
Abstract
Glycosylation is the addition of sugars (glycans) to proteins and lipids and is the most frequent modification of both secreted and membrane-bound proteins. Defective synthesis, assembly or processing of glycans results in a group of disorders known as congenital disorders of glycosylation (CDG). Biochemical testing assesses the level of glycosylation on glycoproteins, such as transferrin, as well as the structure of N-linked or O-linked glycans released from glycoproteins. Molecular CDG single gene and gene panel testing is clinically available for more than 30 CDG-associated genes. An exome sequencing approach is being used to identify the gene defect in individuals with unknown types of CDG (CDG-Ix and CDG-IIx), and can lead to the identification of new CDG genes. Identification of the molecular mechanisms for new subtypes of CDG will provide important building blocks for the development of new treatments and therapies for individuals affected with CDG.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
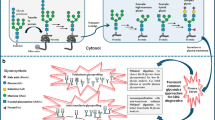
Glycosylation is the addition of sugars (glycans) to proteins and lipids and is the most frequent modification of both secreted and membrane-bound proteins [1]. Two types of protein glycosylation occur, N-linked and O-linked, which differ in the linkage of the oligosaccharide to protein.
Approximately 50 % of all proteins in the human genome are N-glycosylated [2]. In N-glycosylation, an oligosaccharide is assembled, beginning in the cytosol and continued in the endoplasmic reticulum (ER). The sugar chain precursor is assembled on a lipid dolichol pyrophosphate carrier in the ER membrane and consists of two N-acetylglucosamine residues, nine mannose residues, and three glucose residues. Once the assembly process is complete, the oligosaccharyltransferase complex transfers the oligosaccharide to specific asparagine residues on nascent polypeptide chains in the lumen of the ER. After linkage, modification of the glycan begins with the trimming of sugar chains in the ER, and further modification of these sugar chains takes place in the Golgi apparatus. This pathway is well described, and many enzymes are involved in both the assembly and the post-linkage processing of the glycan chain [3]. N-glycosylated proteins are important for a variety of biological processes, including intracellular targeting, cell-cell recognition, protein folding and stability, and immune response [1].
In contrast, O-linked-glycosylation occurs in the Golgi apparatus with sugars added sequentially to serine or threonine amino acids of proteins. O-glycosylated proteins provide a barrier against pathogens, serve as lubricants, provide cushioning and physical integrity to the extracellular matrix, and function as co-receptors for a number of growth factors [1]. Glycosylation of lipids begins within the cytosol and continues in the ER and Golgi apparatus. Glycosylated lipids are involved in signaling, membrane diffusion, and sorting [4]. Hence, the proper development and functioning of multiple organ systems are dependent upon normal glycosylation of proteins and lipids.
Molecular Basis of Disease
An impaired N-glycosylation biosynthesis pathway, due to mutations in genes that encode proteins that function within this pathway or are involved in intracellular protein or nucleotide sugar trafficking between the endoplasmic reticulum and Golgi apparatus, affects multiple organ systems including the brain, heart, bone, endocrine system, immune system, liver, gastrointestinal tract, and vision [1]. Defects within the O-glycosylation pathway mainly affect muscle, bone, cartilage, and the extracellular matrix [1]. Defects in lipid glycosylation primarily affect the nervous system [5, 6].
Defective synthesis, assembly or processing of glycans results in a group of disorders known as congenital disorders of glycosylation (CDG) [1]. The majority of individuals with CDG have symptoms that began in infancy. These symptoms can include severe developmental delay, ataxia, seizures, liver fibrosis, cardiac dysfunction, retinopathy, skeletal abnormalities, and coagulopathies. The liver and intestine are most affected in CDG because these organs consume the most mannose for glycoprotein synthesis. Approximately 20 % of children do not survive beyond 5 years of age due to widespread organ dysfunction and severe infections. Worldwide occurrence of CDG has an estimated prevalence as high as 1 in 20,000. The great variability of symptoms and severity of disease across individuals with CDG makes the diagnosis of these disorders challenging to pediatric health care providers. The majority of CDG types are due to autosomal recessive inheritance. The exceptions include an X-linked N-glycosylation defect and two autosomal dominant O-glycosylation defects.
CDG patients are classified as having either Type I, Type II, or combined Type I and Type II defects, which is defined by serum transferrin analysis. This designation is based on whether the N-glycosylation defect results in either hypo- or mis-glycosylation [7]. Type I CDGs are caused by defects in genes that create the sugar chain precursors or in genes that attach these precursors to proteins and lipids. These defects result in a glycan structure that is partially or totally missing. Type II CDGs are caused by defects in genes that modify the sugar chains after they are attached to proteins and lipids. These defects result in a structurally altered glycan. Combined Type I and Type II defects have recently been reported, making an accurate diagnosis in these patients even more challenging. Most CDG subtypes have been described in only a few individuals; therefore, an understanding of phenotypes for these CDG subtypes is limited.
Currently, efficient treatment is only available for one subtype of CDG, MPI-CDG (CDG-Ib), caused by mutations in the phosphomannose-isomerase (MPI) gene [8]. The defect in MPI-CDG is the inability to convert fructose-6 phosphate to mannose-6-phosphate. The enzyme hexokinase can form mannose-6-phosphate from the administration of oral mannose, thereby bypassing the defect. Two other CDGs that can respond to treatment include SLC35C1-CDG (CDG-IIc) in which fucose is an effective treatment for recurrent infections with hyperleukocystosis, and PIGM-CDG in which butyrate upregulates PIGM transcription, thereby controlling seizures [9, 10]. Only supportive therapy and symptom-based treatment is available for all other CDG subtypes.
Clinical Utility of Testing
Due to the multisystem involvement of CDG, subtype diagnosis based on the clinical phenotype often is not possible. A combination of biochemical and molecular testing is used to identify the CDG subtype. Confirmation of the specific gene defect is important because some life-threatening phenotypes (i.e., cardiac dysfunction) have presented in only certain subtypes of CDG and may develop at any time. Confirmation of the defect allows for close monitoring of different organs for dysfunction and complications that may arise due to coagulation abnormalities. A growing number of individuals have a biochemical diagnosis of CDG without identification of a disease-causing mutation in a known CDG gene. Although molecular testing may miss some mutations in deep intronic or promoter regions of CDG-associated genes, many individuals with negative testing likely have mutations in genes not yet associated with CDG.
Genotype-phenotype correlations are not well described for many subtypes of CDG because only a few individuals have had mutations detected in the majority of CDG-associated genes [11]. Even in the most common CDG subtype, PMM2-CDG (CDG-Ia), individuals with the same mutation have variable severity of the clinical symptoms [12]. Identification of both disease-causing mutations in the affected individual allows carrier and prenatal testing of other family members.
Available Assays
Biochemical Assays
The standard screening for individuals with symptoms of CDG is biochemical testing to assess the level of glycosylation on glycoproteins, such as transferrin, and the structure of N-linked or O-linked glycans released from glycoproteins [13]. Transferrin is an iron-binding protein that is synthesized and metabolized mainly in the liver and is the most sensitive serum glycoprotein marker for CDG. Glycosylation of transferrin can be analyzed using isoelectric focusing (IEF), electrospray ionization mass spectrometry or matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry. Transferrin contains two biantennary N-glycan chains with a total of four terminal sialic acid residues. A normal IEF profile of transferrin isomers consists predominantly of four sialic acids (tetrasialotransferrin). For type I CDG, an abnormal profile is observed with a decreased amount of tetrasialotransferrin and increased amounts of asialo- and disialotransferrin due to some transferrin molecules lacking either one or both N-glycan chains. For type II CDG, an abnormal profile consists of an increase of tri-, di-, mono-, or sometimes asialotransferrin due to a portion of the glycan chains being incomplete. Serum transferrin analysis is relatively of low cost and is available in many clinical laboratories. Transferrin testing is a rapid screen for CDG, but does not identify the specific gene defect.
Serum transferrin screening for CDG has limitations. Several CDG subtypes including MOGS-CDG (CDG-IIb), SLC35C1-CDG (CDG-IIc), and SLC35A1-CDG (CDG-IIf) present with a normal transferrin pattern [1]. Some patients with PMM2-CDG (CDG-Ia) may have a normal transferrin pattern later in life, and analysis of PMM2 enzyme activity may be needed to confirm the diagnosis [14]. These examples demonstrate that a normal transferrin pattern does not necessarily exclude an individual from having CDG, with approximately 25 % of CDG cases estimated to have a normal transferrin pattern [15]. False-positive results can be observed in individuals presenting with liver disease, other metabolic disorders including galactosemia and fructosemia, or alcoholism [16–18]. Transferrin analysis also is not accurate in children less than 6 months of age and may give false-negative results [7]. Transferrin analysis also is reported to have false-positive results in infants less than 3 weeks of age [19].
MALDI-TOF-MS can be used for analysis of N- and O-linked glycan structures. This technique protects glycans from fragmentation and allows for the structural detail of N- and O-linked glycans to be analyzed. This technique has been used to characterize a number of subtypes of CDG that could not be detected by IEF of transferrin and is particularly useful for characterizing Type II defects, combined Type I and Type II defects, and multiple glycosylation defects [20]. Caution needs to be exercised with interpretation of O-glycan MALDI-TOF-MS profiles from patients with cancer or diabetes because these conditions can alter O-glycans at the cellular level [21]. This analysis is available only in a few clinical laboratories.
Enzyme activities of phosphomannomutase (PMM) and phosphomannose isomerase are assessed in patient fibroblasts or leukocytes if the patient is suspected to have a Type I CDG [13]. This will diagnose or rule out two common CDGs, PMM2-CDG (CDG-Ia) (700 individuals worldwide), and MPI-CDG (CDG-Ib) (20 individuals worldwide). Analysis using leukocytes is preferred, especially for PMM enzyme activity, because rapidly dividing fibroblasts can give high PMM residual activity levels [22]. Enzyme activity assays are clinically available only for these two CDG subtypes, and performed by several laboratories worldwide.
Molecular Assays
Since serum transferrin analysis can only determine whether the patient has Type I or Type II CDG, follow up with molecular testing is needed to determine the specific gene defect and to direct testing of additional family members, as needed. If individuals have reduced PMM or MPI enzyme activity, PCR amplification and sequencing of the respective genes can be performed to determine the disease-causing mutations. Single gene testing is currently available for more than 30 genes associated with CDG (Table 8.1). Only a few laboratories provide molecular testing for the more rare CDG subtypes. A comprehensive clinical CDG next-generation sequencing panel is available for comprehensive mutation detection in 38 CDG-associated genes when it is unclear what defect an individual with CDG may have based on phenotype [23]. Array-based comparative genomic hybridization (aCGH) also can be used to detect exon and gene deletions and duplications in these CDG-associated genes.
For the most common CDG, PMM2-CDG (CDG-Ia), over 90 mutations have been identified [8]. The most common mutation is p.Arg141His that leads to almost complete inactivation of the enzyme [12]. This amino acid substitution is seen in about 40 % of compound heterozygous individuals. Another mutation, p.Phe119Leu, is frequently found in affected Northern Europeans with the compound heterozygote genotype (p.Arg141His and p.Phe119Leu), and represents 72 % of mutations in PMM2. Due to the limited number of individuals with defects in the other CDG genes, no common mutations have been identified across individuals.
Interpretation of Results
The majority of mutations identified in CDG patients are missense and nonsense mutations, and small insertions and deletions. Since only a few patients have been identified with defects in each of the CDG genes, the list of mutations in Human Gene Mutation Database is very limited for many CDGs; therefore, a conservative approach should be taken for novel variants identified in CDG genes that have not been previously reported. Interpretation of duplications identified by array CGH is difficult because the duplication may or may not disrupt the function of the encoded protein. The presence of one known disease-causing mutation in a specific CDG gene can assist with the interpretation of aCGH results. Parental studies can be useful in determining whether two variants identified in a single gene are on the same or opposite alleles.
Laboratory Issues
Serum transferrin analysis is prone to both false-positive and false-negative results; therefore, if a patient has a strong clinical indication of CDG, follow-up with additional biochemical or molecular testing is recommended. If biochemical testing indicates an individual has CDG, molecular testing is used to confirm the diagnosis and to identify the disease-causing mutations. Testing for the majority of CDGs is provided by only a few laboratories worldwide. A list of laboratories that test for each CDG subtype is provided on the GeneTests website (http://www.ncbi.nlm.nih.gov/sites/GeneTests/).
Conclusions and Future Directions
Due to the wide spectrum of symptoms and variable severity of CDG, pediatric physicians should be educated to consider glycosylation disorders in patients presenting with multi-organ dysfunction and symptoms that include developmental delay, failure to thrive, liver dysfunction, or neurological involvement. As testing for CDG continues, the number of patients that lack a molecular diagnosis will likely increase, highlighting the need for broader molecular testing to identify the causative gene in these patients. Greater than 40 % of CDG patients are estimated to have an unknown type of CDG (CDG-Ix or CDG-IIx) and lack a molecular diagnosis, with the majority of unsolved cases being CDG-IIx. Approximately, 250–500 genes are estimated to be involved in the process of glycosylation, with the likelihood that defects in a number of these genes will result in CDG. CDG is an ideal candidate syndrome for exome sequencing, and this approach already has successfully identified the gene defect in a previous CDG-Ix patient [24]. New CDG genes identified from exome or genome sequencing can be added to the clinical CDG next-generation sequencing panel to provide a more comprehensive test.
Improved molecular diagnosis of CDG will reduce the number of patients lacking genetic characterization, shorten a patient’s time to diagnosis, facilitate genetic counseling, improve patient management, and facilitate carrier or prenatal testing for other family members. Molecular diagnosis of additional patients with CDG will provide an estimate of the prevalence of each subtype and a greater understanding of the spectrum of phenotypes associated with each subtype. The clinical outcome and natural course for each CDG subtype will also be elucidated. As more patients are identified, the study of genotype/phenotype correlations can be assessed. Identification of new genes also will provide insight into new pathways that are linked to glycosylation. Furthermore, identification of new genes associated with CDG will provide important building blocks for the development of new treatments and therapies for individuals afflicted with different subtypes of CDG.
References
Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7(7):537–51.
Schachter H, Freeze HH. Glycosylation diseases: quo vadis? Biochim Biophys Acta. 2009;1792(9):925–30.
Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr. 2003;162(6):359–79.
Hancock JF. GPI-anchor synthesis: Ras takes charge. Dev Cell. 2004;6(6):743–5.
Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42(10):827–9.
Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36(11):1225–9.
Lefeber DJ, Morava E, Jaeken J. How to find and diagnose a CDG due to defective N-glycosylation. J Inherit Metab Dis. 2011;34(4):849–52. doi:10.1007/s10545-011-9370-0.
Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–8.
Gazit Y, Mory A, Etzioni A, Frydman M, Scheuerman O, Gershoni-Baruch R, et al. Leukocyte adhesion deficiency type II: long-term follow-up and review of the literature. J Clin Immunol. 2010;30(2):308–13.
Almeida A, Layton M, Karadimitris A. Inherited glycosylphosphatidyl inositol deficiency: a treatable CDG. Biochim Biophys Acta. 2009;1792(9):874–80.
Vodopiutz J, Bodamer OA. Congenital disorders of glycosylation-a challenging group of IEMs. J Inherit Metab Dis. 2008;31(2):267–9.
Grunewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim Biophys Acta. 2009;1792(9):827–34.
Marklova E, Albahri Z. Screening and diagnosis of congenital disorders of glycosylation. Clin Chim Acta. 2007;385(1-2):6–20.
Vermeer S, Kremer HP, Leijten QH, Scheffer H, Matthijs G, Wevers RA, et al. Cerebellar ataxia and congenital disorder of glycosylation Ia (CDG-Ia) with normal routine CDG screening. J Neurol. 2007;254(10):1356–8.
Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J Inherit Metab Dis. 2011;34(4):853–8.
Adamowicz M, Pronicka E. Carbohydrate deficient glycoprotein syndrome–like transferrin isoelectric focusing pattern in untreated fructosaemia. Eur J Pediatr. 1996;155(4):347–8.
Charlwood J, Clayton P, Keir G, Mian N, Winchester B. Defective galactosylation of serum transferrin in galactosemia. Glycobiology. 1998;8(4):351–7.
Stibler H, Borg S, Joustra M. Micro anion exchange chromatography of carbohydrate-deficient transferrin in serum in relation to alcohol consumption (Swedish Patent 8400587-5). Alcohol Clin Exp Res. 1986;10(5):535–44.
Clayton P, Winchester B, Di Tomaso E, Young E, Keir G, Rodeck C. Carbohydrate-deficient glycoprotein syndrome: normal glycosylation in the fetus. Lancet. 1993;341.
Faid V, Chirat F, Seta N, Foulquier F, Morelle W. A rapid mass spectrometric strategy for the characterization of N- and O-glycan chains in the diagnosis of defects in glycan biosynthesis. Proteomics. 2007;7(11):1800–13.
Ungar D. Golgi linked protein glycosylation and associated diseases. Semin Cell Dev Biol. 2009;20(7):762–9.
Grunewald S, Schollen E, Van Schaftingen E, Jaeken J, Matthijs G. High residual activity of PMM2 in patients’ fibroblasts: possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am J Hum Genet. 2001;68(2):347–54.
Jones MA, Bhide S, Chin E, Ng BG, Rhodenizer D, Zhang VW, Sun JJ, Tanner A, Freeze HH, Hegde MR. Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation. Genet Med. 2011;13(11):921–32.
Jones MA, Ng BG, Bhide S, Chin E, Rhodenizer D, He P, Losfeld ME, He M, Raymond K, Berry G, Freeze HH, Hegde MR. DDOST mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am J Hum Genet. 2012;90(2):363–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Jones, M.A., Hegde, M.R. (2016). Congenital Disorders of Glycosylation. In: Leonard, D. (eds) Molecular Pathology in Clinical Practice. Springer, Cham. https://doi.org/10.1007/978-3-319-19674-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-19674-9_8
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19673-2
Online ISBN: 978-3-319-19674-9
eBook Packages: MedicineMedicine (R0)