
Abstract
The immune system can be a potent defense mechanism against cancer. Especially CD8+ cytotoxic T lymphocytes (CTL) have a great killing capacity towards tumor cells. However, their potential is often dampened by immune suppressive mechanisms in the tumor microenvironment. Co-inhibitory molecules (CIM) expressed by tumor cells, immune cells and stromal cells in the tumor milieu can severely hamper CD8+ T-cell responses against cancer cells. Today, a variety of co-inhibitory molecules, including PD-1, CTLA-4, LAG3, BTLA, Tim-3 and CD200R, have been implicated in tumor escape from CTL attack. Sustained signaling via these CIM can result in functional exhaustion of T-cells, a process in which the ability to proliferate, secrete cytokines and mediate lysis of tumor cells is sequentially lost. In this chapter, we discuss the influence of co-inhibitory pathways in suppressing CD8+ T-cell function in various immune settings. These include the natural immune surveillance by CTL against tumor cells, or in therapeutic settings like allogeneic stem cell transplantation or chimeric antigen receptor (CAR) T-cell therapy. In addition, we discuss exciting pre-clinical and clinical data of immunotherapeutic approaches interfering with negative co-signaling, either as monotherapy or in conjunction with vaccination strategies. Numerous studies indicate that co-inhibitory signaling limits the clinical benefit of current CTL-based therapies. Therefore, interference with CIM is an attractive immunotherapeutic intervention for cancer therapy.
No potential conflicts of interest were disclosed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
It is evident that both the innate and adaptive immune systems participate in the recognition and clearance of tumor cells by a process known as cancer immunosurveillance. In particular, tumor-reactive CD8+ cytotoxic T-lymphocytes (CTL) are major effectors in the immune response against cancer cells. However, despite the powerful aspects of CTL-mediated immune reactions, too often tumor cells are able to evade immune recognition and destruction. Tumor cells exploit several mechanisms to escape from CTL-mediated immunity, such as disruption of antigen presentation, down-regulation of HLA molecules, recruitment of regulatory T-cells (TREG) and myeloid-derived suppressor cells, as well as secretion of immune suppressive cytokines [1]. In the last decade, another powerful immune suppressive mechanism came into the limelight: the repressive action of co-inhibitory receptors [2].
2 CTL Activation
Activation of T-cells to become CTL effectors, initially requires two signals [3–5]. Firstly, the TCR-CD3 complex needs to interact with the cognate peptide presented in HLA molecules on dendritic cells (DC). However, whether or not the T-cell becomes activated, is predominantly dependent on signaling of either co-stimulatory molecules (CSM) or co-inhibitory molecules (CIM) upon ligation with their corresponding ligands expressed by the APC [6]. The balance between these positive and negative co-signals determines the functionality of T-cells during immunity and tolerance. The stimulatory signal is generally provided by CD28, expressed on the T-cell, interacting with its ligands CD80 and CD86 on the DC. In the absence of co-stimulation the T-cell will become functionally anergic, and thereby tolerant to the antigen, which is one of the physiological mechanisms involved in the elimination of self-reactive T-cells. In addition, ligation of CIM to their corresponding ligands on APC results in T-cell inhibition, and via this natural feedback loop, sustained T-cell activation is prevented and the effector T-cell response resolves. Therefore, the balance in positive and negative co-signals determines the activation state of the T cells during immunity and tolerance.
The kinetics and differentiation of CTL that constitute anti-tumor responses are divided in several stages [7]. First, the CD62L+CCR7+CD45RA+ naive T-cells (TN) encounter the antigen presented by DC. Due to the expression of the selectin CD62L and the chemokine receptor CCR7 these cells home to the secondary lymphoid organs. However, upon this stimulation by the DC, these T-cells clonally expand and loose the expression of CD62L, CCR7, CD28 and CD45RA, while upregulating activation markers such as CD45RO, CD69 and CD25. These effector T-cells (TEFF) subsequently migrate to the target tissues, where they eradicate tumor cells. After the peak of the response, upon which most or all target cells have been destroyed, the contraction phase commences, and most tumor-reactive T-cells will undergo apoptosis. However, a minority of the T-cells will survive to become long-lived memory cells, either effector (TEM) or central memory (TCM). While the TEM reside in the periphery and upon recall show a strong effector response, the TCM have a strong proliferative property, regain expression of CD62L and CCR7, and migrate to the lymph nodes and BM, where they convey a lifelong memory against the antigen of their specificity. During all these activation and differentiation events, signaling through CSM and CIM has a great influence on the functional capacity and differentiation status of CTL.
3 Tumor-Associated Antigens
The immune system can harness a powerful attack against cancer cells. This can be done by cells from the innate immune system, such as NK cells that can attack tumor cells without prior sensitization [8]. Furthermore, the adaptive T-cell immune system has a great potential of recognizing and lysing tumor cells. This is mainly done by CTL, which recognize tumor-associated antigens (TAA) presented by the cancer cells. TAA are overexpressed, or ideally, solely expressed by tumor cells and consequently recognized as foreign, and an effective CD8+ T-cell immune response can be constituted against these antigens. Different classes of TAA exist. One class of TAA is highly overexpressed differentiation genes, such as tyrosinase and gp100 in melanoma, proteins which are differentially expressed at low levels in healthy melanocytes [9].
Another class is the oncofetal antigens, like carcinoembryonic antigen (CEA), which are usually expressed only in the fetal stage, and therefore no immune tolerance against these TAA exists [10]. Furthermore, cancer-testis antigens, including MAGE-A, NY-ESO-1, ACRBP and CTAG1B, can be aberrantly expressed by tumor cells [11, 12]. Since these are also only expressed by fetal tissues and the immune-privileged testes, a prominent immune response can be observed against these TAA. Finally, new antigens caused by de novo mutations in the cancer cells can occur in any gene. Since these mutations result in a true novel epitope, a very strong CTL response can be elicited. Especially in cancers with a high mutation rate, such as melanoma and lung cancer, these novel TAA occur frequently [13].
Altogether, these TAA are the major target in natural CD8+ T-cell tumor surveillance, and form an attractive field for immunotherapy, such as tumor infiltrating lymphocyte (TIL) infusion or DC vaccination loaded with TAA [14]. Vaccination with the most potent APC, i.e. DC, provides a great option for antigen-specific stimulation of tumor-reactive CTL [15]. Currently, DC vaccination is being performed in phase 3 clinical trials against four malignancies, including melanoma, prostate cancer, glioma and renal cell carcinomas [16]. In prostate cancer, vaccination is being performed against the prostate cancer-antigen prostate acid phosphatase (PAP) with the sipuleucel-T treatment [17]. In addition, vaccination with melanoma-antigens has reached promising results [18]. Several parameters of DC vaccination still need to be optimized, such as DC culture, choice of DC subpopulation, the method of loading of tumor antigens, choice of maturation stimuli, and method of administration to the patient [19]. However, in the majority of studies an increase in the median overall survival has been documented, underlining the potential of this therapy [16].
4 Allogeneic Stem Cell Transplantation
Another cancer immunotherapy, based on CTL recognition of antigens expressed on tumor cells, is allogeneic stem cell transplantation (alloSCT). This procedure can still be regarded as one of the most powerful cell-based immunotherapy to date, due to potent graft-versus-tumor (GVT) responses constituted by alloreactive T-cells [20]. These alloreactive CD8+ T-cell responses eradicate the malignant cells upon recognition of polymorphic HLA-presented peptides, known as minor histocompatibility antigens (MiHA). AlloSCT greatly enhanced the cure rate for aggressive hematologic cancers, although many patients still fail to launch effective immune responses and develop relapsed disease. Moreover, a major drawback of alloSCT is the occurrence of graft-versus-host disease (GVHD), a potentially life-threatening complication predominantly caused by alloreactive T-cells recognizing healthy tissues, notably the skin, liver and gastrointestinal tract. Since hemato-restricted MiHA are solely expressed by the redundant patient hematopoietic system including the malignant counterparts, they hold the key to separate GVT from GVHD [21]. In fact, these MiHA are equally immunogenic as de novo TAA or viral epitopes, since the antigens are completely foreign to the donor immune system and immune tolerance has not been initiated. Therefore, alloSCT can be a very powerful and curative cancer immunotherapy.
5 Adoptive T-Cell Transfer
Adoptive transfer of CTL is an appealing means to prevent or treat relapse of the tumor cells, and so far various strategies have been exploited. Nevertheless, specificity is crucial to avoid systemic toxicity. One method to obtain sufficient numbers of T-cells reactive against a TAA or MiHA is via isolation of these cells from the effector repertoire of patients present, followed by a fast expansion protocol [22, 23]. Already in the 1980s, the first studies with tumor-reactive T-cells in mice were performed by the isolation of TIL and subsequent culture and administration, which resulted in remission of cancer [24]. This led to clinical trials in humans, and a response rate near 50 % or more has made TIL administration an established treatment option [25]. A different technique is the isolation and expansion of naive tumor-reactive T-cells from a healthy donor by ex vivo stimulation with peptide-presenting DC [26, 27]. However, this can be a time-consuming and laborious process, especially for overexpressed TAA. The feasibility of both approaches has been demonstrated by several groups [22, 23, 26–28]. Importantly, one phase I trial reported in five out of seven patients with relapsed leukemia a complete, but transient, remission upon adoptive transfer of MiHA-specific T-cells expanded from post-transplant recipient PBMC [28]. Unfortunately, the infused T-cells failed to persist in vivo, which might be due to their terminal TEFF/EM differentiation stage and, consequently, rapid exhaustion of these cells as a result of the extensive in vitro culture protocol. To prevent the exhaustion of these T-cells, a search for the T-cell type with the highest proliferative potential has led to the identification of the stem cell memory T-cell (TSCM) [29, 30]. Although TSCM have experienced antigen-stimulation, they resemble TN in their expression of CD62L and their ability to differentiate into all other T-cell differentiation states. In addition, it was found that by inhibition of either the Akt or Wnt pathway in vitro, it is possible to generate high numbers of tumor-reactive TSCM [30, 31]. Together with their proven excellent anti-tumor effects in murine models, this subtype of CTL holds great promise for future therapies [32].
A third way to efficiently generate high numbers of tumor-reactive CTL with high-affinity TCR is by gene transfer of the antigen-TCR α and β chains into donor T-cells [22, 33]. To prevent the induction of GVHD in patients treated with alloSCT, the TCR genes should preferentially be transferred into donor T-cells with a known specificity that does not recognize and target GVHD-tissues, such as virus-specific T-cells [34]. Another potential complication might be mispairing of the introduced and native TCR chains, thereby generating a new potentially harmful specificity [35]. Efforts are being made to prevent this mispairing, amongst which is the transfer of TCR α and β chains into γδ T-cells. Successful TCR gene transfer and resultant cytolytic competence has been demonstrated for both TAA and MiHA [36–39]. Importantly, with TCR gene transfer the complete MiHA-TCR is introduced into the donor T-cells, therefore matching of the HLA-restriction allele between recipient and donor is no longer required.
A novel therapeutic approach utilizing the power of CTL is chimeric antigen receptor (CAR) T-cells. These CAR consist of an antibody fragment recognizing a tumor antigen expressed on the surface of these T-cells. Ingeniously, to enable T-cell activation, this antibody fragment is coupled to the CD3 ξ-chain, leading to an intracellular activation cascaded upon recognition of the antigen [40]. This chimeric receptor combines the high avidity and specificity of antibodies with the activation of CTL, resulting in highly effective CTL responses. Second and third generation CAR have been engineered to express motives of CSM in the intracellular domain, such as CD28, 4-1BB and OX40. Thereby, in addition to the cytolytic capacity of CAR, also proliferation and survival are sustained. Impressive results have been obtained in clinical trials. Especially CAR recognizing CD19 developed by the June lab, have been able to efficiently lyse cancer cells in patients with high tumor burdens, and have resulted in cure of leukemia patients [41]. After this pioneering work in leukemia, other target antigens are currently being explored in different malignancies, making CAR therapy hold great promise for the future.
Nevertheless, despite the curative potential of the cellular therapies described afore, numerous studies have demonstrated that tumor cells explore immune suppressive mechanisms to dampen tumor-reactive CTL responses, resulting in sub-optimal clinical efficacy. One of the pivotal mechanisms exploited by tumor cells is manipulation of CTL activation, either by enhancing CSM or interfering with CIM signaling. Tumor cells can evade immune control by down-regulating CSM such as CD80 and CD86, and up-regulating various co-inhibitory ligands, thereby limiting the therapeutic potential of current immunotherapies against cancer. This chapter will address the role of CIM in tumor immune evasion from CTL attack, and discuss options to prevent T-cell inhibition without severe adverse effects. We will discuss the role of separate CIM involved in tumor escape from CTL, and subsequently elaborate on combinations of CIM in the tumor setting. Finally, the incorporation of CIM interference in near future anti-cancer immunotherapy will be discussed.
6 Co-inhibitory Molecules in Cancer
A variety of CIM have been implicated in cancer immune escape. Here, we discuss the CIM prominently involved in suppressing anti-tumor immunity.
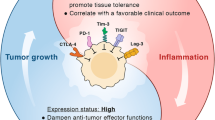
6.1 PD-1
6.1.1 Expression and Function of PD-1
Programmed death 1 (PD-1; CD279) is an immunoreceptor and member of the B7/CD28 family [42]. In 1992, PD-1 was identified on hybridoma T-cells undergoing apoptosis and was believed to be a programmed cell death-induced gene [43]. Further characterization demonstrated that PD-1 is inducibly expressed on stimulated CD8+ T-cells, CD4+ T-cells, B cells and monocytes [44]. PD-1 binds two ligands, PD-L1 (B7-H1; CD274) and PD-L2 (B7-DC; CD273) [45]. While PD-L1 is expressed on various non-lymphoid tissues, PD-L2 expression is mainly restricted to APC, like DC and macrophages [46]. Furthermore, multiple tumor types express PD-L1 and its expression is elevated upon IFN-γ exposure [47]. PD-L1 molecules on tumor cells can deliver negative signals towards PD-1-expressing tumor-reactive CTL, thereby inhibiting anti-tumor immunity [48]. Indeed, PD-L1 expression has been associated with poor prognosis in solid tumors [47].
It has been demonstrated that PD-1 plays a crucial role in T-cell regulation in various immune responses such as peripheral tolerance, autoimmunity, infection and anti-tumor immunity [46]. High PD-1 expression on viral antigen-specific CTL in chronic viral infections was recognized as a hallmark for T-cell dysfunction upon antigen re-encounter [49]. This phenomenon, known as exhaustion, is characterized by the sequential loss of the ability to proliferate, secrete cytokines and kill target cells. Especially in HIV infection, T-cell impairment could be relieved by PD-1 blockade both in vitro and in animal models [50, 51]. It has also been shown that PD-1 strongly attenuates the downstream signaling of the TCR [52]. In an elegant model system, the influence of PD-1 ligation on T-cell triggering was investigated [53]. Engagement of PD-1 raises the threshold of T-cell stimulation by increasing the number of TCR/peptide-MHC complexes needed for activation. It has been reported that exhausted T-cells have elevated expression of multiple CIM and a distinct gene signature, different from anergic cells, resulting in changes in TCR and cytokine signaling pathways [54]. The importance of downstream PD-1 signaling was nicely demonstrated by the identification of an exhaustion-specific gene signature in HIV-specific T-cells [55].
6.1.2 PD-1 in Cancer
PD-1 has been shown to have a prominent suppressive function in anti-cancer immunity. Expression of its ligand PD-L1 on tumor cells conveys a negative signal to tumor-reactive T-cells [56]. In addition, PD-1-expressing TIL present in breast cancer tissue are associated with a poor prognosis [57]. Moreover, in melanoma was shown that PD-1+ TIL were functionally impaired as compared to their PD-1-negative counterparts [58].
The involvement of PD-1 in alloSCT has been investigated both in mouse models and in the human setting. In a study investigating chronic myeloid leukemia (CML), using a retrovirus-induced CML model, it was demonstrated that tumor-specific T-cells can become exhausted [59]. In this model, consisting of PD-1+ tumor-reactive T-cells and PD-L1+ CML cells, exhaustion could be overcome by the administration of either PD-L1 antagonistic antibody or PD-1 deficient T-cells. In accordance, it was shown that the bulk T-cell population from CML patients exhibited increased expression of PD-1 [59]. Also, in the alloSCT setting, high PD-1 expression was observed on alloreactive CTL that specifically recognize hemato-restricted MiHA in myeloid leukemia patients [60]. Furthermore, proliferation of these PD-1+ MiHA-specific CTL by stimulation with MiHA-loaded DC ex vivo was suboptimal, indicating dysfunctional CTL due to PD-1 signaling. Importantly, upon treatment with anti-PD-1 or anti-PD-L1 blocking antibodies ex vivo proliferation of the MiHA-specific CTL was reinvigorated.
These and many more studies have led to clinical trials exploring the potency of PD-1 blocking antibodies, putting the PD-1 pathway in the forefront of anti-tumor therapy. Three antagonistic anti-PD-1 antibodies are currently in advanced clinical trials, i.e. pidilizumab, nivolumab and pembrolizumab (formerly lambrolizumab) (Table 2.1). Furthermore, three anti-PD-L1 antibodies, BMS-936,559, MEDI4736, MPDL3280A and MSB0010718C, are being investigated inclinical trials (Table 2.1). In 2012, exciting reports on the use of the anti-PD-1 nivolumab and the anti-PD-L1 blocking antibody BMS-936,559 in patients with advanced malignancies were published [61, 62]. Response rates upon administration of anti-PD-1 to patients with solid tumors ranged from 18 % to 28 %, depending on tumor type. Importantly, responses were durable, with the majority of patients having responses for over a year. Notably, the therapy was relatively well tolerated and only for one patient a serious adverse event, inflammatory colitis, was reported [63]. Interestingly, also blocking the ligand, PD-L1, could induce durable tumor regression with an objective response rate of 6–17 %, with prolonged responses of over a year. The lack of strong toxic effects in this study provided promise that the PD-1 blockade might have a more subtle effect than the CTLA-4 blockade. This rendered anti-PD-1 antibodies as interesting candidates for cancer therapy and gave rise to more extensive trials. In 2013, the results were reported for the anti-PD-1 antibody pembrolizumab in melanoma [64]. In this more homogenous patient group, a response rate of 38 %, and even 52 % in the highest dose was obtained. As in the previous studies, most responses were durable. These exciting results have encouraged registration if these PD-1 blockers. On July 4th 2014, Ono Pharmaceutical and partner Bristol-Myers Squibb (BMS), gained approval in Japan for nivolumab [65]. Furthermore, Merck aims to receive the First US approval for pembrolizumab in melanoma on October 28th 2014. These approvals open up endless possibilities of using PD-1 antagonists against various malignancies, as well as combining anti-PD-1 antibodies with other treatment modalities.
6.2 CTLA-4
6.2.1 Expression and Function of CTLA-4
Cytotoxic T lymphocyte associated antigen-4 (CTLA-4; CD152), was the first identified CIM, and is partly similar to the co-signaling molecule CD28 [66]. While CD28 is constitutively expressed on the membrane of naïve T-cells, CTLA-4 is primarily localized in intracellular compartments and quickly translocates to the cell membrane upon T-cell activation. The inhibitory function of CTLA-4 was revealed in knockout mice, which showed multi-organ T-cell infiltration leading to lethal lymphoproliferative disease [67]. Like CD28, CTLA-4 has an extracellular domain containing the MYPPPY binding motif, enabling both receptors to interact with CD80 (B7-1) and CD86 (B7-2) expressed by APC. However, the binding affinity of CTLA-4 for these ligands is higher by a factor 10–100, thus outcompeting CD28 and thereby promoting immune inhibition [68].
As CTLA-4 is up-regulated upon TCR ligation, it plays an important role in attenuating effector T-cell activation and maintaining immune homeostasis and central tolerance. In addition, CTLA-4 signaling in immunosuppressive TREG mediates the control of auto-reactive T-cells, as in vivo interference with CTLA-4 on these T-cells elicited pathological autoimmunity [69]. The effect of CTLA-4 interference could either be due to depletion and/or inhibition of TREG. It was shown that TREG-specific CTLA-4 deficiency resulted in down-regulation of CD80/CD86 on APC [70]. This can be explained by endocytosis of APC-derived CD80 and CD86 by TREG [71]. Subsequently, the APC acquires a less stimulatory phenotype, resulting in a lasting inhibitory effect after CTLA-4 ligation. This concept has been investigated further in vivo. Here it was found that TREG can reduce CD80/CD86 expression after encounter with a DC. When CTL are subsequently activated with these hypostimulatory DC, the effector T-cells display enhanced levels of T-cell immunoglobulin and mucin domain 3 (Tim-3) and PD-1. Via this mechanism, CTL function is indirectly attenuated via CTLA-4 signaling [72].
CTLA-4 as such is not a marker of exhausted cells, but elevated levels on viral antigen-specific T-cells correlated with their dysfunction in patients with chronic viral infections, which in turn could be restored by CTLA-4 blockade [73]. Also in metastatic melanoma, high expression of CTLA-4 was correlated to antigen-specific T-cell dysfunction [74]. Moreover, in various CD80 and CD86-positive tumor models, monotherapy with CTLA-4 blocking antibody resulted in elimination of established tumors and long-lasting antitumor immunity [75]. Interestingly, CTLA-4 also has an influence on the motility of CTL. After addition of a CTLA-4 antagonist in a mouse model, it was shown that CTL exhibited increased motility, indicating that CIM blockade does not only restore cytolytic activity, cytokine secretion and proliferation, but could also enhance CTL migration [76]. Although anti-CTLA-4 treatment works in vivo, CTLA-4 blockade in vitro has not been successful in reversing T-cell dysfunction. This can be due to limitations of the in vitro models, as CTLA-4 blockade may exert it’s in vivo action via multiple immune mediators (e.g. effector T-cells, antibody responses, TREG) [77].
All these preclinical findings have stimulated clinical exploration of anti-CTLA-4 blocking antibodies. At the moment, two blocking antibodies exist, ipilimumab and tremelimumab (Table 2.1). Most studies have been performed with ipilimumab in melanoma, and impressively, in these patients the median overall survival almost doubled [78]. In follow-up studies, the effects of CTLA-4 blockade were consistent [79] and in 2011, the FDA and EMA approved ipilimumab treatment for advanced melanoma, thereby paving the way for further exploration of therapies targeting CIM in cancer. Unfortunately, not all studies involving tremelimumab displayed a positive effect on overall survival [80]. Also, for both blocking antibodies, not all patients gained clinical benefit and individual responses are hard to predict. Furthermore, the occurrence of adverse toxic effects upon CTLA-4 blockade were a problem, even leading to death in some cases [80]. However, these were the pioneering studies involving CIM blockade, and by increased clinical awareness and protocols to tackle these immune related complications, severe adverse events have been decreased.
6.2.2 CTLA-4 in Stem Cell Transplantation
Experimental and clinical studies have demonstrated that co-inhibitory molecules hamper T-cell immunity against hematologic cancers in both the autologous and allogeneic settings. This might be due to native expression of CD80 and CD86 on hematologic tumor cells. CTLA-4:CD80/86 interactions also take place between T-cells and hematologic tumor cells. In multiple myeloma (MM) patients, CD86 but not CD80 was expressed by tumor cells, while CTLA-4 was up-regulated on T-cells, which led to anergy of tumor-specific T-cells [81]. Similar to these results, T-cells from chronic lymphocytic leukemia (CLL) patients responded to anti-CD3 activation by a decrease in CD28 and an increase in CTLA-4 expression, resulting in an inhibitory phenotype [82]. In addition to MM, also in acute myeloid leukemia (AML) cells tumor cells were demonstrated to have heterogeneous CD86 expression, but CD80 levels were generally low or absent [60, 83].
The alloreactive T-cell function after alloSCT is also strongly influenced by CIM. The importance of CTLA-4 in modulating allogeneic immune responses has been confirmed by association of certain CTLA-4 genotypes with overall survival and the incidence of leukemia relapse after alloSCT. It was demonstrated that CTLA-4 blockade shortly after alloSCT increased GVHD [84]. However, when anti-CTLA-4 was administered at later time-points after alloSCT, the GVT effect was boosted without signs of GVHD. In patients, ipilimumab administration at late time-points after alloSCT has been explored in one phase I trial [85]. Following a single infusion of the ipilimumab in 29 alloSCT patients with a recurrent or progressive hematological malignancy, three clinical responses were observed. Importantly, no induction or exacerbation of clinical GVHD was reported, although similar to other CTLA-4 blockade trials 14 % of the patients showed organ-specific immune-related adverse events. The lack of GVHD induction is likely attributed to the median interval of 1 year between the last donor T-cell infusion and ipilimumab administration. This provides a window for anti-tumor immunotherapy in the post-alloSCT setting and emphasizes the importance of appropriate timing of CIM blockade.
7 Combining PD-1 and CTLA-4 Blockade
It has been recognized that CTLA-4 and PD-1 exert their role in attenuating T-cell activation at different physiological locations and moments of the immune response. CTLA-4 is mostly involved in the inhibition of CTL priming in the lymph node, while PD-1 seems to limit T-cell proliferation and function in lymphoid tissues as well as in the periphery, i.e. at the tumor site. Therefore, the effects of concurrent PD-1 and CTLA-4 blockade are of great interest. In a mouse tumor model, it was demonstrated that double-positive CD8+ TIL was more dysfunctional than either single PD-1 or CTLA-4 positive CD8+ T-cells. In addition, double PD-1/CTLA-4 blockade led to reversal of TIL dysfunction and subsequent tumor rejection in the majority of mice [86]. Two studies which strengthen the idea of CTLA-4’s role in T-cell priming versus PD-1’s role in peripheral tolerance investigated the TCR repertoire [87, 88]. In patient who had received CTLA-4 blocking antibodies, an increased repertoire of TCR was observed. This indicates that at least part of the effect of CTLA-4 blockade is by an increase in T-cell priming. In contrast, in patients who had been treated with PD-1 blockade, this extended TCR repertoire was not observed, indicating that the clinical efficacy of this treatment is more likely due to reinvigoration of existing CTL responses.
These distinct roles of PD-1 and CTLA-4 warranted combined clinical trials to investigate whether administration of blocking both CIM would have an additive or a synergistic clinical effect. In the first study testing this hypothesis, in an impressive number of 65 % of patients clinical activity was observed, while these were patients with a very poor prognosis [89]. Also at the maximum dosages, 53 % of the patients fulfilled the criteria for the stringent objective responses, all with tumor reduction of 80 % or more. These impressive clinical responses were associated with in grade 3/4 adverse events in 53 % of the patients, which generally were reversible and were not more severe than observed with monotherapy. Also in follow-up data, it was shown that a group of 17 patients who received the optimum combination dose of anti-PD-1 and anti-CTLA-4 showed an overall survival rate of 94 % at 1 year and of 88 % at 2 years, exceeding by far the suboptimal responses typically observed in patients treated with either antibody alone [65]. These results are very promising for the future as optimal combinations of CIM blockade can yield very impressive clinical responses.
8 BTLA
8.1 Expression and Function of BTLA
B and T lymphocyte attenuator (BTLA), (CD272), is an inhibitory receptor with structural similarities to CTLA-4 and PD-1 [90]. BTLA is mainly expressed by immune cells, including T- and B-cells, DC and myeloid cells [91, 92]. In contrast to other B7/CD28 family members, BTLA binds a member of the tumor necrosis factor receptor (TNFR) superfamily, namely herpes virus entry mediator (HVEM) [93]. HVEM is part of an intricate signaling network as it has at least four additional binding partners that distinctively mediate T-cell responses: i.e. CD160, LIGHT (for lymphotoxin-like, exhibits inducible expression, and competes with HSV glycoprotein D for HVEM, a receptor expressed by T lymphocytes), lymphotoxin-α (LT-α) and herpes simplex virus glycoprotein D (gD) [94]. BTLA or CD160 signaling upon HVEM binding results in T-cell inhibition [93, 95]. Interestingly, naïve T-cells express both HVEM and BTLA, and these molecules form a T-cell intrinsic heterodimer complex [96]. Due to formation of this complex, HVEM is unavailable for extrinsic ligands, and no co-stimulatory signal is transduced. In humans, persistent expression of BTLA was observed on EBV- and CMV-specific CD8+ T-cells, which negatively affected T-cell function [97, 98]. Furthermore, high BTLA expression correlated with impaired tumor-reactive T-cell function in melanoma patients [74, 97]. These tumor-specific T-cell responses could be restored in vitro by interference with the BTLA-HVEM pathway in combination with vaccination therapy. In addition, co-expression of BTLA, PD-1 and Tim-3 rendered melanoma-specific CD8+ T-cells highly dysfunctional, which could be reversed by combined blockade of all three CIM [99]. In a mouse tumor vaccination model, blockade of the BTLA/CD160/HVEM pathway caused regression of large tumor masses [100]. These results show that in the right setting BTLA blockade can be of great significance, warranting evaluation of clinical effectiveness. In addition, the effect of a BTLA blocking antibody has been investigated on MiHA-specific T-cell functionality in samples from alloSCT patients [91]. As shown for PD-1, BTLA was also highly expressed on MiHA-specific CTL. Moreover, in the majority of the patients BTLA blockade resulted in increased outgrowth of MiHA-specific CD8+ T-cells. Interestingly, in three patients BTLA blockade effects were more prominent than those of PD-1, indicating that BTLA has a non-redundant function to PD-1, and therefore it holds promise in cancer immunotherapies.
8.2 Tim-3
The co-signaling receptor Tim-3 is expressed on Th1 CD4+ and CD8+ T-cells, and is involved in co-inhibition. In mice, the interaction of Tim-3 with its ligand galectin-9 was demonstrated to prevent in autoimmune diseases and promote malignancies [101]. Furthermore, in HIV [102] and melanoma patients [103], dysfunctional CD8+ T-cells have been shown to co-express Tim-3. In this regard interference with Tim-3 signaling is an interesting treatment option, and enhanced tumor vaccine efficacy has been observed by Tim-3 blockade [104]. Interestingly, both Tim-3 and PD-1 were expressed on a subset of exhausted CD8+ T-cells in a murine AML model, and expression levels increased [105] during tumor progression [106]. While either Tim-3 or PD-L1 blockade alone was not sufficient to improve survival, the combination of the two antagonistic antibodies significantly decreased tumor burden and enhanced survival. Also, in a melanoma vaccination model, the vast majority of vaccination-induced CTL upregulated PD-1 and a minority also upregulated Tim-3. Levels of PD-1 and Tim-3 expression by CTL at the time of vaccine administration correlated inversely with their expansion potential in vivo. Importantly, dual blockade of PD-1 and Tim-3 enhanced the expansion and cytokine production of vaccine-induced CTL in vitro [107]. Also, combining Tim-3 blockade with activation of CD137, a co-stimulatory receptor, conveyed long term protection against ovarian carcinoma in a mouse model [108]. Finally, blocking of Tim-3 and its family member Tim-4 resulted in a better anti-tumor effect against murine melanoma. All these studies show that, although always in conjunction with another co-signaling molecule, Tim-3 can be involved in tumor evasion, making it an attractive partner in combinatorial blockade. In contrast, a stimulatory role for TIM-3 and galectin-9 has been reported in the interaction of CD8+ T-cells and DC [109]. This discrepancy might be explained by the findings that Tim-3 signaling enhances TCR stimulation [96]. T-cell exhaustion may be caused by prolonged TCR signaling via Tim-3, thereby prolonging the effector phase of T-cell activation at the expense of T-cell memory [110]. Therefore, depending on the setting, Tim-3 may act as either a co-stimulatory or a co-inhibitory receptor.
8.3 LAG3
Lymphocyte-activation gene 3 (LAG3; CD223) is a co-inhibitory receptor highly similar to CD4 and therefore also binds HLA class II molecules [111, 112]. LAG3 seems to be non-redundant from PD-1, as both are expressed on distinct populations CTL [113]. Recently, it was demonstrated that PD-1 and LAG3 act synergistically in the onset of autoimmune diseases and tumor escape in mice [114, 115]. In a leukemia model, PD-1 and CTLA-4 were blocked to reverse CTL tolerance. However, also blockade of LAG3 was necessary to fully restore CTL function [116]. Altogether, these results indicate that LAG3, like Tim-3, is a good candidate as an additive blocking target. At the moment, clinical trials are being performed with a blocking antibody and a soluble LAG3 fusion molecule (Table 2.1), and these studies have the potential to add LAG3 to the list of targets in cancer immunotherapy [117].
9 Other Co-inhibitory Players
In addition to the afore discussed molecules, other co-inhibitory players are being studied to characterize their contribution to functional suppression of tumor-reactive T-cell immunity.
In 2011, a new CIM highly similar to PD-1 was discovered by two groups: PD-1H (PD-1 homolog) or VISTA (V-domain Ig suppressor of T-cell activation) [105, 118]. This molecule is broadly expressed on hematopoietic cells and expression levels are further up-regulated on T-cells and APCs following activation. In in vitro studies the interaction with soluble VISTA-Ig fusion protein or VISTA+ APC mediated the suppression of T-cell cytokine production and proliferation, which could be alleviated by blocking antibody treatment [118, 119]. Recently, it was also demonstrated that VISTA can enhance the conversion of naïve T-cells into FoxP3+ T-cells [119]. In vivo, VISTA overexpression on tumor cells strongly hampered protective tumor-reactive T-cell responses. Importantly, VISTA blockade impaired the suppressive function and emergence of TREG, as well as modulated the suppressive tumor micro-environment, thereby, promoting tumor-reactive T-cell immunity [120]. Interestingly, treatment with PD-1H blocking antibody prevented the induction of GVHD in murine alloSCT models, although the mechanism of action has not been elucidated [105]. These data illustrate that PD-1H/VISTA exerts both an immunoregulatory function in the tumor micro-environment, as well as a direct immunosuppressive action on anti-tumor T-cell responses, making it an interesting therapeutic target.
Killer cell lectin-like receptor G1 (KLRG1), also known as CLEC15A or MAFA, is an inhibitory receptor expressed on NK cells and subsets of CD4+ and CD8+ T-cells [121]. It has been demonstrated that interaction with its ligand E-cadherin results in the functional inhibition of KLRG1+ NK cells, thereby preventing effective killing of tumor cells [122, 123]. In T-cells, KLRG1 expression has been mostly studied as a marker of terminal differentiation. KLRG1+ antigen-experienced TEFF/EM cells exhibited preserved capacity to secrete cytokines upon antigen reencounter, but were incapable of proliferation [121]. Importantly, one study demonstrated that interference with KLRG1 signaling, by targeting of E-cadherin with a blocking antibody, results in enhanced TCR-induced proliferation of highly differentiated CD28−CD27− CTL [124]. More studies are warranted to characterize the involvement of KLRG1 signaling in tumor immune escape and the potential of KLRG1 blockade for cancer immunotherapy.
2B4 (i.e. CD244, SLAMf4) is a member of the CD2 subset of the immunoglobulin superfamily, and is expressed on NK cells, monocytes, basophils and eosinophils. Furthermore, 2B4 expression is up-regulated on a subset of CD8+ T-cells following activation [125, 126]. Its binding partner is CD48 [127]. Most functional studies have been performed with NK cells, where 2B4 was demonstrated to have both activating and inhibitory functions [128]. Interestingly, in a murine transplantation model, 2B4 expression was up-regulated on allograft-reactive CD8+ T-cells, but not CD4+ T-cells, following selective CD28 blockade [129]. Preservation of inhibitory signaling via CTLA-4 was required for the up-regulation of 2B4. Subsequent inhibitory signaling via 2B4 reduced expression levels of the co-stimulatory molecule ICOS, and mediated enhanced allograft survival. These results indicate that 2B4 is involved in the control of antigen-specific CTL functionality.
CD200 Receptor (CD200R) is an inhibitory receptor expressed by cells of myeloid and lymphoid origins, including NK cells and T-cells following activation [130]. Its ligand CD200 (OX2) is expressed by diverse cell types, including immune cells, neurons and epithelia. Importantly, overexpression of CD200 by tumor cells has been associated with progression of various solid and hematologic cancers [131]. In a murine leukemia model, CD200Fc suppressed CD4+ and CD8+ T-cell functionality, resulting in loss of protection from tumor growth [132]. Ex vivo studies with human CLL demonstrated that CD200 is involved in the functional suppression of CTL-mediated tumor killing and CD4-mediated suppression of CTL functionality, which could be reverted with CD200 blocking antibody or CD200 siRNA treatment [133]. In a murine model with CD200+ human B-CLL, administration of CD200 blocking antibody resulted in restored T-cell proliferation and tumor control [134]. Moreover, patients with CD200+ AML were found to have reduced numbers of functional NK cells [135], had significantly compromised Th1 memory and CTL memory responses [136], and showed increased numbers of FoxP3+ TREG [137]. CD200 blockade in vitro could recover NK cell and T-cell functionality [135, 136], and is therefore an attractive target for therapy. Interestingly, the first clinical results have already been reported about an anti-CD200 blocking antibody (Table 2.1) [138]. Although it was in a small cohort, promising results were obtained in a study with B-CLL and MM patients: 36 % of patients experienced at least a 10 % reduction in bulky disease and notably, one patient experienced a partial response with a maximum of 71 % reduction in bulky disease.
10 Future Prospects
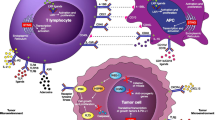
Several therapeutic strategies are being developed to dampen the inhibitory signaling by CIM in order to optimize tumor-reactive CTL immunity (Fig. 2.1). The challenge of interference with immune checkpoints is to boost anti-tumor reactivity, while avoiding adverse events such as systemic toxicity. This can potentially be achieved by combining the alleviation of co-inhibition with other therapeutic options or optimal dosage and timing of antibody administration. Appealing combinations are the simultaneous targeting of multiple co-inhibitory receptors, co-stimulatory agonists in parallel with CIM antagonists, or incorporation in existing cellular therapies. For example, DC vaccination may be applied together with blocking antibodies against CIM to boost CTL-mediated anti-tumor immunity.

To boost tumor-reactive T cell immunity different immunotherapeutic strategies can be exploited as monotherapy or in combination. First, co-inhibitory signaling pathways can be blocked with antagonistic antibodies to prevent and/or alleviate the functional impairment of CTLs. Furthermore, agonistic antibodies targeting co-stimulatory molecules can be applied to further augment CTL functionality. In addition, dendritic cell vaccination can be applied to provide efficient antigen presentation and strong stimulatory signals to tumor-reactive CTLs. Another strategy is the alloSCT in hematological malignancies, which can elicit powerful MiHA-reactive CTL responses. Finally, by adoptive transfer of highly potent TCR-transduced or CAR-transduced (stem-cell like) T cells direct attack of tumor cells can be provoked. The power of these immunotherapeutic approaches can be further intensified by combination with antibodies to interfere with co-inhibitory signaling pathways
CAR T-cells are a promising treatment modality in cancer therapy. Although in second and third generation CAR T-cell constructs a strong co-stimulatory signal is incorporated in the form of CD28, 4-1BB and OX40 intracellular signaling domains, this powerful therapy also seems to be dampened by CIM [139, 140]. In a murine tumor model with Her-2-specific CAR-T-cells, a significant increase in tumor growth inhibition was observed after PD-1 blockade [141]. In addition, the amount of immune suppressive myeloid derived suppressor cells was decreased upon PD-1 blockade, through a yet unknown mechanism. Therefore, CIM blockade in combination with CAR-T-cell therapy may improve the clinical efficacy of this novel therapy.
Although anti-CTLA-4 and anti-PD-1 monotherapies have shown very exciting results, toxic effects of blocking CIM may still be a problem. Approaches that concurrently deliver a tumor-antigen-specific stimulus may lead to less adverse events. These include combination therapies with treatment modalities such as immunomodulatory anti-cancer agents, vaccines, TREG depletion or nanoparticles. Recently, another treatment modality in which an antigen-specific stimulation is combined with an intervention for co-inhibition was explored. PD-L1/L2 silenced MiHA-loaded DC boosted the expansion of MiHA-specific T-cells ex vivo [142], and following these promising results, a clinical trial combining DLI with vaccination of PD-L1/L2 silenced donor DC loaded with hemato-restricted MiHA will start. All clinical studies provide a platform for incorporating blockade of CIM as adjuvant therapy of choice in cancer patients, with numerous options for combination therapies. Importantly, the risk of breaking tolerance systemically by blockade of one CIM could be prevented by using lower levels of multiple blocking antibodies targeting different CIM simultaneously, since together these may boost immune responses in a non-redundant manner. This is stressed by the fact that exhausted T-cells are known to display multiple co-inhibitory receptors [143]. Notably, the impressive results obtained by combining blocking antibodies against PD-1 and CTLA-4 is a perfect example of harnessing the power of these two non-redundant immune checkpoints, and many more combinations need to be investigated in the clinical setting.
After identification of the role of CIM in CD8+ T-cell functions, their significance on T-cell exhaustion was clearly established. However, with time, the notion of CIM as direct markers of dysfunction has been adjusted. Although their negative effect on T-cell functions is evident, expression as such does not qualify a T-cell as exhausted [144]. It has been shown that CIM, most notably PD-1, are also present on healthy cells [145] and that several CIM are up-regulated after T-cell activation [146, 147], while their expression had no direct effect on cytokine production by CTL. The activation-induced up-regulation indicated the physiological role of CIM as a negative feedback loop in CTL effector responses. Moreover, during T-cell differentiation most CIM are also up- or down-regulated [91, 146]. All these results indicate that, although on the whole T-cell population PD-1 expression can be an indicator for exhaustion, expression as such is not a marker of exhaustion on the individual T-cell level. Especially the fact that PD-1 can be an activation marker is demonstrated by a study investigating TIL in melanoma. Here it was shown that PD-1, LAG3 and Tim-3 are the identifying markers for tumor-reactive CTL [148]. The realization that not expression, but signaling via the CIM causes CTL dysfunction, has prompted the investigation of downstream signaling pathways and gene expression in impaired CTL. It was found that exhausted T-cells display a distinct gene signature, different from anergic cells, resulting in changes in TCR and cytokine signaling pathways [54]. Indeed, it was demonstrated that PD-1 downstream signaling results in an exhaustion gene signature in HIV-specific T-cells [55]. Further research into these mechanisms in CTL impairment in the tumor setting can yield novel targets to prevent or reverse exhaustion.
Altogether, CIM play a pivotal role in natural and therapeutic CTL-mediated immunity against cancers. With increasing knowledge of a growing number of CIM, novel mono- and combinatorial treatment options are becoming available. In the end, this can lead to optimized immunotherapy against cancers.
Abbreviations
- AB:
-
Antibody
- ACRBP:
-
Acrosin binding protein
- AG:
-
Antigen
- alloSCT:
-
Allogeneic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- APC:
-
Antigen-presenting cell
- BMS:
-
Bristol-Myers Squibb
- BTLA:
-
B- and T-lymphocyte attenuator
- CAR:
-
Chimeric antigen receptor
- CAR-T:
-
CAR-transduced T-cell
- CCR7:
-
C-C chemokine receptor type 7
- CD:
-
Cluster of differentiation
- CD200R:
-
CD200 Receptor
- CD62L:
-
CD62 ligand
- CEA:
-
Carcinoembryonic antigen
- CIM:
-
Co-inhibitory molecule
- CIM-L:
-
Co-inhibitory ligand
- CIM-R:
-
Co-inhibitory receptor
- CLEC15A:
-
C-type lectin domain family 15 member A
- CLL:
-
Chronic lymphoid leukemia
- CML:
-
Chronic myeloid leukemia
- CMV:
-
Cytomegalovirus
- CSM-L:
-
Co-stimulatory ligand
- CSM-R:
-
Co-stimulatory receptor
- CTAG1B:
-
Cancer/testis antigen 1B
- CTL:
-
Cytotoxic T-lymphocyte
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DC:
-
Dendritic cell
- EBV:
-
Epstein-Barr Virus
- EMA:
-
European Medicines Agency
- Fc:
-
Fragment crystallizable region
- FDA:
-
Food and Drug Administration
- FoxP3:
-
Forkhead box P3
- gD:
-
Herpes simplex virus glycoprotein D
- GVHD:
-
Graft-versus-host-disease
- GVT:
-
Graft-versus-tumor
- Her-2:
-
Human epidermal growth factor receptor 2
- HIV:
-
Human immunodeficiency virus
- HLA:
-
Human lymphocyte antigens
- HVEM:
-
Herpesvirus entry mediator
- ICOS:
-
Inducible T-cell costimulator
- IFN-γ:
-
Interferon gamma
- Ig:
-
Immunoglobulin
- KLRG1:
-
Killer cell lectin-like receptor G1
- LAG3:
-
Lymphocyte-activation gene 3
- LIGHT:
-
Lymphotoxin-like, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for HVEM, a receptor expressed by T lymphocytes
- LT-α:
-
Lymphotoxin-alpha
- MAGE-A:
-
Melanoma-associated antigen
- MHC:
-
Major histocompatibility complex
- MiHA:
-
Minor histocompatibility antigen
- MM:
-
Multiple myeloma
- NK:
-
Natural killer cell
- NY-ESO-1:
-
New York esophageal squamous cell carcinoma-1
- PAP:
-
Prostate acid phosphatase
- PBMC:
-
Peripheral blood mononuclear cells
- PD-1:
-
Programmed cell death 1
- PD-1H:
-
PD-1 homolog
- PD-L1:
-
Programmed death-ligand 1
- PD-L2:
-
Programmed death-ligand 2
- pMHC:
-
Peptide-MHC complex
- siRNA:
-
Small interfering RNA
- SLAMf4:
-
Signaling lymphocyte-activation molecule family member 4
- TAA:
-
Tumor-associated antigen
- TCR:
-
T-cell receptor
- TCR-T:
-
TCR-transduced T-cell
- TEFF :
-
Effector T-cell
- TEM :
-
Effector memory T-cell
- Th1:
-
Helper 1 T-cell
- TIL:
-
Tumor-infiltrating lymphocyte
- Tim-3:
-
T-cell immunoglobulin and mucin domain 3
- Tim-4:
-
T-cell immunoglobulin and mucin domain 4
- TN :
-
Naïve T-cell
- TNFR:
-
Tumor necrosis factor receptor
- TREG :
-
Regulatory T-cell
- TSCM :
-
Stem cell memory T-cell
- VISTA:
-
V-domain Ig suppressor of T-cell activation
- Wnt:
-
Wingless-related integration site
References
Croci DO, Zacarias Fluck MF, Rico MJ, Matar P, Rabinovich GA, Scharovsky OG. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunol Immunother. 2007;56(11):1687–700.
Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4(5):336–47.
Zang X, Allison JP. The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res. 2007;13(18 Pt 1):5271–9.
Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48.
Sharpe AH. Mechanisms of costimulation. Immunol Rev. 2009;229(1):5–11.
Carreno BM, Carter LL, Collins M. Therapeutic opportunities in the B7/CD28 family of ligands and receptors. Curr Opin Pharmacol. 2005;5(4):424–30.
Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev. 2010;236:151–66.
Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, Wang L, Shifrin N, Raulet DH. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. 2014;122:91–128.
Engelhard VH, Bullock TN, Colella TA, Sheasley SL, Mullins DW. Antigens derived from melanocyte differentiation proteins: self-tolerance, autoimmunity, and use for cancer immunotherapy. Immunol Rev. 2002;188:136–46.
Gameiro SR, Jammeh ML, Hodge JW. Cancer vaccines targeting carcinoembryonic antigen: state-of-the-art and future promise. Expert Rev Vaccines. 2013;12(6):617–29.
Meek DW, Marcar L. MAGE-A antigens as targets in tumour therapy. Cancer Lett. 2012;324(2):126–32.
Whitehurst AW. Cause and consequence of cancer/testis antigen activation in cancer. Annu Rev Pharmacol Toxicol. 2014;54:251–72.
Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y, Rosenberg SA. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–52.
Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257(1):56–71.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77.
Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15(7):e257–67.
Westdorp H, Skold AE, Snijer BA, Franik S, Mulder SF, Major PP, Foley R, Gerritsen WR, de Vries IJ. Immunotherapy for prostate cancer: lessons from responses to tumor-associated antigens. Front Immunol. 2014;5:191.
Bol KF, Mensink HW, Aarntzen EH, Schreibelt G, Keunen JE, Coulie PG, de Klein A, Punt CJ, Paridaens D, Figdor CG, de Vries IJ. Long overall survival after dendritic cell vaccination in metastatic uveal melanoma patients. Am J Ophthalmol. 2014;158:939–47.
Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802.
Jenq RR, van den Brink MR. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. 2010;10(3):213–21.
Feng X, Hui KM, Younes HM, Brickner AG. Targeting minor histocompatibility antigens in graft versus tumor or graft versus leukemia responses. Trends Immunol. 2008;29(12):624–32.
Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. 2011;89(3):396–407.
Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332–42.
Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233(4770):1318–21.
Wu R, Forget MA, Chacon J, Bernatchez C, Haymaker C, Chen JQ, Hwu P, Radvanyi LG. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J. 2012;18(2):160–75.
Bleakley M, Otterud BE, Richardt JL, Mollerup AD, Hudecek M, Nishida T, Chaney CN, Warren EH, Leppert MF, Riddell SR. Leukemia-associated minor histocompatibility antigen discovery using T-cell clones isolated by in vitro stimulation of naive CD8+ T cells. Blood. 2010;115(23):4923–33.
Mutis T, Ghoreschi K, Schrama E, Kamp J, Heemskerk M, Falkenburg JH, Wilke M, Goulmy E. Efficient induction of minor histocompatibility antigen HA-1-specific cytotoxic T-cells using dendritic cells retrovirally transduced with HA-1-coding cDNA. Biol Blood Marrow Transplant. 2002;8(8):412–9.
Warren EH, Fujii N, Akatsuka Y, Chaney CN, Mito JK, Loeb KR, Gooley TA, Brown ML, Koo KK, Rosinski KV, Ogawa S, Matsubara A, Appelbaum FR, Riddell SR. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood. 2010;115(19):3869–78.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–7.
Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, Paulos CM, Muranski P, Restifo NP. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15(7):808–13.
van der Waart A, van de Weem N, Maas F, Kramer C, Kester M, Falkenburg F, Schaap N, Jansen J, van der Voort R, Gattinoni L, Hobo W, Dolstra H. Inhibition of Akt-signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood. 2014;124:3490–500.
Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671–84.
Heemskerk MH, Griffioen M, Falkenburg JH. T-cell receptor gene transfer for treatment of leukemia. Cytotherapy. 2008;10(2):108–15.
Heemskerk MH, Hoogeboom M, Hagedoorn R, Kester MG, Willemze R, Falkenburg JH. Reprogramming of virus-specific T cells into leukemia-reactive T cells using T cell receptor gene transfer. J Exp Med. 2004;199(7):885–94.
Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E, Uckert W, Song JY, Haanen JB, Schumacher TN. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16(5):565. -70, 1p.
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9.
Heemskerk MH, Hoogeboom M, de Paus RA, Kester MG, van der Hoorn MA, Goulmy E, Willemze R, Falkenburg JH. Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specific T-cell receptor complexes expressing a conserved alpha joining region. Blood. 2003;102(10):3530–40.
Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–46.
Ochi T, Fujiwara H, Yasukawa M. Application of adoptive T-cell therapy using tumor antigen-specific T-cell receptor gene transfer for the treatment of human leukemia. J Biomed Biotechnol. 2010;2010:521248.
Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012;14(6):405–15.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–95.
Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27(4):195–201.
Freeman GJ. Structures of PD-1 with its ligands: sideways and dancing cheek to cheek. Proc Natl Acad Sci U S A. 2008;105(30):10275–6.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, Zhou J, Li BZ, Shi YH, Xiao YS, Xu Y, Fan J. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15(3):971–9.
Zhang C, Wu S, Xue X, Li M, Qin X, Li W, Han W, Zhang Y. Anti-tumor immunotherapy by blockade of the PD-1/PD-L1 pathway with recombinant human PD-1-IgV. Cytotherapy. 2008;10(7):711–9.
Velu V, Kannanganat S, Ibegbu C, Chennareddi L, Villinger F, Freeman GJ, Ahmed R, Amara RR. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J Virol. 2007;81(11):5819–28.
Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, Amara RR. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206–10.
Kaufmann DE, Walker BD. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol. 2009;182(10):5891–7.
Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014;20(4):265–71.
Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, Freeman GJ, Krogsgaard M, Riley JL. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A. 2013;110(27):E2480–9.
Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–84.
Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, Julg B, Jesneck JL, Brosnahan K, Imam S, Russell K, Toth I, Piechocka-Trocha A, Dolfi D, Angelosanto J, Crawford A, Shin H, Kwon DS, Zupkosky J, Francisco L, Freeman GJ, Wherry EJ, Kaufmann DE, Walker BD, Ebert B, Haining WN. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16(10):1147–51.
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–7.
Muenst S, Soysal SD, Gao F, Obermann EC, Oertli D, Gillanders WE. The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat. 2013;139(3):667–76.
Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–44.
Mumprecht S, Schürch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. 2009;114(8):1528–36.
Norde WJ, Maas F, Hobo W, Korman A, Quigley M, Kester MG, Hebeda K, Falkenburg JH, Schaap N, de Witte TM, van der Voort R, Dolstra H. PD-1/PD-L1 interactions contribute to functional T-cell impairment in patients who relapse with cancer after allogeneic stem cell transplantation. Cancer Res. 2011;71(15):5111–22.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–75.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44.
Sheridan C. First PD-1 inhibitor breezes across finish line. Nat Biotechnol. 2014;32(9):847–8.
Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, Golstein P. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328(6127):267–70.
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–7.
Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, Linsley PS. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med. 1994;180(6):2049–58.
Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–10.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–5.
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LS, Sansom DM. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–3.
Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest. 2014;124(6):2425–40.
Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, Palmer S, Brockman M, Rathod A, Piechocka-Trocha A, Baker B, Zhu B, Le GS, Waring MT, Ahern R, Moss K, Kelleher AD, Coffin JM, Freeman GJ, Rosenberg ES, Walker BD. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. 2007;8(11):1246–54.
Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, Rufer N, Speiser DE. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350–60.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6.
Pentcheva-Hoang T, Simpson TR, Montalvo-Ortiz W, Allison JP. Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol Res. 2014;2:970–80.
Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26(32):5275–83.
Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin Oncol. 2010;37(5):473–84.
Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res. 2011;17:6958–62.
Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, Garbe C, Gogas H, Schachter J, Linette G, Lorigan P, Kendra KL, Maio M, Trefzer U, Smylie M, McArthur GA, Dreno B, Nathan PD, Mackiewicz J, Kirkwood JM, Gomez-Navarro J, Huang B, Pavlov D, Hauschild A. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616–22.
Brown RD, Pope B, Yuen E, Gibson J, Joshua DE. The expression of T cell related costimulatory molecules in multiple myeloma. Leuk Lymphoma. 1998;31(3–4):379–84.
Frydecka I, Kosmaczewska A, Bocko D, Ciszak L, Wolowiec D, Kuliczkowski K, Kochanowska I. Alterations of the expression of T-cell-related costimulatory CD28 and downregulatory CD152 (CTLA-4) molecules in patients with B-cell chronic lymphocytic leukaemia. Br J Cancer. 2004;90(10):2042–8.
Costello RT, Mallet F, Sainty D, Maraninchi D, Gastaut JA, Olive D. Regulation of CD80/B7-1 and CD86/B7-2 molecule expression in human primary acute myeloid leukemia and their role in allogenic immune recognition. Eur J Immunol. 1998;28(1):90–103.
Blazar BR, Taylor PA, Panoskaltsis-Mortari A, Sharpe AH, Vallera DA. Opposing roles of CD28:B7 and CTLA-4:B7 pathways in regulating in vivo alloresponses in murine recipients of MHC disparate T cells. J Immunol. 1999;162(11):6368–77.
Bashey A, Medina B, Corringham S, Pasek M, Carrier E, Vrooman L, Lowy I, Solomon SR, Morris LE, Holland HK, Mason JR, Alyea EP, Soiffer RJ, Ball ED. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood. 2009;113(7):1581–8.
Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73(12):3591–603.
Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B, Koya RC, Graeber TG, Robins H, Ribas A. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. 2014;20(9):2424–32.
Robert L, Harview C, Emerson R, Wang X, Mok S, Homet B, Comin-Anduix B, Koya RC, Robins H, Tumeh PC, Ribas A. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology. 2014;3:e29244.
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–33.
Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4(7):670–9.
Hobo W, Norde WJ, Schaap N, Fredrix H, Maas F, Schellens K, Falkenburg JH, Korman AJ, Olive D, van der Voort R, Dolstra H. B and T lymphocyte attenuator mediates inhibition of tumor-reactive CD8+ T cells in patients after allogeneic stem cell transplantation. J Immunol. 2012;189(1):39–49.
Murphy TL, Murphy KM. Slow down and survive: enigmatic immunoregulation by BTLA and HVEM. Annu Rev Immunol. 2010;28:389–411.
Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6(1):90–8.
Del Rio ML, Lucas CL, Buhler L, Rayat G, Rodriguez-Barbosa JI. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leukoc Biol. 2010;87(2):223–35.
Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol. 2008;9(2):176–85.
Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, D’Souza C, Norris PS, Pfeffer K, Murphy KM, Kronenberg M, Spear PG, Ware CF. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J Immunol. 2009;183(11):7286–96.
Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, Michielin O, Olive D, Speiser DE. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010;120(1):157–67.
Serriari NE, Gondois-Rey F, Guillaume Y, Remmerswaal EB, Pastor S, Messal N, Truneh A, Hirsch I, van Lier RA, Olive D. B and T lymphocyte attenuator is highly expressed on CMV-specific T cells during infection and regulates their function. J Immunol. 2010;185(6):3140–8.
Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Olive D, Kuchroo V, Zarour HM. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72(4):887–96.
Lasaro MO, Sazanovich M, Giles-Davis W, Mrass P, Bunte RM, Sewell DA, Hussain SF, Fu YX, Weninger W, Paterson Y, Ertl HC. Active immunotherapy combined with blockade of a coinhibitory pathway achieves regression of large tumor masses in cancer-prone mice. Mol Ther. 2011;19(9):1727–36.
Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011;32(8):345–9.
Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, Wong JC, Satkunarajah M, Schweneker M, Chapman JM, Gyenes G, Vali B, Hyrcza MD, Yue FY, Kovacs C, Sassi A, Loutfy M, Halpenny R, Persad D, Spotts G, Hecht FM, Chun TW, McCune JM, Kaul R, Rini JM, Nixon DF, Ostrowski MA. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205(12):2763–79.
Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–86.
Lee MJ, Woo MY, Heo YM, Kim JS, Kwon MH, Kim K, Park S. The inhibition of the T-cell immunoglobulin and mucin domain 3 (Tim3) pathway enhances the efficacy of tumor vaccine. Biochem Biophys Res Commun. 2010;402(1):88–93.
Flies DB, Wang S, Xu H, Chen L. Cutting edge: a monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol. 2011;187(4):1537–41.
Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, Murphy WJ, Azuma M, Anderson AC, Kuchroo VK, Blazar BR. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(17):4501–10.
Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, Tarhini AA, Tawbi HA, Kirkwood JM, Moschos S, Wang H, Guillaume P, Luescher IF, Krieg A, Anderson AC, Kuchroo VK, Zarour HM. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer Res. 2014;74(4):1045–55.
Guo Z, Cheng D, Xia Z, Luan M, Wu L, Wang G, Zhang S. Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer. J Transl Med. 2013;11:215.
Nagahara K, Arikawa T, Oomizu S, Kontani K, Nobumoto A, Tateno H, Watanabe K, Niki T, Katoh S, Miyake M, Nagahata S, Hirabayashi J, Kuchroo VK, Yamauchi A, Hirashima M. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J Immunol. 2008;181(11):7660–9.
Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol. 2014;193(4):1525–30.
Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, Auffray C, Triebel F, Piatier-Tonneau D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. 1992;176(2):327–37.
Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, Hercend T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171(5):1393–405.
Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, Hipkiss E, Vignali DA, Pardoll DM, Drake CG. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009;182(11):6659–69.
Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T, Kato Y, Fagarasan S, Muramatsu M, Eto T, Hioki K, Honjo T. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. 2011;208(2):395–407.
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–27.
Berrien-Elliott MM, Jackson SR, Meyer JM, Rouskey CJ, Nguyen TL, Yagita H, Greenberg PD, DiPaolo RJ, Teague RM. Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T-cell tolerance. Cancer Res. 2013;73(2):605–16.
Wang-Gillam A, Plambeck-Suess S, Goedegebuure P, Simon PO, Mitchem JB, Hornick JR, Sorscher S, Picus J, Suresh R, Lockhart AC, Tan B, Hawkins WG. A phase I study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Invest New Drugs. 2013;31(3):707–13.
Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, Lu LF, Gondek D, Wang Y, Fava RA, Fiser A, Almo S, Noelle RJ. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208(3):577–92.
Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O’Connell S, Ceeraz S, Suriawinata AA, Yan S, Ernstoff MS, Noelle R. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74(7):1924–32.
Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, Wang L. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74(7):1933–44.
Voehringer D, Koschella M, Pircher H. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood. 2002;100(10):3698–702.
Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med. 2006;203(2):289–95.
Schwartzkopff S, Grundemann C, Schweier O, Rosshart S, Karjalainen KE, Becker KF, Pircher H. Tumor-associated E-cadherin mutations affect binding to the killer cell lectin-like receptor G1 in humans. J Immunol. 2007;179(2):1022–9.
Henson SM, Franzese O, Macaulay R, Libri V, Azevedo RI, Kiani-Alikhan S, Plunkett FJ, Masters JE, Jackson S, Griffiths SJ, Pircher HP, Soares MV, Akbar AN. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood. 2009;113(26):6619–28.
Garni-Wagner BA, Purohit A, Mathew PA, Bennett M, Kumar V. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J Immunol. 1993;151(1):60–70.
Boles KS, Stepp SE, Bennett M, Kumar V, Mathew PA. 2B4 (CD244) and CS1: novel members of the CD2 subset of the immunoglobulin superfamily molecules expressed on natural killer cells and other leukocytes. Immunol Rev. 2001;181:234–49.
Brown MH, Boles K, van der Merwe PA, Kumar V, Mathew PA, Barclay AN. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J Exp Med. 1998;188(11):2083–90.
Laouar A, Manocha M, Wan M, Yagita H, van Lier RA, Manjunath N. Cutting edge: distinct NK receptor profiles are imprinted on CD8 T cells in the mucosa and periphery during the same antigen challenge: role of tissue-specific factors. J Immunol. 2007;178(2):652–6.
Liu D, Krummey SM, Badell IR, Wagener M, Schneeweis LA, Stetsko DK, Suchard SJ, Nadler SG, Ford ML. 2B4 (CD244) induced by selective CD28 blockade functionally regulates allograft-specific CD8+ T cell responses. J Exp Med. 2014;211(2):297–311.
Rijkers ES, de Ruiter T, Baridi A, Veninga H, Hoek RM, Meyaard L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol Immunol. 2008;45(4):1126–35.
Stumpfova M, Ratner D, Desciak EB, Eliezri YD, Owens DM. The immunosuppressive surface ligand CD200 augments the metastatic capacity of squamous cell carcinoma. Cancer Res. 2010;70(7):2962–72.
Gorczynski RM, Chen Z, Hu J, Kai Y, Lei J. Evidence of a role for CD200 in regulation of immune rejection of leukaemic tumour cells in C57BL/6 mice. Clin Exp Immunol. 2001;126(2):220–9.
Wong KK, Khatri I, Shaha S, Spaner DE, Gorczynski RM. The role of CD200 in immunity to B cell lymphoma. J Leukoc Biol. 2010;88(2):361–72.
Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, Frederickson S, Maruyama T, Wild MA, Nolan MJ, Wu D, Springhorn J, Bowdish KS. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol. 2007;178(9):5595–605.
Coles SJ, Wang EC, Man S, Hills RK, Burnett AK, Tonks A, Darley RL. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia. 2011;25(5):792–9.
Coles SJ, Hills RK, Wang EC, Burnett AK, Man S, Darley RL, Tonks A. Expression of CD200 on AML blasts directly suppresses memory T-cell function. Leukemia. 2012;26(9):2148–51.
Coles SJ, Hills RK, Wang EC, Burnett AK, Man S, Darley RL, Tonks A. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia. 2012;26(9):2146–8.
Mahadevan DLM, Whelden M, Faas SJ, Ulery TL, Kukreja A, Li L, Bedrosian CL, Heffner LT. First-in-human phase I dose escalation study of a humanized anti-CD200 antibody (Samalizumab) in patients with advanced stage B cell chronic lymphocytic leukemia (B-CLL) or multiple myeloma (MM). In: 2nd ASH Annu Meet Exposition 2012, Orlando, FL, 2012.
Morales-Kastresana A, Labiano S, Quetglas JI, Melero I. Better performance of CARs deprived of the PD-1 brake. Clin Cancer Res. 2013;19(20):5546–8.
John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology. 2013;2(10):e26286.
John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–46.
Hobo W, Maas F, Adisty N, de Witte T, Schaap N, van der Voort R, Dolstra H. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood. 2010;116(22):4501–11.
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37.
Speiser DE, Utzschneider DT, Oberle SG, Munz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat Rev Immunol. 2014;14:768–74.
Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, Tata P, Gupta S, Zilliox MJ, Nakaya HI, Pulendran B, Haining WN, Freeman GJ, Ahmed R. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol. 2011;186(7):4200–12.
Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “exhaustion” of human CD8 T Cells. Front Immunol. 2013;4:455.
Haymaker C, Wu R, Bernatchez C, Radvanyi L. PD-1 and BTLA and CD8(+) T-cell “exhaustion” in cancer: “exercising” an alternative viewpoint. Oncoimmunology. 2012;1(5):735–8.
Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, Hanada K, Almeida JR, Darko S, Douek DC, Yang JC, Rosenberg SA. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–59.
Acknowledgments
This work was supported by grants from the Alpe d’HuZes foundation/Dutch Cancer Society (KWF 2011-5041) and (KWF 2012-5410).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Norde, W.J., Hobo, W., Dolstra, H. (2015). Role of Co-inhibitory Molecules in Tumor Escape from CTL Attack. In: Bonavida, B., Chouaib, S. (eds) Resistance of Cancer Cells to CTL-Mediated Immunotherapy. Resistance to Targeted Anti-Cancer Therapeutics, vol 7. Springer, Cham. https://doi.org/10.1007/978-3-319-17807-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-17807-3_2
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17806-6
Online ISBN: 978-3-319-17807-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)