
Abstract
In recent years, DNA sequencing and mass spectrometry technologies have advanced greatly, enabling the collection of more information on the gut microbiome and its metabolome in order to assess the influence of the gut microbiota on human health at a whole-system level. As the gut microbiota has been likened to a functional and measurable organ consisting of prokaryotic cells, which creates the unique gut ecosystem together with the host eukaryotic cells, metagenome and metabolome technologies have demonstrated that the gut microbiota contributes to host overall health status to a great extent. In this chapter, the detailed relationships between gut microbiota and its metabolites like choline, phenols, bile acids and short-chain fatty acids in host health and etiopathogenesis of various metabolic diseases such as obesity, diabetes, atherosclerosis, non-alcoholic fatty liver disease and extraintestinal diseases like multiple sclerosis, chronic kidney disease and autism will be discussed. In addition, therapeutic interventions like probiotic and prebiotic administrations and faecal microbiota transplantations which are recently used in dysbiosis restoration will be reviewed. This unique biology-wide approach of integrating metagenome and metabolome information would aid in the better understanding of the intricate interplay between gut microbiota and host metabolism. We believe that this novel integration of the microbiome, metatranscriptome and metabolome information will lay the way towards an improved holistic understanding of the complex mammalian superorganism. This modelling of the metabolic interactions between lifestyle, dietary habits and the gut microbiota, otherwise known as the “integrated omics-based understanding of the gut ecosystem”, will culminate in the comprehensive interpretation of the role and impact of microbial health potentials, thereby providing exciting novel therapeutic approaches for optimal host health.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 The Gut Microbiota and Metabolomics-Based Integrated Omics Approach
1.1 The Gut Microbiota
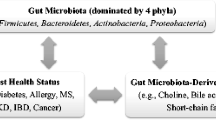
The gut microbiota refers to all the microorganisms inhabiting the gastrointestinal tract. Four bacterial phyla , Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria, dominate the gut microbiota in mammals, and these phyla have been reported to characterize the role of the host metabolism and physiology (Qin et al. 2010). The bacteria populating the gut possess extensive metabolic capabilities (Qin et al. 2010) and amount to about 100 trillion cells, which is approximately three times higher than the total number of cells in the human body (Bianconi et al. 2013). As such, the gut microbiota is often associated to be a functional and measurable organ consisting of prokaryotic cells, which forges with host eukaryotic cells to create a unique gut ecosystem (Fukuda and Ohno 2014). Bacterial communities vary in composition along the digestive tract and evolve within and between individuals over time in accordance to the lifestyle and nutritional status of the host (Xu et al. 2007). It is only in recent years that we have started to comprehend the systemic impact of the gut microbiota on the whole host metabolic repertoire. The gut microbiota is involved in other functions of the body like drug metabolism and toxicity (Clayton et al. 2006), dietary calorific bioavailability (Hooper 2001), immune response (Macpherson 2000) and postsurgical recovery (Kinross et al. 2011). However, more importantly, apart from its obvious role in digestion, gut microbiota has been implicated in maintaining optimal host health and the etiopathogenesis of various metabolic diseases such as obesity (Turnbaugh et al. 2006) and diabetes (Wen et al. 2008; Qin et al. 2010; Wang et al. 2012), intestinal diseases like inflammatory bowel diseases (IBD) (Marchesi et al. 2007) and colonic cancer (Scanlan et al. 2008) and extraintestinal diseases like allergy (Kirjavainen et al. 2002), multiple sclerosis (Berer et al. 2011), chronic kidney disease (Wang et al. 2012) and autism (Finegold 2008) (Fig. 1).

The role of gut microbiota and its metabolites. Four bacterial phyla, Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria, dominate the gut microbiota in mammals, and these phyla have been reported to characterize the role of the host metabolism and physiology. The gut microbiota and its metabolites like choline, bile acids, phenols and short-chain fatty acids are highly implicated in the etiopathogenesis of metabolic diseases, intestinal diseases and extraintestinal diseases, thereby playing a vital role in host health. Therapeutic interventions to improve dysbiosis include faecal microbiota transplantation and probiotic and prebiotic administration. NAFLD non-alcoholic fatty liver disease, IBD inflammatory bowel disease, MS multiple sclerosis, CKD chronic kidney disease, SCFAs short-chain fatty acids
1.2 What Is Metabolomics?
Technological breakthroughs have enabled the simultaneous examination of thousands of genes (genomics), transcripts (transcriptomics), proteins (proteomics), metabolites (metabolomics) and gut microbiota (metagenomics) with high-throughput techniques and analytical tools (Ellis et al. 2007). Since the comprehensive understanding of the organ and systemic metabolism is vital in maintaining health and nutritional status (Nicholson et al. 2012), the rapid advances in DNA sequencing and mass spectrometry (MS) technologies in recent years have enabled the extensive collection of data on the gut microbiome and metabolome to comprehensively evaluate the impact of the gut microbiota on human health (Tringe and Hugenholtz 2008). This has led to major advances in metagenome and metabolome technologies to reveal the important role that the gut microbiota play in contributing to host overall health status.
Two of the most commonly used wide-range metabolomic analytical methods include nuclear magnetic resonance (NMR) and MS, and they are commonly used in the identification of disease biomarkers. By using these approaches, we can accurately identify and have a robust understanding of the metabolites produced by microbiota and host cells in faecal, blood, tissue and urine samples (Dettmer et al. 2007). Methodologies ranging from targeted to untargeted approaches have been reported in screening biochemical pathways involved in central carbon metabolism, glycolysis, tricarboxylic acid cycle, amino acid metabolism, lipid metabolism and selected secondary metabolic pathways (Wenk 2005; Moco et al. 2006; Buescher et al. 2010). These tools allow scientists to comprehend the extent of impact of treatments on the host metabolic profile by the simultaneous analysis of the presence and quantity of thousands of metabolites.
1.3 Using Metabolomics to Understand the Gut Microbiota
Evaluation of the metabolome profile is commonly used to directly compare the metabolism of the gut microbiota and the metabolic outcomes in the host. In an investigation on the systemic influence of administering probiotics or prebiotics or a combination of both in initially germ-free mice colonized with a combination of microbes representing that of a human infant (Martin et al. 2008), it was revealed that dietary supplementation significantly modified the relative composition of gut microbiota community, culminating in systemic changes in the metabolic profiles of different tissues. Prebiotics increased proportions of Bifidobacterium breve, Bifidobacterium longum and Bacteroides distasonis; decreased proportions of Escherichia coli and Clostridium perfringens; and modulated lipid metabolism by decreasing concentrations of glucose and hepatic triglycerides in the plasma (Martin et al. 2008). In another report by Wikoff et al. (2009), the effects of gut microbiota on the host were evaluated via plasma metabolome profile comparison between germ-free and conventionally raised mice. There were many metabolites that were detected only in conventionally raised mice and not in germ-free mice. Additionally, in mice with or without gut microbiota, concentrations of more than a tenth of all metabolites differed by more than 50 % (Wikoff et al. 2009).
The integration between the activities of the gut microbiome and our gene reflects overall human metabolism at the systemic level. Our gastrointestinal tract provides nutrients to cells and tissues via the circulatory system, and likewise, so are the metabolites originating from the gut microbiota. This delicate interplay amongst gut microbiota-derived metabolites, the gut microbiota itself and the host immune system is transmitted through an extensive array of signalling pathways that extend beyond the immune system. The direct chemical interactions between gut microbiota and the host and the immune-mediated signalling mechanisms influence various organs such as the gut, liver, skeletal muscle and the brain, and these complex interrelationships come together mutually to culminate in a series of host–microbe metabolic axes . Within these axes, metabolic reactions can be regulated by gut microbial genomes, resulting in the production of choline, phenols, bile acids and short-chain fatty acids (SCFAs) by both the gut microbiome and host genome that are essential to host health (Aw and Fukuda 2015) (Fig. 2).

The influence of gut microbiota-derived metabolites on host physiology. Choline has been implicated in the pathogenesis of obesity, fatty liver, non-alcoholic fatty liver disease (NAFLD) and cardiovascular diseases. Bile acids are reported to promote hepatocarcinoma development, glucose modulation, obesity, type 2 diabetes and inflammatory diseases. Phenols are involved in inflammatory bowel disease and weight loss. Short-chain fatty acids (SCFAs) have been reported as health-promoting metabolites, and the imbalance of SCFAs mediates type 2 diabetes, obesity, inflammatory and allergic responses
2 Metabolites
2.1 Choline
Choline , which plays a vital role in fat metabolism and synthesis of very-low-density lipoproteins in the liver, is primarily metabolized in the liver and is a vital component of cell membranes mostly obtained from foods like eggs and red meat (Vance 2008). The microbial conversion of dietary choline leads to an altered gut microbial ecology, which results in obesity and liver steatosis both in mice (Henao-Mejia et al. 2012) and in humans (Spencer et al. 2011), and promotes cardiovascular disease in human subjects (Wang et al. 2011). Importantly, it was reported that low concentrations of γ-proteobacteria and high levels of Erysipelotrichia in human faecal microbiota are correlated with hepatic steatosis (Spencer et al. 2011). Dietary choline is converted to trimethylamine (TMA) (Vance 2008) by gut microbial enzymes, which is then further metabolized by the flavin monooxygenase (FMO) system in the liver to produce trimethylamine-N-oxide (TMAO) (Dumas et al. 2006; Prentiss et al. 1961). This phenomenon lowers the levels of choline bioavailability and is implied to trigger non-alcoholic fatty liver disease (NAFLD) in mice (Dumas et al. 2006). In a report investigating the relationship between oral intake of phosphatidylcholine and the involvement of the gut microbiota in proatherogenic TMAO production in humans, plasma TMAO levels were significantly lowered after the administration of antibiotics and then reappeared after withdrawal of antibiotics. Elevated plasma TMAO concentrations were correlated with an increased risk of a major adverse cardiovascular event independently of traditional cardiovascular risk factors, even in low-risk cohorts. As such, the lines of evidence mentioned above support the modulation of gut microbiota as a probable therapeutic approach in relation to these events (Tang et al. 2013). These studies provide evidence behind a potential relationship between the gut microbiota, dietary choline and risk of cardiovascular disease development.
2.2 Phenols
Approximately 50–100 mg of volatile phenols , mainly in the form of 4-cresol and phenol (predominantly as glucuronide and sulfate conjugates), is excreted by humans daily (Bone et al. 1976). The members of the genera Clostridium, Bifidobacterium and Bacteroides are implicated in the production of cresols in mammals. A large diversity of physiological and pathological conditions, ranging from IBD to weight loss, is reported to be correlated with altered levels of urinary 4-cresol metabolites in humans. Additionally, these conditions are also associated with altered gut microbiota composition, particularly, lowered diversity of microbiota due to the loss of Lactobacillus and Bacteroides species in IBD (Ott 2004) and differences in the ratio of Firmicutes and Bacteroidetes members due to weight loss (Ley et al. 2005; Turnbaugh et al. 2006).
2.3 Bile Acids
In the human liver, primary bile acids such as cholic acid and chenodeoxycholic acid are synthesized from cholesterol and then secreted in bile. Primary bile acids are mainly involved in facilitating the metabolism of dietary fat and the absorption of fat-soluble vitamins and cholesterol. Primary bile acids undergo an enterohepatic cycle between the gut and the liver eight times per day, where roughly 90–95 % of the bile acids are reabsorbed by the intestine and returned to the liver, where they are conjugated to taurine in mice and to glycine in humans, to form bile salts (Ridlon et al. 2006; Swann et al. 2011). Some of the remaining primary bile acids are deconjugated and further metabolized to secondary bile acids such as deoxycholic acid (DCA) and lithocholic acid by gut microbes, which belong to the genera Bacteroides, Eubacterium and Clostridium, whilst the others are lost in the faeces (Ridlon et al. 2006; Watanabe et al. 2006). Secondary bile acids are then reabsorbed, mainly by both bile acid transporters in the ileum and passive absorption in the intestine (Watanabe et al. 2006). Aerobic bacteria have been reported to be involved in the biotransformation of a minor portion of bile acids (Ridlon et al. 2006). The induction of senescence-associated secretory phenotype (SASP) in hepatic stellate cells by the enterohepatic circulation of DCA (Friedman 2008) secretes various hepatic inflammatory and tumour-promoting factors, thus facilitating murine hepatocarcinoma development after exposure to chemical carcinogens. More importantly, the suppression of DCA production or reduction in the number of gut microbes efficiently prevents hepatocarcinoma development in obese mice as observed in mice lacking an SASP inducer (Orjalo et al. 2009) or the depletion of senescent hepatic stellate cells. These results indicate that the DCA–SASP axis in hepatic stellate cells is important in obesity-associated hepatocarcinoma development. Moreover, it was also observed that there were signs of SASP in the hepatic stellate cells of hepatocarcinoma arising in patients with non-alcoholic steatohepatitis (Coulouarn et al. 2012), suggesting that there might be a similar pathways responsible for certain aspects of obesity-associated hepatocarcinoma development in humans (Yoshimoto et al. 2013). As the gut microbiota is heavily related to bile acid transformation, conventionally raised rodents have lesser bile acids and a more diverse gut microbiota profile than their germ-free counterparts (Wostmann 1973; Swann et al. 2011). Besides, bile acids also function as signalling molecules and bind to cellular receptors (Watanabe et al. 2006) like the nuclear receptor farnesoid X receptor (FXR) (Prawitt et al. 2011) and the G protein-coupled receptor (GPCR) TGR5 (Thomas et al. 2009). FXR is activated by primary bile acids, whilst TGR5 binds secondary bile acids such as DCA and lithocholic acid. In enteroendocrine L-cells, TGR5 signalling induces secretion of glucagon-like peptide-1 (GLP-1), thereby leading to enhanced hepatic and pancreatic function and improved glucose tolerance in obese mice (Thomas et al. 2009). Both FXR and TGR5 are involved in murine glucose metabolism modulation whereby FXR impairs and TGR5 promotes glucose homeostasis (Prawitt et al. 2011; Thomas et al. 2009). Furthermore, the activation of TGR5 in skeletal muscle and brown adipose tissue has been reported to elevate energy expenditure and protect against diet-induced obesity (Watanabe et al. 2006). As reported by David et al. (2014), in mice that fed on saturated fats from milk and also in humans who consume high-fat diets, the levels of bile-tolerant microorganisms (Alistipes, Bilophila and Bacteroides) were increased and the levels of Firmicutes species that metabolize dietary plant polysaccharides (Roseburia, Eubacterium rectale and Ruminococcus bromii) were decreased. The differences between herbivorous and carnivorous mammals were reflected in the differences in microbial activities. In mice that fed on animal-based diet, the levels of B. wadsworthia were increased, allowing us to draw the correlations between animal-based diets, changes in bile acid concentrations, gut microbiota composition and the increase in the probability of IBD development (David et al. 2014). In light of the above, it has been demonstrated that all major bacterial groups are involved in bile salt hydrolase activity. As such, gut microbiota modulation may control lipid and glucose metabolism via the composition of bile acid pools and the modulation of FXR and TGR5 signalling, thereby playing a role in effective obesity and type 2 diabetes management (Jones et al. 2008).
2.4 Dietary Fibre Fermentation and Short-Chain Fatty Acid Production
When dietary fibre or complex carbohydrates are consumed, they are digested and then fermented mainly in the colon by gut microbiota into SCFAs such as acetate, propionate and butyrate. These SCFAs are recognized by the GPCRs such as GPR41 and GPR43, which are expressed by gut enteroendocrine cells (Samuel et al. 2008). Gut microbiota induces peptide YY expression by L-cells via a GPR41-dependent mechanism as observed in the study where lowered adiposity was observed in conventional Gpr41-deficient mice as compared to conventional wild-type mice, whereas adiposity in germ-free, wild-type and Gpr41-deficient mice were comparable (Samuel et al. 2008). Inflammation can be inhibited by SCFAs via GPR43 signalling in neutrophils (Maslowski et al. 2009; Sina et al. 2009) and insulin signalling in adipocytes (Kimura et al. 2013). In addition, SCFAs also modulate the secretion of the hormone GLP-1, which may improve insulin secretion and suppress diabetes (Tolhurst et al. 2012). In a recent report by Trompette et al. (2014), the composition of both gut and lung microbiota were interestingly changed by dietary fibre content via the alteration of the ratio of Firmicutes to Bacteroidetes. In mice fed with high-fibre diets, SCFA levels were elevated, and they were subsequently protected against allergic inflammation in the lungs, whereas the contrary was observed in mice fed with low-fibre diets. It was observed that in mice that were treated with SCFA propionate, there were increased generation of macrophages and dendritic cell precursors and subsequent seeding of the lungs by dendritic cells of high phagocytic capacity. However, these effects of propionate on allergic inflammation were depended on GPR41 but not GPR43 (Trompette et al. 2014). Other SCFAs such as acetate and propionate are taken up by the liver and used as substrates in gluconeogenesis and lipogenesis (Tremaroli and Backhed 2012). SCFAs can modulate histone deacetylase function by stimulating the sympathetic nervous system, thereby influencing social behaviour in rats (MacFabe et al. 2011). The SCFAs propionate and butyrate, which are generated by the fermentation of soluble fibres, activate intestinal gluconeogenesis that improves glucose and energy homeostasis (De Vadder et al. 2014). Out of all the SCFAs produced from microbial fermentation, butyrate which is mainly produced by species of the order Clostridiales, a dominant order in gut microbiota, is particularly vital as an energy substrate for cellular metabolism in the colonic epithelium (Donohoe et al. 2011). Gut microbiota-derived butyrate activates GPR109a niacin receptor and suppresses both colonic inflammation and carcinogenesis (Singh et al. 2014). In addition, it has also been reported that intestinal gluconeogenesis gene expression is activated by butyrate via a cAMP-dependent mechanism, whilst propionate, a substrate of intestinal gluconeogenesis, activates intestinal gluconeogenesis gene expression through the gut–brain neural circuit involving the fatty acid receptor FFAR3 (De Vadder et al. 2014). In a recent metabolomic-based integrated omics study, it was revealed that in germ-free mice fed with high-fibre diet, there was colonization of Clostridiales which promoted gut microbial fermentation, thereby resulting in the accumulation of luminal SCFAs (Furusawa et al. 2013). Amongst all SCFAs, butyrate induces the in vitro differentiation of regulatory T (Treg) cells. Administration of butyrylated starch induces colonic Treg cell differentiation and attenuates the development of colitis in vivo. Treatment of naïve T cells under Treg-polarizing conditions with butyrate enhances histone H3 acetylation particularly in the promoter and CNS 3 enhancer regions of the Foxp3 gene, the master transcriptional regulator of Treg cells. As such, butyrate derived from dietary fibre fermentation by commensals of the order Clostridiales epigenetically induces the differentiation of colonic Treg cells, which play critical roles in the suppression of inflammatory and allergic responses (Atarashi et al. 2011, 2013; Furusawa et al. 2013). In the view of protection from enteropathogenic infection, using an integrated omics in a simplified model of lethal infection with enterohaemorrhagic Escherichia coli O157:H7, it was revealed by Fukuda et al. (2011) that acetate produced by probiotic bifidobacteria improves intestinal defence by enhancing gut epithelial barrier function (Fukuda et al. 2011). In a study by Okada et al. (2013), microbiota-derived lactate has been reported to be an important factor in the enterocyte hyperproliferation induction in firstly starved then refed mice. During a 12- to 36-h period of starvation, colonic epithelial cell turnover was halted. It then increased 12–24 h after refeeding. As such, the increase in live Lactobacillus murinus, lactate production and dietary fibre content could also enhance epithelial cell proliferation (Okada et al. 2013). Thus, it is evident that SCFAs can influence a range of host processes with significant effects and are thus vital products of microbial fermentation of dietary fibres.
3 Dysbiosis and Metabolic Diseases
The gastrointestinal tract, a complex and well-balanced ecosystem, has commensal microbes and their hosts in a symbiotic relationship under normal conditions. However, qualitative and quantitative changes in the gut microbiota will lead to an imbalance in this equilibrium. The changes in the metabolic activities of the gut microbiota and the changes in their local distribution lead to dysbiosis, which is a condition in which microbial imbalance exerts adverse effects on the host. Various factors like antibiotic consumption, nutritional status and environmental conditions can disrupt microbial stability and are partly responsible for intestinal dysbiosis (Hawrelak and Myers 2004). This leads to various metabolic diseases like obesity, diabetes, chronic kidney disease, atherosclerosis, NAFLD and extraintestinal diseases like multiple sclerosis and autism (Fig. 3).

Dysbiosis-related diseases and therapeutic interventions. Dysbiosis refers to the imbalance between the peacekeeping bacteria and pathobionts, leading to metabolic diseases like obesity, diabetes, atherosclerosis and NAFLD and extraintestinal diseases like multiple sclerosis, chronic kidney disease (CKD) and autism. Ingestion of probiotics and/or prebiotics and faecal microbiota transplantation have been reported to restore symbiosis
3.1 Obesity
It was first suggested in 2005 that obesity could be attributed to gut microbial composition levels as the phylum Bacteroidetes was lower and the phylum Firmicutes was higher in abundance in obese ob/ob mice than their lean littermates although both groups were administered similar diets (Ley et al. 2005). Subsequently, in a study with obese human twins, a correlation was observed between an increase in Firmicutes proportion and a decrease in Bacteroidetes proportion to the enrichment of microbial genes encoding key enzymes involved in carbohydrate metabolism. This is implicated in the host’s ability to digest food and supply energy substrates such as SCFAs (Turnbaugh et al. 2006, 2009). Importantly, the transfer of gut microbiota from both obese mice (Vijay-Kumar et al. 2010) and obese humans (Turnbaugh et al. 2006; Ridaura et al. 2013) into germ-free recipient mice reproduced the obese phenotype. In addition, it has also been reported that bifidobacterial numbers were lower, levels of Staphylococcus aureus were higher (Kalliomäki et al. 2008), levels of Enterobacteriaceae species were significantly higher and levels of Desulfovibrio and Akkermansia muciniphila-like bacteria were significantly lower in obese children than in normal-weight children (Karlsson et al. 2012a). Significant decreases in the number of A. muciniphila, a novel mucin-degrading bacterium that colonizes the mucosal layer constituting of 3–5 % of the bacterial community (Belzer and de Vos 2012), were observed in genetically and high-fat diet-induced obese mice (Everard et al. 2013). Other studies have also shown that this bacterium is inversely correlated with type 1 (Hansen et al. 2012) and type 2 diabetes (Qin et al. 2012) and body weight (Collado et al. 2008; Santacruz et al. 2010; Everard et al. 2011; Karlsson et al. 2012a). Recently, obesity and glucose homeostasis disorders were induced in germ-free mice fed with a high-fat diet that were colonized with endotoxin-producing Enterobacter cloacae B29 strain. This implies that lowering plasma endotoxin levels might be a probable approach in controlling metabolic diseases (Fei and Zhao 2013). In addition, faecal transplantation of gut microbiota from lean healthy donors in human patients presenting with metabolic syndrome resulted in improved insulin sensitivity, which was correlated with an increase in the number of butyrate-producing bacteria, thereby implying that microbial butyrate may be important in promoting this improvement (Vrieze et al. 2012).
3.2 Diabetes
Type 1 and type 2 diabetes development have been correlated with gut microbiota composition. Type 1 diabetes (T1D) is an autoimmune disorder that involves the destruction of pancreatic insulin-producing cells. In studies that were performed in a T1D mouse model, a nonobese diabetic (NOD) mouse, it was revealed that in Myd88 −/− mice, during antibiotic treatment or under germ-free condition, protection against diabetes was diminished (Wen et al. 2008). Bacterial 16S ribosomal RNA gene sequencing also showed variations in the gut microbiota composition in Myd88 −/− NOD mice as compared to their normal littermates. The Firmicutes/Bacteroidetes ratio was lowered, and this indicates that certain bacterial populations were essential in the protection against T1D. However so, it has been recently reported that MYD88 deficiency alone does not remodel gut microbiota composition, and therefore, it is plausible that the genetic background of NOD mice might have influenced the effects of MYD88 (Ubeda et al. 2012). Segmented filamentous bacteria (SFB) have also been reported to protect against T1D (Kriegel et al. 2011) gender specifically as male NOD mice with SFB have lowered incidence of T1D than their female counterparts. Furthermore, the gut microbiota composition in male NOD mice was observed to be different from that in their female counterparts, most probably due to increased testosterone levels in the former, which is associated with T1D protection (Kriegel et al. 2011; Markle et al. 2013). When gut microbiota was transferred from male NOD mice to their female counterparts, the increase in testosterone concentrations and reduced susceptibility to T1D were observed (Markle et al. 2013). In the ob/ob diabetic mice model, it was also observed that a 2-week antibiotic treatment significantly reduced the numbers of both aerobic and anaerobic gut microbes, leading to lower levels of hepatic triglycerides, lower plasma lipopolysaccharide (LPS) concentrations, elevated levels of hepatic glycogen and increased plasma adiponectin concentrations than in their non-treated counterparts (Membrez et al. 2008). In type 2 diabetic (T2D) patients, Larsen et al. (2010) reported significantly lower levels of Firmicutes and Clostridia in male T 2D subjects as compared to nondiabetic control subjects. The same study also reported correlations between plasma glucose concentrations to both the ratios of Bacteroidetes to Firmicutes and of the Bacteroides–Prevotella to the C. coccoides–E. rectale group. As Bacteroidetes and Proteobacteria are composed of gram-negative bacteria and have LPS in their outer membranes, these findings suggest that T2D may be promoted via an endotoxin-induced inflammatory response (Larsen et al. 2010). In a metagenome-wide association study of gut microbiota using data from a total of 345 T2D patients and non-T2D control subjects, most of the genes upregulated in the T2D group were mainly from opportunistic pathogens including Bacteroides caccae, Clostridium hathewayi, Clostridium ramosum, Clostridium symbiosum, Eggerthella lenta and Escherichia coli which have been implicated as causes of underlying human infections, and the genes that were upregulated in the control group were from various butyrate-producing bacterial species like Clostridiales sp. SS3/4, E. rectale, Faecalibacterium prausnitzii, Roseburia intestinalis and Roseburia inulinivorans. The mucin-degrading species A. muciniphila and the sulfate-reducing species Desulfovibrio sp. were also found in abundance in the T2D group (Qin et al. 2012). In another metagenome study conducted in 145 64-year-old European women with normal, deficient or diabetic glucose control, a mathematical model to identify T2D based on gut microbiota metagenomic profile was developed. The same model was also applied to a similar study but using Chinese subjects, and different metagenomic markers in the European T2D and Chinese T2D cohorts were reported (Karlsson et al. 2013). This suggests that age and geographical location may play a role in the variation of gut microbial metagenomic profiles that influence T2D development.
3.3 Atherosclerosis
Host genetic and environmental factors, such as dietary intake and the presence of commensal gut microbes, play important roles in the pathogenesis of atherosclerosis, a vascular disease with a complex pathologic phenotype. Three phospholipid-associated molecules, choline, betaine and TMAO, which are present in the plasma, could be used as biomarkers for predicting the risk of cardiovascular diseases and seemed to promote atherosclerosis (Wang et al. 2011). In another study, Koeth et al. (2013) reported that murine gut microbiota metabolism of dietary l-carnitine, a type of TMA present in large amounts in red meat, also produces TMAO and accelerates the rate of atherosclerosis (Koeth et al. 2013). In addition, vegans produced lesser TMAO than omnivorous human subjects following ingestion of l-carnitine via a microbiota-dependent mechanism (Koeth et al. 2013). There were correlations observed between specific bacterial taxa in human faecal samples, plasma TMAO levels and nutritional status (Koeth et al. 2013). In subjects undergoing cardiac evaluation, concentrations of plasma l-carnitine were predicted to be at higher risk for both prevalent cardiovascular disease and major adverse cardiac events, but only in subjects with coexisting elevated TMAO levels (Koeth et al. 2013). Caecal microbial composition in mice was modified by chronic dietary l-carnitine dietary intervention, and this supplementation also significantly elevated the synthesis of TMA and TMAO and increased the rate of atherosclerosis (Koeth et al. 2013). However, this was not observed when gut microbiota was suppressed (Koeth et al. 2013). In mice with intact gut microbiota, dietary supplementation with TMA, carnitine or choline reduced the rate of reverse cholesterol transport in vivo (Koeth et al. 2013). As such, gut microbiota may contribute to the well-established link between high levels of red meat consumption and cardiovascular disease risk (Koeth et al. 2013). In another study, 16S rRNA gene pyrosequencing of caecal microbiota in mice revealed that the family Prevotellaceae and the genus Prevotella were enriched and positively correlated with the plasma TMAO levels in mice fed with l-carnitine-supplemented diet (Karlsson et al. 2012b). Faecal microbiome metagenomic evaluation also revealed that the genus Collinsella was enriched in symptomatic atherosclerosis patients, whereas the genus Roseburia and Eubacterium were enriched in healthy subjects (Karlsson et al. 2012b). Additionally, metagenomic characterization of the faecal microbiome revealed that genes responsible for peptidoglycan synthesis were upregulated, and phytoene dehydrogenase was depleted in symptomatic atherosclerosis patients (Karlsson et al. 2012b). Accordingly, these patients also presented with lowered serum β-carotene levels (Karlsson et al. 2012b). These findings suggest that characteristic changes in the gut microbiome may be associated with the inflammatory status of symptomatic atherosclerosis patients.
3.4 Non-alcoholic Fatty Liver Disease
NAFLD , one of the most prevalent liver diseases worldwide, is characterized by fat deposition (steatosis) in the liver that is unrelated to excessive alcohol consumption and is usually observed in states of insulin resistance and metabolic syndrome. Non-alcoholic steatohepatitis (NASH), which is the most severe form of NAFLD, affects approximately 10–20 % of all NAFLD patients and is a major cause of hepatic cirrhosis (Moschen et al. 2013). The prevalence of NASH is increasing, and it is often assumed that various genetic, metabolic, inflammatory and environmental factors contribute to its pathogenesis; however, the exact underlying mechanisms remain unknown (Abu-Shanab and Quigley 2010). The progression of NAFLD usually starts from fatty liver to liver cirrhosis and then to hepatocellular carcinoma (Torres et al. 2012; Satapathy and Sanyal 2010). There have been numerous human and animal studies documented to investigate the relationships between the gut microbiota and NAFLD (Cani et al. 2007, 2008; Rivera et al. 2007; Swann et al. 2011; Sabate et al. 2008; Miele et al. 2009; Verdam et al. 2011; Backhed et al. 2004; Cope et al. 2000). Backhed et al. (2004) have reported that in conventional mice, the concentrations of hepatic triglycerides were higher than those in germ-free mice, although the amount of food intake was lower in the former. Colonization of germ-free mice with gut microbiota was associated with a higher rate of monosaccharide absorption from the gut lumen, which promotes de novo fatty acid synthesis and triglyceride production in the liver, which was confirmed by elevated acetyl-CoA carboxylase and fatty acid synthase activities (Backhed et al. 2004). In addition, Cope et al. (2000) have found that microbial fermentation-derived metabolites in the gut, which include ethanol, are key factors in the induction of obesity in mice and may be related to the pathogenesis of fatty liver disease (Cope et al. 2000). As with obesity, Cani et al. (2007, 2008) reported that hepatic Kupffer cell activation in mice contributes to NAFLD progression, and microbial endotoxin-related chronic inflammation involves CD14–TLR4 signalling (Cani et al. 2007, 2008; Rivera et al. 2007). Bile acid metabolism is also regulated by gut microbiota. Swann et al. (2011) have reported that the murine gut microbiota indirectly promotes hepatic steatosis and lipoperoxidation via farnesoid X receptor-mediated signalling, thereby affecting bile acid secretion These animal studies implicated that the gut microbiota, via the production of hepatotoxic ethanol, can induce fatty liver, resulting in an increase in monosaccharide absorption (Backhed et al. 2004), microbial endotoxin-induced low-grade chronic inflammation (Cani et al. 2007, 2008; Rivera et al. 2007), as well as bile acid metabolism (Swann et al. 2011). In human subjects, Sabate et al. (2008) have reported that gut microbial overgrowth in obese patients may be linked to hepatic steatosis, and correspondingly, Wigg et al. (2001) have also reported that half of NASH patients have microbial overgrowth and elevated serum TNF-α levels, suggesting that NASH might be related to imbalance in gut microbial composition and systemic inflammation, despite unaffected intestinal permeability. Miele et al. (2009) have reported that NAFLD development in human subjects is associated with increased intestinal permeability due to microbial overgrowth in the small intestine and disruption of intestinal mucosal tight junctions; small intestinal microbial overgrowth in NAFLD patients might contribute to hepatic fat deposition. Furthermore, Verdam et al. (2011) have shown that chronic endotoxemia in patients is correlated with NAFLD severity, which was also similarly observed in murine models. In addition, NAFLD development in humans might be correlated with dietary choline depletion (Spencer et al. 2011). In studies conducted by Turnbaugh et al., 15 female subjects were placed on well-controlled diets with adjusted choline levels. It was reported that dietary choline deficiency modified gut microbial composition and that the levels of the bacterial classes Gammaproteobacteria and Erysipelotrichi were positively correlated with changes in the liver fat content. The same group also reported that as compared to both healthy controls and steatotic patients, NASH patients had a lower percentage of species of the phylum Bacteroidetes, which was similar to the gut microbial profiles in obese human subjects (Turnbaugh et al. 2006, 2009). Systemic ethanol concentrations were also significantly higher in NASH patients than in the controls, indicating that ethanol-producing microbes might be correlated with NASH pathogenesis (Zhu et al. 2013). Taken together, the results of the above-mentioned findings implicate that the differences in gut microbial profile amongst NAFLD, NASH, obese and healthy controls might serve as a viable diagnostic marker, as well as acting as a target for preventive or therapeutic medicine.
4 Dysbiosis and Extraintestinal Diseases
4.1 Multiple Sclerosis
Numerous reports have implicated the role of intestinal microbiota in the development of various autoimmune central nervous system (CNS) disorders. In mice that exhibited symptoms of experimental autoimmune encephalomyelitis (EAE), broad-spectrum antibiotics were administered orally (Ochoa-Reparaz et al. 2009). In another report documenting a murine model of multiple sclerosis, mice that were immunized with the self-antigen myelin oligodendrocyte glycoprotein (MOG) in complete Freund’s adjuvant (CFA), under germ-free conditions, disease symptoms in both spontaneous EAE and MOG–CFA-induced EAE were diminished (Lee et al. 2011; Berer et al. 2011). Mice monocolonized with SFB related to the genus Clostridium were reported to demonstrate elevated levels of interleukin-17-producing helper T (TH17) cells in both the intestinal lamina propria and the CNS, resulting in severe EAE (Lee et al. 2011). However, it has yet to be confirmed if EAE is a result of the migration of SFB-specific TH17 cells into the CNS or by the expansion of pathogenic autoantigen-specific T cells that are promoted by TH17 cell responses. Conversely, certain populations of commensal bacteria like polysaccharide A (PSA)+ Bacteroides fragilis can attenuate CAS inflammation via the differentiation of Foxp3+ Treg cells, thereby preventing EAE symptoms. As such, the pathogenesis of CNS disorders like multiple sclerosis may be dependent on the balance of the levels of different bacterial strains in the gu t microbiota.
4.2 Chronic Kidney Disease
Due to the fact that the intestinal immunity can be affected to the extent that it is no longer possible to maintain physiological control over the gut microbiota, some extraintestinal non-communicable diseases like chronic kidney disease are associated with gut dysbiosis (Chow and Mazmanian 2010). In chronic kidney disease patients with impaired renal functions, they often present with not only metabolic derangements but also systemic inflammation, and the intestinal microbiota has been increasingly identified as a trigger for immune dysregulation (Schepers et al. 2010; Kotanko et al. 2006). Vaziri et al. (2013) characterized the gut microbiota of uraemic versus non-uraemic rats and in patients and reported that uraemia was associated with an increase in intestinal pathobionts, indicating that the metabolic and haemodynamic alterations in chronic kidney disease influence the composition and role of gut microbiota. Colonic bacteria also generate uraemic toxins like α-phenylacetyl-l-glutamine, 5-hydroxyindole, indoxyl glucuronide, p-cresol sulfate and indoxyl sulfate, all of which contribute to end-stage renal disease (Ranganathan et al. 2006; Aronov et al. 2011). As chronic kidney disease progresses, circulating bacterial endotoxin/LPS levels increase and have been observed to be the highest in patients on kidney dialysis. These LPS levels were comparable to that in patients with liver disease, gut irradiation and decompensated heart failure. As LPS originates from the cell wall component of gram-negative bacteria, microbiota enriched in γ-proteobacteria may be a source of circulating LPS (McIntyre et al. 2011). In a recent report by Wang et al. (2012), rats with experimentally induced uraemia had increased bacterial translocation from the gut into the mesenteric lymph nodes, liver and spleen, and this was associated with elevated levels of serum interleukin-6 and C-reactive protein. In another report, after oral supplementation of nonpathogenic Sporosarcina pasteurii to uraemic rats, this gut microbiota-directed intervention improved uraemic state as exemplified in improved renal function and prolonged lifespan (Ranganathan et al. 2006). In addition, the neutralization of the bacteria-derived uraemic toxin indoxyl sulfate in the gut delayed the progression of chronic kidney disease and cardiovascular disease in uraemic rats. Lastly, when the same indoxyl sulfate-binding agent was administered to pre-dialysis patients, the 5-year survival rate in patients was improved (Niwa 2011). In a recent study by Mishima et al. (2014), the effects of the ClC-2 chloride channel activator lubiprostone (commonly used for the treatment of constipation) on chronic kidney disease were evaluated using an adenine-induced renal failure mouse model. Oral administration of lubiprostone (500 μg/kg per day for 12 days) lowered elevated blood urea nitrogen levels and attenuated tubulointerstitial damage, renal fibrosis and inflammation. Gut microbiome analysis revealed that lubiprostone treatment altered the murine gut microbial composition by recovering the levels of species of the family Lactobacillaceae and genus Prevotella, which were significantly decreased in the renal failure mice. Furthermore, CE-TOFMS-based metabolomics showed that lubiprostone treatment lowered the plasma levels of uraemic toxins, such as indoxyl sulfate, hippurate and trans-aconitate, which are mainly derived from gut microbiota. These results indicate that lubiprostone administration ameliorates chronic kidney disease progression and uraemic toxin accumulation by improvement of the gut ecosystem (Mishima et al. 2014). Although the relationship between gut microbiota and chronic kidney disease is not completely understood, the gut should no longer be overlooked as a potential trigger for chronic kidney disease, and future studies should focus on unravelling the pathogenetic role of gut microbiota in kidney disease and to discover appropriate therapeutic interventions to manipulate the gut microbiota to correct chronic kidney disease-related immune dysregulation and to prevent further health complications.
4.3 Autism
One of the several reported comorbidities in autism spectrum disorder (ASD), a serious neurodevelopmental condition, is gastrointestinal distress (Buie et al. 2010; Coury et al. 2012). It is also correlated with symptom severity (Finegold et al. 2010). There have been numerous studies reporting the altered composition of intestinal microbiota in ASD subjects (Finegold et al. 2010, 2012; Kang et al. 2013; Parracho et al. 2005; Williams et al. 2011, 2012). Bacteroidetes was found at high levels in the severely autistic group, whilst Firmicutes were more predominant in the control group. Small but significant differences in the levels of Actinobacteria and Proteobacteria were also observed. In faecal samples obtained from severely autistic children, both Desulfovibrio species and Bacteroides vulgatus were present in significantly larger numbers than in control subjects (Finegold et al. 2010). In another study by the same author, autistic children were also reported to have higher levels of Lactobacillus species, lower levels Bifidobacterium species and lower levels of total SCFAs including acetate, propionate and valerate (Finegold et al. 2010). In a separate study with autistic children, significantly lower abundances of the genera Prevotella, Coprococcus and unclassified Veillonellaceae were observed (Kang et al. 2013). In addition, it was also reported that there was a correlation between the presence of members belonging to the family Alcaligenaceae in autistic children with autism and who suffered from gastrointestinal dysfunction (Kang et al. 2013). Hsiao et al. (2013) has also recently reported that the oral administration of PSA+ B. fragilis in the offspring of maternal immune activated (MIA) mice corrected gut permeability. This altered gut microbial composition and attenuated defects in ASD associated behavioural and physiological abnormalities in the offspring of MIA mice. Further investigation showed that this treatment with PSA+ B. fragilis also corrected the levels of MIA-induced serum metabolites by restoring the levels of 4-ethylphenylsulfate, which in naïve mice results in certain behaviour abnormalities (Hsiao et al. 2013). These findings implicate relationships between the gut microbiota, metabolome and the brain, providing novel alternative therapeutic opportunities in human neurodevelopmental disorders.
5 Microbial Health Potentials
5.1 Therapeutic Interventions and Dysbiosis
Commonly used approaches in gut microbiota remodelling for maintaining optimal health status in humans include therapeutic interventions such as bacteriotherapy (Borody et al. 2004) and bioecological control (Gibson and Roberfroid 1995; Holmes et al. 2012). The modulation of intestinal microbiota populations by prebiotics, probiotics or synbiotics may also be beneficial to human health (Gibson and Roberfroid 1995; Holmes et al. 2012). Prebiotics are certain food materials such as oligosaccharides that promote the proliferation of probiotics whereas probiotics, typically contained in dairy fermented products such as yogurt, are well known as healthy microbes that, when orally administered in adequate amounts, yields benefits to host health. Probiotics and prebiotics are marketed as health-promoting supplements, and there are numerous publications highlighting the beneficial effects of probiotics and prebiotics in the improvement of the gut environment (Gareau et al. 2010), the prevention of pathogenic infections (Fukuda et al. 2011) and the regulation of immune functions (Round and Mazmanian 2009).
5.2 Therapeutic Implications of Gut Microbiota Modulation
When human baby microbiota-associated mice were treated with the probiotics Lactobacillus paracasei or Lactobacillus rhamnosus and two galactosyl–oligosaccharide prebiotics, the numbers of B. longum and B. breve were increased, whereas the numbers of C. perfringens were lowered. This gut microbiota composition remodelling has resulted in changes in various host metabolic pathways like gluconeogenesis, lipid profiles and amino acid metabolism (Guarner 2009). Conjugated linoleic acid is a naturally occurring isomer of linoleic acid found in ruminant-derived meat and dairy products (Fukuda et al. 2005) and has been reported to protect against colon carcinogenesis, atherosclerosis as well as obesity in mice (West et al. 1998; Fukuda et al. 2006). Lowered relative abundances of both Firmicutes and Clostridium cluster XIVa in the small intestine were observed in mice that were supplemented with L. rhamnosus GG and Lactobacillus sakei NR28. This led to lower body weight gain, decreased fat mass and downregulation of genes involved in lipogenesis (Ji et al. 2012). However, L. acidophilus NCDC13 supplementation in high-fat diet-induced obese mice resulted in the increase in the total number of Bifidobacterium in caecal and faecal content without reducing adiposity (Arora et al. 2012). As reported by Parnell and Reimer (2012), prebiotic fibres have also been implicated in the reduction of the ratio of Firmicutes to Bacteroidetes in obese rats and to ameliorate NAFLD by lowering de novo hepatic lipogenesis. Chitin–glucan supplementation increased the number of bacteria closely related to the Clostridium cluster XIVa including Roseburia spp. Chitin dietary intervention also attenuated fat fain and fat mass development (Neyrinck et al. 2012). Inulin supplementation in obese women increased the levels of Bifidobacterium spp. and F. prausnitzii and decreased the levels of Bacteroides intestinalis, Bacteroides vulgates and Propionibacterium (Dewulf et al. 2013). In another study involving healthy human subjects, the consumption of galactooligosaccharides for 12 weeks increased the levels of several types of Bifidobacterium spp. and decreased in the number of Bacteroides (Davis et al. 2011). The numbers of Bacteroides–Prevotella spp. and Roseburia spp. were restored, and the numbers of Bifidobacterium spp., in particular Bifidobacterium animalis subsp. lactis, were increased in mice that were fed on high-fat diet supplemented with wheat-derived arabinoxylans (Neyrinck et al. 2011). In healthy overweight humans, after oral administration of Lactobacillus gasseri SBT2055, abdominal visceral and subcutaneous fat were lowered (Arora et al. 2012). Besides, infants who were s upplemented with L. rhamnosus GG in formula for half a year reported better growth and larger weight gain (Vendt et al. 2006). In a follow-up study, pre- and postnatal administration of L. rhamnosus GG protected against excessive weight gain in children (Luoto et al. 2010). In addition to the prebiotic and/or probiotic treatment, faecal microbiota transplantation has been reported to be beneficial in antibiotic-associated diarrhoea or Clostridium difficile infection (Borody 2000; Khoruts et al. 2010; van Nood et al. 2013). Faecal microbiota transplantation can also potentially modulate gut microbiota composition in order to improve pathogenesis of various diseases such as chronic gastrointestinal infections, IBD, insulin resistance, multiple sclerosis and idiopathic thrombocytopenic purpura (Smits et al. 2013). Collectively, the improvement of gut microbiota composition by faecal microbiota transplantation or treatment with probiotics and/or prebiotics may be beneficial in the prevention and medical treatment of several dysbiosis-associated disorders (Fig. 3).
6 Conclusions
We strongly advocate for the integration of metagenomic and metabolomic information on a system biology-wide approach as we believe that it is a valuable methodology that would enable us to understand this intricate interplay between gut microbiota and host metabolism to a greater extent. The integration of information derived from microbiomic, metatranscriptomic and metabolomic platforms will lead to an improved comprehensive understanding of the complex mammalian superorganism. The application to integrated omics-based understanding of the metabolic interactions between lifestyle, nutritional habits and gut microbiota promises to provide intriguing novel therapeutic avenues to maintain and promote optimal host health.
References
Abu-Shanab A, Quigley EM (2010) The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 7(12):691–701. doi:10.1038/nrgastro.2010.172
Aronov PA, Luo FJ, Plummer NS, Quan Z, Holmes S, Hostetter TH, Meyer TW (2011) Colonic contribution to uremic solutes. J Am Soc Nephrol 22(9):1769–1776. doi:10.1681/ASN.2010121220
Arora T, Anastasovska J, Gibson G, Tuohy K, Sharma RK, Bell J, Frost G (2012) Effect of Lactobacillus acidophilus NCDC 13 supplementation on the progression of obesity in diet-induced obese mice. Br J Nutr 108(8):1382–1389. doi:10.1017/S0007114511006957
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331(6015):337–341. doi:10.1126/science.1198469
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500(7461):232–236. doi:10.1038/nature12331
Aw W, Fukuda S (2015) Toward the comprehensive understanding of the gut ecosystem via metabolomics-based integrated omics approach. Semin Immunopathol 37(1):5–16. doi:10.1007/s00281-014-0456-2
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101(44):15718–15723. doi:10.1073/pnas.0407076101
Belzer C, de Vos WM (2012) Microbes inside--from diversity to function: the case of Akkermansia. ISME J 6(8):1449–1458. doi:10.1038/ismej.2012.6
Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G (2011) Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479(7374):538–541. doi:10.1038/nature10554
Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, Vitale L, Pelleri MC, Tassani S, Piva F, Perez-Amodio S, Strippoli P, Canaider S (2013) An estimation of the number of cells in the human body. Ann Hum Biol 40(6):463–471. doi:10.3109/03014460.2013.807878
Bone E, Tamm A, Hill M (1976) The production of urinary phenols by gut bacteria and their possible role in the causation of large bowel cancer. Am J Clin Nutr 29(12):1448–1457
Borody T (2000) “Flora Power” – fecal bacteria cure chronic C. difficile diarrhea. Am J Gastroenterol 95(11):3028–3029. doi:10.1111/j.1572-0241.2000.03306.x
Borody T, Warren E, Leis S, Surace R, Ashman O, Siarakas S (2004) Bacteriotherapy using fecal flora toying with human motions. J Clin Gastroenterol 38(6):475–483. doi:10.1097/01.mcg.0000128988.13808.dc
Buescher J, Moco S, Sauer U, Zamboni N (2010) Ultrahigh performance liquid chromatography-tandem mass spectrometry method for fast and robust quantification of anionic and aromatic metabolites. Anal Chem 82(11):4403–4412. doi:10.1021/ac100101d
Buie T, Campbell DB, Fuchs GJ III, Furuta GT, Levy J, Vandewater J, Whitaker AH, Atkins D, Bauman ML, Beaudet AL, Carr EG, Gershon MD, Hyman SL, Jirapinyo P, Jyonouchi H, Kooros K, Kushak R, Levitt P, Levy SE, Lewis JD, Murray KF, Natowicz MR, Sabra A, Wershil BK, Weston SC, Zeltzer L, Winter H (2010) Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: a consensus report. Pediatrics 125(Suppl 1):S1–S18. doi:10.1542/peds.2009-1878C
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56(7):1761–1772. doi:10.2337/db06-1491
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57(6):1470–1481. doi:10.2337/db07-1403
Chow J, Mazmanian SK (2010) A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe 7(4):265–276. doi:10.1016/j.chom.2010.03.004
Clayton TA, Lindon JC, Cloarec O, Antti H, Charuel C, Hanton G, Provost JP, Le Net JL, Baker D, Walley RJ, Everett JR, Nicholson JK (2006) Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 440(7087):1073–1077. doi:10.1038/nature04648
Collado M, Isolauri E, Laitinen K, Salminen S (2008) Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr 88(4):894–899. doi:10.3945/ajcn.2010.29877
Cope K, Risby T, Diehl AM (2000) Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology 119(5):1340–1347
Coulouarn C, Corlu A, Glaise D, Guenon I, Thorgeirsson SS, Clement B (2012) Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res 72(10):2533–2542. doi:10.1158/0008-5472.CAN-11-3317
Coury DL, Ashwood P, Fasano A, Fuchs G, Geraghty M, Kaul A, Mawe G, Patterson P, Jones NE (2012) Gastrointestinal conditions in children with autism spectrum disorder: developing a research agenda. Pediatrics 130(Suppl 2):S160–S168. doi:10.1542/peds.2012-0900N
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505(7484):559–563. doi:10.1038/nature12820
Davis LM, Martinez I, Walter J, Goin C, Hutkins RW (2011) Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PLoS One 6(9), e25200. doi:10.1371/journal.pone.0025200
De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Backhed F, Mithieux G (2014) Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156(1-2):84–96. doi:10.1016/j.cell.2013.12.016
Dettmer K, Aronov PA, Hammock BD (2007) Mass spectrometry based metabolomics. Mass Spectrom Rev 26(1):51–78. doi:10.1002/mas.20108
Dewulf EM, Cani PD, Claus SP, Fuentes S, Puylaert PG, Neyrinck AM, Bindels LB, de Vos WM, Gibson GR, Thissen JP, Delzenne NM (2013) Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 62(8):1112–1121. doi:10.1136/gutjnl-2012-303304
Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, Bultman SJ (2011) The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13(5):517–526. doi:10.1016/j.cmet.2011.02.018
Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK (2006) Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA 103(33):12511–12516. doi:10.1073/pnas.0601056103
Ellis DI, Dunn WB, Griffin JL, Allwood JW, Goodacre R (2007) Metabolic fingerprinting as a diagnostic tool. Pharmacogenomics 8(9):1243–1266
Everard A, Lazarevic V, Derrien M, Girard M, Muccioli G, Neyrinck A, Possemiers S, Van Holle A, François P, de Vos W, Delzenne N, Schrenzel J, Cani P (2011) Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60(11):2775–2786. doi:10.2337/db11-0227
Everard A, Belzer C, Geurts L, Ouwerkerk J, Druart C, Bindels L, Guiot Y, Derrien M, Muccioli G, Delzenne N, de Vos W, Cani P (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110(22):9066–9071. doi:10.1073/pnas.1219451110
Fei N, Zhao L (2013) An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J 7(4):880–884. doi:10.1038/ismej.2012.153
Finegold SM (2008) Therapy and epidemiology of autism--clostridial spores as key elements. Med Hypotheses 70(3):508–511. doi:10.1016/j.mehy.2007.07.019
Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD, Youn E, Summanen PH, Granpeesheh D, Dixon D, Liu M, Molitoris DR, Green JA III (2010) Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 16(4):444–453. doi:10.1016/j.anaerobe.2010.06.008
Finegold SM, Downes J, Summanen PH (2012) Microbiology of regressive autism. Anaerobe 18(2):260–262. doi:10.1016/j.anaerobe.2011.12.018
Friedman SL (2008) Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88(1):125–172. doi:10.1152/physrev.00013.2007
Fukuda S, Ohno H (2014) Gut microbiome and metabolic diseases. Semin Immunopathol 36(1):103–114. doi:10.1007/s00281-013-0399-z
Fukuda S, Furuya H, Suzuki Y, Asanuma N, Hino T (2005) A new strain of Butyrivibrio fibrisolvens that has high ability to isomerize linoleic acid to conjugated linoleic acid. J Gen Appl Microbiol 51(2):105–113. doi:10.1099/mic.0.022921-0
Fukuda S, Suzuki Y, Murai M, Asanuma N, Hino T (2006) Isolation of a novel strain of Butyrivibrio fibrisolvens that isomerizes linoleic acid to conjugated linoleic acid without hydrogenation, and its utilization as a probiotic for animals. J Appl Microbiol 100(4):787–794. doi:10.1111/j.1365-2672.2006.02864.x
Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H (2011) Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469(7331):543–547. doi:10.1038/nature09646
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504(7480):446–450. doi:10.1038/nature12721
Gareau MG, Sherman PM, Walker WA (2010) Probiotics and the gut microbiota in intestinal health and disease. Nat Rev Gastroenterol Hepatol 7(9):503–514. doi:10.1038/nrgastro.2010.117
Gibson G, Roberfroid M (1995) Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr 125(6):1401–1412. doi:10.1079/NRR200479
Guarner F (2009) Prebiotics, probiotics and helminths: the ‘natural’ solution? Dig Dis 27(3):412–417. doi:10.1159/000228582
Hansen CH, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sorensen SJ, Buschard K, Hansen AK (2012) Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia 55(8):2285–2294. doi:10.1007/s00125-012-2564-7
Hawrelak J, Myers S (2004) The causes of intestinal dysbiosis: a review. Altern Med Rev 9(2):180–197
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482(7384):179–185. doi:10.1038/nature10809
Holmes E, Kinross J, Gibson GR, Burcelin R, Jia W, Pettersson S, Nicholson JK (2012) Therapeutic modulation of microbiota-host metabolic interactions. Sci Transl Med 4(137):137rv136. doi:10.1126/scitranslmed.3004244
Hooper LV (2001) Commensal host-bacterial relationships in the gut. Science 292(5519):1115–1118. doi:10.1126/science.1058709
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, Patterson PH, Mazmanian SK (2013) Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155(7):1451–1463. doi:10.1016/j.cell.2013.11.024
Ji YS, Kim HN, Park HJ, Lee JE, Yeo SY, Yang JS, Park SY, Yoon HS, Cho GS, Franz CM, Bomba A, Shin HK, Holzapfel WH (2012) Modulation of the murine microbiome with a concomitant anti-obesity effect by Lactobacillus rhamnosus GG and Lactobacillus sakei NR28. Benefic Microbes 3(1):13–22. doi:10.3920/BM2011.0046
Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR (2008) Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA 105(36):13580–13585. doi:10.1073/pnas.0804437105
Kalliomäki M, Collado M, Salminen S, Isolauri E (2008) Early differences in fecal microbiota composition in children may predict overweight. Am J Clin Nutr 87(3):534–538. doi:10.3945/ajcn.2010.29877
Kang DW, Park JG, Ilhan ZE, Wallstrom G, Labaer J, Adams JB, Krajmalnik-Brown R (2013) Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS One 8(7), e68322. doi:10.1371/journal.pone.0068322
Karlsson CL, Onnerfalt J, Xu J, Molin G, Ahrne S, Thorngren-Jerneck K (2012a) The microbiota of the gut in preschool children with normal and excessive body weight. Obesity 20(11):2257–2261. doi:10.1038/oby.2012.110
Karlsson FH, Fak F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Backhed F, Nielsen J (2012b) Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun 3:1245. doi:10.1038/ncomms2266
Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, Nielsen J, Backhed F (2013) Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498(7452):99–103. doi:10.1038/nature12198
Khoruts A, Dicksved J, Jansson J, Sadowsky M (2010) Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 44(5):354–360. doi:10.1097/MCG.0b013e3181c87e02
Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, Takahashi T, Miyauchi S, Shioi G, Inoue H, Tsujimoto G (2013) The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun 4:1829. doi:10.1038/ncomms2852
Kinross J, Alkhamesi N, Barton R, Silk D, Yap I, Darzi A, Holmes E, Nicholson J (2011) Global metabolic phenotyping in an experimental laparotomy model of surgical trauma. J Proteome Res 10(1):277–287. doi:10.1021/pr1003278
Kirjavainen P, Arvola T, Salminen S, Isolauri E (2002) Aberrant composition of gut microbiota of allergic infants: a target of bifidobacterial therapy at weaning? Gut 51(1):51–55. doi:10.1136/gut.51.1.51
Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL (2013) Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19(5):576–585. doi:10.1038/nm.3145
Kotanko P, Carter M, Levin NW (2006) Intestinal bacterial microflora – a potential source of chronic inflammation in patients with chronic kidney disease. Nephrol Dial Transplant 21(8):2057–2060. doi:10.1093/ndt/gfl281
Kriegel M, Sefik E, Hill J, Wu H, Benoist C, Mathis D (2011) Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA 108(28):11548–11553. doi:10.1073/pnas.1108924108
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sorensen SJ, Hansen LH, Jakobsen M (2010) Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 5(2), e9085. doi:10.1371/journal.pone.0009085
Lee YK, Menezes JS, Umesaki Y, Mazmanian SK (2011) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 108(Suppl 1):4615–4622. doi:10.1073/pnas.1000082107
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102(31):11070–11075. doi:10.1073/pnas.0504978102
Luoto R, Kalliomaki M, Laitinen K, Isolauri E (2010) The impact of perinatal probiotic intervention on the development of overweight and obesity: follow-up study from birth to 10 years. Int J Obes 34(10):1531–1537. doi:10.1038/ijo.2010.50
MacFabe DF, Cain NE, Boon F, Ossenkopp KP, Cain DP (2011) Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: Relevance to autism spectrum disorder. Behav Brain Res 217(1):47–54. doi:10.1016/j.bbr.2010.10.005
Macpherson AJ (2000) A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288(5474):2222–2226. doi:10.1126/science.288.5474.2222
Marchesi J, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F, Wilson I, Wang Y (2007) Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res 6(2):546–551. doi:10.1038/icb.2014.31
Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS (2013) Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339(6123):1084–1088. doi:10.1126/science.1233521
Martin FP, Wang Y, Sprenger N, Yap IK, Lundstedt T, Lek P, Rezzi S, Ramadan Z, van Bladeren P, Fay LB, Kochhar S, Lindon JC, Holmes E, Nicholson JK (2008) Probiotic modulation of symbiotic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol 4:157. doi:10.1038/msb4100190
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461(7268):1282–1286. doi:10.1038/nature08530
McIntyre CW, Harrison LE, Eldehni MT, Jefferies HJ, Szeto CC, John SG, Sigrist MK, Burton JO, Hothi D, Korsheed S, Owen PJ, Lai KB, Li PK (2011) Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol 6(1):133–141. doi:10.2215/CJN.04610510
Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, Corthesy I, Mace K, Chou CJ (2008) Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J 22(7):2416–2426. doi:10.1096/fj.07-102723
Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Masciana R, Forgione A, Gabrieli ML, Perotti G, Vecchio FM, Rapaccini G, Gasbarrini G, Day CP, Grieco A (2009) Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49(6):1877–1887. doi:10.1002/hep.22848
Mishima E, Fukuda S, Shima H, Hirayama A, Akiyama Y, Takeuchi Y, Fukuda NN, Suzuki T, Suzuki C, Yuri A, Kikuchi K, Tomioka Y, Ito S, Soga T, Abe T (2014) Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J Am Soc Nephrol. doi:10.1681/ASN.2014060530
Moco S, Bino RJ, Vorst O, Verhoeven HA, de Groot J, van Beek TA, Vervoort J, de Vos CH (2006) A liquid chromatography-mass spectrometry-based metabolome database for tomato. Plant Physiol 141(4):1205–1218. doi:10.1104/pp. 106.078428
Moschen AR, Kaser S, Tilg H (2013) Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab 24:537–545. doi:10.1016/j.tem.2013.05.009
Neyrinck AM, Possemiers S, Druart C, Van de Wiele T, De Backer F, Cani PD, Larondelle Y, Delzenne NM (2011) Prebiotic effects of wheat arabinoxylan related to the increase in Bifidobacteria, Roseburia and Bacteroides/Prevotella in diet-induced obese mice. PLoS One 6(6), e20944. doi:10.1371/journal.pone.0020944
Neyrinck AM, Possemiers S, Verstraete W, De Backer F, Cani PD, Delzenne NM (2012) Dietary modulation of clostridial cluster XIVa gut bacteria (Roseburia spp.) by chitin-glucan fiber improves host metabolic alterations induced by high-fat diet in mice. J Nutr Biochem 23(1):51–59. doi:10.1016/j.jnutbio.2010.10.008
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S (2012) Host-gut microbiota metabolic interactions. Science 336(6086):1262–1267. doi:10.1126/science.1223813
Niwa T (2011) Role of indoxyl sulfate in the progression of chronic kidney disease and cardiovascular disease: experimental and clinical effects of oral sorbent AST-120. Ther Apher Dial 15(2):120–124. doi:10.1111/j.1744-9987.2010.00882.x
Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, Kasper LH (2009) Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol 183(10):6041–6050. doi:10.4049/jimmunol.0900747
Okada T, Fukuda S, Hase K, Nishiumi S, Izumi Y, Yoshida M, Hagiwara T, Kawashima R, Yamazaki M, Oshio T, Otsubo T, Inagaki-Ohara K, Kakimoto K, Higuchi K, Kawamura YI, Ohno H, Dohi T (2013) Microbiota-derived lactate accelerates colon epithelial cell turnover in starvation-refed mice. Nat Commun 4:1654. doi:10.1038/ncomms2668
Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J (2009) Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA 106(40):17031–17036. doi:10.1073/pnas.0905299106
Ott SJ (2004) Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 53(5):685–693. doi:10.1136/gut.2003.025403
Parnell JA, Reimer RA (2012) Prebiotic fibres dose-dependently increase satiety hormones and alter Bacteroidetes and Firmicutes in lean and obese JCR:LA-cp rats. Br J Nutr 107(4) doi:10.1017/S0007114511003163
Parracho HM, Bingham MO, Gibson GR, McCartney AL (2005) Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J Med Microbiol 54(Pt 10):987–991. doi:10.1099/jmm.0.46101-0
Prawitt J, Abdelkarim M, Stroeve J, Popescu I, Duez H, Velagapudi V, Dumont J, Bouchaert E, van Dijk T, Lucas A, Dorchies E, Daoudi M, Lestavel S, Gonzalez F, Oresic M, Cariou B, Kuipers F, Caron S, Staels B (2011) Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes 60(7):1861–1871. doi:10.2337/db11-0030
Prentiss P, Rosen H, Brown N, Horowitz R, Malm O, Levenson S (1961) The metabolism of choline by the germfree rat. Arch Biochem Biophys 94:424–429. doi:10.1016/0003-9861(61)90069-8
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464(7285):59–65. doi:10.1038/nature08821
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490(7418):55–60. doi:10.1038/nature11450
Ranganathan N, Patel BG, Ranganathan P, Marczely J, Dheer R, Pechenyak B, Dunn SR, Verstraete W, Decroos K, Mehta R, Friedman EA (2006) In vitro and in vivo assessment of intraintestinal bacteriotherapy in chronic kidney disease. ASAIO J 52(1):70–79. doi:10.1097/01.mat.0000191345.45735.00
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341(6150):1241214. doi:10.1126/science.1241214
Ridlon JM, Kang DJ, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47(2):241–259. doi:10.1194/jlr.R500013-JLR200
Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M (2007) Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 47(4):571–579. doi:10.1016/j.jhep.2007.04.019
Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9(5):313–323. doi:10.1038/nri2515
Sabate JM, Jouet P, Harnois F, Mechler C, Msika S, Grossin M, Coffin B (2008) High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: a contributor to severe hepatic steatosis. Obes Surg 18(4):371–377. doi:10.1007/s11695-007-9398-2
Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M, Gordon JI (2008) Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci USA 105(43):16767–16772. doi:10.1073/pnas.0808567105
Santacruz A, Collado MC, Garcia-Valdes L, Segura MT, Martin-Lagos JA, Anjos T, Marti-Romero M, Lopez RM, Florido J, Campoy C, Sanz Y (2010) Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr 104(1):83–92. doi:10.1017/S0007114510000176
Satapathy SK, Sanyal AJ (2010) Novel treatment modalities for nonalcoholic steatohepatitis. Trends Endocrinol Metab 21(11):668–675. doi:10.1016/j.tem.2010.08.003
Scanlan PD, Shanahan F, Clune Y, Collins JK, O’Sullivan GC, O’Riordan M, Holmes E, Wang Y, Marchesi JR (2008) Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ Microbiol 10(3):789–798. doi:10.1111/j.1462-2920.2007.01503.x
Schepers E, Glorieux G, Vanholder R (2010) The gut: the forgotten organ in uremia? Blood Purif 29(2):130–136. doi:10.1159/000245639
Sina C, Gavrilova O, Forster M, Till A, Derer S, Hildebrand F, Raabe B, Chalaris A, Scheller J, Rehmann A, Franke A, Ott S, Hasler R, Nikolaus S, Folsch UR, Rose-John S, Jiang HP, Li J, Schreiber S, Rosenstiel P (2009) G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol 183(11):7514–7522. doi:10.4049/jimmunol.0900063
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, Lee JR, Offermanns S, Ganapathy V (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40(1):128–139. doi:10.1016/j.immuni.2013.12.007
Smits L, Bouter K, de Vos W, Borody T, Nieuwdorp M (2013) Therapeutic potential of fecal microbiota transplantation. Gastroenterology 145(5):946–953
Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA (2011) Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 140(3):976–986. doi:10.1053/j.gastro.2010.11.049
Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, Nicholson JK, Holmes E (2011) Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci USA 108(Suppl 1):4523–4530. doi:10.1073/pnas.1006734107
Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL (2013) Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368(17):1575–1584. doi:10.1056/NEJMoa1109400
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10(3):167–177. doi:10.1016/j.cmet.2009.08.001
Tolhurst G, Heffron H, Lam Y, Parker H, Habib A, Diakogiannaki E, Cameron J, Grosse J, Reimann F, Gribble F (2012) Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61(2):364–371. doi:10.2337/db11-1019
Torres DM, Williams CD, Harrison SA (2012) Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 10(8):837–858. doi:10.1016/j.cgh.2012.03.011
Tremaroli V, Backhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489(7415):242–249. doi:10.1038/nature11552
Tringe S, Hugenholtz P (2008) A renaissance for the pioneering 16s rrna gene. Curr Opin Microbiol 11(5):442–446. doi:10.1016/j.mib.2008.09.011
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ (2014) Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20(2):159–166. doi:10.1038/nm.3444
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444(7122):1027–1031. doi:10.1038/nature05414
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457(7228):480–484. doi:10.1038/nature07540
Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, Khanin R, Pamer EG (2012) Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med 209(8):1445–1456. doi:10.1084/jem.20120504
van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ (2013) Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 368(5):407–415. doi:10.1056/NEJMoa1205037
Vance D (2008) Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr Opin Lipidol 19(3):229–234. doi:10.1097/MOL.0b013e3282fee935
Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ, Ni Z, Nguyen TH, Andersen GL (2013) Chronic kidney disease alters intestinal microbial flora. Kidney Int 83(2):308–315
Vendt N, Grünberg H, Tuure T, Malminiemi O, Wuolijoki E, Tillmann V, Sepp E, Korpela R (2006) Growth during the first 6 months of life in infants using formula enriched with Lactobacillus rhamnosus GG: double-blind, randomized trial. J Hum Nutr Diet 19(1):51–58. doi:10.1159/000185642
Verdam FJ, Rensen SS, Driessen A, Greve JW, Buurman WA (2011) Novel evidence for chronic exposure to endotoxin in human nonalcoholic steatohepatitis. J Clin Gastroenterol 45(2):149–152. doi:10.1097/MCG.0b013e3181e12c24
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328(5975):228–231. doi:10.1126/science.1179721
Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143(4):913–916. doi:10.1053/j.gastro.2012.06.031, e917
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472(7341):57–63. doi:10.1038/nature09922
Wang F, Zhang P, Jiang H, Cheng S (2012) Gut bacterial translocation contributes to microinflammation in experimental uremia. Dig Dis Sci 57(11):2856–2862. doi:10.1007/s10620-012-2242-0
Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439(7075):484–489. doi:10.1038/nature04330
Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV (2008) Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455(7216):1109–1113. doi:10.1038/nature07336
Wenk MR (2005) The emerging field of lipidomics. Nat Rev Drug Discov 4(7):594–610. doi:10.1038/nrd1776
West D, Delany J, Camet P, Blohm F, Truett A, Scimeca J (1998) Effects of conjugated linoleic acid on body fat and energy metabolism in the mouse. Am J Physiol 275(3 Pt 2):667–672. doi:10.1038/sj.ijo.0802304
Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG (2001) The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor α in the pathogenesis of non-alcoholic steatohepatitis. Gut 48:206–211
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 106(10):3698–3703. doi:10.1073/pnas.0812874106
Williams BL, Hornig M, Buie T, Bauman ML, Cho Paik M, Wick I, Bennett A, Jabado O, Hirschberg DL, Lipkin WI (2011) Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS One 6(9), e24585. doi:10.1371/journal.pone.0024585
Williams BL, Hornig M, Parekh T, Lipkin WI (2012) Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. mBio 3(1). doi:10.1128/mBio.00261-11
Wostmann B (1973) Intestinal bile acids and cholesterol absorption in the germfree rat. J Nutr 103(7):982–990
Xu J, Mahowald M, Ley R, Lozupone C, Hamady M, Martens E, Henrissat B, Coutinho P, Minx P, Latreille P, Cordum H, Van Brunt A, Kim K, Fulton R, Fulton L, Clifton S, Wilson R, Knight R, Gordon J (2007) Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol 5(7):1574–1586. doi:10.1371/journal.pbio.0050156
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499(7456):97–101. doi:10.1038/nature12347
Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR (2013) Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57(2):601–609. doi:10.1002/hep.26093
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Aw, W., Fukuda, S. (2015). The Role of Integrated Omics in Elucidating the Gut Microbiota Health Potentials. In: Liong, MT. (eds) Beneficial Microorganisms in Medical and Health Applications. Microbiology Monographs, vol 28. Springer, Cham. https://doi.org/10.1007/978-3-319-23213-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-23213-3_4
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23212-6
Online ISBN: 978-3-319-23213-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)