
Abstract
Statins are the cornerstone of lipid-lowering therapy for dyslipidaemias and prevention of cardiovascular disease. However, residual risk remains a significant challenge, particularly in severe, inherited hypercholesterolaemia. High-intensity statin therapy can be associated with adverse outcomes, most notably myalgia, and does not frequently result in the attainment of therapeutic levels of low-density lipoprotein (LDL) cholesterol and apolipoprotein B (apoB). Combination treatment is therefore required with agents that have complementary mechanisms. Mipomersen is an injectable antisense oligonucleotide, which inhibits apoB messenger RNA. It acts by blocking the translation and synthesis of apoB, resulting in reduced secretion of very-low-density lipoprotein (VLDL) particles and the subsequent production of LDL particles in plasma. There are no known drug interactions between mipomersen and statins. Several phase III studies of mipomersen have shown consistent reductions in plasma LDL cholesterol levels in a range of patient groups with hypercholesterolaemia and on a background of statin treatment. However, there is a concern regarding increased hepatic fat accumulation, as well as adverse effects related to injection site reactions and influenza-like symptoms. Nonetheless, mipomersen has been approved by the US Food and Drug Administration (FDA) for use in homozygous familial hypercholesterolaemia as an orphan drug. Mipomersen can also lower Lp(a) levels, although the cardiovascular benefit of this effect remains unproven. Long-term data are required to justify the net benefits of using mipomersen in patients with severe hypercholesterolaemia.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Statins are the frontline therapy for hypercholesterolaemia and confer significant reductions in cardiovascular mortality and morbidity in both primary and secondary prevention [1]. However, patients with refractory hypercholesterolaemia, in particular those with familial hypercholesterolaemia (FH), have difficulty in achieving their therapeutic targets on statin alone [2]. Currently available lipid-lowering agents, such as bile acid sequestrants, fibrates, ezetimibe and niacin, have limited efficacy as monotherapy or in combination with a statin in lowering low-density lipoprotein (LDL) cholesterol to the target levels in such patients [3]. Several newer agents that lower LDL cholesterol concentration are in the late stages of drug development or being used as orphan drugs [3]. One of these new therapies is mipomersen, which has been approved by the US Food and Drug Administration (FDA), adjunct to lipid-lowering therapy and diet, for the treatment of homozygous FH [4]. We review the efficacy and safety issues of mipomersen and its potential as combination therapy with statins, with emphasis on the management of hypercholesterolaemia in FH.
Hypercholesterolaemia: Dysregulation of Apolipoprotein B-100 (apoB) Metabolism
Understanding the pathophysiology of hypercholesterolaemia requires a brief review of apoB metabolism [5]. ApoB is a key structural and functional component of lipoprotein metabolism. It is involved in the assembly and secretion of very low-density lipoprotein (VLDL) from the liver. Apolipoprotein B-100 (apoB) is coded by the APOB gene and by a single mRNA transcript. ApoB is synthesised in the liver and functions to deliver triglycerides from the liver to the circulation. VLDL and its lipoprotein remnants, intermediate-density lipoprotein (IDL) and LDL, contain a single molecule of apoB per particle. Hence, plasma apoB concentration is an indicator of the total number of atherogenic lipoproteins including Lp(a). Elevated plasma apoB is a risk factor for atherosclerosis and is a predictor of atherosclerotic cardiovascular disease [6].
Owing to the central role of apoB in lipid metabolism, interventions that target apoB metabolism are critical. As indicated later, statins lower LDL cholesterol and apoB by enhancing the clearance of apoB-containing particles. Hence, inhibiting apoB synthesis, and the subsequent production of VLDL and LDL, provides a complimentary approach to statins for reducing elevated levels of LDL cholesterol and apoB. Antisense technology offers a form of treatment whereby a strand of DNA binds to the mRNA produced by the gene of a specific protein and thereby inhibits translation and the production of the protein. An advantage to this approach is the reduced potential of drug interactions, particularly for patients on multiple agents. Dyslipoproteinaemias due to elevated hepatic secretion of apoB may theoretically benefit from this form of therapy.
Severe Familial Hypercholesterolaemia
Patients with severe FH are at high risk of premature coronary artery disease (CAD) owing to elevated LDL cholesterol and apoB concentrations from birth [3]. FH results principally from mutations in the LDL receptor (LDLr) that impair LDL catabolism. Over 1700 mutations in the LDLr have been described worldwide; the severity of the disorder is in part associated with the residual activity of the LDLr. Patients with null mutations show poorer responses to statin treatment [7]. Statins are efficacious in lowering LDL cholesterol in FH. Despite best standard statin treatment, most homozygous and severe heterozygous FH patients do not achieve the recommended plasma concentrations of LDL cholesterol required for abolishing the risk of CAD: LDL cholesterol <2.5 mmol/L (absence of CHD or othermajor risk factors) and <1.8 mmol/L (presence of CHD or other major risk factors) [3]. Bile acid sequestrants, ezetimibe and fibrates are also relevant options as add-on therapy and/or in cases of statin intolerance. Small studies have supported combination treatment in FH [8–11], although there have been no outcome studies to date.
Beyond LDL, increased residual risk of CAD in FH relates to elevated plasma concentrations of lipoprotein(a) [Lp(a)] [12]. Lp(a) is a macromolecular complex assembled from LDL and apolipoprotein (a) (apo[a]). It is a quantitative genetic trait and is a causal and independent risk factor for cardiovascular disease in both the general population and patients with FH [13]. The atherogenicity of Lp(a) may, in part, be mediated by oxidised phospholipids, which associate with small apo(a) isoforms [14]. The apo(a) genes can predict the majority of the variation of Lp(a) levels in plasma [15], with large differences among different ethnic groups [16]. Lp(a) is refractory to lifestyle and standard lipid-lowering therapies. The only potentially effective drug for lowering Lp(a) is niacin, but severe side effects preclude its use [17]. Mipomersen has been shown to reduce plasma Lp(a) in FH patients by 20–30 % [18, 19]. Hence, reduction of Lp(a) with mipomersen presents an additional benefit that complements the LDL-cholesterol-lowering effect of this agent.
Statins
Statins can lower plasma LDL cholesterol by 20–55 % depending on statin type and dose. However, current therapeutic guidelines have lowered the optimal LDL cholesterol target to <1.8 mmol/L for high-risk coronary heart disease [20, 21], emphasising value and use of high-intensity statin therapy. Future guidelines may recommend more stringent targets, especially with clinical trial evidence demonstrating lower risk of cardiovascular events with an LDL cholesterol of <1.3 mmol/L [22]. However, a significant proportion of patients are statin intolerant [23], particularly with higher doses, with side effects including myalgia, myositis, rhabdomyolysis, hepatotoxicity, peripheral neuropathy [24] and new-onset type 2 diabetes [25, 26]. The proportion of statin-associated muscle symptoms is estimated to be between 7 and 29 % [27]. With the exception of dose reduction and re-challenge, there is little evidence to guide the management of statin-intolerant patients [27, 28]. Since statin-related adverse events are dose dependent, high-risk patients on high-intensity statins are a particularly vulnerable group.
The mechanism of action of statins on lipid metabolism fundamentally relies on the decreased conversion of HMG-CoA to mevalonic acid by competitive inhibition of HMG-CoA reductase, a rate-limiting enzyme in hepatic cholesterol synthesis. The resulting reduction in intracellular cholesterol content stimulates LDL receptor synthesis and LDL catabolism [29]. The effect of statins on Lp(a) is modest and inconsistent [13]. Long-term statin use can lower Lp(a) by approximately 20 %, although this does not appear to correlate with changes in carotid atherosclerosis in heterozygous FH subjects [30].
Because statins are not effective in lowering LDL cholesterol to recommended levels and are not generally effective on Lp(a), new therapies have been developed. This is particularly relevant for patients with severe FH, such as those with homozygous and compound heterozygous FH, where LDL receptor functions are either absent or dysfunctional. Mipomersen reduces circulating LDL levels by directly targeting apoB synthesis, an effect that does not require functional LDL receptor activity. By contrast, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition has limited efficacy in patients with no LDL receptor function [31].
Mipomersen
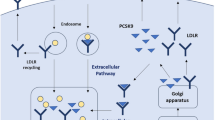
Mipomersen (ISIS-301012, KynamroTM) is a second-generation antisense oligonucleotide (ASO) designed to directly inhibit the synthesis of apoB-100 by targeting its mRNA. Mipomersen is a 2’-O-methoxyethyl chimeric 20-mer oligonucleotide complementary to the coding region of human apoB-100 mRNA, modified to withstand almost all nuclease degradation [32]. Once the apoB ASO binds to the apoB mRNA, its degradation is triggered by ribonuclease H (RNAase H), and protein translation is inhibited. Subsequently, the synthesis of the apoB protein is decreased, with lowering in the level of plasma circulating apoB-containing lipoproteins such as VLDL and LDL (see Fig. 7.1). ASOs are metabolised independently of CYP450, an important advantage in relation to drug interactions [33]. Mipomersen is primarily excreted in the urine after nuclease metabolism [34].

Mechanism of action of statin and mipomersen. (a) Mipomersen specifically binds the apoB mRNA sequence to provide a substrate for RNase H, which hydrolyses the apoB mRNA strand and inhibits apoB synthesis. (b) Statins competitively inhibit HMG-CoA reductase, a rate-limiting enzyme in hepatic cholesterol synthesis, the reduced intracellular cholesterol content induces LDL receptor production and increases LDL catabolism. On the other hand, Mipomersen inhibitors apoB synthesis and reduces the production of atherogenic apoB-containing lipoproteins by the liver (Adapted from [55])
ApoB-100 antisense was originally tested in mice models of hypercholesterolaemia. In LDLr-deficient mice, this antisense therapy lowered LDL cholesterol, consistent with its mechanism of action, and ameliorated atherosclerosis without causing hepatic steatosis [35, 36]. The first human study by Kastelein et al. (2005) showed a maximum of 35 % reduction of LDL cholesterol concentration and 50 % reduction in apoB levels after 4 weeks of multiple-dosing regime in patients with mild dyslipidaemia [37]. However, the majority (72 %) of patients experienced erythema at the injection site. Similarly, another phase I monotherapy trial demonstrated up to 61 % reductions in LDL cholesterol and apoB levels with 300 mg/week doses of mipomersen in subjects with mild-to-moderate hypercholesterolaemia, with injection site reactions experienced at least once in each subject [38]; 18 % of subjects showed consecutive transaminase elevations greater than three times the upper limit of normal. The majority who had increased hepatic transaminase were receiving the 400 mg/week regimen.
Mipomersen has now been evaluated by several phase II and III trials assessing its efficacy, safety, tolerability and utility in patients with severe hypercholesterolaemia as monotherapy (in statin-intolerant subjects) and when combined with statin therapy. These trials continue to demonstrate significant reductions in LDL cholesterol and apoB levels [18, 19, 39–44]. A summary of the trials reported to date is shown in Table 7.1.
Mipomersen for Familial Hypercholesterolaemia
Five mipomersen trials have focused on FH. Akdim et al. (2010) investigated the efficacy of mipomersen (dose range 50–300 mg/week) over a period of 6 weeks in 44 patients with heterozygous FH. Significant reductions in LDL cholesterol were found with the 200 and 300 mg dosing regimens, with maximal reductions 21 and 33 % from baseline, respectively. Extended treatment to 13 weeks with weekly doses of 300 mg mipomersen resulted in 37 % reduction in both LDL cholesterol and apoB [40]. Similarly, Visser et al. (2010) demonstrated, in 21 heterozygous FH patients with a 13-weekly mipomesen regime at a dose of 200 mg/week, a reduction of 22 and 20 % for LDL cholesterol and apoB, respectively [41]. In a randomised trial of 124 heterozygous FH with coronary artery disease, Stein et al. (2012) showed a 28 and 26 % reduction in LDL cholesterol and apoB concentrations, respectively, after 26 weeks of weekly 200 mg mipomersen injections [19]. A trend towards an increase in intrahepatic triglyceride content was found in both studies [19, 41].
In a phase III study involving 51 homozygous FH all on maximally tolerated conventional therapy, mipomersen (200 mg/week) decreased LDL cholesterol by 25 %, and apoB was similarly reduced by 27 % after 26 weeks [18]. In October 2012, the US Food and Drug Administration (FDA) approved mipomersen, adjunct to lipid-lowering therapy and diet, for the treatment of homozygous FH under a Risk Evaluation and Mitigation Strategies (REMS) program. However, Mipomersen was not approved for use in Europe by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) (see detailed reasons under the subsection on adverse reactions).
Finally, the most recent mipomersen report in FH is a 2 year interim analysis of open-label extension trial. The efficacy and safety was similar to previous randomised placebo-controlled trials. 200 mg weekly injections of mipomersen for up to 104 weeks demonstrated 28 % LDL cholesterol and 31 % apoB reductions. In a subgroup of patients who had undergone liver magnetic resonance imaging, there was also an incremental increase in liver fat in the first 6–12 months. However, regression towards baseline with continued mipomersen beyond 1 year denotes metabolic adaptation [45].
Mipomersen for Severe Hypercholesterolaemia
Four trials have studied patients with primary hypercholesterolaemia that was not specifically ascribed to FH; two were carried out on a background of statins and the others as monotherapy. The first of these trials reported that mipomersen (5 weeks, 7 doses of 100–400 mg/week) in hypercholesterolaemic subjects on stable statin therapy was associated with a 21–52 and 19–54 % reduction (across the dose ranges) in plasma LDL cholesterol and apoB concentrations, respectively [39]. In the same study, a subgroup of patients was assigned to 15 doses of 200 mg/week mipomersen over 13 weeks. A 36 % reduction in both LDL cholesterol and apoB levels was shown [39]. In another study, Visser et al. (2012) found that weekly 200 mg administration of mipomersen to high-risk statin-intolerant patients reduced plasma LDL cholesterol and apoB by 47 and 46 %, respectively, after 26 weeks [42]. Liver fat content was significantly increased, with hepatic steatosis confirmed in two subjects who had undergone liver biopsy [42].
McGowan et al. (2012) demonstrated that in severe hypercholesterolaemic patients on maximally tolerated lipid-lowering therapy, 200 mg/week of mipomersen (over 26 weeks) reduced LDL cholesterol and apoB by 36 % [43]. The most recent study by Thomas et al. (2013) randomised 157 high-risk patients with severe hypercholesterolaemia (LDL cholesterol ≥2.6 mmol/l on a maximally tolerated statin dose) to mipomersen and placebo; randomisation was stratified so that a minimum of 40 % of patients in each group would have type 2 diabetes. After 26 weeks of 200 mg weekly mipomersen, LDL cholesterol and apoB levels were lowered by 36 and 37 %, respectively [44]. Elevations in transaminases and liver fat occurred in some patients, but like other studies, these levels returned towards baseline after cessation of treatment.
Mipomersen and Lipoprotein(a)
A recent study in subjects with varying baseline levels of plasma Lp(a) (34.0–56.3 mg/day) from four phase III trials examined the effect of mipomersen on Lp(a). Mipomersen was shown to consistently and significantly lower Lp(a) levels by a median of 26.4 % across patient groups, despite varying baseline Lp(a) levels [46]. The mechanism of Lp(a) lowering by mipomersen remains to be demonstrated but is likely to involve the reduced production of Lp(a) [47]. The cardiovascular benefit of treating elevated Lp(a) is unknown. Clinical trial evidence is needed to determine whether Lp(a) lowering affects cardiovascular outcomes, although this will require a specific Lp(a)-lowering therapy, such as Lp(a) apheresis or apo(a) antisense therapy [47]. Other new agents such as PCSK9 inhibitors, lomitapide and anacetrapib (a CETP inhibitor) also have Lp(a)-lowering effects, with reductions of 31 % (in heterozygous FH) [48], 19 % (in homozygous FH) [49] and 32 % (in heterozygous FH) [50], respectively. The mechanisms of action of these agents on Lp(a) metabolism is also unclear.
Mipomersen: Adverse Reactions, Contraindications, Economics
Mipomesen is not metabolised by enzymes such as CYP450, and pharmacokinetic studies reveal no clinically relevant interactions with the clearance of statins and ezetimibe [51]. However, injection-site reactions occur in the majority of cases, and every patient experiences at least one injection-site reaction [52]. Other side effects associated with mipomersen include mild-to-moderate influenza-like symptoms and hepatic transaminase elevation (alanine transaminase and aspartate transaminase). Table 7.2 summarises these events from four phase III trials.
The main safety concern with mipomersen is increased hepatic steatosis [19, 41, 43]. The negative recommendation by the EMA was based on this. The EMA also noted that a high proportion of patients stopped taking mipomersen within 2 years, owing to side effects, and this applied even in the patients with homozygous FH. This was considered important as mipomersen was intended for long-term treatment of severe hypercholesterolaemia. The long-term consequences of liver toxicity and possible irreversible liver damage still need to be addressed. Additionally, a higher rate of cardiovascular events was observed in those on mipomersen compared with placebo. Hence, in the opinion of the EMA, the potential cardiovascular benefit did not appear to outweigh its cardiovascular risk (Table 7.3).
The current contraindications for use of mipomersen include severe hepatic impairment or active liver disease. In terms of the use by women of reproductive potential, mipomersen is a category B agent, meaning that animal reproduction studies have failed to demonstrate a risk to the fetus. However, there are no adequate studies in pregnant or lactating women. There is as yet no approved indication of use of mipomersen in paediatric patients, although there have been no differences in adverse events in paediatric compared with adult groups [18, 52].
There are other therapeutic approaches to controlling severe refractory hypercholesterolaemia including lomitapide, apheresis, PCSK9 inhibitors and CETP inhibitors. Unlike mipomersen, lomitapide has received orphan drug designation by both the FDA and EMA. Lomitapide taken orally is an inhibitor of CYP3A4, and hence able to interact with a number of drugs (including some statins); it also interacts with drugs that are metabolised by p-glycoprotein (including colchicine, dabigatran, digoxin, sitagliptin, macrolide antibiotic, antifungals and protease inhibitors). By contrast, mipomersen does not exhibit such interactions. Lomitapide is also associated with significant gastrointestinal adverse effects and increases in hepatic fat levels [53]. Mipomersen is expected to cost $176,000/year. In comparison, lomitapide is expected to be more expensive, at an estimated $250,000/year or more and additionally has a pregnancy category X (i.e. positive evidence of human fetal risk based on adverse reaction data, and the risks involved in use of the drug in pregnant women clearly outweigh potential benefits) from the FDA. Furthermore, weekly apheresis costs approximately $208,00/year (excluding costs of travel to apheresis sites) [52]. Based on current data, it is estimated that almost half of the LDL apheresis patients could avoid apheresis with the addition of mipomersen [54].
Ultimately, the long-term outcomes of mipomersen treatment are unclear. Long-term data are required to justify the cardiovascular benefit and hepatic safety profile of mipomersen.
Conclusion
Statins and mipomersen have different pharmacodynamic effects on lipid metabolism which makes the combination rational for the treatment of refractory hypercholesterolaemia. The complementary mechanisms of action, whereby statins increase LDL catabolism and mipomersen inhibits apoB synthesis, provide a good basis for combination treatment. The efficient dose-dependent reduction in plasma LDL cholesterol concentrations achieved by mipomersen therapy is highly significant. However, the risk of hepatic steatosis and injection-site reactions continues to remain a concern that bears on the clinical use of this agent. Studies of longer duration with greater numbers of participants are needed to investigate the significance of the sequelae of hepatic transaminase elevation and hepatic triglyceride accumulation. It is important to investigate whether accumulation of liver fat over time progresses to hepatic inflammation, cirrhosis and liver failure. This is important if mipomersen is extended to more common lipid disorders, such as mixed hyperlipidaemias in the setting of diabetes or insulin resistance that are per se associated with steatohepatitis. Despite the favourable effects of mipomersen on Lp(a), the cardiovascular benefit of treating elevated Lp(a) remains untested. New formulations of mipomersen that do not cause injection-site reactions are essential to increase the acceptability of this form of therapy by patients. The cost of mipomersen also needs to be lowered substantially.
Further studies of combination therapy with ezetimibe bile acid sequestrants, fibrates and including apheresis are required. Balancing the appropriateness of mipomersen therapy in respect of efficacy, acceptability and cost-effectiveness is fundamental and remains to be fully established.
References
Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670–81.
Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209(1):189–94.
Watts GF, Gidding S, Wierzbicki AS, et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol. 2014;171(3):309–25.
Postmarket Drug Safety Information for Patients and Providers – KYNAMRO® (mipomersen sodium). http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM337472.pdf.
Watts G, Chan D, Barrett H. Updating the metabolism of apolipoprotein B-100 containing lipoproteins in dyslipidaemia. In: Toth PP, editor. The year in lipid disorders, vol. 2. Oxford: Clinical Publishing; 2010. p. 118–39.
Benn M. Apolipoprotein B levels, APOB alleles, and risk of ischemic cardiovascular disease in the general population, a review. Atherosclerosis. 2009;206(1):17–30.
Chaves FJ, Real JT, Garcia-Garcia AB, et al. Genetic diagnosis of familial hypercholesterolemia in a South European outbreed population: influence of low-density lipoprotein (LDL) receptor gene mutations on treatment response to simvastatin in total, LDL, and high-density lipoprotein cholesterol. J Clin Endocrinol Metab. 2001;86(10):4926–32.
Wierzbicki AS, Lumb PJ, Cheung J, Crook MA. Fenofibrate plus simvastatin therapy versus simvastatin plus cholestyramine therapy for familial hypercholesterolaemia. QJM. 1997;90(10):631–4.
Kastelein JJP, Akdim F, Stroes ESG, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358(14):1431.
Huijgen R, Abbink EJ, Bruckert E, et al. Colesevelam added to combination therapy with a statin and ezetimibe in patients with familial hypercholesterolemia: a 12-week, multicenter, randomized, double-blind, controlled trial. Clin Ther. 2010;32(4):615–25.
Kawashiri MA, Nohara A, Noguchi T, et al. Efficacy and safety of coadministration of rosuvastatin, ezetimibe, and colestimide in heterozygous familial hypercholesterolemia. Am J Cardiol. 2012;109(3):364–9.
Alonso R, Andres E, Mata N, et al. Lipoprotein (a) levels in Familial Hipercholesterolaemia: an important predictor for cardiovascular disease independent of the type of LDL-receptor mutation. J Am Coll Cardiol. 2014;63(19):1982–9.
Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–53.
Tsimikas S, Witztum JL. The role of oxidized phospholipids in mediating lipoprotein (a) atherogenicity. Curr Opin Lipidol. 2008;19(4):369–77.
Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein (a) gene accounts for greater than 90% of the variation in plasma lipoprotein (a) concentrations. J Clin Invest. 1992;90(1):52–60.
Lanktree MB, Anand SS, Yusuf S, Hegele RA, Investigators S. Comprehensive analysis of genomic variation in the LPA locus and its relationship to plasma lipoprotein(a) in South Asians, Chinese, and European Caucasians. Circ Cardiovasc Genet. 2010;3(1):39–46.
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34(17):1279–91.
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375(9719):998–1006.
Stein EA, Dufour R, Gagne C, et al. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation. 2012;126(19):2283–92.
American Diabetes Association. Standards of medical care in diabetes – 2012. Diabetes Care. 2012;35 Suppl 1:S11–63.
Rydén L, Grant PJ, Anker SD, et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD The Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur Heart J. 2013;34(39):3035–87.
Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining Low-density lipoprotein cholesterol < 50 mg/dl with Rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol. 2011;57(16):1666–75.
Fernandez G, Spatz ES, Jablecki C, Phillips PS. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med. 2011;78(6):393–403.
Mancini GB, Tashakkor AY, Baker S, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol. 2013;29(12):1553–68.
Culver AL, Ockene IS, Balasubramanian R, et al. Statin use and risk of diabetes mellitus in postmenopausal women in the Women’s Health Initiative. Arch Intern Med. 2012;172(2):144–52.
Wang KL, Liu CJ, Chao TF, et al. Statins, risk of diabetes, and implications on outcomes in the general population. J Am Coll Cardiol. 2012;60(14):1231–8.
Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy—European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J. 2015;36:1012–22. ehv043.
Keen HI, Krishnarajah J, Bates TR, Watts GF. Statin myopathy: the fly in the ointment for the prevention of cardiovascular disease in the 21st century? Expert Opin Drug Saf. 2014;13(9):1227–39.
Brown MS, Goldstein JL. A receptor mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47.
Van Wissen S, Smilde TJ, Trip MD, de Boo T, Kastelein JJP, Stalenhoef AFH. Long term statin treatment reduces lipoprotein (a) concentrations in heterozygous familial hypercholesterolaemia. Heart. 2003;89(8):893–6.
Lambert G, Chatelais M, Petrides F, et al. Normalization of low-density lipoprotein receptor expression in receptor defective homozygous familial hypercholesterolemia by inhibition of PCSK9 with alirocumab. J Am Coll Cardiol. 2014;64(21):2299–300.
Gebhard C, Huard G, Kritikou EA, Tardif J-C. Apolipoprotein B antisense inhibition-update on mipomersen. Curr Pharm Des. 2013;19(17):3132–42.
Crooke ST, Geary RS. Clinical pharmacological properties of mipomersen (Kynamro), a second generation antisense inhibitor of apolipoprotein B. Br J Clin Pharmacol. 2013;76(2):269–76.
Yu RZ, Kim T-W, Hong A, Watanabe TA, Gaus HJ, Geary RS. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab Dispos. 2007;35(3):460–8.
Crooke RM, Graham MJ, Lemonidis KM, Whipple CP, Koo S, Perera RJ. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J Lipid Res. 2005;46(5):872–84.
Mullick AE, Fu W, Graham MJ, et al. Antisense oligonucleotide reduction of apoB ameliorated atherosclerosis in LDL receptor-deficient mice. J Lipid Res. 2011;52:885–96. doi:10.1194/jlr.M011791.
Kastelein JJP, Wedel MK, Baker BF, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114(16):1729–35.
Akdim F, Tribble DL, Flaim JD, et al. Efficacy of apolipoprotein B synthesis inhibition in subjects with mild-to-moderate hyperlipidaemia. Eur Heart J. 2011;32(21):2650–9.
Akdim F, Stroes ESG, Sijbrands EJG, et al. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J Am Coll Cardiol. 2010;55(15):1611–8.
Akdim F, Visser ME, Tribble DL, et al. Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia. Am J Cardiol. 2010;105(10):1413–9.
Visser ME, Akdim F, Tribble DL, et al. Effect of apolipoprotein-B synthesis inhibition on liver triglyceride content in patients with familial hypercholesterolemia. J Lipid Res. 2010;51(5):1057–62.
Visser ME, Wagener G, Baker BF, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2012;33(9):1142–9.
McGowan MP, Tardif J-C, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One. 2012;7(11):e49006.
Thomas GS, Cromwell WC, Ali S, Chin W, Flaim JD, Davidson M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2013;62(23):2178–84.
Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur Heart J. 2013;39:566–75.
Santos RD, Raal FJ, Catapano AL, Witztum JL, Steinhagen-Thiessen E, Tsimikas S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein (a) in various populations with hypercholesterolemia results of 4 phase III trials. Arterioscler Thromb Vasc Biol. 2015;35(3):689–99.
Bos S, Yayha R, van Lennep JER. Latest developments in the treatment of lipoprotein (a). Curr Opin Lipidol. 2014;25(6):452–60.
Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385(9965):331–40.
Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40–6.
Kastelein JJP, Besseling J, Shah S, et al. Anacetrapib as lipid-modifying therapy in patients with heterozygous familial hypercholesterolaemia (REALIZE): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet. 2015. http://dx.doi.org/10.1016/S0140-6736(1014)62115-62112.
Yu RZ, Geary RS, Flaim JD, et al. Lack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a 2’-—O-methoxyethyl modified antisense oligonucleotide targeting apolipoprotein B-100 messenger RNA, with simvastatin and ezetimibe. Clin Pharmacokinet. 2009;48(1):39–50.
Bennett LL, Chalk M. Review of Mipomersen Sodium (Kynamro®) for Familial Hypercho-lesterolemia. J Clin Med Res Updat. 2014;1:1–10.
Davis KA, Miyares MA. Lomitapide: a novel agent for the treatment of homozygous familial hypercholesterolemia. Am J Health Syst Pharm. 2014;71(12):1001–8.
Vogt A, Parhofer KG. The potential of mipomersen, an ApoB synthesis inhibitor, to reduce necessity for LDL-apheresis in patients with heterozygous familial hypercholesterolemia and coronary artery disease. Expert Opin Pharmacother. 2013;14(6):691–7.
Hovingh K, Besseling J, Kastelein J. Efficacy and safety of mipomersen sodium (Kynamro). Expert Opin Drug Saf. 2013;12(4):569–79.
Ross JL. Homozygous familial hypercholesterolemia – role of NPs and PAs in achieving optimal outcomes using novel therapeutic interventions. Supplement to Clinician Reviews. 2015:1–9.
KYNAMRO (mipomersen sodium) Product Information. http://www.kynamro.com/families/product-information.aspx.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Pang, J., Chan, D.C., Watts, G.F. (2015). Statins and Mipomersen: Mechanisms of Action and Patient Tolerability. In: Banach, M. (eds) Combination Therapy In Dyslipidemia. Adis, Cham. https://doi.org/10.1007/978-3-319-20433-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-20433-8_7
Publisher Name: Adis, Cham
Print ISBN: 978-3-319-20432-1
Online ISBN: 978-3-319-20433-8
eBook Packages: MedicineMedicine (R0)