
Abstract
Often in the history of medicine, as the field progresses from descriptive to etiologic diagnosis, great strides can be made in the treatment of disease. The current chapter will discuss the 22q11.2 Deletion Syndrome (22q11DS), and argue that knowledge of the syndrome that has emerged in the past several decades, together with rapid progress in applications of genomic analysis to diagnosis of complex neurobehavioral syndromes, is creating the basis for a new understanding of many developmental disabilities. To provide context, it is useful to begin with a historical example, in part abstracted from Pearce’s fascinating account, which illustrates how progressing from description to causation led ultimately to what was arguably one of the great revolutions in twentieth-century psychopharmacology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Autism Spectrum Disorder
- Autism Spectrum Disorder
- Intellectual Disability
- Neurodevelopmental Disorder
- Conotruncal Heart Defect
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
In 1822, Antoine Laurent Jessé Bayle (1799–1858) published his medical thesis “Recherches Sur l’Arachnitis Chronique” [1, 2]. In that work he reported the results of autopsies of patients who had died of a progressive dementing psychosis associated with motor paralysis, an invariably fatal condition that became known in nineteenth-century medicine as general paralysis of the insane (GPI). Bayle concluded that GPI resulted from inflammation of the meninges. This revolutionary concept, that insanity resulted from a physical problem in cerebral tissue, remained controversial for decades. Indeed, at the time, many “alienist” (psychiatric) authorities attributed GPI to weak character- an assertion remarkably reminiscent of the more recent past, when refrigerator mothers were said by some psychiatric experts to cause their children’s autism.
In 1857 Esmarch and Jessen [3] suggested that GPI resulted from syphilis. Although many observers, including Bayle, had noted that GPI patients frequently had histories of syphilis, the causal link between GPI and infection with treponema pallidum was not definitely established until 1913 when Naguchi and Moore [4] cultured the spirochete from the autopsied brains of patients. Fleming’s observation that mold that had contaminated a culture plate destroyed nearby Staphylococcus colonies [5], followed by Florey and Chain’s successful large-scale production of the active principle of the “mold juice,” penicillin [6], led to a highly effective treatment for syphilitic infections. GPI, which once accounted for some 15 % of male psychiatric inpatients and was the leading cause of death in chronic psychiatric hospitals, became so rare that most modern psychiatrists (including the present author) have never treated a single case.
The Emerging Genetics of Neurodevelopmental Disorders
The history of GPI seems relevant to the story of 22q11DS because the latter syndrome is the most common of a newly emerging class of developmental disorders in which a major etiologic contribution can be identified: disorders arising from genomic copy number variants (CNVs). Defined as a segment of DNA at least 1 kilobase in length present in a number differing from that in a reference genome [7], CNVs can occur throughout the human genome. Recurrent CNVs are those that tend to occur de novo in identical or nearly identical form in specific regions of the genome that harbor segmental duplications (or low-copy repeats, LCRs). LCRs are runs of identical DNA sequence of ~10 kilobases or more that reside near each other in the same orientation. These structures pre-dispose to misalignment of sister chromatids during meiosis, leading to duplication of the intervening sequence on one allele, and its duplication on the other, a process known as non-allelic homologous recombination (NAHR) [8]. NAHR in regions containing LCRs generates the de novo production of recurrent CNV in the human population with each new generation. The fact that new recurrent CNVs, some of which clearly associate with low-fecundity neurodevelopmental disorders such as autism spectrum disorders (ASD) and schizophrenia at least partially addresses a question that has dogged evolutionary thought about such disorders for decades. Namely, how do disorders such as ASD and schizophrenia remain so common in the population, when they are highly heritable and clearly reduce reproductive fitness? In the absence of new mutation, evolutionary theory predicts that genes predisposing to lower reproductive fitness should be rapidly removed from populations by purifying selection. However, it is now clear that NAHR in regions of the genome rich in LCRs generates new deleterious mutations that predispose to neurodevelopmental disorders with each generation.
Potential for Treatment
In addition to explaining, at least in part, an evolutionary puzzle, the discovery that CNV-associated disorders are common, accounting for more than 10 % of cases of ASD, multiple congenital anomalies and intellectual disability, has resulted in incorporation of genome-wide testing for CNVs, using an approach called array-based comparative genomic hybridization (aCGH), into the standard of care for evaluation of such disorders [9]. An exciting prospect is that establishing molecular diagnoses can point to specific interventions for specific patients. For example, identification of a 15q13.3 deletion in a patient with intellectual disability, epilepsy and psychosis led the author to alter medication management in an attempt to address the patient’s haploinsufficiency of the CHRNA7 locus, encoding the α7 nicotinic cholinergic receptor, with good clinical effect [10]. An even more exciting example of such personalized medicine was recently discussed on T. Insel’s National Institute of Mental Health Director’s blog (http://www.nimh.nih.gov/about/director/2014/celebrating-science.shtml). In that example, DL Levy and colleagues analyzed the genomes of several members of a family in which some had a schizophrenia-spectrum psychotic illness. The team identified a rare CNV that produced multiple copies of the gene encoding the enzyme glycine dehydrogenase (GLDC), which metabolizes glycine, an essential amino acid that also functions as a modulator of the N-methyl-D-aspartate (NMDA) receptor. For so-called dosage-responsive genes (often, enzyme genes fall into this category), increasing the number of copies of the gene will increase the level of gene product. Thus, the investigators reasoned that the psychosis-affected persons, who all carried greater than two copies of GLDC, might have elevated enzyme activity, leading to a deficiency in glycine that influenced the expression of illness. They performed a double-blind trial of glycine administration in the affected family members. Insel described the patients’ response to the intervention as follows: “…the response was like giving insulin to a person with diabetes—their psychiatric symptoms largely resolved. When the drug was stopped, their symptoms returned. When they received glycine again under non-blind conditions, the same improvements were observed.”
As illustrated by the foregoing examples, expanding knowledge of CNVs and other genomic variants, and applying this knowledge in the clinic with individual patients, promises to revolutionize treatment of neuro-developmental disorders, at least in some cases. A variety of recurrent CNVs are now well established to associate with neuro-developmental disorders [11, 12].
22q11DS: A Molecular Diagnosis Unifying Several Clinical Syndromes
The remainder of this chapter will focus on 22q11DS because it is common, with an estimated prevalence of 1 per 4,000 live births [13], and by far the most deeply studied and well-characterized CNV disorder to date. First, we will review the history of the discovery of 22q11DS, and then summarize some important clinical considerations.
In 1967, Di George and colleagues [14, 15] reported an autopsy series of four infants, all of whom had complete agenesis of the thymus. They noted several other anomalies in these patients, including right-sided aortic arch and dysgenesis of the parathyroids, suggesting abnormal development of derivatives of the third and fourth branchial arches. The infants had exhibited tetany (from hypocalcemia) and severe immune deficiency, and succumbed to infection soon after birth. Several years later, Kinouchi and colleagues [16] published a case series of Japanese children with cardiac outflow malformations (particularly tetralogy of Fallot), a characteristic facial appearance, and hypernasal speech, calling the syndrome conotruncal anomaly face syndrome (CTAF). Although they noted a similarity to the Di George syndrome (DGS), based on thymic involution in some cases, CTAF stood as a distinct diagnosis. Shprintzen and colleagues [17] then reported “a new syndrome,” characterized by cleft palette, ventricular septal defects of the heart, learning disabilities, and typical facies characterized by broad nasal bridge, flattened malar region and elongated face. These authors coined the term velo-cardio-facial syndrome (VCFS), and did not relate the diagnosis to DGS or CTAF.
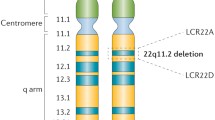
The first clues to a molecular-genetic etiology for DGS came from the observation of de la Chapelle et al [18] that an affected member of a family segregating DGS carried an unbalanced translocation resulting in partial monosomy of chromosome 22, followed by similar observations by Greenberg and colleagues [19] of a distinct unbalanced translocation also affecting chromosome 22. Following accumulation of additional reports of microscopically detectable cytogenetic abnormalities involving chromosome 22 in patients with DGS, and studies of molecular markers on chromosome 22 associated with DGS, definitive evidence emerged when Scambler’s group [20] and Driscoll et al. [21] independently reported submicroscopic deletions in the 22q11 region, detected by fluorescent in situ hybridization (FISH), in samples from both familial and sporadic DGS patients. FISH results from CTAF and VCFS patients soon confirmed 22q11 deletions as occurring in virtually all cases examined [22–24].
Deletions resulting in 22q11DS can vary in size. Approximately 80 % are some 3 megabases in size, encompassing approximately 40 genes, another 15 % are approximately 1.5 Mb in size, and the remainder are smaller. The variability in size of the deletion arises from the presence of four LCRs in the region, with the largest deletions resulting from NAHR involving the two outermost LCRs [25], and the others from other combinations of LCRs [8].
Although the terms DGS, CTAF and VCFS continue to be used, the etiologic unity of these syndromes suggests that 22q11DS is the most appropriate term. As substantial clinical and research experience has accumulated with 22q11DS, it is clear that its phenotypic presentation is extremely variable, and the syndrome is best conceptualized as a multi-system condition. Severity of disability can range from cases affected by multiple life-threatening challenges, intellectual disability and severe neurobehavioral disorders including autism spectrum disorder and schizophrenia to manifestations so mild that a person carrying the deletion only comes to clinical attention after parental “back-testing” reveals he or she is a transmitting parent of a more severely affected child.
The 22q11DS is a multi-system disorder. Once the diagnosis is established, multi-disciplinary evaluation is indicated. The role of the primary-care physician can become challenging, as he or she often must coordinate evaluation and treatment recommendations from multiple specialties. Bassett and colleagues [26] recently published consensus guidelines for evaluation and management of 22q11DS, compiled by a multi-disciplinary group of clinicians and researchers. Table 61.1 is reproduced from that review. It summarizes the wide range of possible medical and behavioral challenges patients with 22q11DS face.
22q11DS: Neurodevelopmental and Neurobehavioral Disorders
Behavioral manifestations in 22q11DS vary widely [27], but are common in children and adults. By the mid-1980s, behavioral difficulties in children with VCFS had been described [28], and psychosis in adolescents with the disorder was reported in 1992 [29]. Pulver and colleagues confirmed that schizophrenia was common in patients with 22q11DS [30] and Karayorgou and colleagues found several previously undiagnosed cases of 22q11DS in a series of patients with idiopathic schizophrenia [31]. The latter study was a landmark because it was the first indication that a small but clinically and epidemiologically significant proportion of the schizophrenia patient population carry 22q11 deletions. It is now clear that the syndrome occurs at a low but non-trival rate in clinically diagnosed schizophrenia patients (~0.5 to 0.75 %, or 30-fold more frequently than in the population at large [32]). Selecting specific phenotypic characteristics prior to molecular testing (e.g., facial dysmorphology, conotruncal heart defects, high arched palate or cleft palate, ID) can substantially increase the diagnostic yield for the deletion in cohorts of patients with SSD [33].
22q11DS: A Diagnostic Odyssey
The medical and anatomic manifestations associated with 22q11DS are manifold. The most common include T-cell related immune disorders, which can associate with frequent infections (e.g., recurrent otitis media). Another common challenge includes velo-palatal malformations, leading to early difficulties with suckling and feeding, including nasal regurgitation of milk or formula. Hypernasal speech (or in infants, crying), with inability to form certain phonemes requiring the build-up of intra-oral pressure (e.g., the sound of the letter, “p”), can be a clue indicating 22q11DS. Perhaps the most dramatic presentation of 22q11DS consist of a variety of cono-truncal heart malformations, including tetralogy of Fallot, transposition of the great vessels, and ventricular-septal defects. Such anomalies often require neonatal surgical intervention. Ruling out 22q11DS by molecular testing (usually with FISH, but increasingly with aCGH) is strongly indicated prior to any neonatal heart repair surgery, because an important clinical decision rests on the presence or absence of the diagnosis. Thus, the risk-benefit balance favors irradiating blood prior to transfusion in the presence of 22q11DS (due to the immune compromise that occurs in the majority of patients), but weighs against it in its absence (due to the degradation of erythrocyte quality inherent in irradiation).
The 22q11DS can be diagnosed at any age, including well into adulthood (the author has some patients initially diagnosed in their 30s). Many families with 22q11DS report years or even decades of “diagnostic odyssey,” as the family and patient seek help from various specialities related to the myriad manifestations of the syndrome. In the author’s clinical experience, diagnoses tend to be established most frequently at one of several ages. First, patients presenting at birth with cardiac anomalies (classic VCFS or CTAF presentation) or severe immune deficiencies or athymia, or tetany and seizures suggestive of hypocalcemia (i.e., the classic DGS presentation) come to clinical attention very early. In later infancy and early childhood, problems with feeding, suckling or swallowing, and then with speech will prompt the alert clinician to consider the diagnosis. In the early school-aged years, behavioral difficulties including learning disabilities, extreme shyness, attention deficits, or poor impulse control, especially in children with facial dysmorphology or other common medical complications, will come to testing. Finally, children, adolescents or adults can present with schizophrenia-spectrum psychotic symptoms. Again, the presence of associated features such as facial dysmorphology, seizure disorder, learning disabilities or ID, a history of heart anomalies, etc., should prompt consideration of testing in the psychiatric setting. The importance of establishing a diagnosis of 22q11DS when present in psychiatric patients can be appreciated when considering, for example, that hypocalcemia is a frequent intermittent difficulty encountered by patients with the syndrome. Low calcium levels lead to reduced seizure thresholds, which can synergize with the epileptogenic effects of anti-psychotic medications to produce seizures. If hypocalcemia is not suspected, such an outcome can lead to discontinuation of the anti-psychotic medication, leading to deleterious mismanagement. The foregoing scenario has been documented with a patient on clozapine [34], which remains the most effective known anti-psychotic medication. Thus, a proper diagnosis, with follow-up testing of ionized calcium levels and treatment with vitamin D and calcium can allow a patient with schizophrenia to continue treatment with an antipschotic medication that otherwise might have been discontinued.
Conclusions
As just summarized, 22q11DS is a diagnostic term that has evolved from a set of syndromes described with emphasis on subsets of features that varied across the patients. Authors described the syndromes from differing points of view centering on overlapping but varying clinical concerns (e.g., immune dysfunction; velo-palatal dysfunction; heart defects; facial dysmorphology). This evolution appears likely to occur in other CNV disorders, since many of them associate with multiple diagnostic categories that are the focus of various specialties. As advances continue in the field of molecular genetics, it should become possible to re-think behaviorally defined syndromes such as autism and schizophrenia, and re-classify at least some groups of patients with these disorders according to molecular etiology. Although still a research tool, whole-genome sequencing is revolutionizing the analysis of developmental disabilities. In a truly astounding recent study, Jiang, Scherer and colleagues [35] recently reported performing whole genome sequencing on 32 patients with autism spectrum disorders and their parents. They identified de novo deleterious sequence variants in six (19 %) patients, and transmitted mutations likely to be deleterious in 10 (31 %) families. Although the causal roles of many of the variants in that study require further study and supportive evidence, the results emphasize the enormous heterogeneity of genetic contributions to neurodevelopmental disorders. Indeed, the autDB database (http://autism.mindspec.org/autdb/Welcome.do), a curated public database of genes potentially relevant to autism, currently lists 573 genes.
A major challenge with regard to applying sequencing to clinical evaluation is the shear enormity and complexity of the information generated by sequencing technologies. However, as improved computational approaches are developed, “digesting” genome-scale sequence data should become more practical. A greater challenge will be to train physicians to use such data to optimal advantage for patients.
References
Pearce JM. Brain disease leading to mental illness: a concept initiated by the discovery of general paralysis of the insane. Eur Neurol. 2012;67(5):272–8.
Bayle ALJ. Recherches sur l’arachnitis chronique. Thèse No. 247, Didot le Jeune, Paris; 1822. (A partial English Translation is available in: Moore M, Solomon HC. Contributions of Haslam, Bayle and Esmarch and Jessen to the history of neurosyphilis: Haslam’s “Observations on insanity,” Bayle’s “Reserche sure l’arachnitis chronique” and Esmarch and Jessen’s “Syphilis und geistesstörung.” Arch Neurol Psychiatr. 1934;32:808–9).
Esmarch F, Jessen W. Syphilis und Geistesstörung. Allg Zeitschr Psychiat. 1857;14:20–32.
Noguchi H, Moore J. A demonstration of Treponema pallidum in the brain in cases of general paralysis. J Exp Med. 1913;17:232–8.
Fleming A. Classics in infectious diseases: on the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae by Alexander Fleming. Reprinted from the Br J Exp Pathol. 1929;10:226–36. Rev Infect Dis. 1980;2(1):129–39.
Florey HW. Use of micro-organisms for therapeutic purposes. BMJ. 1945;2(4427):635–42.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54.
Emanuel BS, Shaikh TH. Segmental duplications: an ‘expanding’ role in genomic instability and disease. Nat Rev Genet. 2001;2(10):791–800.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Cubells J, DeOreo E, Harvey P, Garlow S, Garber K, Adam M, et al. Pharmaco-genetically guided treatment of recurrent rage outbursts in an adult male with 15q13.3 deletion syndrome. Am J Med Genet. 2011;155A(4):805–10.
Moreno-De-Luca D, Cubells JF. Copy number variants: a new molecular frontier in clinical psychiatry. Curr Psychiatr Rep. 2011;13(2):129–37.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–72.
Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112(1 Pt 1):101–7.
Di George A, Lischner H, Dacou C, Arey J. Absence of the thymus. Lancet. 1967;289(7504):1387.
Kirkpatrick Jr JA, DiGeorge AM. Congenital absence of the thymus. Am J Roentgenol Radium Ther Nucl Med. 1968;103(1):32–7.
Kinouchi A, Mori K, Ando M, Takao A. Facial appearance with conotruncal anomalies. Pediatr Jpn. 1976;84:84–9.
Shprintzen RJ, Goldberg RB, Lewin ML, Sidoti EJ, Berkman MD, Argamaso RV, et al. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978;15(1):56–62.
de la Chapelle A, Herva R, Koivisto M, Aula P. A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet. 1981;57(3):253–6.
Greenberg F, Crowder WE, Paschall V, Colon-Linares J, Lubianski B, Ledbetter DH. Familial DiGeorge syndrome and associated partial monosomy of chromosome 22. Hum Genet. 1984;65(4):317–9.
Scambler PJ, Carey AH, Wyse RK, Roach S, Dumanski JP, Nordenskjold M, et al. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10(1):201–6.
Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50(5):924–33.
Driscoll DA, Spinner NB, Budarf ML, McDonald-McGinn DM, Zackai EH, Goldberg RB, et al. Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am J Med Genet. 1992;44(2):261–8.
Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, et al. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet. 1993;30(10):822–4.
Emanuel BS, McDonald-McGinn D, Saitta SC, Zackai EH. The 22q11.2 deletion syndrome. Adv Pediatr. 2001;48:39–73.
Edelmann L, Pandita RK, Morrow BE. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet. 1999;64(4):1076–86.
Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(2):332–9.
Ousley O, Rockers K, Dell ML, Coleman K, Cubells JF. A review of neurocognitive and behavioral profiles associated with 22q11 deletion syndrome: implications for clinical evaluation and treatment. Curr Psychiatr Rep. 2007;9(2):148–58.
Golding-Kushner KJ, Weller G, Shprintzen RJ. Velo-cardio-facial syndrome: language and psychological profiles. J Craniofac Genet Dev Biol. 1985;5(3):259–66.
Shprintzen RJ, Goldberg R, Golding-Kushner KJ, Marion RW. Late-onset psychosis in the velo-cardio-facial syndrome. Am J Med Genet. 1992;42(1):141–2.
Pulver AE, Nestadt G, Goldberg R, Shprintzen RJ, Lamacz M, Wolyniec PS, et al. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J Nerv Ment Dis. 1994;182(8):476–8.
Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R, Borrow J, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci U S A. 1995;92(17):7612–6.
Hoogendoorn ML, Vorstman JA, Jalali GR, Selten JP, Sinke RJ, Emanuel BS, et al. Prevalence of 22q11.2 deletions in 311 Dutch patients with schizophrenia. Schizophr Res. 2008;98(1–3):84–8.
Bassett AS, Chow EW. 22q11 deletion syndrome: a genetic subtype of schizophrenia. Biol Psychiatry. 1999;46(7):882–91.
Caluseriu O, Tayyeb T, Chow E, Bassett AS. Clozapine-associated seizures in a 22q deletion syndrome subtype of schizophrenia. Schizophr Res. 2007;60(1 Suppl 1):70.
Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2014;93(2):249–63.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Cubells, J.F. (2016). 22q11.2 Deletion Syndrome: A Paradigmatic Copy-Number-Variant (CNV) Disorder. In: Rubin, I.L., Merrick, J., Greydanus, D.E., Patel, D.R. (eds) Health Care for People with Intellectual and Developmental Disabilities across the Lifespan. Springer, Cham. https://doi.org/10.1007/978-3-319-18096-0_61
Download citation
DOI: https://doi.org/10.1007/978-3-319-18096-0_61
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-18095-3
Online ISBN: 978-3-319-18096-0
eBook Packages: MedicineMedicine (R0)