
Abstract
Death receptors (DRs) are promising targets for cancer therapies because of their ability to induce apoptosis in cancer cells. These receptors are characterized by an intracellular death domain, which transmits a death signal from their cognate ligands, including TNF-related apoptosis inducing ligand (TRAIL). Currently, multiple clinical trials are underway to evaluate the antitumor activity of recombinant human TRAIL and agonistic antibodies to its receptors DR4 and DR5. Although the products have shown a tolerated safety profile in the completed phase 1 studies, a large number of cancer cell lines are found to be resistant to these agents, raising a concern about their clinical efficacy. This review provides an update of recent advances in understanding the molecular mechanisms involved in cancer cell resistance to DR4/DR5 targeted therapies. This information will be further discussed with respect to combinational strategies to overcome or bypass resistance mechanisms towards a better treatment outcome.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Death receptors
- Apoptosis
- Targeted therapies
- Drug resistance
- Predictive biomarkers
- Combinational therapies
1 Introduction
The cell surface death receptors are promising targets for cancer therapy due to their ability to induce apoptosis in cancer cells. To date, six human death receptors (DRs) have been identified, including Tumor Necrosis Factor Receptor (TNFR) 1, Fas (CD95), DR3, DR4 (TRAIL-R1), DR5 (TRAIL-R2) and DR6. These receptors are characterized by an intracellular death domain that transmits a death signal from their respective cognate ligands including TNFα, FasL, and TNF-related apoptosis inducing ligand (TRAIL/Apo2L). Despite the ability of TNFα and FasL to induce apoptosis in cancer cells, severe toxicities to normal cells leading to hypertension and hepatotoxicity preclude their use in systemic cancer therapy [1, 2]. In contrast, recombinant human TRAIL (rhTRAIL) preferentially induces apoptosis in a variety of tumor cell lines without harming many normal cell types [3, 4]. Moreover, administration of rhTRAIL into mice bearing human tumor xenografts induces significant tumor regression without systemic toxicity. These promising results have led to multiple clinical trials of rhTRAIL and agonistic antibodies to DR4 or DR5 as potential anticancer therapies (Table 10.1). These products have shown a well-tolerated safety profile in the completed Phase I studies [5–9]. However, a significant portion of tumor cell lines as well as primary human tumor cells are found to be resistant to these therapies due to intrinsic or acquired mechanisms [10–14]. Undoubtedly, non-responsive patients will not benefit from the treatments but may still suffer from the potential side effects. An in-depth analysis of resistance mechanisms could facilitate the identification of biomarkers for predicting tumor response to the DR-targeted therapies and aiding in the development of combinational therapies to overcome resistance towards a better clinical outcome of cancer treatment.
2 Apoptosis Signaling Through Death Receptors
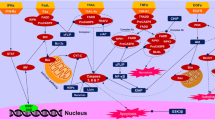
Like other TNF ligands, native TRAIL exists as a homotrimer that cross-links its death receptors (DRs) 4 and/or 5 on the surface membrane of target cells (Fig. 10.1). The agonistic antibodies act in a similar mode. Activation of DR4 and/or DR5 results in the recruitment of the adaptor molecule Fas-associated death domain (FADD) and the procaspase-8 or -10 into a death-inducing signaling complex (DISC). Within the DISC, caspase-8 or -10 is activated by self-processing that subsequently activates the downstream effector caspases such as caspase-3, -6, and -7 in a mitochondrial-independent or -dependent manner. The latter process is linked by caspase-8 mediated truncation of Bid (tBid) [15–17]. The activated caspases propagate apoptotic programming by cleaving a wide range of structural and signaling proteins, ultimately leading to apoptosis of the target cell. TRAIL resistance has been associated with defects in the relevant apoptosis signaling components or regulatory proteins. These factors will be discussed with respect to their functional relevance in cancer resistance: (1) functionality of receptors, (2) availability of caspases, and (3) status of regulatory proteins.

Checkpoints of TRAIL-induced apoptosis signaling pathway. The TRAIL apoptotic signaling pathway is initiated through ligation of TRAIL or agonistic antibodies targeting DR4/5. This ligation induces assembly of a DISC composed of FADD and pro-caspase 8/10. Caspase 8 is activated and released to initiate caspase 3/7 to induce apoptosis. Caspase 8 can also cause the cleavage of Bid to truncated Bid (tBid), linking to the mitochondrial-dependent caspase activation. The key resistance mechanisms include surface deficiency of DR4/5 (R1), upregulated c-FLIP (R2), caspase 8 deficiency (R3), Bcl2 protein family over-expression (R4), inhibitor of apoptosis proteins (R5), caspase 3 deficiency (R6), and mislocalization of the DRs to autophagosomes (R7), the nuclear membrane (R8) or other membrane localization (R9) within the cytoplasm. To achieve better cancer treatment outcomes, combinational therapies can be used to circumvent the specific resistance mechanisms in cancer cells
2.1 Deficiency of Surface Death Receptors
Initiation of a death-signaling cascade relies on the surface expression of DR4 and DR5 for TRAIL or antibody ligation. Deficiency of these receptors on the cell membrane is enough to render cancer cells resistant to TRAIL-induced apoptosis, regardless of the expression status of other signaling proteins. This lack of surface expression is seen in a variety of cancer lines and primary tumor cells, including those derived from breast [14] and oral squamous carcinoma [18]. The absence of death receptors on the surface membrane of the cell does not correlate with receptor total protein levels [14], indicating these receptors are trapped within the cell. Many mechanisms have been implicated in this mislocalization of the death receptors, such as unsuccessful trafficking to the plasma membrane [19–22], increased endocytosis [13, 14, 23–25], and autophagosome localization [26]. Studies have shown in cells with deficient surface expression, the DRs are mainly found in the cytoplasm and the nucleus [19–22, 27, 28]. Our work has shown that both DR4 and DR5 undergo constitutive or ligand-induced internalization in some breast cancer cell lines [12–14]. Constitutive endocytosis may occur through faulty dileucine-based sorting signals, such as EAQC337LL within DR4. DR4/DR5 endocytosis is just beginning to be understood, but may be a mechanism to terminate apoptosis signaling through TRAIL receptors [13].
Autophagy and Ras-dependent events have also been implicated in the surface deficiency of the death receptors. Our work has shown that TRAIL-resistant breast cancer cells have higher levels of basal autophagosomes, with DR4 and DR5 located within LC3-II labeled autophagosomes [26]. This autophagosomal localization may confer TRAIL resistance to the cell, preventing receptor trafficking to the membrane. This resistance due to autophagy has been reported in pancreatic cancer cell lines [29]. Implications of Ras small GTPase in DR-mediated apoptosis come from a study of oral squamous cell carcinomas, which found that TRAIL sensitivity correlated with expression of endogenous Ras [18]. In fact, constitutive expression of active Ras with mutant RasV12 selectively upregulated surface expression of DR5 and restored TRAIL-sensitivity in resistant cell lines. Similar findings were observed in colon cancers where overexpression of H-Ras increased death receptors through a MEK-dependent pathway [30]. Conversely, K-Ras mutations confer resistance to pancreatic and lung cancer cell lines [31]. Our laboratory has recently shown that wild-type H-Ras is upregulated in some cancer cells where it renders cancer resistance to TRAIL and is closely correlated with a deficiency of surface DR4 and DR5 [18]. Mutations in the Ras family are commonly found in tumors and, therefore, further studies on Ras’ effect on TRAIL sensitivity are warranted to better understand the pleiotropic effects Ras family members have on tumor resistance.
DR5 was thought to be the primary receptor for TRAIL leading to apoptosis in various types of cancer cells. At least six anti-DR5 antibodies, which compares with only one anti-DR4 antibody, are currently under development (Table 10.1). Indeed, our studies have shown that DR4 is deficient at a much higher frequency than DR5 [14]. This observation suggests that targeting DR5 would be more beneficial in cancer treatment in several types of cancer lines. Our studies also show that TRAIL requires both DR4 and DR5 for a maximal killing in breast cancer cells. The two death receptors may act synergistically by forming hetero-receptor complexes [14]. The surface deficiency in either receptor would lower the sensitivity of target cells to TRAIL. Paradoxically, other studies show that TRAIL induces apoptosis exclusively through DR4 in cancer cell lines from skin [32], ovary [33], and leukocytes [34] as well as primary cells from chronic lymphocytic leukemia and mantle cell lymphoma [35]. The molecular basis for this preference of TRAIL is not clear but may be due to differences in the functional status of death receptors in a specific tumor.
2.2 Deficiency of Caspase 8 and 10
Inhibition of initiator caspases is another mechanism of preventing death receptor-mediated apoptosis. Cells that lack expression of critical initiator caspases-8 and -10 are found to be resistant to TRAIL-induced apoptosis [36]. For instance, some colon cancer cell lines resist TRAIL-mediated apoptosis by reducing basal procaspase-8 and by increasing active caspase-8 degradation after TRAIL exposure [37]. Alteration of the genetic code, deletion of genetic material, and aberrant gene methylation are also common pathways leading to loss of gene function in human cancers. Many human tumors reduce expression of caspase-8 through hyper-methylation, as was observed in glioma cells with stem cell features [38] in primitive neuroectodermal (PNET) brain tumors [39], small cell lung cancers (SCLC) [40] and in neuroblastomas [41]. The study with PNET tumors found that caspase-8 mRNA and protein expression of caspase-8 could be restored with the DNA methylation inhibitor 5-aza-2′-deoxycytidine [39]. Other studies have demonstrated that interferon (IFN)-γ is capable of restoring caspase-8 expression that is silenced by methylation [40]. The identification of new strategies to overcome methylation of caspase-8 is critical to the promotion of DR-mediated therapies.
2.3 Upregulation of Anti-Apoptosis Proteins
DR-mediated caspase activation is also regulated by intracellular proteins such as c-FLIP, IAPs and Bcl2 family members. Cellular FLICE (FADD-like IL-1-beta-converting enzyme) inhibitory protein (c-FLIP) is a master anti-apoptotic regulator and resistance factor that suppresses death receptors including TNF-α, Fas, and TRAIL. c-FLIP is a family of alternatively spliced variants that primarily consist of long (c-FLIP(L)) and short (c-FLIP(S)) splice variants, and both forms can protect cells from apoptosis [42]. c-FLIP competes with the initiator caspases for binding to FADD due to the high sequence homology to the caspase death domains [43]. The binding of c-FLIP to the death domain of the death receptors prevents DISC formation and subsequent activation of the caspase cascade.
Another mechanism utilized by cancer cells to resist TRAIL-induced apoptosis is upregulation of the inhibitor of apoptosis proteins (IAP). IAP family members are characterized by the presence of a ~70 amino acid motif referred to as the baculovirus IAP Repeat (BIR) domain [44]. These BIR domains mediate IAP binding and caspases inhibition. The most potent IAP is the X chromosome-linked inhibitor of apoptosis (XIAP), which inhibits the function of effector caspases including Caspase-3, -7, and -9 [45]. The inhibitory activity of XIAP is overcome by a protein called second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO). Smac/DIABLO is released from mitochondria during apoptosis and antagonizes XIAP, promoting apoptosis in a positive feedback loop [46].
Apoptosis is also prevented by upregulation of Bcl-2 family members, which include at least 20 proteins, all of which contain one or more conserved Bcl-2 homology (BH) domains. Bcl-2, Bcl-xL, Bcl-w, and Mcl-1 are members of the Bcl-2 family that inhibit apoptosis in response to many cytotoxic agents. Bcl-2 overexpression protects neuroblastoma, glioblastoma, and breast cancer cells from TRAIL-mediated apoptosis by blocking caspase-3, -7, and -9 cleavage as well as cleavage of XIAP [47, 48]. Resistance to TRAIL mediated by Bcl-xL was demonstrated in pancreatic cancer cell lines [49]. These studies demonstrated that TRAIL treated BCL-xL expressing cells resulted in normal caspase-8 cleavage, but suppression of caspase-3 activity and apoptosis, which was abrogated by the administration of antisense oligonucleotides to BCL-xL mRNA [49]. Bcl-2 was also seen to be highly expressed in TRAIL-resistant tissues [50]. The upregulation of anti-apoptotic proteins will aid in cancer survival to TRAIL receptor agonists.
3 Ongoing Clinical Trials Evaluating DR-Targeted Therapies
Multiple clinical trials are underway to evaluate recombinant human TRAIL and agonistic antibodies against DR4 or DR5. Dulanermin (Apo2L/TRAIL/AMG951; Amgen/Genentech) is presently the only recombinant form of human TRAIL in clinical trials. Monoclonal antibodies against DR4 such as mapatumumab (human IgG1; GSK) or DR5 which include lexatumumab (human IgG1; GSK), drozitumab (human IgG1; Genentech/Roche), tigatuzumab (humanized IgG1; Daiichi-Sankyo), conatumumab (human IgG1; Amgen), and LBY135 (chimeric mouse/human IgG1; Novartis) are being developed to in phase I/II clinical studies. Another possible solution is a recombinant adenovirus encoding TRAIL, Ad-TRAIL, is in early development for cancer therapy [51]. A summary of the clinical trials that involved these therapies is found in Table 10.1. Understanding the resistance mechanisms to these drugs will advance the development of combinational therapies to overcome cancer death evasion.
3.1 Major Safety Findings from Phase 1 Studies
Clinical trials have shown death receptor targeting therapeutics do not have substantial toxicity, with the majority of side effects being fatigue and nausea. Dulanermin was given to 71 patients in a Phase 1 trial to patients with advanced cancer [7]. The adverse events (AE) associated with treatment were mostly mild, though two patients with sarcoma (synovial and undifferentiated) experienced serious AEs associated with rapid tumor necrosis. Agonistic anti-DR4 therapy, mapatumumab, was also well tolerated in Phase 1 studies, with no drug-related hepatic or dose-limiting toxicities (DLT) [52, 53]. Mapatumumab was given to 41 patients with advanced cancer and the majority of adverse events were grade 1 (mild) or 2 (moderate) on a 5 point scale [54]. Clinical studies which passed into phase II did not demonstrate an ability to shrink tumors within refractory colon cancer [55] or non-SCLC (NSCLC) [56]. Agonistic antibodies targeting DR5 have also shown tolerance in clinical studies across a variety of tumor types with minor DLT [57, 58]. In one study, lexatumumab was given to 37 patients, resulting in DLTs including asymptomatic elevations of serum amylase, transaminases, and bilirubin [59]. Similar observations were seen for drozitumab [6], tigatuzumab [60], and conatumumab [61].
3.2 Current Combinational Therapies
Survival mechanisms for cancer cells result from multiple pathways, suggesting a combination of therapeutics could potentially work well together to target TRAIL-resistance. In anticipation of tumor resistance to TRAIL signaling pathway targets, the ongoing Phase 2 studies are focused on evaluation of the DR-targeted therapies in combination with classical chemotherapies or other targeted therapies. These include paclitaxel and carboplatin used for NSCLC [62], gemcitabine for advanced pancreatic cancer [63], doxorubicin in unresectable soft tissue sarcomas [64] and FOLFOX for colorectal cancer [65].
The primary mechanisms of action (MOA) for Paclitaxel is inhibition of mitosis by stabilizing microtubules during cell division, while the MOA for platinum compounds like carboplatin is binding DNA, forming crosslinks that affect DNA replication. These compounds may combine well with DR-targeting therapies. Paclitaxel, and other taxanes such as docetaxel, increase the surface expression of DR4/DR5 [66, 67] and decrease AKT activity [68]. Additionally, the platinum family of compounds, such as carboplatin, cisplatin, and oxaliplatin, increase DR4/DR5 [65, 66] and decrease levels of c-FLIP [69]. Current trials have combined Dulanermin with paclitaxel, carboplatin, and bevacizumab in NSCLC and were well tolerated with 58 % progression free survival [70] but in studies combining carboplatin/paclitaxel with tigatuzumab there was no improvement over the efficacy of the chemotherapeutics [71]. Fully understanding the mechanisms through which chemotherapies can enhance DR-induced apoptosis is needed.
Gemcitabine is the standard treatment for patients with advanced pancreatic cancer. Gemcitabine is a nucleoside analog of cytidine but with fluorine atoms replacing the hydrogen atoms on the secondary carbon, resulting in inhibited DNA replication. There is evidence to suggest that gemcitabine may also combine well with DR-targeted therapies. Pyrimidine chemotherapies such as gemcitabine and 5-Fluorouracil reduce the levels of c-FLIP [42] and increase caspase-8 activity [72, 73], both of which are common mechanisms of resistance against DR-induced apoptosis. When gemcitabine was combined with tigatuzumab, 45 % of patients with metastatic pancreatic cancer within a phase II trial showed disease stability [74]. Gemcitabine was also tested in combination with cisplatin and mapatumumab, showing toleration and stable disease progression for a median of 6 months in 25 out of 49 patients [75].
Additionally, there are a series of chemotherapy regimens used for the treatment of colorectal cancer with names such as FOLFOX or FOLFIRI. Combining FOLolinic acid, Fluorouracil, and OXaliplatin is known as FOLFOX, while replacing oxaliplatin with IRInotecan is known as FOLFIRI. Fluorouracil, in particular 5-fluorouracil (5-FU), is a pyrimidine analog that inhibits thymidylate synthase. Folinic acid (leucovorin) augments the function of 5-FU. Oxaliplatin, similar to carboplatin, cross-links DNA preventing replication and transcription. Irinotecan is a topoisomerase inhibitor. The dosage regimens of FOLFOX can be modified and the resulting treatments include names like FOLFOX6 and modified FOLFOX6 (reviewed in [76]). The 5-fluorouracil and oxaliplatin components of FOLFOX are known to sensitize tumors to DR-targeted therapies. Drozitumab combined with both mFOLFOX6 and Bevacizumab resulted in stable disease for three out of eight patients treated [77]. FOLFOX combined with Conatumumab has also been explored in metastatic pancreatic cancer [78].
When treating the apoptotic signaling pathway, combinational therapies can expand to incorporate alternate signaling pathways. Current work is focusing on combining targets against the insulin-like growth factor receptor (IGFR) pathway [79] and the epidermal growth factor receptor (EGFR) pathway [80] with anti-DR5. IGFR agonistic antibody ganitumab with conatumumab showed tumor shrinkage and 36 % stable disease across patients with NSCLC, colorectal cancer, sarcoma, pancreatic cancer, and ovarian cancer [79]. Ganitumab was also combined with conatumumab and FOLFIRI, but resulted in patients with the FOLFIRI and conatumumab demonstrating progression-free survival [81]. Combining conatumumab and soluble TRAIL has shown increased DR clustering, resulting in increased DISC creation [82, 83]. Further studies combining different DR-agonists could yield interesting results for apoptosis induction.
3.3 Alternate Combination Strategies
Studies aim to improve the antitumor activity of DR4/DR5 by rationally designing mixtures that would overcome or bypass the resistance mechanisms within cancer cells. These include combinations with classical chemotherapies such as the ones discussed earlier. To further improve synergistic therapies, the main resistance mechanisms within the cell described earlier should be targeted, such as surface deficiency of DR4 and DR5, increased c-FLIP expression, decreased caspase-8 activity, and overexpression of anti-apoptotic proteins, such as XIAP.
Receptor deficiency on the cell surface is sufficient to render cancer cells resistant to TRAIL-induced apoptosis. Chemotherapies such as cisplatin and carboplatin can be used in combination with DR targeting therapeutics due to their ability to increase the surface expression of DR5 [66]. The proteasome inhibitor bortezomib [84, 85], the anti-melanoma drug ADI-PEG20 [86], p53 activating agents [87], anti-angiogenic therapies such as 3TSR [88], and histone deacetylase inhibitors [89, 90] have shown increases in surface DR expression, sensitizing cells to TRAIL. Therapies which increase DR expression can act synergistically with rhTRAIL or antibodies, dependent on a functional caspase signaling cascade.
c-FLIP is an important target for cancer therapy. Small interfering RNAs (siRNAs) that inhibited the expression of c-FLIP(L) in human cancer cell lines augmented TRAIL-induced DISC recruitment, activation, processing, and release of caspase-8 [91, 92]. The siRNA knockdown of c-FLIP is also postulated to target the tumor initiating cells within breast cancer lines [93]. Additionally, bortezomib is known to reduce c-FLIP expression [94] and sensitize cells to recombinant or immune-mediated TRAIL killing [95]. Small molecule-mediated inhibition of c-FLIP may have a strong therapeutic outlook [96].
Hyper-methylation of caspase-8 is a well-studied mechanism of caspase-8 silencing [38, 40]. Demethylating agents, such as 5-Aza-2′-deoxycytidine (5-dAzaC), are capable of restoring caspase-8 expression caused by hyper-methylation [38, 97]. In fact, treatment of neuroblastoma with a combination of low concentrations of 5-dAzaC and IFN-γ restored caspase-8 expression and sensitized tumors to TRAIL-mediated apoptosis [98]. Overexpression of IFN-γ combined with XIAP inhibitors increased caspase-8 activity in pancreatic cell lines sensitized cancer cells to TRAIL [99].
XIAP inhibitors also present a promising approach to augmenting TRAIL-mediated apoptosis. Small molecule inhibitors of XIAP cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells via enhanced TRAIL-induced activation of caspases, loss of mitochondrial membrane potential, and cytochrome c release in a caspase-dependent manner [100]. Another potential method of reducing XIAP levels is through the sub-toxic doses of roscovitine, a specific inhibitor of Cdc2 and Cdk2 [101]. Roscovitine treated TRAIL-resistant glioma cells reduced their expression of XIAP and survivin, two major inhibitors of caspases, and sensitized the cells to TRAIL-mediated apoptosis. Treatment of selumetinib (therapeutic targeting the MEK pathway) [102] or Dacarbazine [103] both downregulated IAPs and sensitized the cells to TRAIL. Modulation of the pro-apoptotic and anti-apoptotic signaling pathways through chemotherapy and alternative targeted therapeutics will combine well with DR-targeted therapies to induce cancer cell death.
3.4 Predictive Biomarkers for Cancer Response
Successful application of combinational therapies relies on predictive biomarkers of patient response. Biomarkers are currently being evaluated to improve therapeutics targeting the apoptosis signaling pathway [104]. The first indicator of potential responsiveness to DR targeted therapies is expression of DR4 or DR5. However, many studies have demonstrated that sensitivity cannot be predicted based on DR4 or DR5 surface expression alone [18, 27, 105, 106]. Studies from our laboratory have pursued this research topic.
We have identified a gene signature of over 71 over-expressed genes that were predictive of TRAIL sensitivity by examining the genome-wide mRNA expression profiles of 95 human cancer cell lines [11]. The over-expressed genes were dominated by two functionally related gene families: interferon-induced genes and major histocompatibility genes. These data are consistent with the findings that treatment of cancers with interferon sensitizes the tumors to TRAIL-related therapies [98]. Another study approached the identification of biomarkers for TRAIL via mRNA expression and had very interesting results. The mRNA expression profiles of pancreatic, NSCLC, and melanoma cell lines showed that up to 30 % of these tumors had increased expression of GALNT14, a peptidyl O-glycosyltransferase. The investigators were able to increase or decrease TRAIL sensitivity by selectively overexpressing or silencing GALNT14 [27].
Another possibility for identifying biomarkers that indicate apoptosis sensitivity is to look at autophagy. Autophagy and apoptosis have a complex relationship, either being triggered together or developing through mutually exclusive processes. Our studies indicate one role for autophagy is to protect tumor cells from TRAIL-mediated apoptosis. During autophagy, cellular components, including membrane components containing DR4 and DR5, are invaginated into autophagic vesicles (autophagosomes). These autophagosomes fuse with lysosomes to form autolysosomes wherein autophagic cargos are degraded. Our studies indicate that this process also provides tumors with resistance to death receptor-mediated therapies [26]. Breast cancer cell lines with high levels of basal autophagic function in nutrient rich conditions have a high level of TRAIL resistance. Similar levels of basal autophagy were found in pancreatic [107], melanoma [108], and NSCLC [109]. The basal autophagic activity sequesters death receptors into intracellular compartments where they are not exposed to TRAIL and thus are resistant. Our studies indicate that the death receptors were housed in LC3-II labeled autophagosomes, and disruption of the autophagosomes restored surface expression of death receptors and increased sensitivity to TRAIL. Analysis of tumors for LC3-II may provide predictive markers of tumor resistance to TRAIL-related therapies.
Oncogenic proteins, such as Ras GTPases, may also provide unique opportunities to identify biomarkers for TRAIL sensitivity. Many cancer types upregulate the Ras signaling pathway due to gain of function mutations in ras genes themselves or alterations in the proteins that regulate Ras [110–112]. Ras is also shown to promote activation of cell death pathways, in contrast to its best known function of promoting growth [113]. Ras interacts with various downstream effector targets such as MEK, PI3K, and Rho GTPases [114, 115]. Recent evidence suggests that Ras regulates the expression of death receptors and increases TRAIL sensitivity [30, 116, 117]. In fact, transfection of oral squamous cell carcinomas with H-RasV12, a constitutively active H-Ras mutant, increased surface expression of DR5 and sensitivity to either TRAIL or anti-DR5 [18]. These findings support the idea of using Ras as a biomarker for predicting DR sensitivity.
The involvement of Ras in TRAIL sensitivity of tumors is pertinent because of the expansion of growth factor inhibitors currently used to treat cancer. The human epidermal receptor (HER) family of growth factor receptors, including members EGFR and HER2, are important regulators of tumor cell proliferation, survival, angiogenesis, and metastasis [118]. Engagement of an HER by its cognate ligand initiates the Ras signaling pathway. EGFR and HER2 are frequently aberrantly overexpressed or mutated in a wide range of tumors; therefore, these receptors represent attractive targets for cancer treatment. This has resulted in the development of multiple anti-HER therapeutics, including mAbs trastuzumab (anti-HER2), cetuximab (anti-EGFR), and multiple small molecule tyrosine kinases inhibitors targeting EGFR (e.g. gefitnib, erlotnib) and HER2 (e.g. CP-724, 714, M578440).
It has been reported that TRAIL activates the EGRF pathway, and that the cetuximab-mediated sensitization to TRAIL is due to the inhibition of TRAIL-mediated EGFR activation in colorectal cancers [119]. Additionally, the combination of cetuximab with TRAIL resulted in increased clustering of DR4 and FADD into lipid rafts [120], which is known to enhance the function of death receptors [121]. Breast and ovarian cancers treated with trastuzumab are also sensitized to DR-mediated apoptosis. Trastuzumab treatment decreases Akt kinase activation but not mitogen-activated protein kinase activation and sensitizes cell lines to TRAIL [122]. These studies indicate that anti-HER therapeutics, in addition to their direct suppression of tumor growth, also provide favorable conditions for TRAIL-triggered apoptosis.
4 Perspectives
There is great potential in TRAIL-based therapies, yet the limitations of TRAIL receptor agonists as a single agent need to be overcome. The benefits of TRAIL as a targeted therapeutic with mild toxicity are outweighed by the lack of efficacy in clinical trials due to the protein’s short half-life and the cancer cell’s TRAIL resistance mechanisms. Thus, a renewed effort into combinational therapies that counter these resistances allowing DR targeted therapies to initiate apoptosis is needed.
The most effective combinational therapies will overcome DR surface deficiency, loss of the initiator caspases-8 and -10, and overexpression of anti-apoptotic molecules such as c-FLIP and XIAP. Many current combinations use chemotherapies to upregulate DR expression or inhibit anti-apoptotic proteins. The clinical use of combination therapy promotes investigation of molecular interactions of combination components, as not all chemotherapies will improve the efficacy of agonistic antibodies. The effectiveness of DR-targeted therapies will be maximized by optimizing therapies based on the resistances of different cancer types and for individual patients.
The next generation of combination therapies that exploit cellular machinery to augment DR therapies is another exciting realm of future discoveries. The combination of DR-targeted therapies with EGFR inhibitors, such as cetuximab and trastuzumab, offer new potential avenues for cancer treatment now that scientists have a growing understanding of the influence that the EGFR-Ras pathway has on DR-mediated apoptosis. Our recent work demonstrating the high basal level of autophagy causing surface deficiencies of DRs recommend combinational therapies that target the Ras/EGFR pathway in combination with the DR pathway. Chemotherapeutics that inhibit autophagy offer promise by reducing the trafficking of DR4 and DR5 from the surface into autophagosomes. In fact, treatment of cells with hydroxychloroquine, an inhibitor of autophagy, increased caspase-3 activation and DR-mediated apoptosis [123, 124]. Future studies that directly inhibit autophagy with DR targeted therapies are needed to examine the enhancement of apoptotic signaling.
Recent studies analyzing the synergistic effects of combining DR targeted therapeutics that overcome DR signaling pathway deficiencies such as insufficient receptor clustering and DISC formation are clinically relevant [82, 83]. Combining DR-specific therapeutics provides an alternative to chemotherapeutic administration, taking advantage of the benefits of targeted therapeutic and cancer-specific toxicity. This could potentially further the use of rhTRAIL in cancer patients.
Additionally, there is an unmet need for biomarkers that predict tumor sensitivities to TRAIL receptor targeted therapies. Much of the basic science needed to identify biomarkers is available; we are well aware of inducers and inhibitors of apoptosis that generally indicate TRAIL resistance, such as lack of surface DR4/5. However, the apoptotic machinery is redundant, and multiple lines of attack are needed to overcome resistances. Therefore, these biomarkers need additional sophistication than simply surface expression of DR5 or lack of caspase-8. When well designed, combinational drugs could lead to improved outcomes of cancer treatment by circumventing the specific resistance mechanisms in cancer cells.
Abbreviations
- 5-dAzaC:
-
5-Aza-2′-deoxycytidine
- 5-FU:
-
5-Fluorouracil
- AE:
-
Adverse effects
- BH:
-
Bcl-2 homology
- c-FLIP:
-
Cellular FADD-like IL-1 beta-converting enzyme inhibitor protein
- DISC:
-
Death inducing signaling complex
- DLT:
-
Dose limiting toxicities
- DR:
-
Death receptors
- EGFR:
-
Epidermal growth factor receptor
- FADD:
-
Fas-associated death domain
- FOLFIRI:
-
Fololinic acid, fluorouracil, and irinotecan
- FOLFOX:
-
Fololinic acid, fluorouracil, and oxiplatin
- HER:
-
Human epidermal receptor
- IAP:
-
Inhibitor of apoptosis proteins
- IFN:
-
Interferon
- IGFR:
-
Insulin-like growth factor receptor
- MOA:
-
Mechanism of action
- NSCLC:
-
Non-small cell lung cancer
- PNET:
-
Primitive neuroectodermal
- rhTRAIL:
-
Recombinant human TRAIL
- SCLC:
-
Small cell lung cancer
- siRNA:
-
Small interfering RNA
- SMAC/DIABLO:
-
Second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low pI
- tBid:
-
Truncated Bid
- TNFR:
-
Tumor necrosis factor
- TRAIL:
-
TNF-related apoptosis inducing ligand
- XIAP:
-
x Chromosome-linked inhibitor of apoptosis
References
Lenk H, Tanneberger S, Muller U, Ebert J, Shiga T. Phase II clinical trial of high-dose recombinant human tumor necrosis factor. Cancer Chemother Pharmacol. 1989;24:391–2.
Creaven PJ, Plager JE, Dupere S, Huben RP, Takita H, Mittelman A, Proefrock A. Phase I clinical trial of recombinant human tumor necrosis factor. Cancer Chemother Pharmacol. 1987;20:137–44.
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62.
Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, Hooley J, Sherwood S, Pai R, Leung S, Khan L, Gliniak B, Bussiere J, Smith CA, Strom SS, Kelley S, Fox JA, Thomas D, Ashkenazi A. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med. 2001;7:383–5.
Daniel D, Wilson NS. Tumor necrosis factor: renaissance as a cancer therapeutic? Curr Cancer Drug Targets. 2008;8:124–31.
Camidge DR, Herbst RS, Gordon MS, Eckhardt SG, Kurzrock R, Durbin B, Ing J, Tohnya TM, Sager J, Ashkenazi A, Bray G, Mendelson D. A phase I safety and pharmacokinetic study of the death receptor 5 agonistic antibody PRO95780 in patients with advanced malignancies. Clin Cancer Res. 2010;16:1256–63.
Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, Ashkenazi A, Jubb AM, Mendelson DS. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28:2839–46.
Leong S, Cohen RB, Gustafson DL, Langer CJ, Camidge DR, Padavic K, Gore L, Smith M, Chow LQ, von Mehren M, O'Bryant C, Hariharan S, Diab S, Fox NL, Miceli R, Eckhardt SG. Mapatumumab, an antibody targeting TRAIL-R1, in combination with paclitaxel and carboplatin in patients with advanced solid malignancies: results of a phase I and pharmacokinetic study. J Clin Oncol. 2009;27:4413–21.
Roberts NJ, Zhou S, Diaz Jr LA, Holdhoff M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget. 2011;2:739–51.
Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5:a008698.
Chen JJ, Knudsen S, Mazin W, Dahlgaard J, Zhang B. A 71-gene signature of TRAIL sensitivity in cancer cells. Mol Cancer Ther. 2012;11:34–44.
Yoshida T, Zhang Y, Rivera Rosado LA, Zhang B. Repeated treatment with subtoxic doses of TRAIL induces resistance to apoptosis through its death receptors in MDA-MB-231 breast cancer cells. Mol Cancer Res. 2009;7:1835–44.
Zhang Y, Yoshida T, Zhang B. TRAIL induces endocytosis of its death receptors in MDA-MB-231 breast cancer cells. Cancer Biol Ther. 2009;8:917–22.
Zhang Y, Zhang B. TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Mol Cancer Res. 2008;6:1861–71.
Deng Y, Lin Y, Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16:33–45.
Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene. 2001;20:2122–33.
Yin XM. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10:161–7.
Chen JJ, Mikelis CM, Zhang Y, Gutkind JS, Zhang B. TRAIL induces apoptosis in oral squamous carcinoma cells—a crosstalk with oncogenic Ras regulated cell surface expression of death receptor 5. Oncotarget. 2013;4:206–17.
Liu GC, Zhang J, Liu SG, Gao R, Long ZF, Tao K, Ma YF. Detachment of esophageal carcinoma cells from extracellular matrix causes relocalization of death receptor 5 and apoptosis. World J Gastroenterol. 2009;15:836–44.
Leithner K, Stacher E, Wurm R, Ploner F, Quehenberger F, Wohlkoenig C, Balint Z, Polachova J, Olschewski A, Samonigg H, Popper HH, Olschewski H. Nuclear and cytoplasmic death receptor 5 as prognostic factors in patients with non-small cell lung cancer treated with chemotherapy. Lung Cancer. 2009;65:98–104.
Kojima Y, Nakayama M, Nishina T, Nakano H, Koyanagi M, Takeda K, Okumura K, Yagita H. Importin beta1 protein-mediated nuclear localization of death receptor 5 (DR5) limits DR5/tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced cell death of human tumor cells. J Biol Chem. 2011;286:43383–93.
Jin Z, McDonald III ER, Dicker DT, El-Deiry WS. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J Biol Chem. 2004;279:35829–39.
Kohlhaas SL, Craxton A, Sun XM, Pinkoski MJ, Cohen GM. Receptor-mediated endocytosis is not required for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J Biol Chem. 2007;282:12831–41.
Austin CD, Lawrence DA, Peden AA, Varfolomeev EE, Totpal K, De Maziere AM, Klumperman J, Arnott D, Pham V, Scheller RH, Ashkenazi A. Death-receptor activation halts clathrin-dependent endocytosis. Proc Natl Acad Sci U S A. 2006;103:10283–8.
Chen JJ, Shen HC, Rivera Rosado LA, Zhang Y, Di X, Zhang B. Mislocalization of death receptors correlates with cellular resistance to their cognate ligands in human breast cancer cells. Oncotarget. 2012;3:833–42.
Di X, Zhang G, Zhang Y, Takeda K, Rivera Rosado LA, Zhang B. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget. 2013;4:1349–64.
Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF, Totpal K, Huw L, Katta V, Cavet G, Hymowitz SG, Amler L, Ashkenazi A. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13:1070–7.
Haselmann V, Kurz A, Bertsch U, Hubner S, Olempska-Muller M, Fritsch J, Hasler R, Pickl A, Fritsche H, Annewanter F, Engler C, Fleig B, Bernt A, Roder C, Schmidt H, Gelhaus C, Hauser C, Egberts JH, Heneweer C, Rohde AM, Boger C, Knippschild U, Rocken C, Adam D, Walczak H, Schutze S, Janssen O, Wulczyn FG, Wajant H, Kalthoff H, Trauzold A. Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumor cells. Gastroenterology. 2014;146:278–90.
Monma H, Harashima N, Inao T, Okano S, Tajima Y, Harada M. The HSP70 and autophagy inhibitor pifithrin-mu enhances the antitumor effects of TRAIL on human pancreatic cancer. Mol Cancer Ther. 2013;12:341–51.
Drosopoulos KG, Roberts ML, Cermak L, Sasazuki T, Shirasawa S, Andera L, Pintzas A. Transformation by oncogenic RAS sensitizes human colon cells to TRAIL-induced apoptosis by up-regulating death receptor 4 and death receptor 5 through a MEK-dependent pathway. J Biol Chem. 2005;280:22856–67.
Sahu RP, Batra S, Kandala PK, Brown TL, Srivastava SK. The role of K-ras gene mutation in TRAIL-induced apoptosis in pancreatic and lung cancer cell lines. Cancer Chemother Pharmacol. 2011;67:481–7.
Leverkus M, Sprick MR, Wachter T, Denk A, Brocker EB, Walczak H, Neumann M. TRAIL-induced apoptosis and gene induction in HaCaT keratinocytes: differential contribution of TRAIL receptors 1 and 2. J Invest Dermatol. 2003;121:149–55.
Horak P, Pils D, Haller G, Pribill I, Roessler M, Tomek S, Horvat R, Zeillinger R, Zielinski C, Krainer M. Contribution of epigenetic silencing of tumor necrosis factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL resistance and ovarian cancer. Mol Cancer Res. 2005;3:335–43.
Cheng J, Hylander BL, Baer MR, Chen X, Repasky EA. Multiple mechanisms underlie resistance of leukemia cells to Apo2 ligand/TRAIL. Mol Cancer Ther. 2006;5:1844–53.
MacFarlane M, Kohlhaas SL, Sutcliffe MJ, Dyer MJ, Cohen GM. TRAIL receptor-selective mutants signal to apoptosis via TRAIL-R1 in primary lymphoid malignancies. Cancer Res. 2005;65:11265–70.
Crowder RN, El-Deiry WS. Caspase-8 regulation of TRAIL-mediated cell death. Exp Oncol. 2012;34:160–4.
van Geelen CM, Pennarun B, Ek WB, Le PT, Spierings DC, de Vries EG, de Jong S. Downregulation of active caspase 8 as a mechanism of acquired TRAIL resistance in mismatch repair-proficient colon carcinoma cell lines. Int J Oncol. 2010;37:1031–41.
Capper D, Gaiser T, Hartmann C, Habel A, Mueller W, Herold-Mende C, von Deimling A, Siegelin MD. Stem-cell-like glioma cells are resistant to TRAIL/Apo2L and exhibit down-regulation of caspase-8 by promoter methylation. Acta Neuropathol. 2009;117:445–56.
Grotzer MA, Eggert A, Zuzak TJ, Janss AJ, Marwaha S, Wiewrodt BR, Ikegaki N, Brodeur GM, Phillips PC. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene. 2000;19:4604–10.
Hopkins-Donaldson S, Ziegler A, Kurtz S, Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U, Stahel R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003;10:356–64.
Rebbaa A, Chou PM, Emran M, Mirkin BL. Doxorubicin-induced apoptosis in caspase-8-deficient neuroblastoma cells is mediated through direct action on mitochondria. Cancer Chemother Pharmacol. 2001;48:423–8.
Haag C, Stadel D, Zhou S, Bachem MG, Moller P, Debatin KM, Fulda S. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut. 2011;60:225–37.
Safa AR, Day TW, Wu CH. Cellular FLICE-like inhibitory protein (C-FLIP): a novel target for cancer therapy. Curr Cancer Drug Targets. 2008;8:37–46.
Finlay D, Vamos M, Gonzalez-Lopez M, Ardecky RJ, Ganji SR, Yuan H, Su Y, Cooley TR, Hauser CT, Welsh K, Reed JC, Cosford ND, Vuori K. Small-molecule IAP antagonists sensitize cancer cells to TRAIL-induced apoptosis: roles of XIAP and cIAPs. Mol Cancer Ther. 2014;13:5–15.
Cummins JM, Kohli M, Rago C, Kinzler KW, Vogelstein B, Bunz F. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 2004;64:3006–8.
Maas C, Verbrugge I, de Vries E, Savich G, van de Kooij LW, Tait SW, Borst J. Smac/DIABLO release from mitochondria and XIAP inhibition are essential to limit clonogenicity of Type I tumor cells after TRAIL receptor stimulation. Cell Death Differ. 2010;17:1613–23.
Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 2002;21:2283–94.
Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12:228–37.
Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, Bayer E, Walczak H, Kalthoff H, Ungefroren H. Bcl-XL protects pancreatic adenocarcinoma cells against. Oncogene. 2000;19:5477–86.
Subbiah V, Brown RE, Buryanek J, Trent J, Ashkenazi A, Herbst R, Kurzrock R. Targeting the apoptotic pathway in chondrosarcoma using recombinant human Apo2L/TRAIL (dulanermin), a dual proapoptotic receptor (DR4/DR5) agonist. Mol Cancer Ther. 2012;11:2541–6.
Vanoosten RL, Earel Jr JK, Griffith TS. Enhancement of Ad5-TRAIL cytotoxicity against renal cell carcinoma with histone deacetylase inhibitors. Cancer Gene Ther. 2006;13:628–32.
Tolcher AW, Mita M, Meropol NJ, von Mehren M, Patnaik A, Padavic K, Hill M, Mays T, McCoy T, Fox NL, Halpern W, Corey A, Cohen RB. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol. 2007;25:1390–5.
Younes A, Vose JM, Zelenetz AD, Smith MR, Burris HA, Ansell SM, Klein J, Halpern W, Miceli R, Kumm E, Fox NL, Czuczman MS. A phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin's lymphoma. Br J Cancer. 2010;103:1783–7.
Hotte SJ, Hirte HW, Chen EX, Siu LL, Le LH, Corey A, Iacobucci A, MacLean M, Lo L, Fox NL, Oza AM. A phase 1 study of mapatumumab (fully human monoclonal antibody to TRAIL-R1) in patients with advanced solid malignancies. Clin Cancer Res. 2008;14:3450–5.
Trarbach T, Moehler M, Heinemann V, Kohne CH, Przyborek M, Schulz C, Sneller V, Gallant G, Kanzler S. Phase II trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with refractory colorectal cancer. Br J Cancer. 2010;102:506–12.
Greco FA, Bonomi P, Crawford J, Kelly K, Oh Y, Halpern W, Lo L, Gallant G, Klein J. Phase 2 study of mapatumumab, a fully human agonistic monoclonal antibody which targets and activates the TRAIL receptor-1, in patients with advanced non-small cell lung cancer. Lung Cancer. 2008;61:82–90.
Merchant MS, Geller JI, Baird K, Chou AJ, Galli S, Charles A, Amaoko M, Rhee EH, Price A, Wexler LH, Meyers PA, Widemann BC, Tsokos M, Mackall CL. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. J Clin Oncol. 2012;30:4141–7.
Wakelee HA, Patnaik A, Sikic BI, Mita M, Fox NL, Miceli R, Ullrich SJ, Fisher GA, Tolcher AW. Phase I and pharmacokinetic study of lexatumumab (HGS-ETR2) given every 2 weeks in patients with advanced solid tumors. Ann Oncol. 2010;21:376–81.
Plummer R, Attard G, Pacey S, Li L, Razak A, Perrett R, Barrett M, Judson I, Kaye S, Fox NL, Halpern W, Corey A, Calvert H, de Bono J. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res. 2007;13:6187–94.
Forero-Torres A, Shah J, Wood T, Posey J, Carlisle R, Copigneaux C, Luo FR, Wojtowicz-Praga S, Percent I, Saleh M. Phase I trial of weekly tigatuzumab, an agonistic humanized monoclonal antibody targeting death receptor 5 (DR5). Cancer Biother Radiopharm. 2010;25:13–9.
Herbst RS, Kurzrock R, Hong DS, Valdivieso M, Hsu CP, Goyal L, Juan G, Hwang YC, Wong S, Hill JS, Friberg G, LoRusso PM. A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin Cancer Res. 2010;16:5883–91.
Paz-Ares L, Balint B, de Boer RH, van Meerbeeck JP, Wierzbicki R, De SP, Galimi F, Haddad V, Sabin T, Hei YJ, Pan Y, Cottrell S, Hsu CP, RamLau R. A randomized phase 2 study of paclitaxel and carboplatin with or without conatumumab for first-line treatment of advanced non-small-cell lung cancer. J Thorac Oncol. 2013;8:329–37.
Kindler HL, Richards DA, Garbo LE, Garon EB, Stephenson Jr JJ, Rocha-Lima CM, Safran H, Chan D, Kocs DM, Galimi F, McGreivy J, Bray SL, Hei Y, Feigal EG, Loh E, Fuchs CS. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann Oncol. 2012;23:2834–42.
Demetri GD, Le CA, Chawla SP, Brodowicz T, Maki RG, Bach BA, Smethurst DP, Bray S, Hei YJ, Blay JY. First-line treatment of metastatic or locally advanced unresectable soft tissue sarcomas with conatumumab in combination with doxorubicin or doxorubicin alone: a phase I/II open-label and double-blind study. Eur J Cancer. 2012;48:547–63.
Wainberg ZA, Messersmith WA, Peddi PF, Kapp AV, Ashkenazi A, Royer-Joo S, Portera CC, Kozloff MF. A phase 1B study of dulanermin in combination with modified FOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Clin Colorectal Cancer. 2013;12:248–54.
El-Gazzar A, Perco P, Eckelhart E, Anees M, Sexl V, Mayer B, Liu Y, Mikulits W, Horvat R, Pangerl T, Zheng D, Krainer M. Natural immunity enhances the activity of a DR5 agonistic antibody and carboplatin in the treatment of ovarian cancer. Mol Cancer Ther. 2010;9:1007–18.
Nimmanapalli R, Perkins CL, Orlando M, O'Bryan E, Nguyen D, Bhalla KN. Pretreatment with paclitaxel enhances apo-2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of prostate cancer cells by inducing death receptors 4 and 5 protein levels. Cancer Res. 2001;61:759–63.
Asakuma J, Sumitomo M, Asano T, Asano T, Hayakawa M. Selective Akt inactivation and tumor necrosis actor-related apoptosis-inducing ligand sensitization of renal cancer cells by low concentrations of paclitaxel. Cancer Res. 2003;63:1365–70.
Ding L, Yuan C, Wei F, Wang G, Zhang J, Bellail AC, Zhang Z, Olson JJ, Hao C. Cisplatin restores TRAIL apoptotic pathway in glioblastoma-derived stem cells through up-regulation of DR5 and down-regulation of c-FLIP. Cancer Invest. 2011;29:511–20.
Soria JC, Smit E, Khayat D, Besse B, Yang X, Hsu CP, Reese D, Wiezorek J, Blackhall F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J Clin Oncol. 2010;28:1527–33.
Reck M, Krzakowski M, Chmielowska E, Sebastian M, Hadler D, Fox T, Wang Q, Greenberg J, Beckman RA, von Pawel J. A randomized, double-blind, placebo-controlled phase 2 study of tigatuzumab (CS-1008) in combination with carboplatin/paclitaxel in patients with chemotherapy-naive metastatic/unresectable non-small cell lung cancer. Lung Cancer. 2013;82:441–8.
Yi TB, Yang LY. Caspase-8 in apoptosis of hepatoma cell induced by 5-fluorouracil. Hepatobiliary Pancreat Dis Int. 2003;2:98–101.
Yang L, Wu D, Luo K, Wu S, Wu P. Andrographolide enhances 5-fluorouracil-induced apoptosis via caspase-8-dependent mitochondrial pathway involving p53 participation in hepatocellular carcinoma (SMMC-7721) cells. Cancer Lett. 2009;276:180–8.
Forero-Torres A, Infante JR, Waterhouse D, Wong L, Vickers S, Arrowsmith E, He AR, Hart L, Trent D, Wade J, Jin X, Wang Q, Austin T, Rosen M, Beckman R, von Roemeling R, Greenberg J, Saleh M. Phase 2, multicenter, open-label study of tigatuzumab (CS-1008), a humanized monoclonal antibody targeting death receptor 5, in combination with gemcitabine in chemotherapy-naive patients with unresectable or metastatic pancreatic cancer. Cancer Med. 2013;2:925–32.
Mom CH, Verweij J, Oldenhuis CN, Gietema JA, Fox NL, Miceli R, Eskens FA, Loos WJ, de Vries EG, Sleijfer S. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res. 2009;15:5584–90.
Pasetto LM, Jirillo A, Iadicicco G, Rossi E, Paris MK, Monfardini S. FOLFOX versus FOLFIRI: a comparison of regimens in the treatment of colorectal cancer metastases. Anticancer Res. 2005;25:563–76.
Rocha Lima CM, Bayraktar S, Flores AM, MacIntyre J, Montero A, Baranda JC, Wallmark J, Portera C, Raja R, Stern H, Royer-Joo S, Amler LC. Phase Ib study of drozitumab combined with first-line mFOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Cancer Invest. 2012;30:727–31.
Fuchs CS, Fakih M, Schwartzberg L, Cohn AL, Yee L, Dreisbach L, Kozloff MF, Hei YJ, Galimi F, Pan Y, Haddad V, Hsu CP, Sabin A, Saltz L. TRAIL receptor agonist conatumumab with modified FOLFOX6 plus bevacizumab for first-line treatment of metastatic colorectal cancer: a randomized phase 1b/2 trial. Cancer. 2013;119:4290–8.
Tabernero J, Chawla SP, Kindler H, Reckamp K, Chiorean EG, Azad NS, Lockhart AC, Hsu CP, Baker NF, Galimi F, Beltran P, Baselga J. Anticancer activity of the type I insulin-like growth factor receptor antagonist, ganitumab, in combination with the death receptor 5 agonist, conatumumab. Oncol: Target; 2014.
Dolloff NG, Mayes PA, Hart LS, Dicker DT, Humphreys R, El-Deiry WS. Off-target lapatinib activity sensitizes colon cancer cells through TRAIL death receptor up-regulation. Sci Transl Med. 2011;3:86ra50.
Cohn AL, Tabernero J, Maurel J, Nowara E, Sastre J, Chuah BY, Kopp MV, Sakaeva DD, Mitchell EP, Dubey S, Suzuki S, Hei YJ, Galimi F, McCaffery I, Pan Y, Loberg R, Cottrell S, Choo SP. A randomized, placebo-controlled phase 2 study of ganitumab or conatumumab in combination with FOLFIRI for second-line treatment of mutant KRAS metastatic colorectal cancer. Ann Oncol. 2013;24:1777–85.
Tuthill MH, Montinaro A, Zinngrebe J, Prieske K, Draber P, Prieske S, Newsom-Davis T, von Karstedt S, Graves J, Walczak H. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene. 2015;34:2138–44.
Graves JD, Kordich JJ, Huang TH, Piasecki J, Bush TL, Sullivan T, Foltz IN, Chang W, Douangpanya H, Dang T, O'Neill JW, Mallari R, Zhao X, Branstetter DG, Rossi JM, Long AM, Huang X, Holland PM. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell. 2014;26:177–89.
Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, Childs R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66:7317–25.
Ames E, Hallett WH, Murphy WJ. Sensitization of human breast cancer cells to natural killer cell-mediated cytotoxicity by proteasome inhibition. Clin Exp Immunol. 2009;155:504–13.
You M, Savaraj N, Wangpaichitr M, Wu C, Kuo MT, Varona-Santos J, Nguyen DM, Feun L. The combination of ADI-PEG20 and TRAIL effectively increases cell death in melanoma cell lines. Biochem Biophys Res Commun. 2010;394:760–6.
Pishas KI, Neuhaus SJ, Clayer MT, Adwal A, Brown MP, Evdokiou A, Callen DF, Neilsen PM. Pre-activation of the p53 pathway through Nutlin-3a sensitises sarcomas to drozitumab therapy. Oncol Rep. 2013;30:471–7.
Ren B, Song K, Parangi S, Jin T, Ye M, Humphreys R, Duquette M, Zhang X, Benhaga N, Lawler J, Khosravi-Far R. A double hit to kill tumor and endothelial cells by TRAIL and antiangiogenic 3TSR. Cancer Res. 2009;69:3856–65.
Jazirehi AR, Kurdistani SK, Economou JS. Histone deacetylase inhibitor sensitizes apoptosis-resistant melanomas to cytotoxic human T lymphocytes through regulation of TRAIL/DR5 pathway. J Immunol. 2014;192:3981–9.
Earel Jr JK, Vanoosten RL, Griffith TS. Histone deacetylase inhibitors modulate the sensitivity of tumor necrosis factor-related apoptosis-inducing ligand-resistant bladder tumor cells. Cancer Res. 2006;66:499–507.
Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem. 2005;280:19401–9.
Bijangi-Vishehsaraei K, Saadatzadeh MR, Huang S, Murphy MP, Safa AR. 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) targets mRNA of the c-FLIP variants and induces apoptosis in MCF-7 human breast cancer cells. Mol Cell Biochem. 2010;342:133–42.
Piggott L, Omidvar N, Marti PS, Eberl M, Clarkson RW. Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL. Breast Cancer Res. 2011;13:R88.
Sayers TJ, Brooks AD, Koh CY, Ma W, Seki N, Raziuddin A, Blazar BR, Zhang X, Elliott PJ, Murphy WJ. The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003;102:303–10.
Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180:163–70.
Schimmer AD, Thomas MP, Hurren R, Gronda M, Pellecchia M, Pond GR, Konopleva M, Gurfinkel D, Mawji IA, Brown E, Reed JC. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res. 2006;66:2367–75.
Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in neuroblastoma is related to malignancy and resistance to TRAIL-induced apoptosis. Med Pediatr Oncol. 2000;35:608–11.
Fulda S, Debatin KM. 5-Aza-2′-deoxycytidine and IFN-gamma cooperate to sensitize for TRAIL-induced apoptosis by upregulating caspase-8. Oncogene. 2006;25:5125–33.
Buneker CK, Yu R, Deedigan L, Mohr A, Zwacka RM. IFN-gamma combined with targeting of XIAP leads to increased apoptosis-sensitisation of TRAIL resistant pancreatic carcinoma cells. Cancer Lett. 2012;316:168–77.
Fakler M, Loeder S, Vogler M, Schneider K, Jeremias I, Debatin KM, Fulda S. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood. 2009;113:1710–22.
Kim EH, Kim SU, Shin DY, Choi KS. Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene. 2004;23:446–56.
Grazia G, Vegetti C, Benigni F, Penna I, Perotti V, Tassi E, Bersani I, Nicolini G, Canevari S, Carlo-Stella C, Gianni AM, Mortarini R, Anichini A. Synergistic anti-tumor activity and inhibition of angiogenesis by cotargeting of oncogenic and death receptor pathways in human melanoma. Cell Death Dis. 2014;5:e1434.
Engesaeter B, Engebraaten O, Florenes VA, Maelandsmo GM. Dacarbazine and the agonistic TRAIL receptor-2 antibody lexatumumab induce synergistic anticancer effects in melanoma. PLoS One. 2012;7:e45492.
Zoog SJ, Ma CY, Kaplan-Lefko PJ, Hawkins JM, Moriguchi J, Zhou L, Pan Y, Hsu CP, Friberg G, Herbst R, Hill J, Juan G. Measurement of conatumumab-induced apoptotic activity in tumors by fine needle aspirate sampling. Cytometry A. 2010;77:849–60.
Clodi K, Wimmer D, Li Y, Goodwin R, Jaeger U, Mann G, Gadner H, Younes A. Expression of tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors and sensitivity to TRAIL-induced apoptosis in primary B-cell acute lymphoblastic leukaemia cells. Br J Haematol. 2000;111:580–6.
Kontny HU, Hammerle K, Klein R, Shayan P, Mackall CL, Niemeyer CM. Sensitivity of Ewing’s sarcoma to TRAIL-induced apoptosis. Cell Death Differ. 2001;8:506–14.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29.
Lazova R, Klump V, Pawelek J. Autophagy in cutaneous malignant melanoma. J Cutan Pathol. 2010;37:256–68.
Kaminskyy VO, Piskunova T, Zborovskaya IB, Tchevkina EM, Zhivotovsky B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy. 2012;8:1032–44.
Sogabe Y, Suzuki H, Toyota M, Ogi K, Imai T, Nojima M, Sasaki Y, Hiratsuka H, Tokino T. Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int J Oncol. 2008;32:1253–61.
Kuo MY, Jeng JH, Chiang CP, Hahn LJ. Mutations of Ki-ras oncogene codon 12 in betel quid chewing-related human oral squamous cell carcinoma in Taiwan. J Oral Pathol Med. 1994;23:70–4.
Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308.
Overmeyer JH, Maltese WA. Death pathways triggered by activated Ras in cancer cells. Front Biosci (Landmark Ed). 2011;16:1693–713.
Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O'Hagan R, Pantginis J, Zhou H, Horner JW, Cordon-Cardo C, Yancopoulos GD, DePinho RA. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–72.
Li W, Zhu T, Guan KL. Transformation potential of Ras isoforms correlates with activation of phosphatidylinositol 3-kinase but not ERK. J Biol Chem. 2004;279:37398–406.
Nesterov A, Nikrad M, Johnson T, Kraft AS. Oncogenic Ras sensitizes normal human cells to tumor necrosis factor-alpha-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2004;64:3922–7.
Wang Y, Quon KC, Knee DA, Nesterov A, Kraft AS. RAS, MYC, and sensitivity to tumor necrosis factor-alpha-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2005;65:1615–6.
Ocana A, Pandiella A. Targeting HER receptors in cancer. Curr Pharm Des. 2013;19:808–17.
Van SS, Kelly DM, Kyula J, Stokesberry S, Fennell DA, Johnston PG, Longley DB. Src and ADAM-17-mediated shedding of transforming growth factor-alpha is a mechanism of acute resistance to TRAIL. Cancer Res. 2008;68:8312–21.
Xu L, Hu X, Qu X, Hou K, Zheng H, Liu Y. Cetuximab enhances TRAIL-induced gastric cancer cell apoptosis by promoting DISC formation in lipid rafts. Biochem Biophys Res Commun. 2013;439:285–90.
Ouyang W, Yang C, Liu Y, Xiong J, Zhang J, Zhong Y, Zhang G, Zhou F, Zhou Y, Xie C. Redistribution of DR4 and DR5 in lipid rafts accounts for the sensitivity to TRAIL in NSCLC cells. Int J Oncol. 2011;39:1577–86.
Cuello M, Ettenberg SA, Clark AS, Keane MM, Posner RH, Nau MM, Dennis PA, Lipkowitz S. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001;61:4892–900.
Kim WU, Yoo SA, Min SY, Park SH, Koh HS, Song SW, Cho CS. Hydroxychloroquine potentiates Fas-mediated apoptosis of rheumatoid synoviocytes. Clin Exp Immunol. 2006;144:503–11.
Singh K, Sharma A, Mir MC, Drazba JA, Heston WD, Magi-Galluzzi C, Hansel D, Rubin BP, Klein EA, Almasan A. Autophagic flux determines cell death and survival in response to Apo2L/TRAIL (dulanermin). Mol Cancer. 2014;13:70.
Acknowledgment
This work was partly supported by funding from the FDA/CDER Critical Path initiatives.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
No Conflict of Interest/Disclaimer: The comments and discussions in this paper are based on our experimental data and a survey of the related scientific publications. They do not necessarily reflect the official views of the US Food and Drug Administration with respect to the development of the death receptor targeted therapies. The use of product names is for product identification purpose only and does not imply endorsement.
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Twomey, J.D., Hallett, W., Zhang, B. (2015). Overcoming Cancer Cell Resistance to Death Receptor Targeted Therapies. In: Bonavida, B., Chouaib, S. (eds) Resistance of Cancer Cells to CTL-Mediated Immunotherapy. Resistance to Targeted Anti-Cancer Therapeutics, vol 7. Springer, Cham. https://doi.org/10.1007/978-3-319-17807-3_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-17807-3_10
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17806-6
Online ISBN: 978-3-319-17807-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)