
Abstract
Acid-base disorders are a major source of morbidity and mortality among patients in the intensive care unit (ICU). An observational cohort study of 9,799 ICU patients found that nearly two thirds of critically ill patients suffered from acute Metabolic Acidosis. Mortality among those with metabolic acidosis was 45% compared with 25% for those without it. For those with lactic acidosis, the mortality rate was 56% [1]. It is difficult to directly extrapolate these findings to poisoned patients who generally have a significantly lower mortality rate than the average ICU patient. We do know, however, from studies of poisonings with specific substances, such as metformin, ethylene glycol, and methanol, that the presence of severe metabolic acidosis is associated with a relatively poor prognosis [2]. A review of 22 cases of metformin overdose revealed a median pH nadir of 7.30 and median plasma lactate of 10.8 mmol/L among survivors compared with pH 6.71 and median plasma lactate of 35.0 mmol/L among non-survivors [2]. Among 18 ethylene glycol poisoned patients, non-survivors had a mean admission pH of 7.05, compared with 7.31 in survivors [3]. No patient with a pH less than 7.10 survived. Not surprisingly, most of the non-survivors presented to the hospital late after ingestion (from 6 h to > 24 h). In a review of one-time methanol exposures with known time of ingestion, 22 patients presented for care <6 h after ingestion and had an early methanol level. Sixteen of these were acidotic on arrival [4]. Blood methanol concentrations ranged from 10 to 570 mg/dL (3–178 mmol/L), and initial arterial pH ranged from 6.90 to 7.42. All underwent treatment with alcohol dehydrogenase inhibitors (with or without hemodialysis). One patient with pH 6.99 died. Three patients with pH ranging from 7.26 to 7.32 suffered optic neuropathies but survived. One patient with pH 6.90 was described as “alive” on discharge, with the remaining 11 (pH range 7.09–7.42) noted as having a full recovery. In summary, it appears that severe metabolic acidosis is associated with a poor prognosis in representative poisonings and that time of presentation plays a significant role in outcome. A larger study of the prognostic value of acid–base disturbances in poisoning in general among ICU patients would be edifying.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
The constancy of the internal environment is the condition for free and independent life.
Claude Bernard, 1813–1878
Acid–base disorders are a major source of morbidity and mortality among patients in the intensive care unit (ICU). An observational cohort study of 9,799 ICU patients found that nearly two thirds of critically ill patients suffered from acute Metabolic Acidosis. Mortality among those with metabolic acidosis was 45% compared with 25% for those without it. For those with lactic acidosis, the mortality rate was 56% [1]. It is difficult to directly extrapolate these findings to poisoned patients who generally have a significantly lower mortality rate than the average ICU patient. We do know, however, from studies of poisonings with specific substances, such as metformin, ethylene glycol, and methanol, that the presence of severe metabolic acidosis is associated with a relatively poor prognosis [2]. A review of 22 cases of metformin overdose revealed a median pH nadir of 7.30 and median plasma lactate of 10.8 mmol/L among survivors compared with pH 6.71 and median plasma lactate of 35.0 mmol/L among non-survivors [2]. Among 18 ethylene glycol poisoned patients, non-survivors had a mean admission pH of 7.05, compared with 7.31 in survivors [3]. No patient with a pH less than 7.10 survived. Not surprisingly, most of the non-survivors presented to the hospital late after ingestion (from 6 h to >24 h). In a review of one-time methanol exposures with known time of ingestion, 22 patients presented for care <6 h after ingestion and had an early methanol level. Sixteen of these were acidotic on arrival [4]. Blood methanol concentrations ranged from 10 to 570 mg/dL (3–178 mmol/L), and initial arterial pH ranged from 6.90 to 7.42. All underwent treatment with alcohol dehydrogenase inhibitors (with or without hemodialysis). One patient with pH 6.99 died. Three patients with pH ranging from 7.26 to 7.32 suffered optic neuropathies but survived. One patient with pH 6.90 was described as “alive” on discharge, with the remaining 11 (pH range 7.09–7.42) noted as having a full recovery. In summary, it appears that severe metabolic acidosis is associated with a poor prognosis in representative poisonings and that time of presentation plays a significant role in outcome. A larger study of the prognostic value of acid–base disturbances in poisoning in general among ICU patients would be edifying.
In poisoning, alterations of acid–base balance may result from exogenously administered ions, metabolic production of organic acids, impaired mitochondrial function, renal injury, hypoventilation, or inadequate tissue delivery of oxygen because of respiratory or circulatory insufficiency. Poisoned patients may suffer acid–base disturbances due to underlying illness or complications of their hospitalization, including acute or chronic kidney or pulmonary disease, side effects of therapy (crystalloids, diuretics, hyperalimentation, vasopressors), and sepsis. Determining which disorders of acid–base balance are present, and which are due to the toxic exposure versus underlying illness or complications, is not always straightforward. Thus, it is incumbent on critical care physicians caring for poisoned patients to be well versed in the evaluation and management of disorders of acid–base equilibrium.
In the last edition of this text, a “quiet revolution” was ongoing in acid–base disorder analysis, as exemplified by increasing attention to the work of Stewart with respect to electroneutrality and the strong ion gap. In the intervening period, numerous evaluations of the Stewart method have been undertaken, rendering examples of complex acid–base disorders discerned by quantitative acid–base chemistry that might have been missed by the standard approach using base excess. It is fair to say that this issue remains highly controversial, with two very staunch camps, the “bicarbonate-based camp” and the “Stewart camp.” According to Rastegar, the Stewart approach has been largely ignored by nephrologists and renal physiologists while being embraced by anesthesiologists and intensivists [5]. He opines that the Stewart method provides no advantages over the traditional method in terms of prognosis. While numerous small prospective studies have compared these methods, large multicenter comparisons are lacking, and so the argument is not likely to be settled soon. It may be said, however, that the Stewart approach has led even the traditionalists to reexamine the importance of the influence of nonvolatile buffers, such as albumin, as well as the effects of dilution on the standard base excess and on the anion gap, resulting in new corrective formulas even in the “traditional camp.” On the quantitative acid–base side, investigators have attempted to distance themselves from tedious formulas and exhaustive analyses, offering simplified formulas and confining their components to plasma elements that most often undergo significant alteration. Al-Jaghbeer and Kellum argue that the two approaches are complementary and not contradictory [6]. Both approaches to acid–base analysis are discussed in this chapter.
Rapid Review of Key Factors in Acid–Base Equilibrium
Acid–base balance under physiological conditions is the simple sum of the production of organic acids occurring in metabolism and their elimination or neutralization by the body’s buffer systems. The first line of defense is the respiratory buffer system. Carbon dioxide, produced by the metabolism of carbohydrates, forms carbonic acid (H2CO3) when combined with water:
This is considered an “open system” because moderate increases in CO2 are normally eliminated by a compensatory increase in minute volume. Acidosis stimulates respiratory chemoreceptors to produce an increase in ventilation, hence shifting this equation to the right and thus reducing the acidosis. However, severe acidosis may actually impede ventilation and lead to a vicious cycle that may result in death unless rapidly corrected.
Plasma proteins and phosphate stores in the bone serve as a second line of defense against acid–base abnormalities. The important role of these buffer systems will be discussed further with the “strong ion” approach. A strong ion refers to one that generally exists in a dissociated or nearly dissociated form (Na+, K+, Cl−, and others). The kidneys play a critical role in acid–base balance via regulation of strong ions.
All forms of acid–base disorders may be observed in a poisoned patient. However, given its frequent presence in serious poisonings, the majority of this chapter will be devoted to metabolic acidosis.
Toxicant-Induced Respiratory Alkalosis and Acidosis
Respiratory alkalosis and acidosis are direct reflections of increased and decreased ventilation, respectively. Ventilation may be altered through changes in tidal volume, respiratory rate, and gas exchange across the alveoli or by combinations thereof. These disorders may be of central or peripheral origin. Stimulation of the central nervous system may result in tachypnea or hyperpnea and subsequent respiratory alkalosis. Salicylates [7], nicotine [8], and caffeine [9] are examples of agents that stimulate respiratory centers. Opiates decrease the respiratory rate by depressing the central respiratory and brainstem regulatory centers, [10] but also reduce ventilation through peripheral effects, such as non-cardiogenic pulmonary edema and increases in thoracic muscle tone [11]. Benzodiazepines and barbiturates likewise have both central effects (γ-aminobutyric acid-mediated depression in the medulla oblongata) and peripheral effects (muscle weakness resulting in upper airway obstruction and/or respiratory muscle inefficacy); however, the peripheral effects appear to be of greater clinical importance [12]. Inadequate ventilation may also result in inadequate oxygenation and thereby lead to anaerobic glycolysis and lactate production. Thus, combined respiratory and metabolic acidosis is not uncommon in poisoning by toxicants that interfere with ventilation. Clinical diagnosis of these disorders is rather straightforward and based on physical examination and arterial blood gases, particularly PaCO2.
Toxicant-Induced Metabolic Alkalosis
Chemical-induced metabolic alkalosis is relatively rare. The milk-alkali syndrome, characterized by hypercalcemia, metabolic alkalosis, and renal failure, was common in the early to mid-twentieth century, when a common remedy of peptic ulcer disease consisted of hourly administration of milk with a “Sippy” powder (sodium bicarbonate with calcinated magnesia or bismuth subcarbonate) [13]. Nowadays, it is seen most often in elderly women taking calcium supplements for osteoporosis and among patients taking calcium antacids and is rightfully known as calcium-alkali syndrome [13, 14]. It has also been reported in betel nut chewers who use calcium-rich oyster shell paste in its preparation [15]. Laxatives taken in excess can give rise to hypokalemia and metabolic alkalosis [16, 17], as can diuretics [18, 19] and licorice [20, 21]. A non-exhaustive list of toxicants responsible for metabolic alkalosis is found in Table 1.
Toxicant-Induced Metabolic Acidosis
Acute metabolic acidosis is found in 64% of critically ill patients. Its presence is associated with an almost twofold rise in mortality [1]. Al-Jaghbeer and Kellum divide the causes of acidosis into (1) increased anion gap and (2) non-anion gap, consisting of renal, gastrointestinal, and iatrogenic causes (Box 1). While poisoned patients may suffer from any of these causes of metabolic acidosis, we will primarily focus on toxicant-induced etiologies.
Box 1 Causes of Metabolic Acidosis (Modified from Al-Jaghbeer and Kellum [6])
Increased anion gap
-
Renal failure
-
Ketoacidosis
-
Diabetic
-
Alcoholic
-
Starvation
-
Metabolic errors
-
-
Lactic acidosis
-
Toxicants (see also Table 2)
Table 2 Toxicants reported to cause metabolic acidosis -
Methanol
-
Ethylene glycol
-
Paraldehyde 5-oxoproline (pyroglutamic acid)
-
-
Sepsis
Hyperchloremic (non-anion gap)
-
Renal tubular acidosis
-
Gastrointestinal
-
Diarrhea
-
Small bowel/pancreatic drainage
-
-
Iatrogenic
-
Parenteral nutrition
-
Saline
-
Carbonic anhydrase inhibitors
-
Anion-exchange resins
-
Many toxicants provoking acidosis increase the anion gap. Ingested mineral acids (toilet bowl cleaners, battery acid) may serve as exogenous sources of metabolic acidosis. Toxic alcohols and glycols, including methanol and ethylene glycol, are notable causes of metabolic production of organic acids. Interference with mitochondrial function results in lactate production. Anti-retroviral medications and cyanide are well known for their ability to induce lactic acidosis. Agents causing profound cardiovascular collapse, such as chloroquine and colchicine, may give rise to acidosis through diminished perfusion, as well as other mechanisms. In Table 2, the reader is provided with selected references for a non-exhaustive list of toxicants that may induce metabolic acidosis. In-depth discussion of individual toxicants is beyond the scope of this chapter. Related chapters in this text pertaining to specific substances should be consulted.
Clinical Suspicion of Acid–Base Disorders
Vigilance is required to promptly detect and treat acid–base disorders. The finding of altered mental status or abnormal vital signs should always invoke consideration of an acid–base disturbance. Clinical findings may be subtle early in the pathophysiologic process, so habits as elemental as truly counting the respiratory rate may make the difference in early recognition of these disorders. A thorough examination may detect sentinel breath odors (ketones, paraldehyde, and cyanide), altered respiration (hyperpnea, Kussmaul respirations), cyanosis, pressure sores or bullae suggestive of rhabdomyolysis, etc. Clinical signs and symptoms are insensitive, however, so laboratory screening is necessary in all critically ill patients. While point-of-care diagnostic devices have simplified life for the clinician, a multitude of conditions can alter their readings [136–139]. Definitive evaluation of acid–base disorders is based on carefully selected data provided by the chemistry and toxicology laboratories.
Laboratory Diagnostic Tools in Acid–Base Balance
Gaps in Our Knowledge of Acid–Base Disturbances
This double entendre is not original [140] but reflects our incomplete comprehension of acid–base disorders in spite of years of studies and discussions regarding the use of various gaps – base excess, anion, delta, and osmol. Each of these tools, their potential utility, and their shortcomings will be discussed, followed by a discussion of the quantitative approach to acid–base balance, which introduces the more recent strong ion gap.
Given the ubiquity of its use in clinical medicine, the most frequently available laboratory indicator of acidosis is the serum electrolyte panel. For this reason, anion and delta gaps will be discussed before base excess and osmol gaps.
Anion Gap
The serum anion gap (AG) remains a useful but only moderately sensitive tool in the initial evaluation of potential acid–base disturbances. It suffers from numerous limitations, which will be discussed. If the clinician is aware of these limitations and adjusts for them accordingly, the AG provides one of the most rapidly available tools in the evaluation of acid–base disorders.
In its most basic form,
Because the most important anions after chloride and bicarbonate (protein, inorganic phosphate, and sulfate) are not routinely measured in a serum electrolyte panel, a “normal” AG exists. The presence of additional unmeasured anions (often organic acids) creates an “increased” AG. The most commonly used calculation for AG is (using mmol/L units for the anions/cations)
An alternative, though less frequently used, equation adds [K+] to the first term. Extremes of potassium may affect the AG and should be kept in mind; however, the discussion that follows will be based on (Eq. 2). Although its calculation is remarkably simple, its interpretation is not always straightforward.
Because of the body’s requirement for electroneutrality, an increase in unmeasured anions (the AG) should be compensated by an equal decrease in serum bicarbonate or chloride, although in almost all circumstances, the compensation is reflected in a decrease in the bicarbonate concentration [141]. Thus, the first evaluation of the calculated AG should be its relationship to serum bicarbonate. In the simplest case, AG increases in an amount equivalent to the fall in bicarbonate. However, this does not always occur. For example, in the case of diabetic ketoacidosis (DKA), a discordance is often found [142], with AG being smaller than predicted based on the decrease in bicarbonate. Extensive tubular elimination of ketone bodies, along with concomitant retention of chloride, may result in a component of hyperchloremic metabolic acidosis, thus decreasing the AG. The hyperchloremic component of acidosis increases over the course of DKA [143], probably in part due to the large amount of sodium chloride typically administered in DKA. Large-volume fluid resuscitation with isotonic saline can induce acidosis because the equimolar concentrations of sodium and chloride in normal saline (155 mmol/L) will increase plasma chloride to a greater extent than plasma sodium, thus decreasing the serum bicarbonate concentration [144]. A review of several studies on acidosis induced by chloride-rich crystalloids appears in the discussion on treatment [123].
The limitations of the AG fall into two general categories: (1) analytic and (2) physiologic.
Analytic Limitations and Errors
Early published normal ranges of AG values, 12 ± 4 mmol/L [145], continue to be used by many clinicians. However, these values now represent, in many hospitals, a significant overestimate because of changes in technology that occurred in the 1980s. Ion-specific electrode methodology has largely replaced flame photometry for measurement of sodium and chloride, leading to an increase in normal values of chloride of 2–6 mmol/L, with a concomitant decrease in the normally expected AG. Winter and colleagues [146] found a normal AG of approximately 6 mmol/L, with 95% confidence intervals of 3–11 mmol/L in a group of 120 blood donors. This difference from previous normal values may seem small, but as the authors point out, if the true normal AG is 6 mmol/L, a patient with 10 mmol/L of added organic acid and thus an AG of 16 mmol/L would, by generally accepted standards, have a normal AG. In a group of 222 patients with normal renal function and albumin, Sadjadi [147] found an AG of 6.6 ± 2 mmol/L, almost identical to that demonstrated in the study of Winter and coworkers. Failure to take this change into account may result in failure to diagnose acidosis. Using a “normal” range of 12 ± 4 mmol/L, Iberti and colleagues found that 50% of critically ill patients with lactic acidemia in the range of 5–9.9 mmol/L and 79% of those with lactate concentrations 2.5–5 mmol/L had AG values less than 16 mmol/L. Applying the current lower ranges of AG would clearly improve sensitivity. Sadjadi and colleagues more recently retrospectively reviewed the charts of 409 veterans with a glomerular filtration rate ≥ 60 mL/min/1.73 m2 body surface area and serum albumin ≥ 4 g/dL. They found a mean anion gap of 7.2 ± 2 mEq/L. This compares with 299 patients in the same study with lactic acidosis (lactate ≥ 4 mEq/L) and 68 patients with end-stage renal disease on dialysis, in whom the mean anion gaps were 12.5 mEq/L and 12.4 mEq/L, respectively [148]. A word of caution is in order, however. Lolekha points out that to interpret the anion gap accurately, one must use an analyzer-specific reference range. He and his colleagues found slightly differing anion gaps on four auto analyzers, using blood samples from healthy volunteers [149]. Anion gap values may also vary significantly according to the laboratory providing the measurements [150]. Finally, clinicians must also be aware that some laboratories have altered the calibration of the chloride-measuring instrument so that the normal ranges of chloride and AG in those institutions remain closer to “classic” published values. Thus, it is imperative that clinicians discuss this issue with laboratory medicine specialists in their own institutions to ensure that the normal ranges provided by the laboratory have, in fact, been verified in that laboratory.
Kraut and Nagami [151] attempted to determine the sensitivity of the AG in detecting lactic acidosis through a literature review. Unless the AG is corrected for serum albumin, the sensitivity ranged from 39% to 63% for serum lactate greater than 2.5 mmol/L. With correction for albumin, the sensitivity range improved to 75–94% demonstrating two salient points: (1) if the anion gap is to be useful, it should be corrected for serum albumin and (2) if one is truly concerned about lactic acidemia, a serum lactate should be obtained. One last word about the anion gap, which applies to lab tests in general: a single, normal AG is just that – a unique, not analytically infallible, test obtained at a point in time. In contrast, poisonings are dynamic processes which may radically modify the kinetics of absorption, distribution, metabolism, and excretion. Jacob et al. recently reported two cases of life-threatening salicylate poisoning with apparently normal anion gap due to a false increase of measured chloride in the presence of high serum salicylate levels on some analyzers [152]. Herres and colleagues reported a fatal case of salicylate poisoning who presented with a deceptively mild elevation in anion gap and initially undetectable serum salicylate concentration. Failure to repeat the anion gap in a timely manner, to trend salicylate concentrations until they were clearly declining, and to carefully observe the patient in an acute medical setting contributed to his ultimate demise [153].
Physiologic Limitations
Serum albumin. Whereas the potential for abnormal proteins (such as cationic multiple myeloma proteins) to alter the AG is widely appreciated, the critical role of normal proteins such as albumin remains largely ignored. Albumin constitutes the largest component of unmeasured anions under normal physiologic conditions, with inorganic phosphate and sulfates representing most of the remainder. Calculation of the AG without consideration of serum albumin may be justifiable in normal healthy patients, but it is certainly not acceptable in the critically ill. Figge and coworkers found marked hypoalbuminemia in 96% of 152 critically ill patients, with values < 20 g/L (normal range 44 ± 3 g/L) in 49%. Each g/L decrease in serum albumin caused the observed AG to underestimate the total concentration of unmeasured anions by 0.25 mmol/L [154]. A severely malnourished patient may have a significant AG virtually obscured because of the influence of hypoalbuminemia [155]. The AG may be corrected for hypoproteinemia as follows:
If albumin values are reported in grams per deciliter, the factor is 2.5. Hyperalbuminemia from severe dehydration (cholera) may reach significant enough concentrations to contribute to metabolic acidosis [156].
Water excess/deficit. Significant loss or gain of free water will alter serum sodium and chloride by the same percentage but not by the same absolute amount, which will clearly alter the AG [140]. The effect of hyponatremia (water excess) on the AG has been clinically documented [140, 157]. Decaux and Musch compare the electrolytes and anion gap in various states of hypoosmolality (Table 3) [158]. Corrections for the effects of water excess/deficit are taken into account in calculation of the strong ion gap (to be discussed) but have not generally been applied to the AG. A prospective look at the value of such a correction in patients with metabolic acidosis is needed.
Assumptions regarding lactate in the anion gap. The presence of an increased AG acidosis not explained by the presence of ketoacids, renal failure, or historical and laboratory evidence of toxicant ingestion is often assumed to be due to lactate. However, Gabow and colleagues found that a measured increase in lactate of greater than 4 mmol/L was present in only 9 of 21 patients (43%) meeting these criteria, thus illustrating the potential for error in such an assumption [145]. Conversely, Dorwart and Chalmers found normal AGs in 32 of 45 patients with plasma lactate levels between 2.5 and 9.9 mmol/L [159]. Thus, if lactic acidemia is in the differential diagnosis, lactate should be quantified. This is particularly important in poisoning, where measurement of exogenous anions such as oxalate or formate is not always readily available. Quantification of lactate may not only provide clues to the diagnosis but also increase or decrease the suspicion of the presence of other unmeasured anions and should thus be considered an integral part of the laboratory evaluation.
Readers should be aware of other worrisome analytical problems related to lactate. Multiple authors have reported falsely elevated plasma lactate in the setting of glycol poisonings. In the case of ethylene glycol overdose, the glycolate metabolite can cross-react with the recombinant L-lactate oxidase, resulting in a marked false increase in measured lactate [160–165], potentially obscuring the diagnosis of ethylene glycol poisoning. D-lactate, a by-product of propylene glycol, can also be falsely recognized by lactate analyzers as L-lactate [166].
Role of potassium in the anion gap. Serum potassium, as mentioned earlier, is often “discarded” in calculating the AG because the range of serum potassium in most patients is small enough that potassium variations cause only a minimal change in the calculated gap. Nonetheless, there are circumstances (e.g., digitalis and toluene poisoning or acute renal failure) in which extremes of serum potassium may occur and render its consideration in calculation of the AG more important. Furthermore, a reduction in “normal” mean AG (absent the potassium) from 12 to 6 mmol/L renders the variation in potassium of greater importance in terms of percent change in AG. Regardless of whether potassium is included in the calculation used, it exerts an electrical force as a strong ion.
Hyperphosphatemia. Because the contribution of inorganic phosphates to the normal AG is moderate and because normal phosphate concentrations are generally about 1.0 ± 0.2 mmol/L, hypophosphatemia results in a negligible change in the AG. On the other hand, conditions that result in hyperphosphatemia, such as phosphate enema intoxication, may result in an increased AG [167, 168]. Thus, hyperphosphatemia must be added to the differential diagnosis of an increased AG.
Delta Gap
Another method of evaluating acid–base disorders is to compare the change in AG with the change in alkaline reserve [141]. This relationship is expressed as the “delta gap,” which is defined as:
where ΔAG is the observed AG minus the upper normal limit of the AG and ΔHCO3 − is the lower normal HCO3 − minus the observed HCO3 −. The normal range for the delta gap is 0 ± 6. As mentioned previously, the body must maintain electroneutrality, so an increase in unmeasured anions (the AG) should be compensated by an equal decrease in serum bicarbonate [141]. Therefore, for a simple increased AG acidosis , the delta gap should be 0. A significantly positive (greater than +6) delta gap suggests the presence of metabolic alkalosis. A significantly negative (less than −6) delta gap suggests hyperchloremic acidosis. Because normal values for electrolytes (and AGs) vary among hospitals, depending on the methodologies used, it is imperative to calculate these values on the basis of local norms. Wrenn demonstrated through a series of clinical cases that the delta gap can assist in the detection of mixed acid–base disorders that would go unsuspected on the basis of evaluation of the AG alone. It is not foolproof, however, as illustrated by one case in which a patient with a normal AG (4 mmol/L), a normal delta gap (−2 mmol/L), and a bicarbonate concentration of 19 mmol/L actually had a combination of anion gap metabolic acidosis, HCMA, and metabolic alkalosis from volume contraction [141]. While this simple calculation remains a useful screening tool for distinguishing and mixed acid–base disorder from isolated anion gap acidosis [169], one should not rely entirely on either the AG or the delta gap in the evaluation of potential acid–base disorders [140].
Base Excess
Base excess (BE) was the first “gap” to be proposed as a useful approach to the evaluation of acid–base disturbances [170]. It is calculated as follows, but is often provided with the results of the blood gas analyzer:
The multiplier 1.2 takes into account the approximately 25% of buffer capacity not provided by the carbon dioxide/bicarbonate system. HCO3 − is the “standard” bicarbonate, which is the concentration of bicarbonate in plasma when whole blood has been equilibrated at a PaCO2 of 40 mmHg, oxyhemoglobin saturation of 100% and temperature of 37 °C. Once the pH is measured, the standard bicarbonate can be obtained directly from (Eq. 6), the Henderson–Hasselbalch equation:
The normal mean of standard bicarbonate is 22.9 mEq/L, so this value is subtracted in (Eq. 5) from the product to obtain the BE. A BE less than −5 mEq/L is thought to be consistent with metabolic acidosis. Salem and Mujais have warned against the use of this theoretical bicarbonate concentration when calculating the AG (in lieu of total CO2) because rapidly changing conditions in apparent values of pK′ (the pH at which equal concentrations of the acid and base forms of a buffer are present – range: 5.8–6.3) may result in calculated errors in HCO3 − as high as 50% [140]. Others have likewise called attention to this problem [171, 172]. Furthermore, excess heparin in blood gas samples may decrease PaCO2 by up to 25% which also distorts the calculated bicarbonate value [140]. The effects of heparin volume on PaCO2 have recently been shown to vary with syringe volume, needle size, and sample volume [173]. It stands to reason that the BE, determined on the basis of calculated bicarbonate, also risks misinterpretation. In addition to the risk of analytical and calculated errors, the BE is dependent on a number of other factors “assumed” to be normal in this simple calculation: normal water content, electrolytes, and albumin. As pointed out by Balasubramanyan and colleagues, changes in these values will alter the calculated BE independent of changes in lactate, bicarbonate, or unmeasured anions [174].
Quantitative Acid–Base Analysis: The Strong Ion Gap
The strong ion approach to acid–base management appears to be the most fundamentally sound from the perspective of physical chemistry [175]. This method, originally described by Stewart [176] and refined by Fencl and Leith [177], relies on the fact that systems operate under a number of restraints imposed by physical laws that must always and simultaneously be met:
-
1.
Electroneutrality must always exist: the sum of all positive charges must always be equal to the sum of all negative charges.
-
2.
Dissociation equilibria of all incompletely dissociated substances must always be satisfied.
-
3.
Mass is conserved; that is, the total concentration of an incompletely dissociated substance can always be accounted for as the sum of the concentrations of its dissociated and undissociated forms.
The hydrogen ion concentration in blood is held within a very tight range, 36–43 nmol/L, because this range is critical to the maintenance of appropriate protein function (enzymes, pumps, etc.) and thus cellular function [175]. The source of hydrogen ions is the dissociation of water. Dissociation of water into hydrogen and hydroxyl ions is determined by three independent determinants (each can be changed independently of the others): the strong ion difference [SID], PaCO2, and the total weak acid concentration (ATOT).
The first independent variable is [SID], or the net electrical charge difference of the strong ions (i.e., those that are completely or nearly completely dissociated). This variable comprises the strong cations (Na+, K+, Ca2+, Mg2+) and the strong anions (Cl− and other strong anions of low pKa such as lactate and ketoacids), which are likewise almost completely dissociated at physiologic pH. The second independent variable is PaCO2, which, of course, varies with ventilation. The third independent variable is ATOT, the sum of weak acids [HA] and their anions [A−]. The weak acids are primarily composed of proteins (mainly albumin) and inorganic phosphates [Pi−]. Kellum and coworkers [178] point out that weak acids [HA] without their anions are not an independent variable because their equilibrium with A− changes with alterations in [SID] and PaCO2. Looking at Fig. 1, one observes that the [SID] can also be calculated by adding the [HCO3 −] to the [Alb−] + [Pi−]. The central tenet of the strong ion approach is that neither [H +] nor HCO 3 − can change without a change in one or more of these three independent variables. Neither H+ nor HCO3 − is a strong ion. Strong ions cannot be created or destroyed to satisfy electrical neutrality, but hydrogen ions can be generated or consumed by changes in water dissociation [178]. Likewise, bicarbonate production or consumption is a result of changes in the three independent variables. Thus, the “classic” approach of looking at HCO3 − is in a sense backward, in that we are examining the end result rather than the underlying cause of the disturbance. Acid–base disturbances may be classified according to the underlying change in these three independent variables (Table 4).

Electroneutrality must be maintained in blood plasma; thus, the sum of positive charges must equal the sum of negative charges. Hydroxyl, carbonate, and protons are not shown because their concentrations are in the nanomolar to micromolar range. [SIG] represents unidentified strong anions (lactate, sulfate, keto acids, others). [SID], strong ion difference (Adapted from Fencl et al. [155])
Figure 1 demonstrates that [SID] is equivalent to the following:
[HCO3 −] is obtained from arterial blood gas measurements, and the effects of albumin (g/L) and inorganic phosphate (mmol/L) are based on their direct measurement in serum. This formula is a simplification and uses factors of 0.28 and 1.8 to correct for the actual charge (in mEq) provided by albumin and inorganic phosphates. If your laboratory provides [Pi] in mg/dL, the factor for [Pi] is 0.6 rather than 1.8.
The strong ion gap [SIG], sometimes expressed as [XA−], is comparable to the anion gap in that it consists of strong anions other than Cl− (lactate, ketoacids, and other organic anions such as toxic metabolites or sulfate), some of which are not readily measured in plasma. [SIG] is thus derived as
where [SID] is obtained from (Eq. 7). Water excess or deficit, determined on the basis of an abnormal [Na+], will alter [Cl−] and [SIG], both of which must be corrected:
Correction of chloride allows one to determine the plasma excess or deficit in chloride.
Although this series of steps is a bit more demanding in time and cost than is the calculation of AG, it allows recognition of complex acid–base disturbances that might be missed by simple examination of BE or AG [155]. Through Monte Carlo simulation, Antsey estimated the 95% confidence interval of normal [SIG] to be 3.9 ± 6.4 mEq/L [179]. Gunnerson and colleagues have shown that the [SIG] is substantially higher in stable ICU patients compared with healthy controls, possibly representing occult acid–base disorders or simply metabolic differences among the critically ill. In this small study of 15 healthy adults and 15 stable adult patients just before ICU discharge, the mean [SIG] was 1.4 ± 1.8 mEq/L versus 5.1 ± 2.9 mEq/L, respectively [180]. Like the anion gap, it appears the [SIG] varies from one laboratory to another and from one instrument to another, so that local norms must be established.
There are now several approaches to using quantitative acid–base chemistry clinically. In the first approach, one calculates the strong ion gap [SIG], which, similar to the AG, identifies the presence of unmeasured anions. This multi-equation approach has been criticized as too complex for the bedside such that simplified equations have been developed. Examples of these will follow. The second approach provides corrections to BE based on strong ion theory. Each approach has been shown to provide advantages over the use of the classic AG and BE.
Fencl and colleagues [155] compared the Stewart method to classic methodology (calculation of the anion gap and base excess) in a study involving nine healthy subjects ([SIG] mean ± S. D. = 8 ± 2) and 152 intensive care unit patients. 96% of patients had serum albumin concentrations less than or equal to three standard deviations below the mean of the control subjects. In 20 and 22 patients, respectively, the base excess and plasma bicarbonate were normal. The Stewart method detected complex acid–base disturbances among many of these, in some cases grave. The authors conclusively demonstrated that reliance on the AG (particularly if not corrected for albumin) or BE results in false interpretation of the acid–base status of critically ill patients. The strong ion approach, though requiring a few more direct laboratory measurements, is comprehensive and thus more sensitive.
Durward et al. obtained arterial blood samples on admission and at 24 h in 85 children undergoing surgery for congenital heart disease. An elevated strong ion gap (>3 mEq/L) was present in 41% of children on admission and 52% at 24 h. Both the strong ion gap and lactate increased with surgical complexity. [SIG] was superior to lactate in predicting mortality [181].
Moviat et al. conducted a prospective, observational multicenter study in Dutch ICU patients. 137 of 312 consecutive patients (44%) who had normal pH, normal base excess, and normal PaCO2 underwent arterial blood sampling. Strong ion gap was calculated from three consecutive arterial blood samples. These patients were found to have mixed acid–base disorders due to hyperchloremia and hypoalbuminemia, in spite of normal blood gas parameters [182].
Zheng and colleagues [183] studied 78 patients with metabolic acidosis with or without acute kidney injury (AKI). They analyzed physicochemical parameters at 24 and 72 h, at 1 week, 1 month, and 3 months on survival after AKI. Mortality was higher in AKI group with higher anion gaps and strong ion gaps. The AG was strongly associated with mortality at 1 and 3 months after acute kidney injury. However, an elevated [SIG] most strongly predicted mortality at 24 h, 72 h, 1 week, 1 month, and 3 months post acute kidney injury.
Applying Strong Ion Theory to the Base Excess
The corrected base excess approach to using clinical quantitative acid–base chemistry is illustrated by Balasubramanyan and colleagues [174]. They used three equations of Fencl and Leith [177] to calculate a corrected BE that takes into account the fact that free water, changes in chloride concentration, and albumin all affect BE. First, the effect on BE caused by free water (BEfw) is calculated as
Changes in BE accounted for by chloride are calculated as
where Cl− corr is the corrected chloride: Cl− × 140/Na+. Finally, the effect of albumin on BE (BEalb) is calculated as
where albumin is reported in grams per deciliter.
The “classic” BE, as calculated from plasma bicarbonate, is really the sum of these three components plus a component attributable to unmeasured anions, BEua:
If one subtracts the first three from reported BE, one obtains BEua or BE caused by unmeasured anions:
In his study of 255 children in whom arterial blood gases, electrolytes, and albumin were measured simultaneously, Balasubramanyan demonstrated that BEua predicts increases in plasma lactate concentration better than BE and, furthermore, that it is a better predictor of mortality than either BE or the lactate concentration [174].
Story et al. [184] further simplified the Fencl–Stewart methodology based on the premise that a change in the sodium-chloride component of the [SID] will change the base excess directly. Assuming a normal median sodium of 140 mEq/L and a normal median chloride of 102 mEq/L, the median difference is 38 mmol/L. The simplified version of the equation for sodium minus chloride effect on base excess is thus
The albumin likewise has a direct effect on the base excess as the principal contributor to plasma total weak acid concentration. The simplified Fencl–Stewart equation for albumin effect is as follows:
This was tested prospectively in a non-randomized ICU study of 300 patient samples. In the samples, they compared the agreement between the more complex Fencl–Stewart equations and their simplified versions using Bland–Altman analyses. They concluded that the simplified equations agreed well with the more complex equations. The authors proposed analysis of blood gases using the following four steps: (1) obtain standard base excess (mmol/L) from a blood gas machine; (2) calculate sodium-chloride effect (mEq/L) = [Na+] – [Cl−] -38; (3) calculate albumin effect (mEq/L) = 0.25 × [42 – albumin (g/L)]; and (4) calculate unmeasured ion effect:
Employing these four steps, Ahmed and colleagues performed a randomized, prospective, interventional trial among 300 patients with “abdominal sepsis.” Patients were randomized to receive 20 mL/kg of either normal saline or Ringers lactate over 30 min. Patients in the normal saline arm had a significant drop in pH and base excess, whereas Ringers lactate patients did not. See Fig. 2. This was largely explained by the sodium-chloride effect on base excess. Dilution of serum albumin caused a partial correction of acidosis, creating a metabolic alkalosis. This mixed acid-based disorder would not have been recognized by the standard base excess approach or the anion gap approach but was detected by the Fencl–Stewart method [185].

Infusion of electrolyte solutions of varying SID will, according to the strong ion theory, variably alter the pH, base excess, and the concentrations of the strong ions themselves. These panels represent the effects of infusion of 20 ml/kg of either normal saline (NS) or Ringer’s lactate (RL) over 30 min. NS has an [SID] of 0 mmol/L ([Na+]−{Cl−] = 154 – 154 = 0 mmol/L), whereas RL has an SID of 28 mmol/L ([Na+] + [K+] + [Ca2+] − [Cl−] = 130 + 4 + 3 – 109 = 28 mmol/L). Panel A. Changes in pH following infusion of normal saline or Ringer’s lactate. Panel B. Changes in serum base excess, sodium, chloride, and anion gap following infusion of normal saline or Ringer’s lactate. Panel C. Changes in standard base excess, sodium minus chloride effect, albumin effect, and unmeasured anion effect on base excess following infusion of normal saline or Ringer’s lactate (Adapted from Ahmed et al. [185] doi: 10.4103/2229-5151.119203. PubMed PMID: 24404459; PubMed Central PMCID: PMC3883200. Copyright: © International Journal of Critical Illness and Injury Science. This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited)
The studies of Balasubramanyan [174], Fencl [155], and their colleagues provide evidence that critical care patients warrant an aggressive evaluation of acid–base status.
Osmol Gap
Osmolality is an expression of the number of particles in a given weight of solvent. Thus, each molecule of a substance, regardless of its molecular weight, contributes exactly the same as a molecule of another substance to osmolality. The vast majority of circulating osmols consist of sodium (and its associated anions), glucose, and urea. The presence of unanticipated osmols (osmol gap) is a potentially useful signal of the presence of a toxic alcohol. The true utility of the osmol gap in poisoned patients has been questioned, perhaps unfairly, on the basis of poor sensitivity [186]. However, in true clinical circumstances, the sensitivity of the osmol gap for “toxic alcohol” poisoning is 90% [187]. The osmol gap is calculated as
where MO is the measured osmolality and CO the calculated (anticipated) osmolarity. The small difference between osmolality and osmolarity is ignored in the calculation of the osmol gap. There are normally a limited number of unmeasured osmols, so a small gap is expected. In the context of a poisoned patient, the OG is most commonly measured in an attempt to determine whether substantial concentrations of circulating exogenous osmols are present. Generally, the presence of excess measured osmols in a poisoned patient will be due to alcohols or glycols, such as ethanol, methanol, isopropanol, ethylene glycol , or propylene glycol, the last often present because of its widespread use in therapeutically administered medications. Since alcohols are volatile, MO for them should be determined by freezing-point depression to ensure that they are not liberated during determination of osmolality. This is not as much of a concern with glycols. The relative contributions of several molecules of interest to MO are indicated in Table 5. It should be emphasized that the figures in this table are theoretical and should be used only as a rough guide to expected changes in osmolality or, conversely, plasma concentration. Osmolality is measured in mOsm/kg H2O. Numerous formulas have been used for calculation of this gap, and one popular formula for the predicted serum osmolality, proposed by Dorwart and Chalmers, [159] follows:
where concentrations of sodium, glucose, and blood urea nitrogen (BUN) are expressed in mmol/L.
where sodium is expressed in mmol/L and glucose and urea in mg/dL [159].
Purssell and colleagues [189] derived an osmol gap formula using linear regression with the blood ethanol concentration taken into account and then validated it in convenience samples from 128 patients. This formula is based on that of Mahon et al. [190], who used a multiplier of 2 for Na+ rather than 1.86. Purssell added a correction factor of 1.25 in SI units for the contribution to osmolality by ethanol. Given the frequency with which ethanol is found in poisoned patients, its presence should be confirmed and included in the calculation. Thus, adding the Purssell ethanol correction factor, one arrives at a formula for CO, corrected for measured blood ethanol (COcorrected):
If mass units (mg/dL) are used for the latter three, the following formula is used:
Two more recent studies have studied the effect of ethanol on the osmol gap [191, 192]. Carstairs and colleagues gave ten healthy volunteers up to 140 mL 100% ethanol in sugar-free soda (n = 8) or plain sugar-free soda (n = 2). The Dorwart formula was used. The osmol gap was divided by the blood ethanol to determine the mean coefficient of ethanol’s contribution to serum osmolality, yielding 4.25 (95% CI, 4.13–4.38). This divisor is smaller than that predicted by molecular weight of ethanol, indicating that it contributes more to total osmolality than predicted for an ideal solute [191]. The study has been criticized for proposing a mean coefficient when the underlying individual subjects had coefficients that were substantially inconsistent [193]. In a much larger study of 603 emergency department patients, Garrard and colleagues retrospectively reviewed patients who had concurrent ethanol, basic metabolic panel, and measured serum osmolality results available [192]. The median ethanol concentration was 166 mg/dL (36 mmol/L). The mean osmol gap was 47, with a significant proportional relationship between ethanol and osmol gap. The authors concluded that the contribution of ethanol to the osmol gap is defined by the ethanol concentration (mg/dL)/4.0 [192].
Dozens of formulas of varying performance have been proposed for the plasma osmolality calculation. Not surprisingly skepticism about the value of the osmolal gap as a predictive tool for the presence of toxic alcohols has arisen [186]. Nonetheless, many hospitals do not have access to immediate confirmation for the presence and quantity of toxic alcohols in the blood. They thus depend on the anion and osmol gaps for determining the likelihood of a toxic alcohol exposure.
Khajuria and Krahn [188], in an effort to derive a formula that can be used in normal patients, as well as in those who are hyperglycemic and/or intoxicated, performed a series of in vitro experiments and tested several formulas for the calculated osmol gap. They demonstrated that glucose and alcohols don’t accurately predict osmolality on the basis of their molecular weights alone. The experiments demonstrated the need for correction factors for glucose, ethanol, methanol, and ethylene glycol of 1.15, 1.20, 1.07, and 1.00, respectively, in order to accurately predict osmolality. The authors then tested two formulas against data from 37 healthy volunteers (without ethanol) and 129 emergency department patients with mean ethanol concentrations of 41.5 ± 27.0 mmol/L. Both formulas predicted osmolality well, in the presence or absence of ethanol. One of the two formulas, a modification of the Dorwart formula [159], with potassium added follows:
This formula yielded a mean osmol gap of 0.77 ± 3.80 mOsm/kg in healthy volunteers (without ethanol). The mean osmol gap in patients with ethanol present was −0.2 ± 5.00 mOsm/kg, with a reference interval of −8.04–6.50 mOsm/kg [188], substantially narrower than that reported by Hoffman and colleagues (−5–15 mOsm/kg, using a different formula) [186]. This formula and that of Siervo (see below) utilize urea in SI units rather than BUN. However, there is 1 mmol of nitrogen in 1 mmol of urea, so the conversion is the same as usual if BUN is employed (i.e., divide by 2.8 to obtain mmol/L from mg/dL):
One notes that the correction factor for ethanol in this case resolves to 4.6/1.2 = 3.8, almost precisely the correction factor proposed by Purssell [189].
Siervo and colleagues [194] subsequently compared 38 predictive equations, including the Khajuria formula (minus the ethanol correction), in a group of 186 frail older people with and without diabetes. They found four equations which showed reasonable agreement with measured osmolality, but that the formula with narrowest limits of agreement was the following:
\( \mathrm{C}\mathrm{O} = 1.86\left[{\mathrm{Na}}^{+}+{\mathrm{K}}^{+}\right] + 1.15\left[\mathrm{Glu}\right] + \left[\mathrm{Urea}\right] + 14 \). One immediately notes that this is the same formula as that of Khajuria (Eq. 22a) minus the correction for ethanol.
Martin-Calderon and colleagues [195] independently compared 14 calculated osmolality equations, comparing goodness of fit of the calculation with osmolality measured by freezing-point depression among 146 healthy volunteers (96 males/50 females). They warned that the original Dorwart–Chalmer equation should not be used for osmolality calculations. Like Siervo, they did not include ethanol in their calculations. The best fit was obtained by the same equation adjusted for use of mass units:
* BUN/2.8 was substituted for urea/6 for use in the USA, where measurement of urea is uncommon. Thus, while prospective confirmation of its validity in a wide variety of patients with normal values, hyperglycemia, elevated blood ethanol, and/or toxic alcohols is needed, this formula (adding the correction for ethanol) clearly looks promising.
One other issue of importance to the calculation of the osmol gap is that of “pseudohyponatremia” associated with hyperglycemia. Pseudohyponatremia is an accurate description of the volume exclusion effect of very elevated triglycerides or protein when sodium is measured by indirect methods. The move to specific ion electrodes has largely eliminated this issue. The decrease in sodium seen with hyperglycemia is not pseudohyponatremia, but rather a shift of water from the intracellular space to the plasma induced by the osmotic effect of glucose, such that the sodium in the vascular compartment is indeed decreased by dilution. The specific ion electrode for sodium provides accurate measurements even in the presence of elevated glucose; thus, there is no reason to “correct” the sodium for elevated glucose in calculating the osmol gap [196].
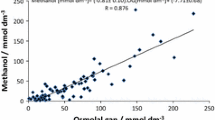
So, what is one to do with these different equations for calculated osmols? First, inquire from your laboratory whether they have determined the normal osmol gap in your patient population. As Purssell warns, the calculation of the osmolal gap at the bedside without knowledge of how the calculation works in that environment is poor practice and subject to serious error [189]. Of the published equations, the Khajuria (Eqs. 22a and 22b) appear to offer very close correlation with measured osmols. In the end, it is important to remember that assuming the presence or absence of a toxic alcohol on the basis of the osmol gap, regardless of formula, is not ideal. It is imperative to understand that the range of “normal” osmol gaps will vary significantly, depending on the calculation chosen and on laboratory equipment used for the analysis. The osmol gap appears to be most reliable when it exceeds 25 mOsm/kg [197], although a more recent paper reports excellent sensitivity down to 10 mOsm/kg [187]. Because of variations in the results (and normal values) obtained depending on the formula used, the relatively small number of osmols produced by a toxic concentration of ethylene glycol (a blood concentration of 25 mg/dL [4 mmol/L] is equivalent to only ≈4 mOsm/kg see Table 5), and the possibility of missing even severe very early or late manifestations of ethylene glycol or methanol toxicity, the osmol gap should be interpreted with caution. Small or absent anion or osmol gaps may be obtained after severe toxic alcohol poisoning, depending on the extent of absorption and metabolism that have taken place since the ingestion. Therefore, both anion and osmol gaps must be viewed with prudence and do not completely rule out such poisonings. If any clinical suspicion exists, a direct measurement of the possible offending alcohols or glycols should be obtained and strong consideration given to initiation of blockade of alcohol dehydrogenase (ADH). When the osmol gap is elevated in the case of methanol poisoning, the osmol gap may be followed to assess treatment progress. Jacobsen and colleagues have reported on 28 patients from a recent methanol outbreak, in whom the osmol gap predicted in a linear fashion the methanol concentration, with a correlation coefficient of 0.94 [198]. This same group demonstrated good correlation between serum methanol and osmol gaps during hemodialysis, concluding that, in the absence of serum methanol analyses, the osmol gap is useful to assess indication for and duration of dialysis in methanol poisoned patients [199].
An unvalidated algorithm in which the various gaps are used for evaluation of acid–base disorders is presented in Fig. 3. Suggestions for use of the anion and delta gaps are found in Figs. 4 and 5, respectively. A side-by-side comparison of the Stewart and modified anion gap–base excess methodologies is found in Fig. 6.

An unvalidated algorithm (level of evidence III) for the evaluation of acid–base disorders based on clinical suspicion of a disorder or patient acuity. While simple acid–base evaluation, using classic laboratory studies and calculation of “gaps,” may suffice where suspicion of acid–base derangements and acuity is low, quantitative “strong ion” analysis is indicated where there is high suspicion of acid–base disorder or high acuity. Failure to clarify the etiology of the disorder with simple gaps and ABG analysis should also indicate the need for quantitative acid–base analysis. See text and Fig. 6 for details on calculation. Abbreviations: ABG arterial blood gas, AG anion gap, ALB − serum albumin, AMS altered mental status, BE base excess, LOE level of evidence, Pi inorganic phosphate, [SIG] strong ion gap. Level of evidence: Corrected anion gap-II-2, Delta gap-III, [SIG]-I, BE corrected-II-1

Evaluation of the anion gap (AG). *AG is calculated as [Na+] − ([Cl−] + [HCO3 −]). The normal range of AG is 3–11 mmol/L but will vary according to individual hospital laboratories. **Correction for hypoalbuminemia: AG observed + 0.25 × ([Albumin, nL] − [Albumin observed]) = AG corrected, where normal albumin is considered to be 45 g/L. ***Correction for hyperphosphatemia: AG observed − 0.32 × ([Phosphate observed] − [Phosphate, nL]) = AG corrected, where the upper normal phosphate level is considered to be 5 mg/dL. If phosphate is reported in mmol/L, simply drop the multiplier of 0.32. ABG, arterial blood gases; [SIG], strong ion gap. Level of evidence: Corrected anion gap-II-2

Evaluation of the delta gap. *The delta gap is calculated as Δgap = ΔAG − ΔHCO3 −, where ΔAG = observed AG − the upper normal limit of the AG and ΔHCO3 − = lower normal HCO3 − – observed HCO3 −. The normal range for the delta gap is 0 ± 6. **Albert formula: Expected PaCO2 = 1.5 [HCO3 −] + 8 + 2. ***See text and Fig. 6 for calculation of [SIG] (XA−) (Adapted from Wrenn [141]). Level of evidence: Delta gap-III

Evaluation of patients with perceived complex acid–base disorders. Both Stewart physicochemical methods and modifications of the anion gap and base excess methodology have been proposed in recent years. The choice of methodology remains controversial, so multiple methods of acid–base analysis are provided here. Abbreviations: Alb −albumin, ABG arterial blood gas, AG c corrected anion gap, BUN blood urea nitrogen, [SID] strong ion difference, [SIG] strong ion gap, UA unmeasured anions. Level of evidence: Corrected anion gap-II-2; Delta gap-III; [SIG]-I; BE corrected-II-1
Arterial Blood Gases
Interpretation of arterial blood gases in a poisoned patient does not differ significantly from that in other patients and is therefore not discussed in detail here. It is worthwhile, however, to note that according to the strong ion theory, the change in pH induced by a change in PaCO2 will vary depending on [SID] and ATOT, thus indicating that PaCO2 is important not only in respiratory acidosis but also in metabolic acidosis and reinforcing the importance of evaluation of all three independent variables [176]. The compensation formulas for simple acid–base disorders proposed by Narins and Emmett [200] are provided in Table 6. The caveats discussed above with regard to “standard” bicarbonate hold.
Treatment of Acid–Base Disorders
The sine qua non of therapy for acid–base disorders is correction of the underlying cause of the disorder. Attention to the patency of the airway and adequacy of ventilation and oxygenation cannot be overemphasized.
In terms of metabolic derangements, the strong ion theory significantly simplifies the task of correction. A careful evaluation of strong ions will determine the need for electrolyte replacement, so reflexive administration of large volumes of solutions that alter [SID], such as sodium chloride, is avoided in the treatment of metabolic acidosis. Hypoperfusion persisting in spite of correction of vascular water and electrolyte deficits may require the use of vasoactive substances. It must be appreciated that their use may often worsen acidosis via the production of lactate, so careful attention to the risk–benefit ratio of their use is of utmost clinical interest [201, 202].
Since the advent of the Stewart approach, increasing attention has been directed at correction of strong ion differences through the use of various crystalloid solutions. Morgan and colleagues studied the relationship between the strong ion difference of a diluting crystalloid and its acid–base effects on in vitro blood dilution [203]. They tested three solutions of varying chloride content (120, 110, and 100 mmol/L ([SID] equals 20, 30, and 40 mEq/L respectively), as well as 0.9% saline ([SID] equals 0) and Hartmann solution ([SID] equals −4 mEq/L). With the exception of the solution of 40 mEq/L [SID], for which [SID] did not change, plasma [SID] decreased during hemodilution. For solutions with higher [SID], base excess increased during hemodilution. The relationship between hemoglobin concentration and both plasma [SID] and a whole blood base excess were linear. Linear regression revealed the [SID] of crystalloid producing a 0 base excess/hemoglobin concentration slope during blood dilution (in other words no acid–base change) was 23.7 mEq/L. This same group demonstrated in rats undergoing normovolemic hemodilution that there is a linear relationship between the crystalloid [SID] and the postdilutional metabolic acid–base status [204]. The [SID] of a crystalloid balance for normovolemic hemodilution is 24 mEq/L, identical to the standard bicarbonate.
Omron and Omron performed clinical simulations of modeled acid–base and fluid compartment parameters in a 70 kg test participant at standard physiologic state [205]. Comparing simulated infusions of up to 10 L of normal saline ([SID] equals 0), Ringers lactate ([SID] equals 28), Plasma-Lyte 148 ([SID] = 50), half normal saline +75 mEq sodium HCO3 − ([SID] = 75) and 0.15 mole/L sodium ([SID] = 150) with a hypothetical crystalloid solution of [SID] = 24.5 mEq/L, the authors demonstrated that a crystalloid [SID] equivalent to standard-state actual HCO3 − (24.5 mEq/L) results in a neutral metabolic acid–base status or standardized base excess approximately equal to 0 mEq/L for infusions up to 10 L. The larger the infused volume, the greater the displacement of SBE from 0 mEq/L when crystalloid [SID] and actual HCO3 − are discordant. The lower the [SID] relative to actual HCO3 −, the greater the standard base deficit; the greater the crystalloid [SID] relative to actual HCO3 −, the greater the SBE. A1 L normal saline infusion resulted in metabolic acidosis, whereas the same infusion of Ringers lactate resulted in metabolic alkalosis.
In 2013, Morgan [123] reviewed the subject of the “balanced” crystalloid in the context of quantitative acid–base chemistry. Noting that the [SID] of isotonic saline is 0 (equal concentrations of sodium and chloride), Morgan makes the case that administration of large volumes of saline over a relatively short period of time forces reductions in [SID], leading to hyperchloremic metabolic acidosis, while at the same time reducing Atot through dilution of plasma proteins and phosphate, which gives rise to metabolic alkalosis. The net result however is metabolic acidosis. Admittedly, saline-induced acidosis is relatively mild to moderate. Nonetheless, he underscores that there are data suggesting that hyperchloremic metabolic acidosis is pro-inflammatory, causing multiorgan dysfunction. He reminds us that reduced chloride exposure may lessen kidney injury with a possible mortality benefit [206]. To achieve exact balance from an acid–base perspective, it is sufficient to reduce the strong ion difference to 24 mEq/L. This can be done by replacing chloride in an isotonic saline solution with HCO3 or a substitute such as acetate. He points out that Ringer’s acetate solution is as close to a balanced solution that exists currently, both from acid–base and tonicity perspectives. He suggests that a large, randomized clinical trial is necessary to establish definitively the benefits of using a balanced crystalloid solution [123].
Raghunathan and colleagues carried out a retrospective cohort study of patients admitted with sepsis, comparing those who received only “no-balanced fluids” (i.e., isotonic saline with or without dextrose 5% [SID] = 0) against patients who received some (widely variable) quantity of “balanced” solutions such as Ringers lactate in 53,448 patients among 360 US hospitals [207]. While the patients treated with balanced fluids were younger and less likely to have heart disease or chronic renal failure, they were more likely to receive mechanical ventilation, invasive monitoring, colloids, steroids, and larger crystalloid volumes. The authors then applied propensity matching to arrive at two comparison groups of 3,396 matched subjects. Receipt of balanced fluids was associated with lower in-hospital mortality (19.6% vs. 22.8%) with mortality progressively decreasing among patients receiving larger proportions of balanced fluids [207]. While these findings are of interest, there are clearly methodological issues. Thus, a large randomized controlled trial of well-defined balanced versus unbalanced fluids is clearly needed.
The comparison of Ringers lactate to normal saline and abdominal sepsis patients by Ahmed and colleagues has already been cited. Figure 2 demonstrates significant differences in pH and base excess induced by relatively modest (20 mL/kg) infusion volumes of 0 [SID] solutions such as normal saline.
The specific (i.e., other than supportive) treatment of acid–base disorders remains controversial. For many years, the cornerstone of treatment of metabolic acidosis, with the exception of DKA, was the administration of bicarbonate, sometimes in very large quantities. Correction of abnormal pH (excess H+) via administration of bicarbonate appears logical on the surface. However, Graf and colleagues [208] pointed out evidence that administration of bicarbonate may actually worsen intracellular acidosis, which led to proposals by some that we should “ban bicarbonate” in the treatment of acidosis [209]. Whether this rapid swing of the pendulum in the opposite direction was warranted depends on the belief that all metabolic acidosis is alike and should therefore be treated similarly. One might question, however, whether the acidosis produced by metabolic conversion of methanol is equivalent to the lactic acidosis produced in septic shock. Kellum has pointed out that when treatment is based on [SID], the need for bicarbonate therapy becomes clear [175]. If the disorder involved is characterized by decreased or normal sodium, administration of sodium in the form of bicarbonate may in fact be warranted. However, it is in reality the sodium concentration (the strong ion) that is being addressed. If one accepts the Stewart theory, bicarbonate will actually be determined by the three independent variables (strong ions, PaCO2, and weak acids/salts). Only by increasing sodium relative to chloride can sodium bicarbonate “repair” metabolic acidosis. Thus, when sodium is already increased, there is no role for bicarbonate therapy [175]. Other considerations make the use of sodium bicarbonate potentially appropriate in metabolic acidosis of toxic origin. Alkalinization of the urine has a demonstrated beneficial effect on the distribution of salicylates and barbiturates by hastening elimination and diminishing CNS penetration. Sodium administration (as bicarbonate or hypertonic saline) appears to be effective as well in the treatment of cyclic antidepressant-related dysrhythmias, which may be associated with metabolic acidosis [210–212]. In summary, although bicarbonate should no longer be viewed as the “universal antidote” for metabolic acidosis, its use should not be summarily abandoned in all cases of acidosis. Kraut and Madias have recently provided recommendations for calculation of bicarbonate requirements in severe lactic acidosis (pH ≤ 7.10), diabetic ketoacidosis (pH ≤ 7.0), and non-gap metabolic acidosis (pH ≤ 7.20) when attempts at correcting the underlying disorder have been unsuccessful:
The authors further recommend that, in order to minimize potential complications of bicarbonate administration, therapy should be initiated based on calculation using bicarbonate space of 50% body weight (kg); if this is not successful in achieving desired serum [HCO3 −], administer larger quantities of bicarbonate based on bicarbonate space calculated from the above formula [213].
The use of the strong ion approach allows us to not only recognize the often multiple underlying causes of acid–base disorders but also correct them. Hypoalbuminemia contributing to severe metabolic alkalosis may warrant replacement therapy. Chloride excess leading to acidosis may be addressed by hemofiltration or the use of a weak base such as tris(hydroxymethyl)aminomethane (THAM, or tromethamine). The presence of a strong ion gap (excess unmeasured anions) calls for therapy to reduce their production or facilitate their removal [175].
THAM has been proposed as an alternative to sodium bicarbonate, because it reduces arterial hydrogen ion concentration without producing CO2 and penetrates cells easily, reducing intracellular acidosis [214]. Weber and colleagues studied THAM in 12 patients undergoing permissive hypercapnia for ARDS [215]. Patients who received THAM experienced less myocardial depression and lessened effects of hypercapnia on arterial pressure and mean pulmonary arterial pressure. Marfo and colleagues reported the case of a patient with severe lactic acidosis secondary to highly active antiretroviral therapy successfully treated with THAM [216]. Several studies have shown promising results in selected patient populations [217, 218], but additional studies are needed. Kraut and Madias recommend consideration of THAM in patients with lactic acidosis, diabetic ketoacidosis, or non-gap metabolic acidosis in patients when CO2 retention is present or incipient. They estimate THAM requirements using the following formula:
where base deficit = desired serum [HCO3 −] – actual serum [HCO3 −] [213].
Finally, extracorporeal treatments may be of interest in acidosis of toxic etiology [219–221]. Hemodialysis can correct not only the plasma bicarbonate (the result of the underlying disturbance in strong ion imbalance) but also some strong ion abnormalities (hyperkalemia). Furthermore, it has the capacity to remove a number of endogenous (lactate, pyruvate) and exogenous (methanol and metabolites, ethylene glycol and metabolites) compounds of low molecular weight and limited volume of distribution.
Based on admittedly limited data (mostly case reports), the Extracorporeal Treatments in Poisoning (EXTRIP) workgroup has recommended extracorporeal removal of metformin in the case of severe poisoning (plasma lactate > 20 mmol/L [180 mg/dl], pH ≤ 7.0, shock, failure of standard support measures, and decreased level of consciousness) [222]. EXTRIP has also recently released recommendations for extracorporeal treatment for valproic acid (VPA) poisoning. Intermittent hemodialysis is recommended for valproic acid poisoning when at least one of the following criteria for severe poisoning is present: VPA concentration >1,300 mg/L (9,000 μmol/L), the presence of cerebral edema, or shock; suggestions for extracorporeal treatment include a VPA concentration >900 mg/L (6,250 μmol/L), coma or respiratory depression requiring mechanical ventilation, acute hyperammonemia, or pH ≤ 7.10 [223]. Note, however, that some degree of hyperammonemia is common with even minor VPA toxicity. Thus, the hyperammonia should be significant if that is the only reason the patient is being considered for extracorporeal drug elimination. Cessation of extracorporeal treatment (ECTR) is indicated when clinical improvement is apparent or the serum VPA concentration is between 50 and 100 mg/L (350–700 μmol/L). Intermittent hemodialysis is the preferred ECTR in VPA poisoning. If hemodialysis is not available, then intermittent hemoperfusion or continuous renal replacement therapy is an acceptable alternative. EXTRIP has recommended extracorporeal treatment for acetaminophen (APAP) poisoning in the case of mitochondrial dysfunction as reflected by early development of altered mental status and severe metabolic acidosis preceding liver failure [224]. They point out that extracorporeal treatment should be reserved for those rare situations when the efficacy of N-acetylcysteine (NAC) cannot be definitively demonstrated. Specific recommendations for extracorporeal treatment include: an APAP concentration over 1,000 mg/L (6,600 μmol/L) if NAC is not administered, signs of mitochondrial dysfunction and an APAP concentration over 700 mg/L (4,630 mmol/L) if NAC is not administered, and signs of mitochondrial dysfunction and an APAP concentration over 900 mg/L (5,960 mmol/L) if NAC is administered (level of evidence [LOE] = III). Intermittent hemodialysis (HD) is the preferred ECTR modality in APAP poisoning [LOE] = III.
Summary
Acid–base disorders in poisoned patients may be of multiple toxic and nontoxic etiologies. A careful history and physical examination are necessary to suspect the disorder, and a thorough laboratory evaluation is required to distinguish the probable etiology. Reliance on simple calculations such as anion or osmol gaps may result in errors, including missed diagnosis. A systematic approach using multiple diagnostic tools, including the quantitative (strong ion) method, will decrease the likelihood of errors. Treatment is largely supportive. Bicarbonate or THAM therapy is useful in selected cases but should be based on a quantitative analysis of acid–base derangements rather than being a rote response to abnormal laboratory values. Hemodialysis may also be of benefit in selected patients, again relying on the patient’s clinical condition as a guide to therapy.
References
Gunnerson KJ, Saul M, He S, Kellum JA. Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients. Crit Care. 2006;10(1):R22.
Dell’Aglio DM, Perino LJ, Kazzi Z, Abramson J, Schwartz MD, Morgan BW. Acute metformin overdose: examining serum pH, lactate level, and metformin concentrations in survivors versus nonsurvivors: a systematic review of the literature. Ann Emerg Med. 2009;54(6):818–23.
Tanasescu A, Macovei RA, Tudosie MS. Outcome of patients in acute poisoning with ethylene glycol – factors which may have influence on evolution. J Med Life. 2014;7(Spec No. 3):81–6.
Kostic MA, Dart RC. Rethinking the toxic methanol level. J Toxicol Clin Toxicol. 2003;41(6):793–800.
Rastegar A. Clinical utility of Stewart’s method in diagnosis and management of acid–base disorders. Clin J Am Soc Nephrol. 2009;4(7):1267–74.
Al-Jaghbeer M, Kellum JA. Acid–base disturbances in intensive care patients: etiology, pathophysiology and treatment. Nephrol Dial Transplant. 2014;30(7):1104–11.
O’Malley GF. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin North Am. 2007;25(2):333–46. abstract viii.
Fernandez R, Larrain C, Zapata P. Acute ventilatory and circulatory reactions evoked by nicotine: are they excitatory or depressant? Respir Physiol Neurobiol. 2002;133(3):173–82.
Karapetian GK, Engels HJ, Gretebeck KA, Gretebeck RJ. Effect of caffeine on LT, VT and HRVT. Int J Sports Med. 2012;33(7):507–13.
Kolarzyk E, Targosz D, Pach D, Misiolek L. Nervous regulation of breathing in opiate dependent patient. Part I. Respiratory system efficiency and breathing regulation in the first stage of controlled abstinence. Przegl Lek. 2000;57(10):531–5.
Campbell C, Weinger MB, Quinn M. Alterations in diaphragm EMG activity during opiate-induced respiratory depression. Respir Physiol. 1995;100(2):107–17.
Gueye PN, Lofaso F, Borron SW, Mellerio F, Vicaut E, Harf A, et al. Mechanism of respiratory insufficiency in pure or mixed drug-induced coma involving benzodiazepines. J Toxicol Clin Toxicol. 2002;40(1):35–47.
Patel AM, Adeseun GA, Goldfarb S. Calcium-alkali syndrome in the modern era. Nutrients. 2013;5(12):4880–93.
Patel AM, Goldfarb S. Got calcium? Welcome to the calcium-alkali syndrome. J Am Soc Nephrol. 2010;21(9):1440–3.
Wu KD, Chuang RB, Wu FL, Hsu WA, Jan IS, Tsai KS. The milk-alkali syndrome caused by betelnuts in oyster shell paste. J Toxicol Clin Toxicol. 1996;34(6):741–5.
Muller-Lissner SA. Adverse effects of laxatives: fact and fiction. Pharmacology. 1993;47 Suppl 1:138–45.
Tatsumi H, Masuda Y, Imaizumi H, Kuroda H, Yoshida S, Kyan R, et al. A case of cardiopulmonary arrest caused by laxatives-induced hypermagnesemia in a patient with anorexia nervosa and chronic renal failure. J Anesth. 2011;25(6):935–8.
Chiang WF, Yan MT, Wu TJ, Lin SH. A hypokalaemic woman with nephrocalcinosis: rebirth of old knowledge. Ann Clin Biochem. 2013;50(Pt 2):176–9.
Miller JL, Schaefer J, Tam M, Harrison DL, Johnson PN. Ethacrynic Acid continuous infusions in critically ill pediatric patients. J Pediatr Pharmacol Ther. 2014;19(1):49–55.
Isaia GC, Pellissetto C, Ravazzoli M, Tamone C. Acute adrenal crisis and hypercalcemia in a patient assuming high liquorice doses. Minerva Med. 2008;99(1):91–4.
Onishi A, Muto S, Homma S, Inaba T, Shuto R, Kusano E, et al. Pseudoaldosteronism with increased serum cortisol associated with pneumonia, hypouricemia, hypocalcemia, and hypophosphatemia. Clin Nephrol. 2010;74(5):403–8.
Chrispal A, Boorugu H, Prabhakar AT, Moses V. Amikacin-induced type 5 Bartter-like syndrome with severe hypocalcemia. J Postgrad Med. 2009;55(3):208–10.
Arroyo M, Fenves AZ, Emmett M. The calcium-alkali syndrome. Proc (Baylor Univ Med Cent). 2013;26(2):179–81.
Olveira Fuster G, Mancha Doblas I, Vazquez San Miguel F, de Antonio EI, CSE F. Surreptitious intake of diuretics as the cause of pseudo-Bartter’s syndrome: apropos of a case and differential diagnosis. An Med Interna. 1996;13(10):496–9.
Ricci Z, Haiberger R, Pezzella C, Garisto C, Favia I, Cogo P. Furosemide versus ethacrynic acid in pediatric patients undergoing cardiac surgery: a randomized controlled trial. Crit Care. 2015;19(1):2.
Lenzenhuber E, Muller C, Rommelspacher H, Spies C. Gamma-hydroxybutyrate for treatment of alcohol withdrawal syndrome in intensive care patients. A comparison between with two symptom-oriented therapeutic concepts. Anaesthesist. 1999;48(2):89–96.
Chou CL, Chen YH, Chau T, Lin SH. Acquired bartter-like syndrome associated with gentamicin administration. Am J Med Sci. 2005;329(3):144–9.
Shetty AK, Rogers NL, Mannick EE, Aviles DH. Syndrome of hypokalemic metabolic alkalosis and hypomagnesemia associated with gentamicin therapy: case reports. Clin Pediatr (Phila). 2000;39(9):529–33.
Centers for Disease Control, Prevention. Infant metabolic alkalosis and soy-based formula – United States. 1979. MMWR Morb Mortal Wkly Rep. 1996;45(45):985–8.
Herrmann U, Schwille PO, Schwarzlaender H, Berger I, Hoffmann G. Citrate and recurrent idiopathic calcium urolithiasis. A longitudinal pilot study on the metabolic effects of oral potassium sodium citrate administered as short-, medium- and long-term to male stone patients. Urol Res. 1992;20(5):347–53.
Smith GI, Jeukendrup AE, Ball D. Sodium acetate induces a metabolic alkalosis but not the increase in fatty acid oxidation observed following bicarbonate ingestion in humans. J Nutr. 2007;137(7):1750–6.
Adeva-Andany MM, Fernandez-Fernandez C, Mourino-Bayolo D, Castro-Quintela E, Dominguez-Montero A. Sodium bicarbonate therapy in patients with metabolic acidosis. Scientific World Journal. 2014;2014:627673.
Ichai C, Orban JC, Fontaine E. Sodium lactate for fluid resuscitation: the preferred solution for the coming decades? Crit Care. 2014;18(4):163.
Geara AS, Parikh A, Rekhtman Y, Rao MK. The case mid R: metabolic alkalosis in a patient with cystic fibrosis. Kidney Int. 2012;81(4):421–2.
Gee P, Richardson S, Woltersdorf W, Moore G. Toxic effects of BZP-based herbal party pills in humans: a prospective study in Christchurch, New Zealand. N Z Med J. 2005;118(1227):U1784.
Balikova M. Nonfatal and fatal DOB (2,5-dimethoxy-4-bromoamphetamine) overdose. Forensic Sci Int. 2005;153(1):85–91.
Ben-Abraham R, Szold O, Rudick V, Weinbroum AA. “Ecstasy” intoxication: life-threatening manifestations and resuscitative measures in the intensive care setting. Eur J Emerg Med. 2003;10(4):309–13.
Greene SL, Dargan PI, O’Connor N, Jones AL, Kerins M. Multiple toxicity from 3,4-methylenedioxymethamphetamine (“ecstasy”). Am J Emerg Med. 2003;21(2):121–4.
Liss DB, Paden MS, Schwarz ES, Mullins ME. What is the clinical significance of 5-oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–27.
Kassamali R, Sica DA. Acetazolamide: a forgotten diuretic agent. Cardiol Rev. 2011;19(6):276–8.
Muraki K, Inoue Y, Ohta I, Kondo K, Matayoshi Y, Kamei T. Massive rhabdomyolysis and acute renal failure after acetonitrile exposure. Intern Med. 2001;40(9):936–9.
Foley RJ. Inhaled industrial acetylene. A diabetic ketoacidosis mimic. JAMA. 1985;254(8):1066–7.
Budris WA, Roxe DM, Duvel JM. High anion gap metabolic acidosis associated with aminocaproic acid. Ann Pharmacother. 1999;33(3):308–11.
Lan KC, Lin YF, Yu FC, Lin CS, Chu P. Clinical manifestations and prognostic features of acute methamphetamine intoxication. J Formos Med Assoc. 1998;97(8):528–33.
Gerard JM, Luisiri A. A fatal overdose of arginine hydrochloride. J Toxicol Clin Toxicol. 1997;35(6):621–5.
Chang S, Lamm SH. Human health effects of sodium azide exposure: a literature review and analysis. Int J Toxicol. 2003;22(3):175–86.
Schwarz ES, Wax PM, Kleinschmidt KC, Sharma K, Chung WM, Cantu G, et al. Multiple poisonings with sodium azide at a local restaurant. J Emerg Med. 2014;46(4):491–4.
Corradi F, Brusasco C, Palermo S, Belvederi G. A case report of massive acute boric acid poisoning. Eur J Emerg Med. 2010;17(1):48–51.
Benaissa ML, Megarbane B, Borron SW, Baud FJ. Is elevated plasma lactate a useful marker in the evaluation of pure carbon monoxide poisoning? Intensive Care Med. 2003;29(8):1372–5.
Kao LW, Nanagas KA. Carbon monoxide poisoning. Med Clin North Am. 2005;89(6):1161–94.
Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011;30(8):836–43.
Sokal JA, Kralkowska E. The relationship between exposure duration, carboxyhemoglobin, blood glucose, pyruvate and lactate and the severity of intoxication in 39 cases of acute carbon monoxide poisoning in man. Arch Toxicol. 1985;57(3):196–9.
Paillet-Loilier M, Cesbron A, Le Boisselier R, Bourgine J, Debruyne D. Emerging drugs of abuse: current perspectives on substituted cathinones. Subst Abuse Rehabil. 2014;5:37–52.
Szerlip HM, Singer I. Hyperchloremic metabolic acidosis after chlorine inhalation. Am J Med. 1984;77(3):581–2.
Bin Salih S, Al Qahtani M, Al Anazi T, Al Hussein M, Al Hayyan H, Al Modaimegh H. Metabolic acidosis and generalized seizures secondary to citalopram overdose: a case report. J Clin Pharm Ther. 2010;35(4):479–82.
Hoffman RS. Treatment of patients with cocaine-induced arrhythmias: bringing the bench to the bedside. Br J Clin Pharmacol. 2010;69(5):448–57.
Allam S, Noble JS. Cocaine-excited delirium and severe acidosis. Anaesthesia. 2001;56(4):385–6.
Baud FJ, Barriot P, Toffis V, Riou B, Vicaut E, Lecarpentier Y, et al. Elevated blood cyanide concentrations in victims of smoke inhalation. N Engl J Med. 1991;325(25):1761–6.
Bismuth C, Baud F, Borron S, Scherrmann J. Antibodies proposed as therapeutic agents. Arch Toxicol Suppl. 1996;18:321–32.
Baud FJ, Borron SW, Megarbane B, Trout H, Lapostolle F, Vicaut E, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044–50.
Borron SW, Baud FJ. Antidotes for acute cyanide poisoning. Curr Pharm Biotechnol. 2012;13(10):1940–8.
Huntington S, Heppner J, Vohra R, Mallios R, Geller RJ. Serious adverse effects from single-use detergent sacs: report from a U.S. statewide poison control system. Clin Toxicol (Phila). 2014;52(3):220–5.
Beuhler MC, Gala PK, Wolfe HA, Meaney PA, Henretig FM. Laundry detergent “pod” ingestions: a case series and discussion of recent literature. Pediatr Emerg Care. 2013;29(6):743–7.
Wilson KC, Reardon C, Farber HW. Propylene glycol toxicity in a patient receiving intravenous diazepam. N Engl J Med. 2000;343(11):815.
Moon JM, Chun BJ. Clinical characteristics of patients after dicamba herbicide ingestion. Clin Toxicol (Phila). 2014;52(1):48–53.
Dragovic G, Jevtovic D. The role of nucleoside reverse transcriptase inhibitors usage in the incidence of hyperlactatemia and lactic acidosis in HIV/AIDS patients. Biomed Pharmacother. 2012;66(4):308–11.
Dlamini J, Ledwaba L, Mokwena N, Mokhathi T, Orsega S, Tsoku M, et al. Lactic acidosis and symptomatic hyperlactataemia in a randomized trial of first-line therapy in HIV-infected adults in South Africa. Antivir Ther. 2011;16(4):605–9.
Borron SW, Baud FJ, Garnier R. Intravenous 4-methylpyrazole as an antidote for diethylene glycol and triethylene glycol poisoning: a case report. Vet Hum Toxicol. 1997;39(1):26–8.
Vassiliadis J, Graudins A, Dowsett RP. Triethylene glycol poisoning treated with intravenous ethanol infusion. J Toxicol Clin Toxicol. 1999;37(6):773–6.
Brent J. Fomepizole for the treatment of pediatric ethylene and diethylene glycol, butoxyethanol, and methanol poisonings. Clin Toxicol (Phila). 2010;48(5):401–6.
Kraut JA, Xing SX. Approach to the evaluation of a patient with an increased serum osmolal gap and high-anion-gap metabolic acidosis. Am J Kidney Dis. 2011;58(3):480–4.
Moses V, Peter JV. Acute intentional toxicity: endosulfan and other organochlorines. Clin Toxicol (Phila). 2010;48(6):539–44.
Sharma RK, Kaul A, Gupta A, Bhadauria D, Prasad N, Jain A, et al. High anion gap refractory metabolic acidosis as a critical presentation of endosulfan poisoning. Indian J Pharmacol. 2011;43(4):469–71.
Allison MG, McCurdy MT. Alcoholic metabolic emergencies. Emerg Med Clin North Am. 2014;32(2):293–301.
Brent J. Fomepizole for ethylene glycol and methanol poisoning. N Engl J Med. 2009;360(21):2216–23.
Ghannoum M, Hoffman RS, Mowry JB, Lavergne V. Trends in toxic alcohol exposures in the United States from 2000 to 2013: a focus on the use of antidotes and extracorporeal treatments. Semin Dial. 2014;27(4):395–401.
Gualtieri JF, DeBoer L, Harris CR, Corley R. Repeated ingestion of 2-butoxyethanol: case report and literature review. J Toxicol Clin Toxicol. 2003;41(1):57–62.
Hung T, Dewitt CR, Martz W, Schreiber W, Holmes DT. Fomepizole fails to prevent progression of acidosis in 2-butoxyethanol and ethanol coingestion. Clin Toxicol (Phila). 2010;48(6):569–71.
Osterhoudt KC. Fomepizole therapy for pediatric butoxyethanol intoxication. J Toxicol Clin Toxicol. 2002;40(7):929–30.
Nitter-Hauge S. Poisoning with ethylene glycol monomethyl ether. Report of two cases. Acta Med Scand. 1970;188(4):277–80.
Bedichek E, Kirschbaum B. A case of propylene glycol toxic reaction associated with etomidate infusion. Arch Intern Med. 1991;151(11):2297–8.
Ganesh A, Audu P. Hyperosmolar, increased-anion-gap metabolic acidosis and hyperglycemia after etomidate infusion. J Clin Anesth. 2008;20(4):290–3.
McConnel JR, Ong CS, McAllister JL, Gross TG. Propylene glycol toxicity following continuous etomidate infusion for the control of refractory cerebral edema. Neurosurgery. 1996;38(1):232–3.
Van de Wiele B, Rubinstein E, Peacock W, Martin N. Propylene glycol toxicity caused by prolonged infusion of etomidate. J Neurosurg Anesthesiol. 1995;7(4):259–62.
Moyle G. Toxicity of antiretroviral nucleoside and nucleotide analogues: is mitochondrial toxicity the only mechanism? Drug Saf. 2000;23(6):467–81.
Honkoop P, Scholte HR, de Man RA, Schalm SW. Mitochondrial injury. Lessons from the fialuridine trial. Drug Saf. 1997;17(1):1–7.
Arnaudo JP, Maheut H, Martin B, Hesse JY. Reversible ketoacidosis and hyperglycemia after absorption of flumequine. Effect of high doses in a non-diabetic adult. Nouv Presse Med. 1980;9(9):636.
Pandey CK, Agarwal A, Baronia A, Singh N. Toxicity of ingested formalin and its management. Hum Exp Toxicol. 2000;19(6):360–6.
Dalus D, Mathew AJ, Pillai SS. Formic acid poisoning in a tertiary care center in South India: a 2-year retrospective analysis of clinical profile and predictors of mortality. J Emerg Med. 2013;44(2):373–80.
Yang CC, Ger J, Li CF. Formic acid: a rare but deadly source of carbon monoxide poisoning. Clin Toxicol (Phila). 2008;46(4):287–9.
Kamijo Y, Takai M, Fujita Y, Hirose Y, Iwasaki Y, Ishihara S. A multicenter retrospective survey on a suicide trend using hydrogen sulfide in Japan. Clin Toxicol (Phila). 2013;51(5):425–8.
Asif MJ, Exline MC. Utilization of hyperbaric oxygen therapy and induced hypothermia after hydrogen sulfide exposure. Respir Care. 2012;57(2):307–10.
Madiwale T, Liebelt E. Iron: not a benign therapeutic drug. Curr Opin Pediatr. 2006;18(2):174–9.
Chang TP, Rangan C. Iron poisoning: a literature-based review of epidemiology, diagnosis, and management. Pediatr Emerg Care. 2011;27(10):978–85.
Osterhoudt KC, Henretig FM. A 16-year-old with recalcitrant seizures. Pediatr Emerg Care. 2012;28(3):304–6.
Minns AB, Ghafouri N, Clark RF. Isoniazid-induced status epilepticus in a pediatric patient after inadequate pyridoxine therapy. Pediatr Emerg Care. 2010;26(5):380–1.
Riker RR, Fraser GL. Adverse events associated with sedatives, analgesics, and other drugs that provide patient comfort in the intensive care unit. Pharmacotherapy. 2005;25(5 Pt 2):8S–18.
Zosel A, Egelhoff E, Heard K. Severe lactic acidosis after an iatrogenic propylene glycol overdose. Pharmacotherapy. 2010;30(2):219.
Pillai U, Hothi JC, Bhat ZY. Severe propylene glycol toxicity secondary to use of anti-epileptics. Am J Ther. 2014;21(4):e106–9.
Adebamiro A, Perazella MA. Recurrent acute kidney injury following bath salts intoxication. Am J Kidney Dis. 2012;59(2):273–5.
Chan P, Chen JH, Lee MH, Deng JF. Fatal and nonfatal methamphetamine intoxication in the intensive care unit. J Toxicol Clin Toxicol. 1994;32(2):147–55.
Roberts DM, Yates C, Megarbane B, Winchester JF, Maclaren R, Gosselin S, et al. Recommendations for the role of extracorporeal treatments in the management of acute methanol poisoning: a systematic review and consensus statement. Crit Care Med. 2015;43(2):461–72.
Pearson JM, Hargraves TL, Hair LS, Massucci CJ, Frazee 3rd CC, Garg U, et al. Three fatal intoxications due to methylone. J Anal Toxicol. 2012;36(6):444–51.
Eizadi-Mood N. Nalidixic acid overdose and metabolic acidosis. CJEM. 2006;8(2):78.
Dhongade RK, Kavade SG, Damle RS. Neem oil poisoning. Indian Pediatr. 2008;45(1):56–7.
Meeran M, Murali A, Balakrishnan R, Narasimhan D. “Herbal remedy is natural and safe” – truth or myth? J Assoc Physicians India. 2013;61(11):848–50.
Hottinger DG, Beebe DS, Kozhimannil T, Prielipp RC, Belani KG. Sodium nitroprusside in 2014: a clinical concepts review. J Anaesthesiol Clin Pharmacol. 2014;30(4):462–71.
Lipper B, Bell A, Gaynor B. Recurrent hypotension immediately after seizures in nortriptyline overdose. Am J Emerg Med. 1994;12(4):452–3.
Ballesteros S, Martinez MA, Ballesteros MA, de la Torre CS, Rodriguez-Borregan JC. A severe case of olanzapine overdose with analytical data. Clin Toxicol (Phila). 2007;45(4):412–5.
Zadik Z, Blachar Y, Barak Y, Levin S. Organophosphate poisoning presenting as diabetic ketoacidosis. J Toxicol Clin Toxicol. 1983;20(4):381–5.
Haddad LM, Dimond KA, Schweistris JE. Phenol poisoning. JACEP. 1979;8(7):267–9.
Todorovic V. Acute phenol poisoning. Med Pregl. 2003;56 Suppl 1:37–41.
Hainer V, Aldhoon-Hainerova I. Tolerability and safety of the new anti-obesity medications. Drug Saf. 2014;37(9):693–702.
Caravati EM. Metabolic abnormalities associated with phosphoric acid ingestion. Ann Emerg Med. 1987;16(8):904–6.
Fernandez OU, Canizares LL. Acute hepatotoxicity from ingestion of yellow phosphorus-containing fireworks. J Clin Gastroenterol. 1995;21(2):139–42.
Woolf AD, Ebert TH. Toxicity after self-poisoning by ingestion of potassium chloroplatinite. J Toxicol Clin Toxicol. 1991;29(4):467–72.
Bismuth C, Baud FJ, Djeghout H, Astier A, Aubriot D. Cyanide poisoning from propionitrile exposure. J Emerg Med. 1987;5(3):191–5.
Scolnick B, Hamel D, Woolf AD. Successful treatment of life-threatening propionitrile exposure with sodium nitrite/sodium thiosulfate followed by hyperbaric oxygen. J Occup Med. 1993;35(6):577–80.
Mirrakhimov AE, Voore P, Halytskyy O, Khan M, Ali AM. Propofol infusion syndrome in adults: a clinical update. Crit Care Res Pract. 2015;2015:260385.
Lawson-Smith P, Jansen EC, Hyldegaard O. Cyanide intoxication as part of smoke inhalation – a review on diagnosis and treatment from the emergency perspective. Scand J Trauma Resusc Emerg Med. 2011;19:14.
Orbegozo Cortes D, Rayo Bonor A, Vincent JL. Isotonic crystalloid solutions: a structured review of the literature. Br J Anaesth. 2014;112(6):968–81.
Guidet B, Soni N, Della Rocca G, Kozek S, Vallet B, Annane D, et al. A balanced view of balanced solutions. Crit Care. 2010;14(5):325.
Morgan TJ. The ideal crystalloid – what is “balanced”? Curr Opin Crit Care. 2013;19(4):299–307.
Lactic Acidosis International Study Group. Risk factors for lactic acidosis and severe hyperlactataemia in HIV-1-infected adults exposed to antiretroviral therapy. AIDS. 2007;21(18):2455–64.
McLeod HL, Baker Jr DK, Pui CH, Rodman JH. Somnolence, hypotension, and metabolic acidosis following high-dose teniposide treatment in children with leukemia. Cancer Chemother Pharmacol. 1991;29(2):150–4.
Charytan D, Jansen K. Severe metabolic complications from theophylline intoxication. Nephrology (Carlton). 2003;8(5):239–42.
Manzanares W, Hardy G. Thiamine supplementation in the critically ill. Curr Opin Clin Nutr Metab Care. 2011;14(6):610–7.
Tormoehlen LM, Tekulve KJ, Nanagas KA. Hydrocarbon toxicity: a review. Clin Toxicol (Phila). 2014;52(5):479–89.
Dell’Orto VG, Belotti EA, Goeggel-Simonetti B, Simonetti GD, Ramelli GP, Bianchetti MG, et al. Metabolic disturbances and renal stone promotion on treatment with topiramate: a systematic review. Br J Clin Pharmacol. 2014;77(6):958–64.
Scheulen ME, Hilger RA, Oberhoff C, Casper J, Freund M, Josten KM, et al. Clinical phase I dose escalation and pharmacokinetic study of high-dose chemotherapy with treosulfan and autologous peripheral blood stem cell transplantation in patients with advanced malignancies. Clin Cancer Res. 2000;6(11):4209–16.
Hemstreet BA. Antimicrobial-associated renal tubular acidosis. Ann Pharmacother. 2004;38(6):1031–8.
Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323–38.
Watson ID, McBride D, Paterson KR. Fatal xylenol self-poisoning. Postgrad Med J. 1986;62(727):411–2.
Tripuraneni NS, Smith PR, Weedon J, Rosa U, Sepkowitz D. Prognostic factors in lactic acidosis syndrome caused by nucleoside reverse transcriptase inhibitors: report of eight cases and review of the literature. AIDS Patient Care STDS. 2004;18(7):379–84.
Arenas-Pinto A, Grant AD, Edwards S, Weller IV. Lactic acidosis in HIV infected patients: a systematic review of published cases. Sex Transm Infect. 2003;79(4):340–3.
Wilson BJ, Cowan HJ, Lord JA, Zuege DJ, Zygun DA. The accuracy of pulse oximetry in emergency department patients with severe sepsis and septic shock: a retrospective cohort study. BMC Emerg Med. 2010;10:9.
Pretto JJ, Roebuck T, Beckert L, Hamilton G. Clinical use of pulse oximetry: official guidelines from the Thoracic Society of Australia and New Zealand. Respirology. 2014;19(1):38–46.
Weaver LK, Churchill SK, Deru K, Cooney D. False positive rate of carbon monoxide saturation by pulse oximetry of emergency department patients. Respir Care. 2013;58(2):232–40.
Chang KC, Orr J, Hsu WC, Yu L, Tsou MY, Westenskow DR, et al. Accuracy of CO2 monitoring via nasal cannulas and oral bite blocks during sedation for esophagogastroduodenoscopy. J Clin Monit Comput. 2015.
Salem MM, Mujais SK. Gaps in the anion gap. Arch Intern Med. 1992;152(8):1625–9.
Wrenn K. The delta (delta) gap: an approach to mixed acid–base disorders. Ann Emerg Med. 1990;19(11):1310–3.
Adrogue HJ, Wilson H, Boyd 3rd AE, Suki WN, Eknoyan G. Plasma acid–base patterns in diabetic ketoacidosis. N Engl J Med. 1982;307(26):1603–10.
Brivet F, Bernardin M, Dormont J. Hyperchloremic acidosis in metabolic acidosis with anion gap excess. Comparison with diabetic ketoacidosis. Presse Med. 1991;20(9):413–7.
Skellett S, Mayer A, Durward A, Tibby SM, Murdoch IA. Chasing the base deficit: hyperchloraemic acidosis following 0.9% saline fluid resuscitation. Arch Dis Child. 2000;83(6):514–6.
Gabow PA, Kaehny WD, Fennessey PV, Goodman SI, Gross PA, Schrier RW. Diagnostic importance of an increased serum anion gap. N Engl J Med. 1980;303(15):854–8.
Winter SD, Pearson JR, Gabow PA, Schultz AL, Lepoff RB. The fall of the serum anion gap. Arch Intern Med. 1990;150(2):311–3.
Sadjadi SA. A new range for the anion gap. Ann Intern Med. 1995;123(10):807.
Sadjadi SA, Manalo R, Jaipaul N, McMillan J. Ion-selective electrode and anion gap range: what should the anion gap be? Int J Nephrol Renov Dis. 2013;6:101–5.
Lolekha PH, Vanavanan S, Teerakarnjana N, Chaichanajarernkul U. Reference ranges of electrolyte and anion gap on the Beckman E4A, Beckman Synchron CX5, Nova CRT, and Nova Stat Profile Ultra. Clin Chim Acta. 2001;307(1–2):87–93.
Paulson WD, Roberts WL, Lurie AA, Koch DD, Butch AW, Aguanno JJ. Wide variation in serum anion gap measurements by chemistry analyzers. Am J Clin Pathol. 1998;110(6):735–42.
Kraut JA, Nagami GT. The serum anion gap in the evaluation of acid–base disorders: what are its limitations and can its effectiveness be improved? Clin J Am Soc Nephrol. 2013;8(11):2018–24.
Jacob J, Lavonas EJ. Falsely normal anion gap in severe salicylate poisoning caused by laboratory interference. Ann Emerg Med. 2011;58(3):280–1.
Herres J, Ryan D, Salzman M. Delayed salicylate toxicity with undetectable initial levels after large-dose aspirin ingestion. Am J Emerg Med. 2009;27(9):1173 e1–3.
Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807–10.
Fencl V, Jabor A, Kazda A, Figge J. Diagnosis of metabolic acid–base disturbances in critically ill patients. Am J Respir Crit Care Med. 2000;162(6):2246–51.
Wang F, Butler T, Rabbani GH, Jones PK. The acidosis of cholera. Contributions of hyperproteinemia, lactic acidemia, and hyperphosphatemia to an increased serum anion gap. N Engl J Med. 1986;315(25):1591–5.
Decaux G, Schlesser M, Coffernils M, Prospert F, Namias B, Brimioulle S, et al. Uric acid, anion gap and urea concentration in the diagnostic approach to hyponatremia. Clin Nephrol. 1994;42(2):102–8.
Decaux G, Musch W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175–84.
Dorwart WV, Chalmers L. Comparison of methods for calculating serum osmolality form chemical concentrations, and the prognostic value of such calculations. Clin Chem. 1975;21(2):190–4.
Brindley PG, Butler MS, Cembrowski G, Brindley DN. Falsely elevated point-of-care lactate measurement after ingestion of ethylene glycol. CMAJ. 2007;176(8):1097–9.
Fijen JW, Kemperman H, Ververs FF, Meulenbelt J. False hyperlactatemia in ethylene glycol poisoning. Intensive Care Med. 2006;32(4):626–7.
Jorens PG. Falsely elevated lactate and ethylene glycol. Clin Toxicol (Phila). 2009;47(7):691.
Manini AF, Hoffman RS, McMartin KE, Nelson LS. Relationship between serum glycolate and falsely elevated lactate in severe ethylene glycol poisoning. J Anal Toxicol. 2009;33(3):174–6.
Verelst S, Vermeersch P, Desmet K. Ethylene glycol poisoning presenting with a falsely elevated lactate level. Clin Toxicol (Phila). 2009;47(3):236–8.
Megarbane B, Resiere D. Toxicological analysis is mandatory to interpret elevation in blood lactate concentration in toxic alcohol poisoning. Ann Fr Anesth Reanim. 2014;33(5):368–9.
Jorens PG, Demey HE, Schepens PJ, Coucke V, Verpooten GA, Couttenye MM, et al. Unusual D-lactic acid acidosis from propylene glycol metabolism in overdose. J Toxicol Clin Toxicol. 2004;42(2):163–9.
Marraffa JM, Hui A, Stork CM. Severe hyperphosphatemia and hypocalcemia following the rectal administration of a phosphate-containing Fleet pediatric enema. Pediatr Emerg Care. 2004;20(7):453–6.
Hsu HJ, Wu MS. Extreme hyperphosphatemia and hypocalcemic coma associated with phosphate enema. Intern Med. 2008;47(7):643–6.
Wiener SW. Toxicologic acid–base disorders. Emerg Med Clin North Am. 2014;32(1):149–65.
Astrup P, Jorgensen K, Andersen OS, Engel K. The acid–base metabolism. A new approach. Lancet. 1960;1(7133):1035–9.
O’Leary TD, Langton SR. Calculated bicarbonate or total carbon dioxide? Clin Chem. 1989;35(8):1697–700.
Rosan RC, Enlander D, Ellis J. Unpredictable error in calculated bicarbonate homeostasis during pediatric intensive care: the delusion of fixed pK’. Clin Chem. 1983;29(1):69–73.
Kume T, Sisman AR, Solak A, Tuglu B, Cinkooglu B, Coker C. The effects of different syringe volume, needle size and sample volume on blood gas analysis in syringes washed with heparin. Biochem Med (Zagreb). 2012;22(2):189–201.
Balasubramanyan N, Havens PL, Hoffman GM. Unmeasured anions identified by the Fencl-Stewart method predict mortality better than base excess, anion gap, and lactate in patients in the pediatric intensive care unit. Crit Care Med. 1999;27(8):1577–81.
Kellum JA. Metabolic acidosis in the critically ill: lessons from physical chemistry. Kidney Int Suppl. 1998;66:S81–6.
Stewart PA. Modern quantitative acid–base chemistry. Can J Physiol Pharmacol. 1983;61(12):1444–61.
Fencl V, Leith DE. Stewart’s quantitative acid–base chemistry: applications in biology and medicine. Respir Physiol. 1993;91(1):1–16.
Kellum JA, Kramer DJ, Pinsky MR. Strong ion gap: a methodology for exploring unexplained anions. J Crit Care. 1995;10(2):51–5.
Anstey CM. An assessment of the population variance of the strong ion gap using Monte Carlo simulation. Anaesth Intensive Care. 2009;37(6):983–91.
Gunnerson KJ, Srisawat N, Kellum JA. Is there a difference between strong ion gap in healthy volunteers and intensive care unit patients? J Crit Care. 2010;25(3):520–4.
Durward A, Tibby SM, Skellett S, Austin C, Anderson D, Murdoch IA. The strong ion gap predicts mortality in children following cardiopulmonary bypass surgery. Pediatr Crit Care Med. 2005;6(3):281–5.
Moviat M, van den Boogaard M, Intven F, van der Voort P, van der Hoeven H, Pickkers P. Stewart analysis of apparently normal acid–base state in the critically ill. J Crit Care. 2013;28(6):1048–54.
Zheng CM, Liu WC, Zheng JQ, Liao MT, Ma WY, Hung KC, et al. Metabolic acidosis and strong ion gap in critically ill patients with acute kidney injury. Biomed Res Int. 2014;2014:819528.
Story DA, Morimatsu H, Bellomo R. Strong ions, weak acids and base excess: a simplified Fencl-Stewart approach to clinical acid–base disorders. Br J Anaesth. 2004;92(1):54–60.
Ahmed SM, Maheshwari P, Agarwal S, Nadeem A, Singh L. Evaluation of the efficacy of simplified Fencl-Stewart equation in analyzing the changes in acid base status following resuscitation with two different fluids. Int J Crit Illn Inj Sci. 2013;3(3):206–10.
Hoffman RS, Smilkstein MJ, Howland MA, Goldfrank LR. Osmol gaps revisited: normal values and limitations. J Toxicol Clin Toxicol. 1993;31(1):81–93.
Lynd LD, Richardson KJ, Purssell RA, Abu-Laban RB, Brubacher JR, Lepik KJ, et al. An evaluation of the osmole gap as a screening test for toxic alcohol poisoning. BMC Emerg Med. 2008;8:5.
Khajuria A, Krahn J. Osmolality revisited – deriving and validating the best formula for calculated osmolality. Clin Biochem. 2005;38(6):514–9.
Purssell RA, Pudek M, Brubacher J, Abu-Laban RB. Derivation and validation of a formula to calculate the contribution of ethanol to the osmolal gap. Ann Emerg Med. 2001;38(6):653–9.
Mahon WA, Holland J, Urowitz MB. Hyperosmolar, non-ketotic diabetic coma. Can Med Assoc J. 1968;99(22):1090–2.
Carstairs SD, Suchard JR, Smith T, Simon LV, Kalynych CJ, Shimada M, et al. Contribution of serum ethanol concentration to the osmol gap: a prospective volunteer study. Clin Toxicol (Phila). 2013;51(5):398–401.
Garrard A, Sollee DR, Butterfield RC, Johannsen L, Wood A, Bertholf RL. Validation of a pre-existing formula to calculate the contribution of ethanol to the osmolar gap. Clin Toxicol (Phila). 2012;50(7):562–6.
Sud P, Nelson LS, Bouchard M, Lee WW. Contribution of serum ethanol concentration to the osmol gap: a prospective volunteer study. Clin Toxicol (Phila). 2013;51(8):810.
Siervo M, Bunn D, Prado CM, Hooper L. Accuracy of prediction equations for serum osmolarity in frail older people with and without diabetes. Am J Clin Nutr. 2014;100(3):867–76.
Martin-Calderon JL, Bustos F, Tuesta-Reina LR, Varona JM, Caballero L, Solano F. Choice of the best equation for plasma osmolality calculation: comparison of fourteen formulae. Clin Biochem. 2015;48(7–8):529–33.
Porter WH. Invalid correction of “falsely” elevated osmol gap. J Emerg Med. 2007;32(3):311–2. author reply 2–3.
Buckley NA, Whyte IM, Dawson AH. Osmolal gap. J Toxicol Clin Toxicol. 1994;32:93–5.
Hovda KE, Hunderi OH, Rudberg N, Froyshov S, Jacobsen D. Anion and osmolal gaps in the diagnosis of methanol poisoning: clinical study in 28 patients. Intensive Care Med. 2004;30(9):1842–6.
Hunderi OH, Hovda KE, Jacobsen D. Use of the osmolal gap to guide the start and duration of dialysis in methanol poisoning. Scand J Urol Nephrol. 2006;40(1):70–4.
Narins RG, Emmett M. Simple and mixed acid–base disorders: a practical approach. Medicine (Baltimore). 1980;59(3):161–87.
Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med. 2011;39(3):450–5.
Heckmann M, Trotter A, Pohlandt F, Lindner W. Epinephrine treatment of hypotension in very low birthweight infants. Acta Paediatr. 2002;91(5):566–70.
Morgan TJ, Venkatesh B, Hall J. Crystalloid strong ion difference determines metabolic acid–base change during in vitro hemodilution. Crit Care Med. 2002;30(1):157–60.
Morgan TJ, Venkatesh B, Hall J. Crystalloid strong ion difference determines metabolic acid–base change during acute normovolaemic haemodilution. Intensive Care Med. 2004;30(7):1432–7.
Omron EM, Omron RM. A physicochemical model of crystalloid infusion on acid–base status. J Intensive Care Med. 2010;25(5):271–80.
Shaw AD, Bagshaw SM, Goldstein SL, Scherer LA, Duan M, Schermer CR, et al. Major complications, mortality, and resource utilization after open abdominal surgery: 0.9% saline compared to Plasma-Lyte. Ann Surg. 2012;255(5):821–9.
Raghunathan K, Shaw A, Nathanson B, Sturmer T, Brookhart A, Stefan MS, et al. Association between the choice of IV crystalloid and in-hospital mortality among critically ill adults with sepsis*. Crit Care Med. 2014;42(7):1585–91.
Graf H, Leach W, Arieff AI. Evidence for a detrimental effect of bicarbonate therapy in hypoxic lactic acidosis. Science. 1985;227(4688):754–6.
Forsythe SM, Schmidt GA. Sodium bicarbonate for the treatment of lactic acidosis. Chest. 2000;117(1):260–7.
Liebelt EL. Targeted management strategies for cardiovascular toxicity from tricyclic antidepressant overdose: the pivotal role for alkalinization and sodium loading. Pediatr Emerg Care. 1998;14(4):293–8.
Mackway-Jones K. Towards evidence based emergency medicine: best BETs from the Manchester Royal Infirmary. Alkalinisation in the management of tricyclic antidepressant overdose. J Accid Emerg Med. 1999;16(2):139–40.
Woolf AD, Erdman AR, Nelson LS, Caravati EM, Cobaugh DJ, Booze LL, et al. Tricyclic antidepressant poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 2007;45(3):203–33.
Kraut JA, Madias NE. Treatment of acute metabolic acidosis: a pathophysiologic approach. Nat Rev Nephrol. 2012;8(10):589–601.
Gehlbach BK, Schmidt GA. Bench-to-bedside review: treating acid–base abnormalities in the intensive care unit – the role of buffers. Crit Care. 2004;8(4):259–65.
Weber T, Tschernich H, Sitzwohl C, Ullrich R, Germann P, Zimpfer M, et al. Tromethamine buffer modifies the depressant effect of permissive hypercapnia on myocardial contractility in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1361–5.
Marfo K, Garala M, Kvetan V, Gasperino J. Use of Tris-hydroxymethyl aminomethane in severe lactic acidosis due to highly active antiretroviral therapy: a case report. J Clin Pharm Ther. 2009;34(1):119–23.
Kallet RH, Jasmer RM, Luce JM, Lin LH, Marks JD. The treatment of acidosis in acute lung injury with tris-hydroxymethyl aminomethane (THAM). Am J Respir Crit Care Med. 2000;161(4 Pt 1):1149–53.
Nahas GG, Sutin KM, Fermon C, Streat S, Wiklund L, Wahlander S, et al. Guidelines for the treatment of acidaemia with THAM. Drugs. 1998;55(2):191–224.
Heaney D, Majid A, Junor B. Bicarbonate haemodialysis as a treatment of metformin overdose. Nephrol Dial Transplant. 1997;12(5):1046–7.
Ledebo I. Acid–base correction and convective dialysis therapies. Nephrol Dial Transplant. 2000;15 Suppl 2:45–8.
Sabeel AI, Kurkus J, Lindholm T. Intensified dialysis treatment of ethylene glycol intoxication. Scand J Urol Nephrol. 1995;29(2):125–9.
Calello DP, Liu KD, Wiegand TJ, Roberts DM, Lavergne V, Gosselin S, et al. Extracorporeal treatment for metformin poisoning: systematic review and recommendations from the extracorporeal treatments in poisoning workgroup. Crit Care Med. 2015;43:1716.
Ghannoum M, Laliberte M, Nolin TD, MacTier R, Lavergne V, Hoffman RS, et al. Extracorporeal treatment for valproic acid poisoning: systematic review and recommendations from the EXTRIP workgroup. Clin Toxicol (Phila). 2015;53(5):454–65.
Gosselin S, Juurlink DN, Kielstein JT, Ghannoum M, Lavergne V, Nolin TD, et al. Extracorporeal treatment for acetaminophen poisoning: recommendations from the EXTRIP workgroup. Clin Toxicol (Phila). 2014;52(8):856–67.
Acknowledgment
This chapter is dedicated to the memory of Professor Chantal Bismuth, whose contributions to medical toxicology are innumerable and lasting. With her sharply analytical mind, incisive wit, and charming smile, she mentored hundreds of toxicologists-in-training, challenged the status quo, and brought focus to many nebulous concepts in toxicology. She will be missed.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Grading System for Levels of Evidence Supporting Recommendations in Critical Care Toxicology, 2nd Edition
-
I
Evidence obtained from at least one properly randomized controlled trial.
-
II-1
Evidence obtained from well-designed controlled trials without randomization.
-
II-2
Evidence obtained from well-designed cohort or case-control analytic studies, preferably from more than one center or research group.
-
II-3
Evidence obtained from multiple time series with or without the intervention. Dramatic results in uncontrolled experiments (such as the results of the introduction of penicillin treatment in the 1940s) could also be regarded as this type of evidence.
-
III
Opinions of respected authorities, based on clinical experience, descriptive studies, and case reports, or reports of expert committees.
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this entry
Cite this entry
Borron, S.W. (2017). Acid–Base Balance in the Poisoned Patient. In: Brent, J., et al. Critical Care Toxicology. Springer, Cham. https://doi.org/10.1007/978-3-319-17900-1_67
Download citation
DOI: https://doi.org/10.1007/978-3-319-17900-1_67
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17899-8
Online ISBN: 978-3-319-17900-1
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences