15 years, 57 ± 15 years, 57.6 ± 14 years, and 58.3 ± 15 years, respectively (p < 0.001). In contrast, the age at genetic testing in PI*ZZ individuals was not significantly different for the years 2004–2009.
Origin of the Sample
Samples originated from US Postal Regions Northeast, Midwest, South, and West in 13.2 %, 18.9 %, 55 %, and 12.9 % of the cases, respectively. The age at genetic diagnosis of PI*ZZ was not significantly different among regions.
Reasons for AATD Testing
Patients were tested predominantly for respiratory diseases (68.2 %) (Tables 5.2 and 5.3). Heterozygous and homozygous for PI*Z had a higher proportion of individuals with family history of respiratory diseases than subjects without any Z allele (p < 0.001).
Interpretation of the National Detection Program
In this study, which included a large number of patients who underwent AATD targeted detection in the USA, we observed a higher frequency of AATD alleles compared with population screening studies in the USA. We noted that patients were genetically tested at a relatively advanced age. Homozygous and heterozygous variants of PI*Z were diagnosed at a younger age, a difference more noticeable in current smokers. Interestingly, no correlation was found between age at genetic diagnosis and gender, race, year of genetic testing, and origin of the sample in the USA in PI ZZ subjects.
Targeted detection is a tool to increase AATD recognition that has a higher yield and lower cost than population screening. Using the algorithm described in Fig. 5.1, we found that 16 % of the samples had a genotype other than PiMM, a figure almost twice the prevalence observed in the general population (8.8 %) [24]. In the present assessment, the detection rate for PI ZZ was 1 %, which increased to 1.1 % (401/37,731) and 1.71 % (645/37,731) if Z null/rare and PI SZ genotypes were sequentially included. When compared with general screening, the detection rate of PI ZZ individuals increased approximately 48 times with targeted detection. A review of the literature reveals a broad degree of detection rates for PI ZZ, ranging from 0.2 to 6.6 % [11, 17, 25–27]. This variation is likely to result from the different criteria used for testing of potential AATD subjects.
The use of the real-time polymerase chain reaction for genotyping, as the first test in our algorithm, allows the detection of the two most common deficiency alleles found in AATD individuals in a rapid and efficient way. If used alone, this test may result in the misclassification of rare deficiency alleles. For this reason, we measured AAT protein DBS levels in patients homozygous and heterozygous for PI*Z and PI*S, for confirmation and to avoid misclassification of a second deficient allele. In the event that AAT protein level was low in DBS, we perform PI typing as a reflex testing. In PI MM individuals, we only performed AAT plasma levels in those subjects considered to be at high risk (less than 50 years of age, presence of emphysema, and/or suggestive family history). The routine measurement of AAT plasma levels in all PI MM subjects would not be cost-effective, as a minuscule proportion of these subjects would have severe AATD. To prove this concept, we analyzed data from the Alpha-one coded testing (ACT) trial (unpublished) on 5670 individuals with genotype PI*MM who had measurement of DBS AAT protein level. In this study, only 27 patients (0.4 %) had an AAT DBS level below or equal to 0.6 μM (a conservative cutoff).
A similar approach to diagnosis was validated by other studies [28, 29] that used genotyping and AAT protein serum concentrations with reflex IEF if results are discordant. Snyder et al. [28] found that 4 % of the samples were discordant for genotype and AAT protein serum concentration, requiring reflex PI typing. Bornhorst et al. [29] observed that by adding genotyping, AAT protein serum concentration, and reflex PI typing, 1.6 % of the samples with two deficient alleles were misclassified..
Most of the patients who had PI ZZ genotype were 50 years of above, of white race, and had a history of cigarette smoking. AATD is largely under-recognized and there are significant diagnostic delays [4], due to the fact that many individuals do not have significant clinical impairment, especially if they do not smoke. This may in part explain why more than 50 % of AATD individuals were diagnosed after the age of 50 years. Other explanations are the relatively low percentage of patients wo were tested for family history of respiratory diseases including AATD (17 %) and the relatively low rate of current smokers (10.6 %). The mean age at the time of inclusion of AATD individuals in different registries ranged from 46.1 to 51 years [2, 27, 30, 31], though this result may be biased as patients may have been diagnosed several years prior to the inclusion in these registries. PI ZZ individuals were diagnosed at a younger age than PiMM subjects. Similarly, Lieberman et al. [32] found that COPD individuals homozygous for PI ZZ were younger (55.9 ± 9.8 years) than PI MM subjects (64 ± 8.7 years, p <0.001).
We observed a balanced gender distribution in PI ZZ patients, although more women were tested than men. Other studies found a slight male predominance (52.3–55 %), particularly in index cases [2, 30, 33].
Homozygous PI*Z individuals were only of Caucasian, Hispanic, or mixed race, even though a significant number of patients were of African-American, Asian-Pacific, and American-Indian race. In these latter races, few individuals were heterozygous for PI*Z. This marked white race predominance was observed in other studies, ranging from 96.2 to 99.2 % [2, 31]. de Serres et al. described that the highest risk for AATD is found in Caucasians, followed by Hispanics and African Americans. The lower prevalence is among Mexican Americans and Asians [34].
Homozygous or heterozygous individuals for PI*Z had less percentage of current smokers and more never-smoker individuals than the overall population studied. These findings are similar to the smoking status of other studies (current smoker ranged from 2.1 to 8.3 % [2, 12, 31]. In our study, PI ZZ smokers were diagnosed at a younger age than nonsmokers or ex-smokers, a finding supported by other author that demonstrated that nonsmoking AATD individuals have a delay of symptoms and most of them a normal life span [33, 35–38]. Interestingly, the duration of smoking also influenced the age at the time of testing, since individuals who smoked more than 10 years were diagnosed 7.4 (95 % CI: 2.6) years before than subjects who smoked less than for this duration of time (p = 0.005).
The number of subjects tested in our program has increased during the 6-year study period. The age at the time of testing has remained stable at around 57–58 years after an initial increase. Contrary to what one would have expected after the release of the ATS/ERS consensus statement in 2003 [1], we did not observe a reduction in the age of the subjects tested for the deficiency.
More than 50 % of the samples were originated in US Postal Region III, as we are the referral center for the State of Florida AATD detection program. We did not find a statistical difference in the prevalence of AATD in the samples tested when divided by Regions. Furthermore, the age of individuals tested from the four US Postal Regions was similar.
More than half of the individual were tested due to respiratory symptoms. In a small percentage (2.4 %) of the samples, a family history of respiratory disease and AATD was volunteered. This percentage is lower than the one reported by other studies [2, 30]. We did not find a correlation between reason for testing and age at the time of genetic diagnosis.
There are limitations to this study. The results only apply to individuals who underwent targeted detection and are not representative of all individuals with the disease. Symptomatic individuals are more likely to undergo testing than asymptomatic subjects, leading to ascertainment bias. Single rare deficiency alleles were not detected by our algorithm and few homozygous null/rare individuals could have been missed. More than 50 % of the samples originated in US Region IV (other Regions were less represented), and a family history of AATD was not separated from a family history of respiratory diseases (COPD or emphysema). In spite of these limitations, this study provides a valuable insight into the prevalence and demographic and clinical characteristics of severe AATD in patients who undergo targeted screening in the USA.
The results of this study support the use of targeted detection programs to identify non-index and index individuals with severe AATD who can benefit from genetic counseling, risk-reduction behaviors, and/or augmentation treatment. Furthermore, the program provides opportunities to increase the awareness about this largely unrecognized disease.
Abbreviations
- AAT:
-
Alpha-1 antitrypsin
- AATD:
-
Alpha-1 antitrypsin deficiency
- FEV1 :
-
Forced expiratory volume in one second
- COPD:
-
Chronic obstructive pulmonary disease
References
American Thoracic Society, European Respiratory Society. American Thoracic Society/European respiratory society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:818–900.
McElvaney NG, Stoller JK, Buist AS, Prakash UB, Brantly ML, Schluchter MD, Crystal RD, Alpha 1-Antitrypsin Deficiency Registry Study Group. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute registry of alpha 1-antitrypsin deficiency. Chest. 1997;111(2):394–403.
Tobin MJ, Cook PJ, Hutchison DC. Alpha 1 antitrypsin deficiency: the clinical and physiological features of pulmonary emphysema in subjects homozygous for Pi type Z. A survey by the British Thoracic Association. Br J Dis Chest. 1983;77(1):14–27.
Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest. 2005;128(4):1989–94.
Strange C, Stoller JK, Sandhaus RA, Dickson R, Turino G. Results of a survey of patients with alpha-1 antitrypsin deficiency. Respiration. 2006;73(2):185–90.
Brantly M. Efficient and accurate approaches to the laboratory diagnosis of alpha1-antitrypsin deficiency: the promise of early diagnosis and intervention. Clin Chem. 2006;52(12):2180–1.
Strange C, Dickson R, Carter C, Carpenter MJ, Holladay B, Lundquist R, Brantly ML. Genetic testing for alpha1-antitrypsin deficiency. Genet Med. 2004;6(4):204–10.
O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr. 1978;92(6):1006–10.
Silverman EK, Miletich JP, Pierce JA, Sherman LA, Endicott SK, Broze Jr GJ, Campbell EJ. Alpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis. 1989;140(4):961–6.
Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med. 2009;103(3):335–41.
de la Roza C, Rodríguez-Frías F, Lara B, Vidal R, Jardí R, Miravitlles M. Results of a case-detection programme for alpha1-antitrypsin deficiency in COPD patients. Eur Respir J. 2005;26(4):616–22.
Brantly ML, Paul LD, Miller BH, Falk RT, Wu M, Crystal RG. Clinical features and history of the destructive lung disease associated with alpha-1-antitrypsin deficiency of adults with pulmonary symptoms. Am Rev Respir Dis. 1988;138(2):327–36.
Brantly M. Alpha 1-antirypsin genotypes and phenotypes. In: Crystal RG, editor. Alpha 1-antitrypsin deficiency. New York: Marcel Dekker; 1996. p. 45–59.
Dahl M, Tybjaerg-Hansen A, Lange P, Vestbo J, Nordestgaard BG. Change in lung function and morbidity from chronic obstructive pulmonary disease in alpha1-antitrypsin MZ heterozygotes: a longitudinal study of the general population. Ann Intern Med. 2002;136(4):270–9.
Hersh CP, Dahl M, Ly NP, Berkey CS, Nordestgaard BG, Silverman EK. Chronic obstructive pulmonary disease in alpha1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax. 2004;59(10):843–9.
Dahl M, Hersh CP, Ly NP, Berkey CS, Silverman EK, Nordestgaard BG. The protease inhibitor PI*S allele and COPD: a meta-analysis. Eur Respir J. 2005;26(1):67–76.
World Health Organization. Alpha1-antitrypsin deficiency: memorandum from WHO meeting. Bull World Health Organ. 1997;75:397–415.
Census Bureau Region and Division Codes. Accessed on April 2010. http://www.census.gov/popest/geographic/codes02.html
Brantly ML, Wittes JT, Vogelmeier CF, Hubbard RC, Fells GA, Crystal RG. Use of a highly purified alpha 1-antitrypsin standard to establish ranges for the common normal and deficient alpha 1-antitrypsin phenotypes. Chest. 1991;100(3):703–8.
Brantly M. Laboratory diagnosis of alpha1 antitrypsin deficiency. In: Crystal RG, editor. Alpha 1-antitrypsin deficiency. New York: Marcel Dekker; 1996. p. 211–25.
Ferrarotti I, Scabini R, Campo I, Ottaviani S, Zorzetto M, Gorrini M, Luisetti M. Laboratory diagnosis of alpha1-antitrypsin deficiency. Transl Res. 2007;150(5):267–74.
Laurell CB, Sveger T. Mass screening of newborn Swedish infants for alpha antitrypsin deficiency. Am J Hum Genet. 1975;27(2):213–7.
de Serres FJ, Blanco I, Fernández-Bustillo E. Genetic epidemiology of alpha-1 antitrypsin deficiency in North America and Australia/New Zealand: Australia, Canada, New Zealand and the United States of America. Clin Genet. 2003;64(5):382–97.
de Serres FJ. Alpha-1 antitrypsin deficiency is not a rare disease but a disease that is rarely diagnosed. Environ Health Perspect. 2003;111(16):1851–4.
Gelmont D, Chung KC, Felinczak BS, Campbell EJ. Prevalence of alpha1-antitrypsin deficiency (AATD) in patients with COPD in a nationwide screening program. Am J Respir Crit Care Med. 2007;165:A987.
Campbell EJ. Alpha1-antitrypsin deficiency: incidence and detection program. Respir Med. 2000;94(Suppl C):S18–21.
Luisetti M, Massi G, Massobrio M, Guarraci P, Menchicchi FM, Beccaria M, Balbi B. A national program for detection of alpha 1-antitrypsin deficiency in Italy. Gruppo IDA. Respir Med. 1999;93(3):169–72.
Snyder MR, Katzmann JA, Butz ML, Wiley C, Yang P, Dawson DB, Halling KC, Highsmith WE, Thibodeau SN. Diagnosis of alpha-1-antitrypsin deficiency: an algorithm of quantification, genotyping, and phenotyping. Clin Chem. 2006;52(12):2236–42.
Bornhorst JA, Procter M, Meadows C, Ashwood ER, Mao R. Evaluation of an integrative diagnostic algorithm for the identification of people at risk for alpha1-antitrypsin deficiency. Am J Clin Pathol. 2007;128(3):482–90.
Stockley RA, Luisetti M, Miravitlles M, Piitulainen E, Fernandez P, Alpha One International Registry (AIR) Group. Ongoing research in Europe: alpha one international registry (AIR) objectives and development. Eur Respir J. 2007;29(3):582–6.
Stoller JK, Brantly M, Fleming LE, Bean JA, Walsh J. Formation and current results of a patient-organized registry for alpha(1)-antitrypsin deficiency. Chest. 2000;118(3):843–8.
Lieberman J, Winter B, Sastre A. Alpha 1-antitrypsin Pi-types in 965 COPD patients. Chest. 1986;89(3):370–3.
Seersholm N, Kok-Jensen A, Dirksen A. Survival of patients with severe alpha 1-antitrypsin deficiency with special reference to non-index cases. Thorax. 1994;49(7):695–8.
de Serres FJ, Blanco I, Fernández-Bustillo E. Ethnic differences in alpha-1 antitrypsin deficiency in the United States of America. Ther Adv Respir Dis. 2010;4(2):63–70.
Piitulainen E, Tornling G, Eriksson S. Effect of age and occupational exposure to airway irritants on lung function in non-smoking individuals with alpha 1-antitrypsin deficiency (PI ZZ). Thorax. 1997;52(3):244–8.
Larsson C. Natural history and life expectancy in severe alpha1-antitrypsin deficiency Pi Z. Acta Med Scand. 1978;204(5):345–51.
Janus ED, Phillips NT, Carrell RW. Smoking, lung function, and alpha 1-antitrypsin deficiency. Lancet. 1985;1(8421):152–4.
Hutchison DC, Tobin MJ, Cook PJ. Alpha 1 antitrypsin deficiency: clinical and physiological features in heterozygotes of Pi type SZ. A survey by the British Thoracic Association. Br J Dis Chest. 1983;77(1):28–34.
Acknowledgment
We greatly appreciate the outstanding and invaluable work performed daily by all members of the Alpha 1 Detection laboratory.
Funding
Alpha-1 Foundation
Disclosure
Adriano R. Tonelli, M.D., has no conflict of interest to disclose
Farshid Rouhani, M.S., has no conflict of interest to disclose
Pam Schreck, R.N., M.S.N., has no conflict of interest to disclose
Mark L. Brantly, M.D., receives grant support from Grifols Alpha-1-Foundation, Kamada Ltd, and National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tonelli, A.R., Rouhani, F., Brantly, M.L. (2016). United States Targeted Detection Program for Alpha-1 Antitrypsin Deficiency. In: Wanner, A., Sandhaus, R. (eds) Alpha-1 Antitrypsin. Respiratory Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-23449-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-23449-6_5
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-23448-9
Online ISBN: 978-3-319-23449-6
eBook Packages: MedicineMedicine (R0)