
Abstract
Epstein–Barr nuclear antigen 1 (EBNA1) plays multiple important roles in EBV latent infection and has also been shown to impact EBV lytic infection. EBNA1 is required for the stable persistence of the EBV genomes in latent infection and activates the expression of other EBV latency genes through interactions with specific DNA sequences in the viral episomes. EBNA1 also interacts with several cellular proteins to modulate the activities of multiple cellular pathways important for viral persistence and cell survival. These cellular effects are also implicated in oncogenesis, suggesting a direct role of EBNA1 in the development of EBV-associated tumors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Epstein–Barr nuclear antigen 1 (EBNA1) is expressed in all forms of EBV latency in proliferating cells and was the first reported EBV latency protein (Reedman and Klein 1973). EBNA1 has been extensively studied and shown to have multiple important roles in EBV infection. These include contributions to both the replication and mitotic segregation of EBV episomes that lead to stable persistence of EBV episomes in latent infection. EBNA1 also activates the transcription of other EBV latency genes important for cell immortalization. These functions require EBNA1 binding to specific DNA elements in the EBV latent origin of DNA replication ( oriP) . In recent years, it has become apparent that EBNA1 functions are not limited to its roles on EBV episomes but rather that EBNA1 also alters the cellular environment in multiple ways that contribute to cell survival and proliferation and viral persistence. EBNA1 lacks enzymatic activities but is able to affect many processes due to interactions with a variety of cellular proteins. This chapter reviews the multiple functions and mechanisms of action of EBNA1.
2 EBNA1 Functions at EBV Genomes
2.1 DNA Replication
The origin of latent DNA replication, termed oriP (for plasmid origin), was identified by screening EBV DNA fragments for the ability to enable the replication and stable maintenance of plasmids in human cells that were latently infected with EBV (Yates et al. 1984). Subsequent studies showed that the only viral protein required for the replication of oriP plasmids was EBNA1 (Yates et al. 1985). Both EBV episomes and oriP plasmids were found to replicate once per cell cycle, mimicking cellular replication and providing a good model system for human DNA replication (Yates and Guan 1991; Sternas et al. 1990). Note that oriP is not the only origin of replication for EBV episomes, as replication forks have also been found to initiate from a poorly defined region outside of oriP that appears to be independent of EBNA1 (Little and Schildkraut 1995; Norio et al. 2000; Ott et al. 2011).
OriP contains two functional elements: the dyad symmetry (DS) element and the family of repeats (FR) (Reisman et al. 1985) (Fig. 1). The DS contains four EBNA1 recognition sites, two of which are located within a 65-bp DS sequence (Reisman et al. 1985; Rawlins et al. 1985). The DS element is the origin of replication within oriP (Gahn and Schildkraut 1989) and has been shown to be both essential and sufficient for plasmid replication in the presence of EBNA1 (Wysokenski and Yates 1989; Harrison et al. 1994; Yates et al. 2000). Efficient replication from the DS element requires all four EBNA1 binding sites; however, a low level of DNA replication can be achieved with only two adjacent EBNA1 sites, provided that the 3-bp spacing between these sites is maintained (Koons et al. 2001; Harrison et al. 1994; Yates et al. 2000; Atanasiu et al. 2006; Bashaw and Yates 2001; Lindner et al. 2008). The FR element consists of 20 tandem copies of a 30-bp sequence, each of which contains an EBNA1 binding site (Rawlins et al. 1985; Reisman et al. 1985). The primary function of the FR element is in the mitotic segregation and transcriptional activation functions of EBNA1 as discussed below, although in some cell lines the FR also appears to be required for replication from the DS (Hodin et al. 2013). In addition, when bound by EBNA1, the FR can affect DNA replication by inhibiting the passage of replication forks, forming a major pause site (Gahn and Schildkraut 1989; Dhar and Schildkraut 1991; Norio and Schildkraut 2001, 2004; Ermakova et al. 1996).

Schematic representation of oriP. Organization of the oriP DS and FR elements showing genome nucleotide coordinates and EBNA1 binding sites (gray boxes). For the DS element, the positions of the four EBNA1 binding sites and 65-bp dyad symmetry sequence (arrows) are indicated
The DNA replication activity of EBNA1 requires its DNA binding domain as well as additional sequences in the N-terminal half of EBNA1 (Fig. 2) (Yates and Camiolo 1988; Van Scoy et al. 2000; Kim et al. 1997; Kirchmaier and Sugden 1997; Mackey and Sugden 1999; Shire et al. 1999; Ceccarelli and Frappier 2000; Wu et al. 2002). Single localized deletions or mutations that disrupt EBNA1 DNA replication function have not been identified; rather, this function appears to involve redundant contributions of at least two EBNA1 regions (amino acids 8–67 and 325–376; Fig. 2). These N-terminal sequences can tether EBNA1 to cellular chromosomes (see Sect. 2.2), and the finding that they can be functionally replaced by a nucleosome interacting sequence suggests that chromosome tethering contributes to the replication function of EBNA1 (Hodin et al. 2013). In addition, deletion of EBNA1 amino acids 61–83 or 395–450, or point mutation of G81 or G425 within these regions, has been found to increase replication efficiency (Holowaty et al. 2003c; Wu et al. 2002; Deng et al. 2005). These point mutations disrupt EBNA1 binding to tankyrase, suggesting that tankyrase negatively regulates replication by EBNA1, possibly through the poly-ADP ribosylation of EBNA1 (Deng et al. 2005). In addition, the EBNA1 395–450 region is responsible for binding and recruiting to oriP the host ubiquitin-specific protease 7 (USP7) , suggesting that USP7 may negatively regulate replication (Holowaty et al. 2003c). This contention was further supported by a study showing that the latent origin binding proteins of other gamma-herpesviruses also recruit USP7 to their origins and that disruption of this interaction increases DNA replication (Jager et al. 2012). Therefore, negative regulation of replication by USP7 appears to be a conserved feature of gamma-herpesvirus replication.

Organization of the EBNA1 protein. Positions of some of the key elements of EBNA1 are indicating, including the two Gly–Arg-rich regions, variable Gly–Ala repeat, USP7 and CK2 binding sites, and DNA binding and dimerization domain (black), along with flanking and core subcomponents of the DNA binding and dimerization domain. Amino acid numbers are indicated below
EBNA1 is bound to the oriP elements throughout the cell cycle, indicating that EBNA1 binding to the DS is not sufficient to activate DNA replication (Hsieh et al. 1993; Niller et al. 1995; Ritzi et al. 2003). While EBNA1 is the only EBV protein involved in latent-phase DNA replication, it lacks any enzymatic activities, including DNA helicase and origin melting activities present in the origin binding proteins of some viruses (Frappier and O’Donnell 1991b). Therefore, EBV depends heavily on host cellular proteins to replicate its episomes . Several studies have shown that the cellular origin recognition complex (ORC) and minichromosome maintenance (MCM) complex are associated with the DS element of oriP, implicating them in the initiation and licensing of EBV DNA replication (Schepers et al. 2001; Chaudhuri et al. 2001; Dhar et al. 2001). A functional role for ORC in oriP plasmid replication was shown by the failure of these plasmids to stably replicate in a cell line containing a hypomorphic ORC2 mutation (Dhar et al. 2001). EBV replication was also found to be inhibited by geminin, a protein that inhibits rereplication from cellular origins by interacting with Cdt1 (Dhar et al. 2001). This suggests that Cdt1 loads the MCM complexes on EBV origins, as it does on cellular origins.
EBNA1 has been shown to be important for ORC recruitment to the DS (Schepers et al. 2001; Dhar et al. 2001; Julien et al. 2004). In addition, EBNA1 was found to interact with Cdc6 and this interaction increased ORC recruitment to the DS in vitro (Moriyama et al. 2012). Interestingly, ORC is not recruited by EBNA1 bound to the FR element, suggesting that the DS DNA sequence or arrangement of the EBNA1 binding sites are important for ORC recruitment (Schepers et al. 2001; Chaudhuri et al. 2001; Dhar et al. 2001; Moriyama et al. 2012). ORC recruitment by EBNA1 was initially reported to involve EBNA1N-terminal sequences including the Gly–Arg-rich regions, and in vitro studies suggested that this interaction was mediated by RNA molecules (Norseen et al. 2008; Moriyama et al. 2012). However, a second study found that, in the presence of Cdc6, EBNA1 could recruit ORC to the DS in an RNA-independent manner (Moriyama et al. 2012). In addition, it was recently reported that the EBNA1 DNA binding domain is sufficient for ORC recruitment to the DS (Hodin et al. 2013). EBNA1 may also facilitate the recruitment of telomere repeat binding factor 2 (TRF2) to site in the DS (Deng et al. 2002, 2003; Moriyama et al. 2012). TRF2 then appears to contribute to ORC recruitment to the DS in conjunction with EBNA1 (Julien et al. 2004; Atanasiu et al. 2006) and also affects the timing of replication in S phase through recruitment of additional proteins (Zhou et al. 2009, 2010).
EBNA1 has also been found to recruit template activating factor Iβ (TAF-Iβ also called SET) to both the DS and FR elements, through a direct interaction with the 325–376 Gly–Arg-rich region of EBNA1 (Holowaty et al. 2003b; Wang and Frappier 2009). TAF-Iβ appears to negatively regulate replication from oriP as TAF-Iβ depletion was found to increase oriP plasmid replication, while TAF-Iβ overexpression inhibited it (Wang and Frappier 2009). Since TAF-Iβ is a nucleosome-associated protein that can recruit either histone acetylases or deacetylases (Seo et al. 2001; Shikama et al. 2000), TAF-Iβ may negatively regulate replication from oriP by affecting the chromatin structure of the origin.
2.2 Mitotic Segregation
In latency, EBV episomes are present at low copy numbers that are stably maintained in proliferating cells. This stable maintenance requires a mechanism to ensure even partitioning of the episomes to the daughter cells during cell division. The mitotic segregation or partitioning of the EBV episomes requires two viral components, EBNA1 and the oriP FR element (Lupton and Levine 1985; Krysan et al. 1989; Lee et al. 1999). EBNA1 and the FR can also confer stability on a variety of constructs when combined with heterologous origin sequences (Krysan et al. 1989; Kapoor et al. 2001; Simpson et al. 1996). EBNA1 binding to its multiple recognition sites in the FR is crucial for its segregation function, as is the central Gly–Arg-rich region of EBNA1 (325–376) (Shire et al. 1999).
EBNA1 functions in segregation by tethering the EBV episomes to the cellular mitotic chromosomes. EBNA1, EBV episomes, and oriP-containing constructs have all been found to associate with mitotic chromosomes (Harris et al. 1985; Delecluse et al. 1993; Simpson et al. 1996; Grogan et al. 1983; Petti et al. 1990), and the association of oriP plasmids with mitotic chromosomes was shown to depend on the EBNA1-chromosome interaction (Kanda et al. 2001; Kapoor et al. 2005). In addition, EBNA1 mutants that are nuclear but defective in mitotic chromosome attachment fail to partition oriP plasmids (Hung et al. 2001; Shire et al. 1999; Wu et al. 2000). EBNA1 and EBV episomes are not localized to particular regions of mitotic chromosomes, but rather are widely distributed over the chromosomes, leading to the initial suggestion that EBNA1 and EBV episomes interact randomly with chromosomes (Harris et al. 1985). However, subsequent studies have indicated that initial pairing of EBV episomes on sister chromatids may ensure their equal distribution to the daughter cells and that this pairing may stem from the catenation of the newly replicated EBV plasmids (Delecluse et al. 1993; Kanda et al. 2007; Dheekollu et al. 2007; Nanbo et al. 2007). In addition, the FR element has been found to direct EBV genomes to chromatin regions with histone modifications typical of active chromatin (Deutsch et al. 2010).
Studies with EBNA1 deletion mutants showed that the central Gly–Arg-rich region of EBNA1 (amino acids 325–376) was critical for chromosome attachment and that N-terminal sequences (8–67) also contribute to this interaction (Wu et al. 2000, 2002; Shire et al. 1999, 2006; Marechal et al. 1999; Hung et al. 2001; Kanda et al. 2013). Interestingly, fusion proteins in which these EBNA1 regions have been replaced by other chromosome binding sequences are also able to support oriP plasmid maintenance (Hung et al. 2001; Sears et al. 2003). Both the central Gly–Arg repeat of EBNA1 and sequences spanning the smaller Gly–Arg-rich N-terminal sequence (amino acids 33–53) can cause proteins to associate with mitotic chromosomes when fused to them (Hung et al. 2001; Marechal et al. 1999; Sears et al. 2004). However, deletion of the N-terminal Gly–Arg sequence within EBNA1 does not affect EBNA1’s ability to maintain oriP plasmids or to associate with mitotic chromosomes, indicating that it is the central Gly–Arg-rich region that is normally used by EBNA1 for chromosome interactions and segregation (Nayyar et al. 2009; Wu et al. 2002). This region contains a repeated GGRGRGGS sequence that is phosphorylated on the serines and methylated by PRMT1 or PRMT5 on the arginine residues (Laine and Frappier 1995; Shire et al. 2006).
The segregation of viral genomes by attachment to cellular chromosomes is not unique to EBV but is a strategy also used by Kaposi sarcoma associated herpesvirus (KSHV) and papillomavirus. In each case, the viral origin binding (LANA for KSHV and E2 for papillomavirus) tethers the viral plasmid to the cellular chromosome through interactions with one or more cellular proteins (You 2010; Krithivas et al. 2002; Barbera et al. 2006; Parish et al. 2006). For EBNA1, interactions with the cellular protein, EBP2 , appear to be important for metaphase chromosome attachment and segregation function (Wu et al. 2000; Shire et al. 1999; Kapoor and Frappier 2003, 2005). EBP2 is largely nucleolar in interphase but redistributes to the chromosomes in mitosis (Wu et al. 2000). The EBNA1 325–376 region critical for chromosome attachment mediates EBP2 binding, and there is a close correspondence between the effect of EBNA1 mutations on EBP2 and metaphase chromosome interactions (Shire et al. 1999; Wu et al. 2000, 2002; Shire et al. 2006; Nayyar et al. 2009). In addition, EBP2 depletion in various cell lines, including the EBV-positive C666-1 nasopharyngeal carcinoma (NPC) cells, resulted in redistribution of EBNA1 from the metaphase chromosomes to the soluble cell fraction and a corresponding release of oriP plasmids from the chromosomes (Kapoor et al. 2005). EBP2 was also found to enable EBNA1 to segregate plasmids in budding yeast by facilitating EBNA1 attachment to the yeast mitotic chromosomes (Kapoor et al. 2001; Kapoor and Frappier 2003).
Detailed studies on the timing of chromosome association in human cells showed that EBNA1 associates with the chromosomes earlier in mitosis than EBP2 and that EBNA1 and EBP2 only associate on the chromosomes in metaphase to telophase (Nayyar et al. 2009). This suggests that EBNA1 initially contacts the chromosomes by an EBP2-independent mechanism and that subsequent interactions with EBP2 in mid-to-late mitosis might be important to maintain EBNA1 on chromosomes. The initial chromosome contact could involve direct DNA binding or interactions with chromosome-associated RNA molecules, since the 325–376 and N-terminal arginine-rich regions have been found to have some capacity to interact with DNA and RNA in vitro, and drugs that bind G-quadruplex RNA have been reported to decrease the mitotic chromosome association of EBNA1 (Sears et al. 2004; Norseen et al. 2008, 2009; Snudden et al. 1994). In addition, FRET analysis identified an interaction between EBNA1 and EBP2 in the nucleoplasm and nucleolus in interphase suggesting additional roles for this interaction, including the possibility that the EBNA1-EBP2 interaction in interphase is important for EBNA1-chromosome interactions in mitosis (Jourdan et al. 2012). This possibility is reminiscent of findings for bovine papillomavirus segregation, in which an interphase interaction between the viral E2 protein and host ChlR1 protein is required in order for E2 to associate with mitotic chromosomes and segregate papillomavirus genomes (Feeney et al. 2011; Parish et al. 2006).
2.3 EBV Transcriptional Activation
Another function of EBNA1 at EBV episomes is in transcriptional activation. EBNA1 can act as a transcriptional activator when bound to the oriP FR element, enhancing the expression of reporter genes on FR-containing plasmids in a distance-independent manner (Lupton and Levine 1985; Reisman and Sugden 1986). The EBNA1-bound FR was also shown to activate expression from the viral Cp and LMP promoters, suggesting a role for EBNA1 in inducing the expression of the EBNA and LMP EBV latency genes in latent infection (Sugden and Warren 1989; Gahn and Sugden 1995). The EBNA1 residues required for transcriptional activation have been mapped to the 65–83 N-terminal sequence (Wu et al. 2002; Kennedy and Sugden 2003) as well as to the central Gly–Arg-rich region (residues 325–376) also required for segregation function (Yates and Camiolo 1988; Ceccarelli and Frappier 2000; Wang et al. 1997; Van Scoy et al. 2000). EBNA1 requires both of these regions to activate transcription, as deletion of either one abrogates the transcriptional activation function of EBNA1 (Wu et al. 2002; Ceccarelli and Frappier 2000). A Δ61–83 EBNA1 mutant was found to be fully active for replication and segregation functions, indicating that transcriptional activation is a distinct EBNA1 function (Wu et al. 2002). Similar conclusions were reached with a Δ65–89 EBNA1 mutant in the context of an infectious EBV, where EBNA1 Δ65–89 was shown to be defective in activating expression of the EBNA genes from the Cp promoter, but still supported stable plasmid replication (Altmann et al. 2006). EBV containing the Δ65–89 EBNA1 was also shown to be severely impaired in the ability to transform cells, indicating the importance of EBNA1-mediated transcriptional activation for EBV infection (Altmann et al. 2006).
Two cysteine residues within the N-terminal transactivation sequence (at positions 79 and 82) have been shown to be important for transactivation activity and to mediate an interaction with zinc (Aras et al. 2009). There is also evidence that the transcriptional activity of EBNA1 is zinc-dependent, suggesting that a zinc-dependent structure formed in the N-terminal transactivation region mediates the activity of this sequence (Aras et al. 2009). The 61–83 region also mediates an interaction with Brd4 (Lin et al. 2008), a cellular bromodomain protein that interacts with acetylated histones to regulate transcription (Wu and Chiang 2007). Within the EBV genome, Brd4 was shown to be preferentially localized to the EBNA1-bound FR enhancer element (Lin et al. 2008). Furthermore, Brd4 depletion inhibited EBNA1-mediated transcriptional activation, suggesting that EBNA1 uses Brd4 to activate transcription (Lin et al. 2008). Interestingly, an interaction between Brd4 and papillomavirus E2 proteins (the functional equivalent to EBNA1) has been shown to be important for transcriptional activation by E2 (Schweiger et al. 2006; McPhillips et al. 2006; Ilves et al. 2006), suggesting that EBNA1 and E2 may use common mechanisms to activate transcription. Whether or not the EBNA1–Brd4 interaction is zinc-dependent remains to be determined.
The EBNA1 325–376 region mediates interactions with several cellular proteins, some of which have been implicated in the transcriptional activity of EBNA1. For example, P32/TAP, which interacts with Arg-rich sequences, has been detected at oriP by chromatin immunoprecipitation, and its C-terminal region has some ability to activate a reporter gene when fused to the GAL4 DNA binding domain (Van Scoy et al. 2000; Wang et al. 1997). However, it is not clear whether P32/TAP is important for EBNA1-mediated transcriptional activation. The related nucleosome assembly proteins, NAP1, TAF-Iβ (also called SET), and nucleophosmin, also interact with the EBNA1 325–376 sequence and are known to affect transcription in multiple ways (Holowaty et al. 2003c; Wang and Frappier 2009; Park and Luger 2006; Malik-Soni and Frappier 2012, 2013). A role for NAP1, TAF-Iβ, and nucleophosmin in EBNA1-mediated transcriptional activation is supported by the finding that each protein is recruited to the FR element by EBNA1 and that EBNA1 transactivation activity is decreased upon depleting any of these proteins (Wang and Frappier 2009; Malik-Soni and Frappier 2013). Depletion of nucleophosmin had the biggest effect on EBNA1-mediated transcriptional activation, suggesting, either that the EBNA1-nucleophosmin interaction is the most important for transcription function, or that nucleophosmin is more limiting in the cell than NAP1 or TAF-Iβ (Malik-Soni and Frappier 2013). Note that another histone chaperone protein, nucleolin, was also recently reported to contribute to EBNA1 functions including transactivation, but this appears to be due to an effect on EBNA1 binding to oriP (Chen et al. 2014). As a whole, the data suggest that recruitment of both nucleosome assembly proteins and Brd4 are important for transcriptional activation by EBNA1, reflecting the requirement for the two transcriptional activation sequences.
It is expected that transcriptional activation by EBNA1 will involve changes to histone modifications and this may include ubiquitylation of histone H2B. This is suggested by the finding that EBNA1 binds to a complex of USP7 and GMP synthetase that functions to deubiquitylate H2B and recruits it to the FR (Sarkari et al. 2009). USP7 depletion results in increased levels of monoubiquitylated H2B at the FR and decreased transcriptional activation, suggesting that monoubiquitylation of H2B inhibits EBNA1-mediated transcriptional activation. In keeping with this result, an EBNA1 mutant defective in USP7 binding has decreased ability to activate transcription (Holowaty et al. 2003c).
2.4 Autoregulation
In addition to interactions with the oriP FR and DS elements, EBNA1 was found to bind a third region of the EBV genome near the Qp promoter that is used to express EBNA1 in the absence of other EBNAs (Jones et al. 1989; Sample et al. 1992; Nonkwelo et al. 1996). EBNA1 binding to two recognition sites located downstream of Qp was reported to repress EBNA1 expression from Qp (Sample et al. 1992). Since EBNA1 has lower affinity for these sites than either the DS or FR elements, EBNA1 would only bind the Qp sites when its levels are high enough to saturate the FR and DS elements, providing a feedback mechanism to shut off EBNA1 expression when EBNA1 levels are high (Jones et al. 1989; Ambinder et al. 1990). While EBNA1 was initially thought to inhibit expression from Qp by repressing transcription, a more recent study found that EBNA1 acts post- or co-transcriptionally to inhibit the processing of primary transcripts (Yoshioka et al. 2008).
3 EBNA1–DNA Interactions
3.1 Interactions with the EBV Genome
EBNA1 specifically recognizes an 18-bp palindromic sequence present in multiple copies in the oriP DS and FR elements as well as in the BamHI-Q fragment containing the Qp promoter (Rawlins et al. 1985; Jones et al. 1989; Ambinder et al. 1990, 1991; Frappier and O’Donnell 1991b; Shah et al. 1992). Sequence variation within the multiple copies of this palindrome results in different affinities of EBNA1 for the FR, DS, and BamHI-Q regions and for individual sites within these regions (Ambinder et al. 1990; Summers et al. 1996). EBNA1 has highest affinity for the FR and DS regions and remains bound to these sites throughout the cell cycle (Hsieh et al. 1993; Niller et al. 1995; Ritzi et al. 2003).
EBNA1 interacts with its recognition sites through its C-terminal domain (amino acids 459 and 607; Fig. 2), which also mediates the dimerization of EBNA1 (Ambinder et al. 1991; Chen et al. 1993; Summers et al. 1996; Shah et al. 1992). EBNA1 forms very stable homodimers both in solution and when bound to its recognition sites (Frappier and O’Donnell 1991b; Ambinder et al. 1991; Shah et al. 1992). The crystal structure of the DNA binding and dimerization domain was determined both in solution and bound to the EBNA1 consensus binding site (Bochkarev et al. 1995, 1996). The structure showed that dimerization was mediated by residues 504–604 (referred to as the core domain), which form an eight-stranded antiparallel β-barrel, comprised of four strands from each monomer and two α-helices per monomer (Fig. 3). This core domain is strikingly similar to the structure of the DNA binding domain of the E2 protein of papillomavirus, despite a complete lack of sequence homology (Edwards et al. 1998; Hegde et al. 1992). Residues 461–503 flank the core domain (flanking domain) and are comprised of an α-helix oriented perpendicular to the DNA and an extended chain that tunnels along the base of the minor groove of the DNA (Fig. 3). Both the helix and the extended chain make sequence-specific DNA contacts. In addition, a direct role of the core domain in DNA recognition was suggested by analogy to the E2 DNA binding domain and later confirmed by mutational analyses (Cruickshank et al. 2000). Combined, the structural and biochemical studies indicate that the core and flanking domains of EBNA1 work together to load EBNA1 on its recognition site, likely through a two-step DNA binding mechanism. In keeping with this model, thermodynamic and kinetic analyses of the EBNA1 DNA binding domain–DNA interaction revealed two DNA association and dissociation events (Oddo et al. 2006). In addition, the ability of EBNA1 to bind its recognition sites, both in vitro and in vivo, was found to be greatly stimulated by USP7 through its interaction with EBNA1 amino acids close to the flanking domain (442–448; Fig. 2) (Sarkari et al. 2009), suggesting that this USP7 interaction may facilitate the DNA loading of the flanking domain.

Crystal structure of the EBNA1 DNA binding and dimerization domain bound to DNA. The core and flanking components of the DNA binding and dimerization domain are shown in green and yellow, respectively. EBNA1 amino acid numbers are indicated. Reprinted with permission from Bochkarev et al. (1996)
The interaction of the EBNA1 DNA binding and dimerization domain with a single recognition site causes the DNA to be smoothly bent and causes localized regions of helical overwinding and underwinding (Bochkarev et al. 1996). The overwinding is caused by the EBNA1 flanking domain residues that traverse along the minor groove (amino acids 463–468) (Bochkarev et al. 1998; Summers et al. 1997) and this results in the increased sensitivity of one T residue within the DS sites to permanganate oxidation (Frappier and O’Donnell 1992; Hearing et al. 1992; Hsieh et al. 1993; Summers et al. 1997). EBNA1 dimers assemble cooperatively on adjacent sites in the DS (Summers et al. 1996; Harrison et al. 1994), and this is predicted to induce additional changes in the DNA structure (such as unwinding), in order to accommodate the closely packed dimers (Bochkarev et al. 1996). The strict requirement for the 3-bp spacing that separates neighboring sites in the DS for origin function suggests that the proper interaction between the EBNA1 dimers bound to these sites is crucial for the initiation of DNA replication, possibly because of the DNA structural changes that it imparts (Bashaw and Yates 2001; Harrison et al. 1994). Interactions of EBNA1 dimers on the multiple sites within the DS and FR elements likely also contribute to the pronounced bending of these elements that have been observed and to the appearance of EBNA1 as a large single complex on each element (Frappier and O’Donnell 1991a; Goldsmith et al. 1993; Bashaw and Yates 2001).
EBNA1 complexes bound to the DS and FR elements of oriP can also interact with each other cause the looping out of the intervening DNA (when interaction occur within an oriP molecule) and the linking of multiple oriP molecules (when interactions occur between oriP molecules) (Frappier and O’Donnell 1991a; Goldsmith et al. 1993; Su et al. 1991; Middleton and Sugden 1992). The DNA looping and linking interactions stabilize EBNA1 binding to the DS and involve homotypic interactions mediated by two different regions of EBNA1; a stable interaction mediated by amino acids 327–377 and a less stable interaction mediated by residues 40–89 (Frappier et al. 1994; Laine and Frappier 1995; Mackey et al. 1995; Mackey and Sugden 1999; Avolio-Hunter and Frappier 1998). The looping/linking interactions of EBNA1 are not restricted to EBNA1 complexes formed on the DS or FR elements but also occur between single EBNA1 dimers bound to distant recognition sites (Goldsmith et al. 1993). The contribution of DNA looping and linking to EBNA1 functions remains unclear but the amino acids required for these interactions overlap with those required for EBNA1 replication, segregation, and transcriptional activation functions (Mackey and Sugden 1999; Wu et al. 2002; Shire et al. 1999).
In vivo EBV genomes are assembled into nucleosomes with a spacing similar to that in cellular chromatin (Shaw et al. 1979; Dyson and Farrell 1985). Since nucleosomes tend to inhibit sequence-specific DNA interactions, the ability of EBNA1 to bind its site in the DS in the context of a nucleosome was examined. Surprisingly, EBNA1 was able to access its recognition sites within the nucleosome and destabilized the nucleosome structure such that the histones could be displaced from the DNA (Avolio-Hunter et al. 2001). Efficient assembly of EBNA1 on the FR and DS elements was also observed on larger oriP templates containing physiologically spaced nucleosomes (Avolio-Hunter and Frappier 2003). The disruption of the DS-nucleosome by EBNA1 required all four recognition sites in the DS and was intrinsic to the DNA binding and dimerization domain of EBNA1 (Avolio-Hunter et al. 2001). The ability of EBNA1 to destabilize nucleosomes might be important for initiating DNA replication, a process known to be sensitive to nucleosome positioning. In addition, the ability of EBNA1 to access its sites within a nucleosome is likely to be important at times when chromatin is established prior to EBNA1 expression, for example, when latently infected resting cells (which do not express EBNA1) switch to proliferating forms of latency in which EBNA1 is expressed.
3.2 Interactions with Cellular DNA Sequences
The fact that EBNA1 can activate transcription, when bound to the EBV FR element, has prompted several studies to determine whether EBNA1 might also interact with specific sequences in cellular DNA to affect cellular gene expression. Chromatin IP (ChIP) experiments performed for EBNA1 from EBV-positive lymphoblastoid cell lines, followed by promoter array analysis, identified several EBNA1-associated DNA fragments, some of which were confirmed to be directly bound by EBNA1 in vitro (Dresang et al. 2009). While this approach identified a new EBNA1 recognition sequence (distinct from those in oriP), EBNA1 binding to this sequence did not activate reporter gene expression, so the significance of these EBNA1-cellular DNA interactions is not clear. ChIP combined with deep sequencing was also used to determine EBNA1 binding sites in B cells, identifying many EBNA1-associated sites, several of which were close to transcriptional start sites for cellular genes (Lu et al. 2010). The expression of some of these cellular genes was decreased upon EBNA1 depletion and induced by EBNA1 expression, suggesting that EBNA1 may affect their transcription. Like the previous study, these EBNA1 sites differed from those in oriP, but some were similar in sequence to those identified by Dresang et al. (2009). In addition, a cluster of high-affinity EBNA1 binding sites was identified on chromosome 11 between the divergent FAM55D and FAM55B genes, although the expression of these genes was not affected by EBNA1 (Lu et al. 2010). Canaan et al. (2009) conducted microarray experiments to compare cellular transcripts in B cells and 293 cells with and without EBNA1 and identified a small percentage of transcripts that were affected by EBNA1. In addition, EBNA1 was found to ChIP to most of these gene promoters, suggesting that it directly regulated them. However, whether or not EBNA1 bound directly to these promoters or was recruited through protein interactions was not determined.
The transcriptional activation function of EBNA1 on the EBV genome requires EBNA1 binding to multiple tandem recognition sites in the FR (Wysokenski and Yates 1989), and therefore, it seems unlikely that EBNA1 binding to any single recognition site would be sufficient to activate cellular transcription. To increase the probability of identifying functionally relevant EBNA1 interactions with cellular DNA, D’Herouel et al. (2010) used nearest neighbor position weight matrices to identify repeated EBNA1 binding sites in the human genome. The sites they identified had considerable overlap with those found by Dresang et al. (2009). Although the significance of the repeated EBNA1 sites that they identified remains to be determined, it is interesting that they include weak binding sites near the c-Jun and ATF promoters, which were previously shown to be activated by and associated with EBNA1 in NPC cells (O’Neil et al. 2008).
By comparing cell cycle-specific transcripts from EBV-negative B cells with and without EBNA1 expression, Lu et al. (2011) identified survivin (an inhibitor of apoptosis) as an EBNA1 target gene. EBNA1 increased the levels of survivin transcripts and protein and was shown to associate with the survivin promoter. Induction of survivin protein and transcripts required the EBNA1 residues 65–89 containing the N-terminal transcriptional activation sequence (Wu et al. 2002; Kennedy and Sugden 2003), suggesting that EBNA1 was activating the transcription of the survivin gene. However, since activation of the survivin promoter by EBNA1 involves the Sp1 binding sites, EBNA1 may be recruited to the promoter through the Sp1 host protein, as opposed to binding directly to the DNA. Similarly, Owen et al. (2010) found that EBNA1 increased the level of TFIIIC and ATF-2 transcripts and was associated with their promoter regions, consistent with a direct role in transcriptional activation. Finally, EBNA1 was recently reported to induce the expression of cellular let-7a microRNAs (miRNA) in nasopharyngeal and gastric carcinoma cells (Mansouri et al. 2014). EBNA1 increased the level of let-7a primary transcripts in a manner dependent on its N-terminal transactivation sequence, suggesting that EBNA1 directly induces their transcription. However, whether or not this involves a direct interaction of EBNA1 with DNA sequences regulating these primary transcripts remains to be determined.
Presumably any of the above direct interactions of EBNA1 with specific DNA sites would be mediated by the EBNA1 DNA binding domain. However, there have also been reports of less specific interactions of EBNA1 with DNA or chromatin through its Gly–Arg-rich regions , which resemble AT hooks (Sears et al. 2004; Coppotelli et al. 2013). In addition, EBNA1 has been reported to decondense heterochromatin through its Gly–Arg-rich sequences (Coppotelli et al. 2013). However, it is unclear whether this effect involves the association of the Gly–Arg sequences with DNA, chromatin-associated proteins (including the nucleosome assembly proteins known to bind to them), or another mechanism.
4 Cellular Effects of EBNA1
In addition to the roles of EBNA1 at the EBV genome , numerous reports suggest that EBNA1 directly contributes to cell proliferation and survival typical of latent EBV infection. The first implications came from the observations that EBNA1 is the only EBV protein expressed in all EBV-positive tumors and latency types in proliferating cells and is sometimes the only EBV protein expressed. EBNA1 was subsequently shown to be important for efficient B-cell immortalization by EBV (Hume et al. 2003; Altmann et al. 2006) and for the continued proliferation of some EBV-positive tumor cells (Kennedy et al. 2003; Hong et al. 2006; Yin and Flemington 2006). EBNA1 expression in various EBV-negative cancer cells has also been found to increase tumorigenicity (Sheu et al. 1996; Cheng et al. 2010; Kube et al. 1999; Kaul et al. 2007). In addition, EBNA1 expression in the B-cell compartment of a transgenic mouse has been reported to be sufficient to induce B-cell lymphomas (Wilson et al. 1996; Tsimbouri et al. 2002). However, these results were not reiterated in a second independent transgenic mouse study, suggesting that secondary events might contribute to the development of EBNA1-induced lymphomas (Kang et al. 2005, 2008). Nonetheless, the body of evidence indicates that EBNA1 contributes to oncogenesis, likely due to multiple effects on cellular proteins as discussed below and summarized in Fig. 4.

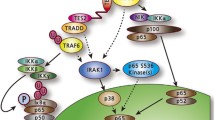
Summary of EBNA1 cellular effects. The cellular proteins whose functions or levels are affected by EBNA1 are shown, where arrows represent positive regulation and blunted lines represent negative regulation. Associated cellular processes are also indicated
4.1 USP7 Interaction
Proteomics methods identified several cellular proteins that are bound by EBNA1, including an interaction with the cellular ubiquitin-specific protease USP7 [also called HAUSP (Holowaty et al. 2003c; Malik-Soni and Frappier 2012)]. USP7 was originally discovered as a binding partner of the ICP0 protein from herpes simplex virus type 1 and has since been shown to be targeted by proteins from several different herpesviruses (Everett et al. 1997; Salsman et al. 2012; Lee et al. 2011; Jager et al. 2012). USP7 has been reported to bind and regulate several cellular proteins including p53 and Mdm2 (an E3 ubiquitin ligase for p53), which USP7 stabilizes by removing the polyubiquitin chains that normally signal degradation (Li et al. 2002, 2004; Cummins et al. 2004; Nicholson and Suresh Kumar 2011; Frappier and Verrijzer 2011). EBNA1, p53, and Mdm2 compete for the same binding pocket in the N-terminal TRAF domain of USP7; however, EBNA1 was found to outcompete p53 or Mdm2 due to its higher affinity for USP7 (Holowaty et al. 2003a; Saridakis et al. 2005; Sheng et al. 2006). The EBNA1 region just N-terminal to the DNA binding domain was identified as the USP7 binding site, and a subsequent crystal structure of this EBNA1 peptide bound to the USP7 TRAF domain showed that EBNA1 amino acids 442–448 contact USP7 (Fig. 2) (Holowaty et al. 2003a; Saridakis et al. 2005; Sheng et al. 2006; Hu et al. 2006).
In theory, EBNA1 could destabilize either p53 or Mdm2 by blocking their interaction with USP7, resulting in opposite effects on p53 levels. In vivo EBNA1 has not been reported to lower Mdm2 levels, but has been confirmed to lower p53 levels at least in some cell backgrounds. For example, expression of EBNA1 but not a USP7-binding mutant of EBNA1 in U2OS cells was shown to reduce the accumulation of p53 in response to DNA damage and subsequent apoptosis (Saridakis et al. 2005). Similarly, EBNA1 expression in CNE2 NPC cells decreased the accumulation of p53 in response to DNA damage (Sivachandran et al. 2008), and the presence of EBNA1 or EBV in AGS or SCM1 gastric carcinoma cells decreased the steady-state levels of p53 (Sivachandran et al. 2012a; Cheng et al. 2010). This suggests that EBNA1 could promote cell survival by modulating p53 in EBV-infected epithelial cells.
4.2 Effects on PML Nuclear Bodies
Promyelocytic leukemia (PML) nuclear bodies (also called ND10s) are nuclear foci for which PML tumor suppressor proteins form the structural basis. PML bodies are important for several cellular processes, including apoptosis, DNA repair, senescence, and p53 activation by acetylation (Salomoni et al. 2008; Bernardi and Pandolfi 2007; Takahashi et al. 2004; Guo et al. 2000; Wang et al. 1998; Pearson et al. 2000), and their loss has been associated with the development and/or progression of several tumors (Gurrieri et al. 2004; Salomoni et al. 2008). In addition, PML nuclear bodies suppress lytic viral infection as part of the innate antiviral response (Geoffroy and Chelbi-Alix 2011; Everett and Chelbi-Alix 2007; Reichelt et al. 2011). To counter this defense, many viruses encode proteins that disrupt PML nuclear bodies either by interfering with PML protein interactions need to form the bodies or by inducing the degradation of the PML proteins (Everett 2001).
EBNA1 was found to induce the loss of PML nuclear bodies in both NPC and gastric carcinoma cells, by inducing the degradation of the PML proteins (Sivachandran et al. 2008, 2012a). Consistent with known PML functions, EBNA1 expression in these cells was also found to decrease DNA repair efficiency, p53 acetylation, and apoptosis in response to DNA damaging agents (Sivachandran et al. 2008, 2012a). The results suggest that, as a result of EBNA1-induced PML loss, cells expressing EBNA1 are more likely to survive with DNA damage, which would be expected to contribute to the development of carcinomas. Importantly, these observations in cell lines appear to hold true in vivo, as a comparison of EBV-positive and EBV-negative gastric carcinoma tumor biopsies showed that PML levels were greatly reduced by the presence of EBV, presumably due to the action of EBNA1 (Sivachandran et al. 2012a).
The mechanism by which EBNA1 induces the degradation of PML proteins involves EBNA1 binding to both USP7 and the host casein kinase 2 (CK2) and recruitment of these proteins to the PML nuclear bodies (Sivachandran et al. 2008, 2010). EBNA1 was found to preferentially interact with PML isoform IV over the other five nuclear PML isoforms, and therefore, EBNA1 may localize to PML nuclear bodies through interactions with PML IV (Sivachandran et al. 2008, 2012b). EBNA1 mutants that fail to bind either USP7 or CK2 can still associate with PML bodies but do not induce their loss (Sivachandran et al. 2008, 2010). Similarly, wild-type EBNA1 does not affect PML nuclear bodies when USP7 or CK2 is depleted. In keeping with these observations, USP7 was subsequently shown to negatively regulate PML proteins (even in the absence of EBV or EBNA1), by a mechanism that is independent of its ubiquitin cleavage activity (Sarkari et al. 2011).
The interaction of EBNA1 with CK2 involves a direct interaction of EBNA1 amino acids 387–394 with the binding pocket in the β-regulatory subunit of CK2 and this interaction requires EBNA1 to be phosphorylated at S393 (Sivachandran et al. 2010; Cao et al. 2014). CK2 was previously identified as a negative regulator of PML and was shown to phosphorylate PML proteins at a particular serine residue that triggers polyubiquitylation and subsequent degradation (Scaglioni et al. 2006, 2008). Through its interaction with CK2, EBNA1 was shown to increase CK2-mediated phosphorylation of PML, which is expected to increase PML polyubiquitylation (Sivachandran et al. 2010). Since CK2 is involved in many cellular processes, it is possible that the interaction of EBNA1 with CK2 also affects additional pathways.
4.3 Modulation of Signaling Pathways
EBNA1 has been reported to affect several signaling pathways. First, EBNA1 expression in three different carcinoma cell lines was found to increase the expression of STAT1 (Wood et al. 2007; Kim and Lee 2007). EBNA1 was subsequently shown to enhance STAT1 phosphorylation and nuclear localization in response to IFNγ (Wood et al. 2007). Second, EBNA1 expression was found to decrease the expression of TGF-β1-responsive genes suggesting that EBNA1 interferes with TGF-β signaling (Wood et al. 2007). This effect may be due to increased turnover of SMAD2 in the presence of EBNA1, resulting in decreased levels of SMAD complexes needed for TGF-β1-induced transcription (Wood et al. 2007; Flavell et al. 2008). Third, using NF-κB reporter plasmids in carcinoma cell lines, EBNA1 was found to inhibit NF-κB activity and DNA binding (Valentine et al. 2010). Additional experiments showed that the levels, nuclear localization, and phosphorylation of the p65 NF-κB subunit were all reduced in the presence of EBNA1 as was the phosphorylation of the p65 kinase, IKKα/β (Valentine et al. 2010). How EBNA1 elicits any of the above effects is presently unclear as no physical interaction has been detected between EBNA1 and STAT1, SMAD2, p65, or IKKα/β.
4.4 Induction of Oxidative Stress
EBV infection is associated with increased oxidative stress (Lassoued et al. 2008; Cerimele et al. 2005) and this may be at least partly due to EBNA1 expression. Stable or transient EBNA1 expression in B-cell lines was found to increase levels of reactive oxygen species (ROS) , DNA damage foci and dysfunctional, uncapped telomeres, and these EBNA1 effects were decreased by ROS scavengers (Gruhne et al. 2009; Kamranvar and Masucci 2011). In addition, EBNA1 was found to increase the expression of the NOX2 NADPH oxidase which might account for the ROS induction (Gruhne et al. 2009). Similarly, a comparison of the nuclear proteome in NPC cells with and without EBNA expression showed that EBNA1 increased the levels of several oxidative stress response proteins including the antioxidants superoxide dismutase 1 and peroxiredoxin 1, known to be induced by ROS (Cao et al. 2011). Further studies confirmed that, in the presence of EBNA1, ROS levels were elevated and that NOX1 and NOX2 transcripts were increased (Cao et al. 2011). Therefore, EBNA1 appears to have multiple effects on the oxidative stress response, although the mechanisms of these effects are not yet known.
4.5 Effects on Noncoding RNA
EBNA1 expression has been reported to increase the transcript and protein levels of the RNA polymerase III transcription factor TFIIIC and, in keeping with this finding, increased the expression of several cellular pol III-transcribed genes (Owen et al. 2010). In the same study, EBNA1 was also found to induce the expression of ATF-2, a pol II transcription factor known to contribute to the expression of EBV EBER noncoding RNAs. The result prompted examination of the effect of EBNA1 on EBER levels and confirmed that EBNA1 induces EBER expression.
The effects of EBNA1 on cellular miRNAs have also been examined, using high-throughput sequencing to compare miRNA levels in two different EBV-negative NPC cell lines with and without transient EBNA1 expression (Mansouri et al. 2014). A small percentage of miRNAs were found to be consistently affected by EBNA1 expression in the two cell lines; in particular, let-7 family miRNAs were increased by EBNA1. Further studies on let-7a miRNA in both EBV-positive and EBV-negative carcinoma cell lines confirmed that EBNA1 upregulates let-7a as well as its primary transcripts and that this required the EBNA1 61–83 transcriptional activation sequence (Mansouri et al. 2014). This induction of let-7a was shown to result in a corresponding decrease in the let-7a target, Dicer. The decreased Dicer levels did not result in global impairment of miRNA biogenesis, but rather the decreased Dicer and increased let-7a levels promoted EBV latency by inhibiting EBV reactivation. In addition, in another study EBNA1 overexpression was reported to decrease levels of miR-200a and miR-200b and this was suggested to contribute to induction of the epithelial–mesenchymal transition (EMT) (Wang et al. 2014).
4.6 Effects on Metastatic Potential
A comparison of the nuclear proteomes of NPC cells with and without EBNA1 expression found that EBNA1 increased the nuclear levels of Nm23-H1, stathmin 1, and maspin, all of which have been found to be contribute to metastases (Cao et al. 2011). This effect on Nm23-H1 corroborated a previous study that showed that EBNA1 co-immunoprecipitated with Nm23-H1 from lymphoid cells and cause it to relocalize to the nucleus (Murakami et al. 2005). This interaction required EBNA1 amino acids 65–89, which are also important for transcriptional activation (Murakami et al. 2005). Nm23-H1 is a known suppressor of metastasis and cell migration, and EBNA1 was shown to counteract the ability of Nm23-H1 to suppress cell migration both in vitro and in a nude mouse model (Murakami et al. 2005; Kaul et al. 2007). The results suggest that EBNA1 contributes to the metastatic potential of EBV tumors. This contention is supported by a report by Sheu et al. (1996) that EBNA1 expression in HONE-1 NPC cells increased tumor metastases in nude mice. In addition, a recent study showed that EBNA1 expression in CNE NPC cell lines decreased expression of epithelial cell markers and increased expression of mesenchymal cell markers, consistent with induction of EMT (Wang et al. 2014). In keeping with this finding, EBNA1 expression increased the migration and colony formation of CNE1 cells, providing further support for a role of EBNA1 in metastasis.
5 Immune Evasion
While most of the EBV latency proteins elicit a strong immune response, cells that only express EBNA1 (referred to as latency I or EBNA1-only program) largely avoid immune detection (Babcock et al. 2000; Munz 2004; Khanna et al. 1995). This is due to inefficient presentation of EBNA1 peptides on MHC class I molecules (Blake et al. 1997). Reduced EBNA1 presentation has been attributed to the central Gly–Ala repeat of EBNA1 which varies in size in different EBV isolates (~230 amino acid long in the B95-8 strain ; Fig. 2). Removal of this repeat has been shown to restore EBNA1 presentation, while the addition of the Gly–Ala repeat to EBNA4 inhibits its recognition by cytotoxic T lymphocytes (Blake et al. 1997; Levitskaya et al. 1995).
Initially, it was thought that the Gly–Ala repeat inhibited EBNA1 presentation by interfering with its proteasomal processing. This was based on the studies in which insertion of the Gly–Ala repeat in other proteins inhibited their degradation (Levitskaya et al. 1997; Sharipo et al. 1998; Heessen et al. 2002; Dantuma et al. 2000; Zhang and Coffino 2004). However, the deletion of the Gly–Ala repeat from EBNA1 was not found to affect EBNA1 turnover, as EBNA1 is extremely stable with or without the Gly–Ala repeat (Daskalogianni et al. 2008). In addition, Tellam et al. (2004) found that EBNA1 turnover varied considerably in different cell backgrounds but that these rates did not correspond to the level of MHC I-restricted presentation of EBNA1 peptides. Moreover, EBNA1 presentation was shown to derive from newly synthesized protein, and the primary contribution of the Gly–Ala repeat on the presentation of EBNA1 peptides was found to be due to inhibition of its own translation (Tellam et al. 2004; Yin et al. 2003). This supported a model where the MHC I-restricted presentation of EBNA1 occurs through the generation of defective ribosomal products (DRiPs) which are reduced by the presence of the Gly–Ala repeat (Fahraeus 2005).
Further studies confirmed that MHC class I presentation and CTL recognition corresponded to the rate of EBNA1 translation and the levels of DRiPs and that all were inhibited by the Gly–Ala repeat (Tellam et al. 2007; Apcher et al. 2009, 2010; Tellam et al. 2008). The Gly–Ala repeat was also reported to interfere with the initiation of translation (Apcher et al. 2010). The importance of the translation elongation rate was further supported by the recent finding that the RNA encoding the Gly–Ala repeats forms clusters of G-quadruplexes that affect both translation and antigen presentation (Murat et al. 2014). Specifically, antisense oligonucleotides that destabilize these G-quadruplexes were shown to increase translation, ribosome association, and antigen presentation, while stabilization of G-quadruplexes by pyridostatin treatment inhibited translation. Since the EBNA1 protein has been reported to bind G-quadruplex RNA (Norseen et al. 2009), it is possible that EBNA1 may autoregulate its translation by binding to its own mRNA during translation, but this remains to be determined. It has also been reported that Hsp90 inhibitors decrease EBNA translation suggesting a role for Hsp90 in this process; however, the mechanism of this effect is unclear (Sun et al. 2010).
Unlike MHC class I presentation, EBNA1 elicits a strong CD4 response due to efficient MHC class II processing (Munz et al. 2000; Leen et al. 2001). The MHC class II-mediated presentation of EBNA1 results, at least in part, from the ability of endogenous EBNA1 to undergo lysosomal processing after entering the autophagy pathway (Paludan et al. 2005). In keeping with this conclusion, inhibition of autophagy decreased recognition of EBNA1 by CD4+ T cells (Paludan et al. 2005). Subsequently, the range of EBNA1 CD4 epitopes generated was shown to be affected by the cellular localization of EBNA1, with nuclear localization limiting the displayed epitopes due to decreased accessibility to the macroautophagy pathway relative to cytoplasmic EBNA1 (Leung et al. 2010). How EBNA1-expressing cells are able to persist in the body despite recognition by CD4+ T cells is unclear.
6 EBNA1 in Lytic EBV Infection
The above roles of EBNA1 all pertain to latent EBV infection. However, EBNA1 is also expressed in lytic infection from a lytic cycle-specific promoter (Fp) , suggesting that it also contributes to productive infection (Brink et al. 2001; Lear et al. 1992; Schaefer et al. 1995). The contributions of EBNA1 to viral reactivation to the lytic cycle were examined in EBV-positive AGS gastric carcinoma cells (Sivachandran et al. 2012b). EBNA1 silencing was found to increase the frequency with which EBV spontaneously entered the lytic cycle, suggesting that EBNA1 can suppress reactivation. This may be due to ability of EBNA1 to induce let7 family miRNAs (including let-7a), which results in downregulation of Dicer, as both upregulation of let-7a and downregulation of Dicer have been shown to inhibit EBV reactivation (Iizasa et al. 2010; Mansouri et al. 2014).
EBNA1 has also been found to play a role in EBV lytic infection. When the lytic cycle is chemically induced, EBNA1 silencing decreases lytic gene expression and viral genome amplification indicating that EBNA1 can promote lytic infection. However, EBNA1 does not positively contribute to lytic infection in cells depleted in PML proteins, suggesting that the role of EBNA1 in lytic infection is in overcoming suppression by PML proteins. In keeping with this interpretation, PML proteins and nuclear bodies were found to suppress lytic infection by EBV (Sivachandran et al. 2012b; Sides et al. 2011).
7 Conclusion
In summary, EBNA1 makes multiple contributions to EBV infection due to its ability to interact with specific DNA sequences and multiple cellular proteins. Latency contributions include the replication and mitotic segregation of EBV episomes, contributions to viral transcription, and multiple effects on cellular proteins and pathways that promote cell survival and proliferation. In addition, EBNA1 impacts the reactivation of EBV and contributes to EBV lytic infection by overcoming suppression by PML nuclear bodies . This combination of diverse functions makes EBNA1 a key protein for EBV infection.
References
Altmann M, Pich D, Ruiss R, Wang J, Sugden B, Hammerschmidt W (2006) Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc Natl Acad Sci USA 103(38):14188–14193
Ambinder RF, Shah WA, Rawlins DR, Hayward GS, Hayward SD (1990) Definition of the sequence requirements for binding of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA. J Virol 64:2369–2379
Ambinder RF, Mullen M, Chang Y, Hayward GS, Hayward SD (1991) Functional domains of Epstein-Barr nuclear antigen EBNA-1. J Virol 65:1466–1478
Apcher S, Komarova A, Daskalogianni C, Yin Y, Malbert-Colas L, Fahraeus R (2009) mRNA translation regulation by the Gly-Ala repeat of Epstein-Barr virus nuclear antigen 1. J Virol 83(3):1289–1298
Apcher S, Daskalogianni C, Manoury B, Fahraeus R (2010) Epstein Barr virus-encoded EBNA1 interference with MHC class I antigen presentation reveals a close correlation between mRNA translation initiation and antigen presentation. PLoS Pathog 6(10):e1001151. doi:10.1371/journal.ppat.1001151
Aras S, Singh G, Johnston K, Foster T, Aiyar A (2009) Zinc coordination is required for and regulates transcription activation by Epstein-Barr nuclear antigen 1. PLoS Pathog 5(6):e1000469. doi:10.1371/journal.ppat.1000469
Atanasiu C, Deng Z, Wiedmer A, Norseen J, Lieberman PM (2006) ORC binding to TRF2 stimulates OriP replication. EMBO Rep 7(7):716–721
Avolio-Hunter TM, Frappier L (1998) Mechanistic studies on the DNA linking activity of the Epstein-Barr nuclear antigen 1. Nucl Acids Res 26:4462–4470
Avolio-Hunter TM, Frappier L (2003) EBNA1 efficiently assembles on chromatin containing the Epstein-Barr virus latent origin of replication. Virol 315:398–408
Avolio-Hunter TM, Lewis PN, Frappier L (2001) Epstein-Barr nuclear antigen 1 binds and destabilizes nucleosomes at the viral origin of latent DNA replication. Nucl Acids Res 29:3520–3528
Babcock GJ, Hochberg D, Thorley-Lawson DA (2000) The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13:497–506
Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM (2006) The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311(5762):856–861
Bashaw JM, Yates JL (2001) Replication from oriP of Epstein-Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J Virol 75:10603–10611
Bernardi R, Pandolfi PP (2007) Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8:1006–1016
Blake N, Lee S, Redchenko I, Thomas W, Steven N, Leese A, Steigerwald-Mullen P, Kurilla MG, Frappier L, Rickinson A (1997) Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-ALA) containing protein requires exogenous processing. Immunity 7:791–802
Bochkarev A, Barwell J, Pfuetzner R, Furey W, Edwards A, Frappier L (1995) Crystal structure of the DNA binding domain of the Epstein-Barr virus origin binding protein EBNA1. Cell 83:39–46
Bochkarev A, Barwell J, Pfuetzner R, Bochkareva E, Frappier L, Edwards AM (1996) Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin binding protein, EBNA1, bound to DNA. Cell 84:791–800
Bochkarev A, Bochkareva E, Frappier L, Edwards AM (1998) 2.2A structure of a permanganate-sensitive DNA site bound by the Epstein-Barr virus origin binding protein, EBNA1. J Mol Biol 284:1273–1278
Brink AA, Meijer CJ, Nicholls JM, Middeldorp JM, van den Brule AJ (2001) Activity of the EBNA1 promoter associated with lytic replication (Fp) in Epstein-Barr virus associated disorders. Mol Pathol 54(2):98–102
Canaan A, Haviv I, Urban AE, Schulz VP, Hartman S, Zhang Z, Palejev D, Deisseroth AB, Lacy J, Snyder M, Gerstein M, Weissman SM (2009) EBNA1 regulates cellular gene expression by binding cellular promoters. Proc Natl Acad Sci USA 106(52):22421–22426. doi:10.1073/pnas.0911676106 (0911676106 [pii])
Cao JY, Mansouri S, Frappier L (2011) Changes in the nasopharyngeal carcinoma nuclear proteome induced by the EBNA1 protein of Epstein-Barr virus reveal potential roles for EBNA1 in metastasis and oxidative stress responses. J Virol. doi:10.1128/JVI.05648-11 (JVI.05648-11 [pii])
Cao JY, Shire K, Landry C, Gish GD, Pawson T, Frappier L (2014) Identification of a novel protein interaction motif in the regulatory subunit of casein kinase 2. Mol Cell Biol 34(2):246–258. doi:10.1128/MCB.00968-13MCB.00968-13 ([pii])
Ceccarelli DFJ, Frappier L (2000) Functional analyses of the EBNA1 origin DNA binding protein of Epstein-Barr virus. J Virol 74:4939–4948
Cerimele F, Battle T, Lynch R, Frank DA, Murad E, Cohen C, Macaron N, Sixbey J, Smith K, Watnick RS, Eliopoulos A, Shehata B, Arbiser JL (2005) Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr Virus (EBV)-positive versus EBV-negative Burkitt’s lymphoma. Proc Natl Acad Sci USA 102(1):175–179. doi:10.1073/pnas.0408381102 (0408381102 [pii])
Chaudhuri B, Xu H, Todorov I, Dutta A, Yates JL (2001) Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proc Natl Acad Sci USA 98:10085–10089
Chen M-R, Middeldorp JM, Hayward SD (1993) Separation of the complex DNA binding domain of EBNA-1 into DNA recognition and dimerization subdomains of novel structure. J Virol 67:4875–4885
Chen YL, Liu CD, Cheng CP, Zhao B, Hsu HJ, Shen CL, Chiu SJ, Kieff E, Peng CW (2014) Nucleolin is important for Epstein-Barr virus nuclear antigen 1-mediated episome binding, maintenance, and transcription. Proc Natl Acad Sci USA 111(1):243–248. doi:10.1073/pnas.13218001111321800111 ([pii])
Cheng TC, Hsieh SS, Hsu WL, Chen YF, Ho HH, Sheu LF (2010) Expression of Epstein-Barr nuclear antigen 1 in gastric carcinoma cells is associated with enhanced tumorigenicity and reduced cisplatin sensitivity. Int J Oncol 36(1):151–160
Coppotelli G, Mughal N, Callegari S, Sompallae R, Caja L, Luijsterburg MS, Dantuma NP, Moustakas A, Masucci MG (2013) The Epstein-Barr virus nuclear antigen-1 reprograms transcription by mimicry of high mobility group A proteins. Nucleic Acids Res 41(5):2950–2962. doi:10.1093/nar/gkt032 ([pii])
Cruickshank J, Davidson A, Edwards AM, Frappier L (2000) Two domains of the Epstein-Barr virus origin DNA binding protein, EBNA1, orchestrate sequence-specific DNA binding. J Biol Chem 275:22273–22277
Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B (2004) Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428:486–487
D’Herouel AF, Birgersdotter A, Werner M (2010) FR-like EBNA1 binding repeats in the human genome. Virology 405(2):524–529. doi:10.1016/j.virol.2010.06.040 (S0042-6822(10)00422-8 [pii])
Dantuma NP, Heessen S, Lindsten K, Jellne M, Masucci MG (2000) Inhibition of proteasomal degradation by the gly-Ala repeat of Epstein-Barr virus is influenced by the length of the repeat and the strength of the degradation signal. Proc Natl Acad Sci USA 97(15):8381–8385
Daskalogianni C, Apcher S, Candeias MM, Naski N, Calvo F, Fahraeus R (2008) Gly-Ala repeats induce position- and substrate-specific regulation of 26 S proteasome-dependent partial processing. J Biol Chem 283(44):30090–30100
Delecluse H-J, Bartnizke S, Hammerschmidt W, Bullerdiek J, Bornkamm GW (1993) Episomal and integrated copies of Epstein-Barr virus coexist in Burkitt’s lymphoma cell lines. J Virol 67:1292–1299
Deng Z, Lezina L, Chen C-J, Shtivelband S, So W, Lieberman PM (2002) Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol Cell 9:493–503
Deng Z, Atanasiu C, Burg JS, Broccoli D, Lieberman PM (2003) Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate replication of Epstein-Barr virus OriP. J Virol 77(22):11992–12001
Deng Z, Atanasiu C, Zhao K, Marmorstein R, Sbodio JI, Chi NW, Lieberman PM (2005) Inhibition of Epstein-Barr virus OriP function by tankyrase, a telomere-associated poly-ADP ribose polymerase that binds and modifies EBNA1. J Virol 79(8):4640–4650
Deutsch MJ, Ott E, Papior P, Schepers A (2010) The latent origin of replication of Epstein-Barr virus directs viral genomes to active regions of the nucleus. J Virol 84(5):2533–2546. doi:10.1128/JVI.01909-09 (JVI.01909-09 [pii])
Dhar V, Schildkraut CL (1991) Role of EBNA-1 in arresting replication forks at the Epstein-Barr virus oriP family of tandem repeats. Mol Cell Biol 11:6268–6278
Dhar SK, Yoshida K, Machida Y, Khaira P, Chaudhuri B, Wohlschlegel JA, Leffak M, Yates J, Dutta A (2001) Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287–296
Dheekollu J, Deng Z, Wiedmer A, Weitzman MD, Lieberman PM (2007) A role for MRE11, NBS1, and recombination junctions in replication and stable maintenance of EBV episomes. PLoS ONE 2(12):e1257
Dresang LR, Vereide DT, Sugden B (2009) Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J Virol 83(7):2930–2940. doi:10.1128/JVI.01974-08 (JVI.01974-08 [pii])
Dyson PJ, Farrell PJ (1985) Chromatin structure of Epstein-Barr virus. J Gen Virol 66(Pt 9):1931–1940
Edwards AM, Bochkarev A, Frappier L (1998) Origin DNA-binding proteins. Curr Opin Struct Biol 8:49–53
Ermakova O, Frappier L, Schildkraut CL (1996) Role ot the EBNA-1 protein in pausing of replication forks in the Epstein-Barr virus genome. J Biol Chem 271:33009–33017
Everett RD (2001) DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20(49):7266–7273
Everett RD, Chelbi-Alix MK (2007) PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89(6–7):819–830
Everett R, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J (1997) A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J 16:1519–1530
Fahraeus R (2005) Do peptides control their own birth and death? Nat Rev Mol Cell Biol 6(3):263–267
Feeney KM, Saade A, Okrasa K, Parish JL (2011) In vivo analysis of the cell cycle dependent association of the bovine papillomavirus E2 protein and ChlR1. Virology 414(1):1–9. doi:10.1016/j.virol.2011.03.015 (S0042-6822(11)00140-1 [pii])
Flavell JR, Baumforth KR, Wood VH, Davies GL, Wei W, Reynolds GM, Morgan S, Boyce A, Kelly GL, Young LS, Murray PG (2008) Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood 111(1):292–301
Frappier L, O’Donnell M (1991a) Epstein-Barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of Epstein-Barr virus. Proc Natl Acad Sci USA 88:10875–10879
Frappier L, O’Donnell M (1991b) Overproduction, purification and characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J Biol Chem 266:7819–7826
Frappier L, O’Donnell M (1992) EBNA1 distorts oriP, the Epstein-Barr virus latent replication origin. J Virol 66:1786–1790
Frappier L, Verrijzer CP (2011) Gene expression control by protein deubiquitinases. Curr Opin Genet Dev 21(2):207–213. doi:10.1016/j.gde.2011.02.005 (S0959-437X(11)00048-7 [pii])
Frappier L, Goldsmith K, Bendell L (1994) Stabilization of the EBNA1 protein on the Epstein-Barr virus latent origin of DNA replication by a DNA looping mechanism. J Biol Chem 269:1057–1062
Gahn TA, Schildkraut CL (1989) The Epstein-Barr virus origin of plasmid replication, oriP, contains both the initiation and termination sites of DNA replication. Cell 58:527–535
Gahn T, Sugden B (1995) An EBNA1 Dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstien-Barr virus LMP gene. J Virol 69:2633–2636
Geoffroy MC, Chelbi-Alix MK (2011) Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31(1):145–158. doi:10.1089/jir.2010.0111
Goldsmith K, Bendell L, Frappier L (1993) Identification of EBNA1 amino acid sequences required for the interaction of the functional elements of the Epstein-Barr virus latent origin of DNA replication. J Virol 67:3418–3426
Grogan EA, Summers WP, Dowling S, Shedd D, Gradoville L, Miller G (1983) Two Epstein-Barr viral nuclear neoantigens distinguished by gene transfer, serology and chromosome binding. Proc Natl Acad Sci USA 80:7650–7653
Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG (2009) The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci USA 106(7):2313–2318. doi:10.1073/pnas.0810619106 (0810619106 [pii])
Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP (2000) The function of PML in p53-dependent apoptosis. Nat Cell Biol 2(10):730–736
Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, Verbel DA, Cordon-Cardo C, Pandolfi PP (2004) Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst 96(4):269–279
Harris A, Young BD, Griffin BE (1985) Random association of Epstein-Barr virus genomes with host cell metaphase chromosomes in Burkitt’s lymphoma-derived cell lines. J Virol 56:328–332
Harrison S, Fisenne K, Hearing J (1994) Sequence requirements of the Epstein-Barr virus latent origin of DNA replication. J Virol 68(3):1913–1925
Hearing J, Mulhaupt Y, Harper S (1992) Interaction of Epstein-Barr virus nuclear antigen 1 with the viral latent origin of replication. J Virol 66:694–705
Heessen S, Leonchiks A, Issaeva N, Sharipo A, Selivanova G, Masucci MG, Dantuma NP (2002) Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc Natl Acad Sci USA 99(3):1532–1537
Hegde RS, Grossman SR, Laimins LA, Sigler PB (1992) Crystal structure at 1.7 Å of the bovine papillomavirus-1 E2 DNA-binding protein bound to its DNA target. Nature 359:505–512
Hodin TL, Najrana T, Yates JL (2013) Efficient replication of Epstein-Barr virus-derived plasmids requires tethering by EBNA1 to host chromosomes. J Virol 87(23):13020–13028. doi:10.1128/JVI.01606-13JVI.01606-13 ([pii])
Holowaty MN, Sheng Y, Nguyen T, Arrowsmith C, Frappier L (2003a) Protein interaction domains of the ubiqutin specific protease, USP7/HAUSP. J Biol Chem 278:47753–47761
Holowaty MN, Zeghouf M, Wu H, Tellam J, Athanasopoulos V, Greenblatt J, Frappier L (2003b) Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J Biol Chem 278(32):29987–29994. doi:10.1074/jbc.M303977200M303977200 ([pii])
Holowaty MN, Zeghouf M, Wu H, Tellam J, Athanasopoulos V, Greenblatt J, Frappier L (2003c) Protein profiling with Epstein-Barr nuclear antigen 1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J Biol Chem 278:29987–29994
Hong M, Murai Y, Kutsuna T, Takahashi H, Nomoto K, Cheng CM, Ishizawa S, Zhao QL, Ogawa R, Harmon BV, Tsuneyama K, Takano Y (2006) Suppression of Epstein-Barr nuclear antigen 1 (EBNA1) by RNA interference inhibits proliferation of EBV-positive Burkitt’s lymphoma cells. J Cancer Res Clin Oncol 132(1):1–8
Hsieh D-J, Camiolo SM, Yates JL (1993) Constitutive binding of EBNA1 protein to the Epstein-Barr virus replication origin, oriP, with distortion of DNA structure during latent infection. EMBO J 12:4933–4944
Hu M, Gu L, Li M, Jeffrey PD, Gu W, Shi Y (2006) Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol 4(2):e27
Hume S, Reisbach G, Feederle R, Delecluse H-J, Bousset K, Hammerschmidt W, Schepers A (2003) The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci 100:10989–10994
Hung SC, Kang M-S, Kieff E (2001) Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc Natl Acad Sci USA 98:1865–1870
Iizasa H, Wulff BE, Alla NR, Maragkakis M, Megraw M, Hatzigeorgiou A, Iwakiri D, Takada K, Wiedmer A, Showe L, Lieberman P, Nishikura K (2010) Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J Biol Chem 285(43):33358–33370. doi:10.1074/jbc.M110.138362M110.138362 ([pii])
Ilves I, Maemets K, Silla T, Janikson K, Ustav M (2006) Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J Virol 80(7):3660–3665
Jager W, Santag S, Weidner-Glunde M, Gellermann E, Kati S, Pietrek M, Viejo-Borbolla A, Schulz TF (2012) The ubiquitin-specific protease USP7 modulates the replication of Kaposi’s sarcoma-associated herpesvirus latent episomal DNA. J Virol 86(12):6745–6757. doi:10.1128/JVI.06840-11 (JVI.06840-11 [pii])
Jones CH, Hayward SD, Rawlins DR (1989) Interaction of the lymphocyte-derived Epstein-Barr virus nuclear antigen EBNA-1 with its DNA-binding sites. J Virol 63:101–110
Jourdan N, Jobart-Malfait A, Dos Reis G, Quignon F, Piolot T, Klein C, Tramier M, Coppey-Moisan M, Marechal V (2012) Live-cell imaging reveals multiple interactions between Epstein-Barr virus nuclear antigen 1 and cellular chromatin during interphase and mitosis. J Virol 86(9):5314–5329. doi:10.1128/JVI.06303-11 (JVI.06303-11 [pii])
Julien MD, Polonskaya Z, Hearing J (2004) Protein and sequence requirements for the recruitment of the human origin recognition complex to the latent cycle origin of DNA replication of Epstein-Barr virus oriP. Virology 326(2):317–328
Kamranvar SA, Masucci MG (2011) The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 25(6):1017–1025. doi:10.1038/leu.2011.35leu201135 ([pii])
Kanda T, Otter M, Wahl GM (2001) Coupling of mitotic chromosome tethering and replication competence in Epstein-Barr virus-based plasmids. Mol Cell Biol 21:3576–3588
Kanda T, Kamiya M, Maruo S, Iwakiri D, Takada K (2007) Symmetrical localization of extrachromosomally replicating viral genomes on sister chromatids. J Cell Sci 120(Pt 9):1529–1539
Kanda T, Horikoshi N, Murata T, Kawashima D, Sugimoto A, Narita Y, Kurumizaka H, Tsurumi T (2013) Interaction between basic residues of Epstein-Barr virus EBNA1 protein and cellular chromatin mediates viral plasmid maintenance. J Biol Chem 288(33):24189–24199. doi:10.1074/jbc.M113.491167M113.491167 ([pii])
Kang MS, Lu H, Yasui T, Sharpe A, Warren H, Cahir-McFarland E, Bronson R, Hung SC, Kieff E (2005) Epstein-Barr virus nuclear antigen 1 does not induce lymphoma in transgenic FVB mice. Proc Natl Acad Sci USA 102(3):820–825
Kang MS, Soni V, Bronson R, Kieff E (2008) Epstein-Barr Virus Nuclear Antigen 1 does not cause lymphoma in C57BL/6J mice. J Virol 82:4180–4183
Kapoor P, Frappier L (2003) EBNA1 partitions Epstein-Barr virus plasmids in yeast by attaching to human EBNA1-binding protein 2 on mitotic chromosomes. J Virol 77:6946–6956
Kapoor P, Frappier L (2005) Methods for measuring the replication and segregation of Epstein-Barr virus-based plasmids. Methods Mol Biol 292:247–266
Kapoor P, Shire K, Frappier L (2001) Reconstitution of Epstein-Barr virus-based plasmid partitioning in budding yeast. EMBO J 20:222–230
Kapoor P, Lavoie BD, Frappier L (2005) EBP2 plays a key role in Epstein-Barr virus mitotic segregation and is regulated by aurora family kinases. Mol Cell Biol 25(12):4934–4945
Kaul R, Murakami M, Choudhuri T, Robertson ES (2007) Epstein-Barr virus latent nuclear antigens can induce metastasis in a nude mouse model. J Virol 81(19):10352–10361
Kennedy G, Sugden B (2003) EBNA-1, a bifunctional transcriptional activator. Mol Cell Biol 23(19):6901–6908
Kennedy G, Komano J, Sugden B (2003) Epstein-Barr virus provide a survival factor to Burkitt’s lymphomas. Proc Natl Acad Sci 100:14269–14274
Khanna R, Burrows SR, Moss DJ (1995) Immune regulation in Epstein-Barr virus-associated diseases. Microbiol Rev 59(3):387–405
Kim HS, Lee MS (2007) STAT1 as a key modulator of cell death. Cell Signal 19(3):454–465. doi:10.1016/j.cellsig.2006.09.003 (S0898-6568(06)00269-5 [pii])
Kim AL, Maher M, Hayman JB, Ozer J, Zerby D, Yates JL, Lieberman PM (1997) An imperfect correlation between DNA replication activity of Epstein-Barr virus nuclear antigen 1 (EBNA1) and binding to the nuclear import receptor, Rch1/importin α. Virology 239:340–351
Kirchmaier AL, Sugden B (1997) Dominant-negative inhibitors of EBNA1 of Epstein-Barr virus. J Virol 71:1766–1775
Koons MD, Van Scoy S, Hearing J (2001) The replicator of the Epstein-Barr virus latent cycle origin of DNA replication, oriP, is composed of multiple functional elements. J Virol 75:10582–10592
Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD (2002) Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J Virol 76:11596–11604
Krysan PJ, Haase SB, Calos MP (1989) Isolation of human sequences that replicate autonomously in human cells. Mol Cell Biol 9:1026–1033
Kube D, Vockerodt M, Weber O, Hell K, Wolf J, Haier B, Grasser FA, Muller-Lantzsch N, Kieff E, Diehl V, Tesch H (1999) Expression of Epstein-Barr virus nuclear antigen 1 is associated with enhanced expression of CD25 in the hodgkin cell line L428. J Virol 73:1630–1636
Laine A, Frappier L (1995) Identification of Epstein-Barr nuclear antigen 1 protein domains that direct interactions at a distance between DNA-bound proteins. J Biol Chem 270:30914–30918
Lassoued S, Ben Ameur R, Ayadi W, Gargouri B, Ben Mansour R, Attia H (2008) Epstein-Barr virus induces an oxidative stress during the early stages of infection in B lymphocytes, epithelial, and lymphoblastoid cell lines. Mol Cell Biochem 313(1–2):179–186. doi:10.1007/s11010-008-9755-z
Lear AL, Rowe M, Kurilla MG, Lee S, Henderson S, Kieff E, Rickinson AB (1992) The Epstein-Barr virus (EBV) nuclear antigen 1 BamHI F promoter is activated on entry of EBV-transformed B cells into the lytic cycle. J Virol 66(12):7461–7468
Lee MA, Diamond ME, Yates JL (1999) Genetic evidence that EBNA-1 is needed for efficient, stable latent infection by Epstein-Barr virus. J Virol 73(4):2974–2982
Lee HR, Choi WC, Lee S, Hwang J, Hwang E, Guchhait K, Haas J, Toth Z, Jeon YH, Oh TK, Kim MH, Jung JU (2011) Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat Struct Mol Biol 18(12):1336–1344. doi:10.1038/nsmb.2142nsmb.2142 ([pii])
Leen A, Meij P, Redchenko I, Middeldorp J, Bloemena E, Rickinson A, Blake N (2001) Differential immunogenicity of Epstein-Barr virus latent-cycle proteins for human CD4(+) T-helper 1 responses. J Virol 75(18):8649–8659
Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS (2010) Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci USA 107(5):2165–2170. doi:10.1073/pnas.09094481070909448107 ([pii])
Levitskaya J, Coram M, Levitsky V, Imreh S, Steigerwald-Mullen P, Klein G, Kurilla MG, Masucci MG (1995) Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375:685–688
Levitskaya J, Shapiro A, Leonchiks A, Ciechanover A, Masucci MG (1997) Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci USA 94:12616–12621
Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416(6881):648–653
Li M, Brooks CL, Kon N, Gu W (2004) A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell 13(6):879–886. doi:10.1016/S1097276504001571 ([pii])
Lin A, Wang S, Nguyen T, Shire K, Frappier L (2008) The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J Virol 82(24):12009–12019
Lindner SE, Zeller K, Schepers A, Sugden B (2008) The affinity of EBNA1 for its origin of DNA synthesis is a determinant of the origin’s replicative efficiency. J Virol 82(12):5693–5702
Little RD, Schildkraut CL (1995) Initiation of latent DNA replicatoon in the Epstein-Barr virus genome can occur at sites other than the genetically defined origin. Mol Cell Biol 15:2893–2903
Lu F, Wikramasinghe P, Norseen J, Tsai K, Wang P, Showe L, Davuluri RV, Lieberman PM (2010) Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1). Virol J 7:262. doi:10.1186/1743-422X-7-262 (1743-422X-7-262 [pii])
Lu J, Murakami M, Verma SC, Cai Q, Haldar S, Kaul R, Wasik MA, Middeldorp J, Robertson ES (2011) Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 410(1):64–75. doi:10.1016/j.virol.2010.10.029 (S0042-6822(10)00676-8 [pii])
Lupton S, Levine AJ (1985) Mapping of genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol Cell Biol 5:2533–2542
Mackey D, Sugden B (1999) The linking regions of EBNA1 are essential for its support of replication and transcription. Mol Cell Biol 19:3349–3359
Mackey D, Middleton T, Sugden B (1995) Multiple regions within EBNA1 can link DNAs. J Virol 69:6199–6208
Malik-Soni N, Frappier L (2012) Proteomic profiling of EBNA1-Host Protein Interactions in latent and lytic Epstein-Barr virus infections. J Virol 86(12):6999–7002. doi:10.1128/JVI.00194-12 (JVI.00194-12 [pii])
Malik-Soni N, Frappier L (2013) Nucleophosmin contributes to the transcriptional activation function of the Epstein-Barr virus EBNA1 Protein. J Virol. doi:10.1128/JVI.02521-13 (JVI.02521-13 [pii])
Mansouri S, Pan Q, Blencowe BJ, Claycomb JM, Frappier L (2014) Epstein-Barr Virus EBNA1 protein regulates viral latency through effects on let-7 MicroRNA and dicer. J Virol 88(19):11166–11177. doi:10.1128/JVI.01785-14JVI.01785-14 ([pii])
Marechal V, Dehee A, Chikhi-Brachet R, Piolot T, Coppey-Moisan M, Nicolas J (1999) Mapping EBNA1 domains involved in binding to metaphse chromosomes. J Virol 73:4385–4392
McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA (2006) Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol 80(19):9530–9543
Middleton T, Sugden B (1992) EBNA1 can link the enhancer element to the initiator element of the Epstein-Barr virus plasmid origin of DNA replication. J Virol 66:489–495
Moriyama K, Yoshizawa-Sugata N, Obuse C, Tsurimoto T, Masai H (2012) Epstein-Barr Nuclear Antigen 1 (EBNA1)-dependent recruitment of origin recognition complex (Orc) on oriP of Epstein-Barr virus with purified proteins: stimulation by Cdc6 through its direct interaction with EBNA1. J Biol Chem 287(28):23977–23994. doi:10.1074/jbc.M112.368456 (M112.368456 [pii])
Munz C (2004) Epstein-barr virus nuclear antigen 1: from immunologically invisible to a promising T cell target. J Exp Med 199(10):1301–1304
Munz C, Bickham KL, Subklewe M, Tsang ML, Chahroudi A, Kurilla MG, Zhang D, O’Donnell M, Steinman RM (2000) Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J Exp Med 191(10):1649–1660
Murakami M, Lan K, Subramanian C, Robertson ES (2005) Epstein-Barr virus nuclear antigen 1 interacts with Nm23-H1 in lymphoblastoid cell lines and inhibits its ability to suppress cell migration. J Virol 79(3):1559–1568
Murat P, Zhong J, Lekieffre L, Cowieson NP, Clancy JL, Preiss T, Balasubramanian S, Khanna R, Tellam J (2014) G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat Chem Biol 10(5):358–364. doi:10.1038/nchembio.1479nchembio.1479 ([pii])
Nanbo A, Sugden A, Sugden B (2007) The coupling of synthesis and partitioning of EBV’s plasmid replicon is revealed in live cells. EMBO J 26(19):4252–4262
Nayyar VK, Shire K, Frappier L (2009) Mitotic chromosome interactions of Epstein-Barr nuclear antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2). J Cell Sci 122(Pt 23):4341–4350
Nicholson B, Suresh Kumar KG (2011) The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys 60(1–2):61–68. doi:10.1007/s12013-011-9185-5
Niller HH, Glaser G, Knuchel R, Wolf H (1995) Nucleoprotein complexes and DNA 5′-ends at oriP of Epstein-Barr virus. J Biol Chem 270:12864–12868
Nonkwelo C, Skinner J, Bell A, Rickinson A, Sample J (1996) Transcription start site downstream of the Epstein-Barr virus (EBV) Fp promoter in early-passage Burkitt lymphoma cells define a fourth promoter for expression of the EBV EBNA1 protein. J Virol 70:623–627
Norio P, Schildkraut CL (2001) Visualization of DNA replication on individual Epstein-Barr virus episomes. Science 294:2361–2364
Norio P, Schildkraut CL (2004) Plasticity of DNA replication initiation in Epstein-Barr virus episomes. PLoS Biol 2(6):e152
Norio P, Schildkraut CL, Yates JL (2000) Initiation of DNA replication within oriP is dispensable for stable replication of the latent Epstein-Barr virus chromosome after infection of established cell lines. J Virol 74:8563–8574
Norseen J, Thomae A, Sridharan V, Aiyar A, Schepers A, Lieberman PM (2008) RNA-dependent recruitment of the origin recognition complex. Embo J 27(22):3024–3035. doi:10.1038/emboj.2008.221 (emboj2008221 [pii])
Norseen J, Johnson FB, Lieberman PM (2009) Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J Virol 83(20):10336–10346. doi:10.1128/JVI.00747-09 (JVI.00747-09 [pii])
O’Neil JD, Owen TJ, Wood VH, Date KL, Valentine R, Chukwuma MB, Arrand JR, Dawson CW, Young LS (2008) Epstein-Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J Gen Virol 89(Pt 11):2833–2842. doi:10.1099/vir.0.2008/003392-0 (89/11/2833 [pii])
Oddo C, Freire E, Frappier L, de Prat-Gay G (2006) Mechanism of DNA recognition at a viral replication origin. J Biol Chem 281(37):26893–26903
Ott E, Norio P, Ritzi M, Schildkraut C, Schepers A (2011) The dyad symmetry element of Epstein-Barr virus is a dominant but dispensable replication origin. PLoS ONE 6(5):e18609. doi:10.1371/journal.pone.0018609PONE-D-10-06467 ([pii])
Owen TJ, O’Neil JD, Dawson CW, Hu C, Chen X, Yao Y, Wood VH, Mitchell LE, White RJ, Young LS, Arrand JR (2010) Epstein-Barr virus-encoded EBNA1 enhances RNA polymerase III-dependent EBER expression through induction of EBER-associated cellular transcription factors. Mol Cancer 9:241. doi:10.1186/1476-4598-9-2411476-4598-9-241 ([pii])
Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C (2005) Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307(5709):593–596. doi:10.1126/science.1104904 (1104904 [pii])
Parish JL, Bean AM, Park RB, Androphy EJ (2006) ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol Cell 24(6):867–876
Park YJ, Luger K (2006) Structure and function of nucleosome assembly proteins. Biochem Cell Biol 84(4):549–558
Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406(6792):207–210
Petti L, Sample C, Kieff E (1990) Subnuclear localization and phosphorylation or Epstein-Barr virus latent infection nuclear proteins. Virology 176:563–574
Rawlins DR, Milman G, Hayward SD, Hayward GS (1985) Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA1) to clustered sites in the plasmid maintenance region. Cell 42:859–868
Reedman BM, Klein G (1973) Cellular localization of an Epstein-Barr virus (EBV)-associated complement-fixing antigen in producer and non-producer lymphoblastoid cell lines. Int J Cancer 11:499–520
Reichelt M, Wang L, Sommer M, Perrino J, Nour AM, Sen N, Baiker A, Zerboni L, Arvin AM (2011) Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog 7(2):e1001266. doi:10.1371/journal.ppat.1001266
Reisman D, Sugden B (1986) trans Activation of an Epstein-Barr viral transcripitonal enhancer by the Epstein-Barr viral nuclear antigen 1. Mol Cell Biol 6:3838–3846
Reisman D, Yates J, Sugden B (1985) A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol Cell Biol 5:1822–1832
Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, Hammerschmidt W, Schepers A (2003) Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J Cell Sci 116(Pt 19):3971–3984
Salomoni P, Ferguson BJ, Wyllie AH, Rich T (2008) New insights into the role of PML in tumour suppression. Cell Res 18(6):622–640
Salsman J, Jagannathan M, Paladino P, Chan PK, Dellaire G, Raught B, Frappier L (2012) Proteomic profiling of the human cytomegalovirus UL35 gene products reveals a role for UL35 in the DNA repair response. J Virol 86(2):806–820. doi:10.1128/JVI.05442-11JVI.05442-11 ([pii])
Sample J, Henson EBD, Sample C (1992) The Epstein-Barr virus nuclear protein 1 promoter active in type I latency is autoregulated. J Virol 66:4654–4661
Saridakis V, Sheng Y, Sarkari F, Holowaty MN, Shire K, Nguyen T, Zhang RG, Liao J, Lee W, Edwards AM, Arrowsmith CH, Frappier L (2005) Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell 18(1):25–36
Sarkari F, Sanchez-Alcaraz T, Wang S, Holowaty MN, Sheng Y, Frappier L (2009) EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog 5(10):e1000624. doi:10.1371/journal.ppat.1000624
Sarkari F, Wang X, Nguyen T, Frappier L (2011) The herpesvirus associated ubiquitin specific protease, USP7, is a negative regulator of PML proteins and PML nuclear bodies. PLoS ONE 6(1):e16598. doi:10.1371/journal.pone.0016598
Scaglioni PP, Yung TM, Cai LF, Erdjument-Bromage H, Kaufman AJ, Singh B, Teruya-Feldstein J, Tempst P, Pandolfi PP (2006) A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126(2):269–283
Scaglioni PP, Yung TM, Choi SC, Baldini C, Konstantinidou G, Pandolfi PP (2008) CK2 mediates phosphorylation and ubiquitin-mediated degradation of the PML tumor suppressor. Mol Cell Biochem 316:149–154
Schaefer BC, Strominger JL, Speck SH (1995) The Epstein-Barr virus BamHI F promoter is an early lytic promoter: lack of correlation with EBNA 1 gene transcription in group 1 Burkitt’s lymphoma cell lines. J Virol 69(8):5039–5047
Schepers A, Ritzi M, Bousset K, Kremmer E, Yates JL, Harwood J, Diffley JFX, Hammerschmidt W (2001) Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J 20:4588–4602
Schweiger MR, You J, Howley PM (2006) Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol 80(9):4276–4285
Sears J, Kolman J, Wahl GM, Aiyar A (2003) Metaphase chromosome tethering is necessary for the DNA synthesis and maintenance of oriP plasmids but is insufficient for transcription activation by Epstein-Barr nuclear antigen 1. J Virol 77(21):11767–11780
Sears J, Ujihara M, Wong S, Ott C, Middeldorp J, Aiyar A (2004) The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J Virol 78(21):11487–11505
Seo S-B, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D (2001) Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the Set oncoprotein. Cell 104:119–130
Shah WA, Ambinder RF, Hayward GS, Hayward SD (1992) Binding of EBNA-1 to DNA creates a protease-resistant domain that encompasses the DNA recognition and dimerization functions. J Virology 66:3355–3362
Sharipo A, Imreh M, Leonchiks A, Imreh S, Masucci MG (1998) A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat Med 4(8):939–944
Shaw J, Levinger L, Carter C (1979) Nucleosomal structure of Epstein-Barr virus DNA in transformed cell lines. J Virol 29:657–665
Sheng Y, Saridakis V, Sarkari F, Duan S, Wu T, Arrowsmith CH, Frappier L (2006) Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat Struct Mol Biol 13(3):285–291
Sheu LF, Chen A, Meng CL, Ho KC, Lee WH, Leu FJ, Chao CF (1996) Enhanced malignant progression of nasopharyngeal carcinoma cells mediated by the expression of Epstein-Barr nuclear antigen 1 in vivo. J Pathol 180(3):243–248
Shikama N, Chan HM, Krstic-Demonacos M, Smith L, Lee CW, Cairns W, La Thangue NB (2000) Functional interaction between nucleosome assembly proteins and p300/CREB-binding protein family coactivators. Mol Cell Biol 20(23):8933–8943
Shire K, Ceccarelli DFJ, Avolio-Hunter TM, Frappier L (1999) EBP2, a human protein that interacts with sequences of the Epstein-Barr nuclear antigen 1 important for plasmid maintenance. J Virol 73:2587–2595
Shire K, Kapoor P, Jiang K, Hing MN, Sivachandran N, Nguyen T, Frappier L (2006) Regulation of the EBNA1 Epstein-Barr virus protein by serine phosphorylation and arginine methylation. J Virol 80(11):5261–5272
Sides MD, Block GJ, Shan B, Esteves KC, Lin Z, Flemington EK, Lasky JA (2011) Arsenic mediated disruption of promyelocytic leukemia protein nuclear bodies induces ganciclovir susceptibility in Epstein-Barr positive epithelial cells. Virology 416(1–2):86–97. doi:10.1016/j.virol.2011.04.005S0042-6822(11)00173-5 ([pii])
Simpson K, McGuigan A, Huxley C (1996) Stable episomal maintenance of yeast artificial chromosomes in human cells. Mol Cell Biol 16:5117–5126
Sivachandran N, Sarkari F, Frappier L (2008) Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog 4(10):e1000170. doi:10.1371/journal.ppat.1000170
Sivachandran N, Cao JY, Frappier L (2010) Epstein-Barr virus nuclear antigen 1 Hijacks the host kinase CK2 to disrupt PML nuclear bodies. J Virol 84(21):11113–11123
Sivachandran N, Dawson CW, Young LS, Liu FF, Middeldorp J, Frappier L (2012a) Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J Virol 86(1):60–68. doi:10.1128/JVI.05623-11 (JVI.05623-11[pii])
Sivachandran N, Wang X, Frappier L (2012b) Functions of the Epstein-Barr virus EBNA1 Protein in viral reactivation and lytic infection. J Virol 86(11):6146–6158. doi:10.1128/JVI.00013-12 (JVI.00013-12 [pii])
Snudden DK, Hearing J, Smith PR, Grasser FA, Griffin BE (1994) EBNA1, the major nuclear antigen of Epstein-Barr virus, resenbles ‘RGG’ RNA binding proteins. EMBO J 13:4840–4848
Sternas L, Middleton T, Sugden B (1990) The average number of molecules of Epstein-Barr nuclear antigen 1 per cell does not correlate with the average number of Epstein-Barr virus (EBV) DNA molecules per cell among different clones of EBV-immortalized cells. JVirol 64:2407–2410
Su W, Middleton T, Sugden B, Echols H (1991) DNA looping between the origin of replication of Epstein-Barr virus and its enhancer site: stabilization of an origin complex with Epstein-Barr nuclear antigen 1. Proc Natl Acad Sci USA 88:10870–10874
Sugden B, Warren N (1989) A promoter of Epstein-Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J Virol 63(6):2644–2649
Summers H, Barwell JA, Pfuetzner RA, Edwards AM, Frappier L (1996) Cooperative assembly of EBNA1 on the Epstein-Barr virus latent origin of replication. J Virol 70:1228–1231
Summers H, Fleming A, Frappier L (1997) Requirements for EBNA1-induced permanganate sensitivity of the Epstein-Barr virus latent origin of DNA replication. J Biol Chem 272:26434–26440
Sun X, Barlow EA, Ma S, Hagemeier SR, Duellman SJ, Burgess RR, Tellam J, Khanna R, Kenney SC (2010) Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism. Proc Natl Acad Sci USA 107(7):3146–3151. doi:10.1073/pnas.0910717107 (0910717107 [pii])
Takahashi Y, Lallemand-Breitenbach V, Zhu J, de The H (2004) PML nuclear bodies and apoptosis. Oncogene 23(16):2819–2824
Tellam J, Connolly G, Green KJ, Miles JJ, Moss DJ, Burrows SR, Khanna R (2004) Endogenous presentation of CD8+ T cell epitopes from Epstein-Barr virus-encoded nuclear antigen 1. J Exp Med 199(10):1421–1431
Tellam J, Fogg MH, Rist M, Connolly G, Tscharke D, Webb N, Heslop L, Wang F, Khanna R (2007) Influence of translation efficiency of homologous viral proteins on the endogenous presentation of CD8+ T cell epitopes. J Exp Med 204(3):525–532
Tellam J, Smith C, Rist M, Webb N, Cooper L, Vuocolo T, Connolly G, Tscharke DC, Devoy MP, Khanna R (2008) Regulation of protein translation through mRNA structure influences MHC class I loading and T cell recognition. Proc Natl Acad Sci USA 105(27):9319–9324
Tsimbouri P, Drotar ME, Coy JL, Wilson JB (2002) bcl-xL and RAG genes are induced and the response to IL-2 enhanced in EmuEBNA-1 transgenic mouse lymphocytes. Oncogene 21(33):5182–5187
Valentine R, Dawson CW, Hu C, Shah KM, Owen TJ, Date KL, Maia SP, Shao J, Arrand JR, Young LS, O’Neil JD (2010) Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol Cancer 9:1. doi:10.1186/1476-4598-9-1 (1476-4598-9-1 [pii])
Van Scoy S, Watakabe I, Krainer AR, Hearing J (2000) Human p32: a coactivator for Epstein-Barr virus nuclear antigen-1-mediated transcriptional activation and possible role in viral latent cycle DNA replication. Virology 275:145–157
Wang S, Frappier L (2009) Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J Virol 83(22):11704–11714. doi:10.1128/JVI.00931-09 (JVI.00931-09 [pii])
Wang Y, Finan JE, Middeldorp JM, Hayward SD (1997) P32/TAP, a cellular protein that interacts with EBNA-1 of Epstein-Barr virus. Virology 236:18–29
Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, Pandolfi PP (1998) PML is essential for multiple apoptotic pathways. Nat Genet 20(3):266–272
Wang L, Tian WD, Xu X, Nie B, Lu J, Liu X, Zhang B, Dong Q, Sunwoo JB, Li G, Li XP (2014) Epstein-Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer 120(3):363–372. doi:10.1002/cncr.28418
Wilson JB, Bell JL, Levine AJ (1996) Expression of Epstein-Barr virus nuclar antigen-1 induces B cell neoplasia in transgenic mice. EMBO 15:3117–3126
Wood VH, O’Neil JD, Wei W, Stewart SE, Dawson CW, Young LS (2007) Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFbeta signaling pathways. Oncogene 26(28):4135–4147
Wu SY, Chiang CM (2007) The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem 282(18):13141–13145. doi:10.1074/jbc.R700001200 (R700001200 [pii])
Wu H, Ceccarelli DFJ, Frappier L (2000) The DNA segregation mechanism of the Epstein-Barr virus EBNA1 protein. EMBO Rep 1:140–144
Wu H, Kapoor P, Frappier L (2002) Separation of the DNA replication, segregation, and transcriptional activation functions of Epstein-Barr nuclear antigen 1. J Virol 76(5):2480–2490
Wysokenski DA, Yates JL (1989) Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J Virol 63:2657–2666
Yates JL, Camiolo SM (1988) Dissection of DNA replication and enhancer activation functions of Epstein-Barr virus nuclear antigen 1. Cancer Cells 6:197–205
Yates JL, Guan N (1991) Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J Virol 65:483–488
Yates JL, Warren N, Reisman D, Sugden B (1984) A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci USA 81:3806–3810
Yates JL, Warren N, Sugden B (1985) Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313:812–815
Yates JL, Camiolo SM, Bashaw JM (2000) The minimal replicator of Epstein-Barr virus oriP. J Virol 74:4512–4522
Yin Q, Flemington EK (2006) siRNAs against the Epstein Barr virus latency replication factor, EBNA1, inhibit its function and growth of EBV-dependent tumor cells. Virology 346(2):385–393
Yin Y, Manoury B, Fahraeus R (2003) Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 301(5638):1371–1374
Yoshioka M, Crum MM, Sample JT (2008) Autorepression of Epstein-Barr virus nuclear antigen 1 expression by inhibition of pre-mRNA processing. J Virol 82(4):1679–1687
You J (2010) Papillomavirus interaction with cellular chromatin. Biochim Biophys Acta 1799(3–4):192–199. doi:10.1016/j.bbagrm.2009.09.009 (S1874-9399(09)00114-X [pii])
Zhang M, Coffino P (2004) Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. J Biol Chem 279(10):8635–8641. doi:10.1074/jbc.M310449200M310449200 ([pii])
Zhou J, Snyder AR, Lieberman PM (2009) Epstein-Barr virus episome stability is coupled to a delay in replication timing. J Virol 83(5):2154–2162
Zhou J, Deng Z, Norseen J, Lieberman PM (2010) Regulation of Epstein-Barr virus origin of plasmid replication (OriP) by the S-phase checkpoint kinase Chk2. J Virol 84(10):4979–4987. doi:10.1128/JVI.01300-09 (JVI.01300-09 [pii])
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Frappier, L. (2015). EBNA1. In: Münz, C. (eds) Epstein Barr Virus Volume 2. Current Topics in Microbiology and Immunology, vol 391. Springer, Cham. https://doi.org/10.1007/978-3-319-22834-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-22834-1_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22833-4
Online ISBN: 978-3-319-22834-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)