
Abstract
Many new agents have been introduced in the treatment of acute myeloid leukemia (AML), and around 80 % of AML achieve complete remission (CR). However, a considerable number of patients relapse, which is mainly associated with drug resistance. Gemtuzumab ozogamicin (GO) is a conjugate of a cytotoxic agent, a calicheamicin derivative, linked to a recombinant humanized monoclonal antibody (mAb) directed against the CD33 antigen, which is expressed on leukemia cells from more than 90 % of patients with AML. GO was approved with promising results from phase I and II studies. However, the initial phase III study failed to confirm the merit of GO compared to conventional chemotherapies. One of the reasons is explained by the drug resistance acquired in leukemia cells before and during the treatments. Several resistance mechanisms against GO have been proposed. Among them, the most important resistant mechanism is the multidrug resistant (MDR) P-glycoprotein (P-gp). Some MDR modifiers removed the resistance of GO in vitro. However, one of the MDR modifiers, cyclosporine A (CyA), did not improve the response rate or survival, despite considerable number of adverse effects. Several investigators have reported promising results with the use of GO in acute promyelocytic leukemia (APL), which commonly expresses a larger amount of CD33 and a lower amount of P-gp than that of AML. Recent results show the efficacy of GO in a favorable risk of AML, such as core binding factor leukemia and APL. Another calicheamicin immunoconjugate, inotuzumab ozogamicin (IO), also introduced in B cell malignancies, provides us with promising results. However, IO also reportedly has similar resistant mechanisms to GO.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Calichamicin
- Immunoconjugate
- Gemtuzumab ozogamicin
- Inotuzumab ozogamicin
- Acutemyeloid leukemia
- B cell malignancies
- P-glycoprotein
7.1 Introduction
Acute myeloid leukemia (AML) , one of the most representative hematological malignancies [1], constitutes approximately 25–30 % of adult leukemias in the Western countries. The age-adjusted incidence rate of AML is approximately 3–4 per 100,000 people, and the incidence increases with aging. AML is characterized by the clonal proliferation of hematopoietic precursor cells and impairment of normal hematopoiesis. Many agents have been introduced in the treatment of AML, and around 80 % of AML cases achieve complete remission (CR) [2, 3]. However, a considerable number of patients relapse, and as a result, the 5-year-overall survival (OS) and disease-free survival (DFS) remain at around 40 and 20 %, respectively. The reasons have been explored mainly by the genomic methods, which showed that AML was genetically more heterogeneous than expected. Moreover, the specificity of molecular diagnosis does not necessarily result in a specific molecular targeted therapy. Several promising agents have failed to win through randomized trials in AML [4, 5]. Monoclonal antibody therapy against CD33 was also introduced and developed despite such a background.
Gemtuzumab ozogamicin (GO) , whose development code was CMA676, is a conjugate of a calicheamicin derivative and a recombinant humanized antibody (IgG4) directed against the CD33 antigen [6]. Calicheamicin is a highly potent anti-tumor antibiotic [7–10], which binds to DNA, breaks double-stranded DNA, and induces cell death. It is classified under the same category as toxin-conjugated antibody against surface antigen of tumor cells. Here, we try to understand the action and resistant mechanism of calicheamicin immune-conjugates by GO. In addition, we introduce several means to overcome the drug resistance.
7.2 CD33
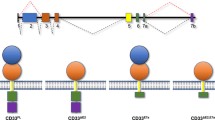
The CD33 antigen, a 67-kDa trans-membrane glycoprotein, belongs to the immunoglobulin (Ig) superfamily subgroup of sialic acid-binding Ig-like lectins (siglecs). [6, 11, 12]. It consists of two Ig-like extracellular domains and two cytoplasmic domains, [13] which have tyrosine residues similar to the immune-receptor tyrosine-based inhibitory motifs. Several protein tyrosine phosphatase inhibitors or the bridge formation by immunoglobulins result in phosphorylation of the tyrosine. While the molecular reaction stream after the phosphorylation of the tyrosine and the precise function of CD33 have not been well elucidated, it has been thought to be associated with cell adhesion and interaction. It could suppress cell proliferation and function, and induce apoptosis in vitro [14], but these functions have not been clarified in vivo.
CD33 is normally expressed on myelocyte and myelomonocytic precursor cells, as well as mature myeloid lineage cells, macrophages, monocytes, and dendritic cells [15–17]. The amount of CD33 reaches highest in promyelocytes and myelocytes, and decreases with maturation of the myeloid lineage. CD33 is also expressed on erythroblasts, megakaryoblasts, and Kupffer cells at some level, [11, 12] but not on normal hematopoietic stem cells and lymphocytes [18, 19].
Eighty to 90 % of AML are reportedly considered as CD33-positive [17, 20–22]. The amount of CD33 on AML cells is estimated at 10,000–20,000 copies/cell, which is 3–5 times more than normal bone marrow cells [23]. CD33 is sometimes determined on acute lymphoblastic leukemia (ALL), but the amount is relatively smaller (5–26 %) than AML [22, 24] and differs among the ALL subtypes. These facts suggest that CD33 is a useful target for the development of therapeutic agents for AML and limited ALL.
Fluorescence conjugated with anti-CD33 antibody, hP67.8, which was detected on the cell surface just after incubation, moved to intracellular location after 3–5 h and disappeared after 24 h [25]. The data supports that CD33 is rapidly internalized after anti-CD33 antibody binding, and then moved to the lysosome where the immunoconjugates undergodegradation and quenching of the fluorochrome. The internalization process indicated that antibody-cytotoxic agent complexes can effectively be taken up by CD33 positive leukemia cells. Consequently, radio- and toxin-conjugated anti-CD33 antibodies have been developed, such as conjugates of radioisotopes, calicheamicin , gelonin, and ricin [26–29]. Of these, GO has drawn attention with the encouraging results.
Many surface antigens are reportedly co-expressed on CD33-positive AML cells [24]. However, only CD34 reportedly relates to the efficacy of GO. In the previous study, GO was less effective on CD34-positive leukemia cells, even when they expressed a sufficient amount of CD33; this effect was independent of the amount of CD34 [30]. Sievers et al [31] reported in their clinical study that the expression of CD34 was associated with a shorter survival after treatment with GO. These might be explained by that CD34-positive cells have more defensive mechanisms including P-glycoprotein (P-gp) than CD34-negative cells.
7.2.1 Gentuzumab Ozogamicin (GO)
GO is a humanized IgG4 anti-CD33 monoclonal antibody (hP67.6) conjugated to NAc-gamma calicheamicin DMH, a hydrazide derivative of calicheamicin (Fig. 7.1) [32]. Approximately half of antibodies are conjugated by calicheamicin, with an average load of 4–6 molecules of calicheamicin per antibody. Calicheamicin, a hydrophobic enediyne antibiotic agent, was first isolated from the actinomycete Micromonospora echiospora ssp. Calichensis [7, 8]. The hydrazone function in the AcBut linker, which links the antibody and calicheamicin, releases calicheamicin divertive from its conjugated state under acidic conditions.

GO is a humanized IgG4 anti-CD33 monoclonal antibody (hP67.6) conjugated to NAc-gamma calicheamicin DMH, a hydrazide derivative of calicheamicin
After GO binds to CD33 on the cells, CD33-antibody complexes are rapidly internalized and transferred into lysosomes [25]. The calicheamicin derivative is released via hydrolysis in the acid environment of the lysosome. Then it moves to the nucleus, and binds to the minor groove of DNA in a sequence-specific manner. It cleaves single and double-stranded DNAs by the removal of specific hydrogen atoms from the deoxyribose rings of DNAs [9]. DNA damage leads to apoptotic or non-apoptotic cell death due to mitochondrial damage [33–35]. Naito et al [36] observed cell morphology after the incubation of GO by video-microscopy, which revealed some cells exhibited apoptotic changes, while the remaining cells showed non-apoptotic features. The cytotoxic mechanism of GO is the same as that of free calicheamicin, except for the internalization via CD33. Cells incubated with calicheamicin undergo either temporary or permanent cell cycle arrest depending on the concentration [31, 36]. Transient G2/M arrest was observed prior to the increase of the hypodiploid portion in cell lines incubated with GO. Several molecular pathways, such as Chk1 and Chk2 phosphorylation and caspase 3, reportedly played roles in this process [37].
Cells expressing higher levels of CD33 were reportedly more susceptible to GO [38]. On the other hand, several patients with CD33-negative leukemia have also responded to GO [39]. Several studies have tried to explain the efficacy of GO on CD33-negative leukemia. One proposed explanation is that GO is partially moved into cell by CD33-independent endocytosis [39]. Another is that CD33-negative leukemia cells may have a sub-threshold low amount of CD33, which reacts substantially with GO [40].
7.2.2 GO Monotherapy, Phase I Study
In a phase I study conducted in the U.S., 40 patients with relapsed or refractory (relapsed/refractory) AML were treated by GO (0.25–9 mg/m2) [41]. Leukemia cells were eliminated from the blood and bone marrow of 8 (20 %) of the 40 patients. Neutrophil counts recovered in five of these eight patients, but platelet count recovered in only three. Patients who achieved complete remission (CR) without recovering the platelet count more than 100 × 109/L were entered to the concept of CR with thrombocytopenia (CRp), which has been subsequently used in the evaluation of GO.
7.2.3 Phase II Study
Phase II trials with GO were started at a dose of 9 mg/m2 (2-week intervals for two doses) [42]. A total of 142 patients with AML in first relapse were enrolled in the study. Of these, 30 % achieved overall response (OR), including CR and CRp. The median relapse-free survival (RFS) was 5.3 months [43]. Grade 3 or 4 bilirubinemia was observed in 23 %, and hepatic transaminitis in 17 %. Hepatic sinusoid obstructed syndrome (SOS) was observed in seven patients (3 %), and three of these were fatal. Five patients, who received hematopoietic stem cell transplantation (HSCT) before the treatment of GO, did not have apparent SOS. However, 3 of 27 patients, who received HSCT after the treatment of GO, died of SOS. Based on these results, the Food and Drug Administration of U.S. approved GO for relapsed CD33-positive AML in patients 60 years of age or older [45].
7.2.4 Drug Resistance via P-glycoprotein
MDR is a phenomenon in which malignant cells acquire cross-resistance to a variety of unrelated cytotoxic drugs. P-gp, one of the most potent MDR mechanisms, is a membrane glycoprotein that actively pumps cytotoxic agents out from cells, and decreases intracellular drug accumulation [44, 45]. Various agents have been introduced to overcome P-gp-associated drug resistance . They include calcium blocker, quinidine, cyclosporine, cepharantin, carotenoids and soforth. Naito et al [36] analyzed the cytotoxic effect of GO on NOMO-1 and NB4 cell lines as well as their multidrug resistant sublines, NOMO-1/MDR and NB4/MDR. They analyzed it by a video-microscopic system, DNA fragmentation, dye exclusion and 3H-thymidine uptake after analysis of CD33, CD34 and P-gp expressions. A concentration-dependent cytotoxic effect of GO was observed in cell lines that expressed CD33. Sensitive cells were temporally arrested at the G2/M phase of the cell cycle before undergoing morphological changes. GO was not effective on the multidrug-resistant sublines compared with the parental cell lines. MDR modifiers, MS209 and PSC833, restored the cytotoxic effect of GO in P-gp-expressing sublines. They concluded that calicheamicin derivatives, which are internalized with GO via CD33 and detached from GO in lysosomes, could be pumped out by P-gp from the cells (Fig. 7.2) [36]. Matsui et al [31] continuously analyzed the in vitro effects of GO on leukemia cells from 27 AML patients in relation to the amount of P-gp, MDR-associated protein 1 (MRP1), CD33 and CD34. The effect of GO, estimated by the amount of hypodiploid portion on the cell cycle, was inversely related to the amount of P-gp estimated by the MRK16 monoclonal antibody, and to the P-gp function assessed by intracellular rhodamine-123 accumulation in the presence of MDR modifiers. They showed that MDR modifiers reversed GO resistance in P-gp-expressing CD33+leukemia cells. GO was less effective on CD33+CD34+ than CD33+CD34− cells. Interestingly, similar results were obtained in studies using inotuzumab ozogamicin (IO) , a calicheamicin-conjugated anti-CD22 antibody, for lymphoid malignancies [46, 47]. It will, herein, subsequently be described in detail. Another study showed the cells that were persistently exposed to low-dose GO acquired resistance to GO and expressed P-gp [48]. GO-sensitiveHL-60 cells, which were persistently exposed to low concentrations of GO, changed to GO-resistant HL-60(HL-60/GOR) cells. P-gp was significantly expressed in HL-60/GOR cells, but not in parental HL-60 cells.

After GO binds to CD33 on the cells, CD33-antibody complexes are internalized and transferred into lysosomes, in which calicheamicin is detached. Intracellularly released calicheamicin derivatives are pumped out via P-gp in multidrug-resistant cells. MDR modifiers recover the effect of GO
These in vitro results were confirmed imperviously mentioned phase I studies of GO [40, 41]. Good responders were more frequently observed in leukemia patients characterized by low dye efflux in vitro. Any kind of screening tests for P-gp before the treatment of GO might be helpful to have a better clinical outcome. Naito et al [36] suggested that the combination use of GO and MDR modifiers may be an ideal therapeutic approach for P-gp-expressing leukemia, assuming that the hematologic and non-hematologic toxicities are not worsened. This idea has been tried clinically in relapsed/refractory AML.
7.2.5 GO Treatment with MDR Modifier, CyA
Cyclosporin A (CyA), which has been easily-available and widely used as an immunosuppressant, has a considerable effect as an MDR modifier on the other hand. It has, in fact, been administered as an adjunct to GO-containing chemotherapy in the treatment of AML (Table 7.1) [49–51]. Apostolidou et al [49] treated with GO (6 mg/m2 on day 6), cytosine arabinoside (Ara-C)(1 g/m2 on days 1–5), liposome-encapsulated daunorubicin (DNR) (75 mg/m2 on days 6–8) and CyA (on day 6) (MDAC regimen)for 11 patients with relapsed/refractory AML. One (9 %) patient achieved a transient CR, and one achieved CRp. Grade 3/4 toxicities included sepsis in 7 patients(63 %); hyperbilirubinemia in 6 (54 %), and mucositis in 3 (27 %).
Tsimberidou et al [50] evaluated the efficacy and toxicity of a combination regimen of GO (6 mg/m2 on day 1),fludarabine (15 mg/m2 on days 2–6), AraC (0.5 g/m2 on days 2–6) and CyA (6 mg/kg on days 1 and 2) (MFAC regimen) in 59 patients with previously untreated AML, refractory anemia with excess blasts (RAEB), or RAEB in transformation (RAEBT): 39 patients (66 %) were AML and 20 patients (34 %) were RAEB/RAEBT. CR was achieved in 27 patients (46 %) and CRp was achieved in patient (2 %). The 1-year OS was 38 % and the event-free survival (EFS) in patients with CR/CRp was 27 %. Grade 3/4 toxicity included hyperbilirubinemia in 31 % and transaminitis in 7 % of the patients. Four patients (7 %) developed SOS. They conducted a Phase II study of the MFAC regimen in 32 patients with resistant/relapsed AML [51]. Nine (28 %) patients achieved CR, and 2 (6 %) CRp. The 1-year OS was 19 %. Fourteen patients (44 %) developed grade 3/4 hyperbilirubinemia, 6 (18 %) grade 3/4 hepatic transaminitis, and 3 (9 %) SOS.
CyA did not improve the response rate nor survival, although SOS was observed in a considerable number of patients. The unsuccessful attempt of the treatment may be explained by the possibility that CyA ablates the function of P-gp, which is widely distributed across critical organ systems, resulting in increased adverse effects, and that the clinical outcome from the P-gp negative cases assumed influence on the non-significance of the results [52]. Several transporters other than P-gp have also been suggested. MRP1, another well-known transporter protein, is sometimes expressed in AML [53]. However, the clinical importance of MRP1 was relatively limited among the mechanisms of resistance to GO [54]. Other transporters reportedly have further limited effects.
7.2.6 Drug Resistance Other Than P-glycoprotein
The roles of βcl-2 and βcl-x, anti-apoptotic proteins, in the resistance to GO have been reported [55, 56]. GO induced proapoptotic activation of Bak and Bax and stress-activated protein kinase in sensitive AML cells, but not in resistant ones, KG1a AML cells. The effect of GO was enhanced by βcl-2 antisense oligonucleotide, oblimersen sodium, but reduced by over-expression of βcl-2 and βcl-x. Bax, Bak and stress-activated protein kinase may play a role in resistance to GO [57]. The resistance mechanism is not specific for GO, but considerable. Oblimersen (7 mg/kg, days 1–7 and 15–21) was administered with GO (9 mg/m2 on days 4 and 18) in 48 elderly patients with relapsed AML (Table 7.1) [55]. Twelve patients (25 %) achieved OR. The median OS for all patients enrolled was 2.3 months. Grade 3/4 toxicities were sepsis (12 %) urinary tract infection (8 %), pneumonia (6 %) and respiratory events (31 %) .
The peripheral benzodiazepine receptors (pBzRs) locate in the multiprotein mitochondrial pore complex which regulates mitochondrial membrane potential. Bcl-2 and related anti-apoptotic proteins block apoptosis by keeping the pores closed, but pBzR ligands promote the opening of pores and induce apoptosis. The pBzRs ligand, PK11195, increased the sensitivity of AML cells to standard chemotherapeutics both by inhibiting P-gp and by promoting mitochondrial apoptosis [56]. It increased the sensitivity to GO in AML cells in vitro.
Rosen et al [58] reported that the activation of survival signaling pathways, such as PI3K/AKT, MEK/ERK and JAK/STAT, is reportedly associated with GO resistance in vitro in AML cells. An AKT inhibitor, MK-2206, restored the resistance of GO and calicheamicin in resistant AML cells.
The transport of GO into the bone marrow may be important for intensifying the effect of GO [27, 38]. An excess of circulating CD33-positive cells decreased the effect of GO, and resulted in worse outcomes [26, 59]. GO may be spent in the circulation before it reaches the bone marrow [31, 32, 36]. This suggests that GO might be made more effective by the reduction of CD33 in peripheral blood by proceeding chemotherapy [46]. Therefore, GO is often managed several days after the start of induction chemotherapy. However, we understand that a high blast cell count is equally an adverse prognostic factor in leukemias treated with other anti-leukemic agents.
Several agents may also enhance the effect of GO. G-CSF increased the effect of GO, and induced AML cells to enter G2/M and a hypodiploid phase [60, 61]. Valproic acid, a histone deacetylase inhibitor, strengthened the effect of GO [62]. However, the synergistic effect of GO with these agents has not been confirmed in clinical studies. Clinically, multiple mechanisms may simultaneously arise in the development of resistance to GO.
7.2.7 Phase III Study with GO for AML and Disappearance from the Market
The Southwest Oncology Group (SWOG) studyS0106 reported the benefit and toxicity of adding GO to standard therapy in 627 patients with de novo AML [63]. Patients were randomized to receive induction therapy with DNR (45 mg/m2 on days 1–3) and AraC (100 mg/m2 on days 1–7) and GO (6 mg/m2 on day 4) (AD+GO) or standard induction therapy with DNR (60 mg/m2on days 1–3) and AraC (100 mg/m2 on days 1–7) (AD). After patients achieved CR, they received consolidation therapy with 3 courses of high dose AraC (HiDAC). Patients in remission were re-randomized to the treatment of GO (5 mg/m2 every 28 days, 3 doses) or observation. The OR rate was 74 % in both induction arms. The RFS was not significantly different between two arms. Adverse effects were significantly increased in the AD+GO arm. The results of SWOG-S0106 triggered Pfizer Corp. to voluntarily withdraw GO from the market in 2010.
7.2.8 Subsequent Phase III Study for AML
In a subsequent study, 238 patients with de novo AML and an intermediate karyotype were treated with standard chemotherapy with or without GO [64]. GO (6 mg/m2) was added to standard 3 + 7 induction, and to a consolidation of mitoxantrone (MIT) and AraC. The CR rate and early death rate were not different between both groups. Grade 3/4 hepatic toxicities were increased in the GO arm. The EFS and the OS were not changed in both treatment arms. In patients who did not receive HSCT, EFS was significantly higher in the GO arm (54 vs 27 %) while OS was not improved.
In the MRC-AML15 trial, 1113 patients with de novo AML, excluding APL, were randomly assigned to receive either of the following 3 induction treatments: DNR and AraC; DNR, etoposide (ETP) and AraC; or fludarabine, IDA, AraC and G-CSF; with or without GO (3 mg/m2) [65]. After achieving remission, 948 patients were randomly assigned to GO (3 mg/m2) in combination with amsacrine, AraC and ETP or HiDAC (1.5 g or 3 g/m2). The CR rate or the OS were not significantly different between both groups. Survival benefit of GO was observed in patients with favorable cytogenetics, but not in patients with high-risk cytogenetics. GO did not increase toxicity.
In other results from the UK and Denmark, 1115 patients with AML or high-risk MDS were randomly assigned to receive induction chemotherapy with either DNR (50 mg/m2 on days 1, 3, 5) and AraC (100 mg/m2 twice a day on days 1–10) or DNR and clofarabine (20 mg/m2 on days 1–5), with or without GO (3 mg/m2) [66]. The OR rates were not different between both groups. GO did not increase toxicity and mortality. Three-year cumulative incidence of relapse was significantly lower, and 3-year OS was significantly better in the patients treated with GO.
Two hundred and seventy-eight elderly patients with de novo AML received DNR (60 mg/m2 on days 1–3) and AraC (200 mg/m2 for 7 days) without (control group) or with GO (3 mg/m2 on days 1, 4, and 7) [67]. The OR rate was not different between the two groups. The 2-year-EFS, OS, and RFS were significantly improved by the addition of GO. GO did not increase the risk of death from toxicity.
These recent results demonstrated some advantage for patients treated with GO. In addition, induction mortality was not increased in these studies. Efficacy was observed, typically in patients with favorable-risk, and sometimes in intermediate-risk. The reason for this has not been elucidated. However, multiple resistant mechanisms observed in high-risk could explain it.
7.2.9 The Efficacy of GO for Acute Promyelocytic Leukemia (APL)
APL, which is classified asAML-M3 in the FAB classification system and as APL with t(15;17)(q22;q12) and PML-RARA transcript within myeloid malignancies according to the World Health Organization (WHO) classification system [68]. This disease is characterized by differentiation arrest in myeloid precursor cells and their uncontrolled proliferation. All-trans retinoic acid (ATRA) has dramatically decreased these complications, and around 90 % of newly-diagnosed patients achieved CR and more than 60 % survived long-term with subsequent post-remission chemotherapy [69–72] . While ATRA combined with chemotherapy has been the standard treatment for patients with APL, approximately 20 % undergo relapse [73–75]. Several salvage therapies, including tamibarotene (Am80), arsenic trioxide (ATO), and stem cell transplantation, have been introduced for the treatment of APL [76, 77]. GO was also administered to APL, and the successful outcome of this therapy has been reported for patients with newly diagnosed or relapsed APL [78–80].
Several reasons have been proposed to explain the efficacy of GO for APL [81, 82]. First, a large amount of CD33 is commonly expressed on the surface of APL cells. Second, the level of P-gp on the surface of APL cells is significantly lower than that of AML. Third, APL cells are highly sensitive for free calicheamicin. Lo-Coco et al [79] reported that 14 of 16 patients with molecularly relapsed APL achieved molecular remission (MR) after GO monotherapy (6 mg/m2at 2-week intervals for three doses). Of 14 responders, seven (50 %) remained in sustained MR for a median of 15 months. GO was administered again in two patients with relapse, and both obtained a new MR.
Another study reported that two patients in a third morphologic relapse with a considerable number of APL cells were treated by GO monotherapy (9 mg/m2 on days 1 and 15) and achieved CR [80]. One of the patients was treated with consolidation chemotherapy, but the other was not. Both patients had a considerably long CR. GO may represent another treatment option if stem cell transplantation is not being considered in APL.
Aribi et al [81] reported the efficacy of a combination therapy consisting of ATO, ATRA and GO in eight patients with APL in first relapse. Patients were treated with ATO until CR, and then received the consolidation therapy including ATO, ATRA and GO (9 mg/m2) once a month for 10 months. The second CR was longer than the first CR in 75 %. Moreover, all patients achieved MR. Grade 3/4 non-hematological toxicities were not observed. These reports show that GO is effective for APL patients with molecularly relapsed and advanced relapsed forms of the disease. These data also support the use of GO treatment for APL, which usually have low levels of P-gp and high levels of CD33.
7.3 CD22
CD22, a 140 kD a transmembrane sialo-adhesion glycoprotein, is widely distributed in mature B cells.[83–85] CD22 is a member of the Ig super-family and has seven extracellular Ig-like domains, which mediate cell adhesion tosialic-acid-bearing ligands. The cytoplasmic regions of CD22 have the immune receptor tyrosine activation motifs (ITAM) and tyrosine inhibitory motifs (ITIM). CD22 ITAMs are phosphorylated after BCR activation, and enhance the recruitment of protein tyrosine phosphatases to CD22. The CD22-associated phosphatases then dephosphorylate BCR components resulting in the attenuation of BCR signaling. The function of CD22 is reportedly to modulate the B-cell antigen receptor (BCR) signaling and to regulate cell-cell interactions. The activation of CD22 by ligand binding and cross-linking send negative signals and result in cytotoxicity for B-cell lymphoma [86–89].
7.3.1 Inotuzuma Bozogamicin
Calicheamicin conjugated antibody-targeted chemotherapy strategy has been also applied to B cell malignancies . Because the expression of CD22 is restricted to the B cell lineage and CD22 has a characteristic of internalising molecules, anti-CD22 antibody can be used for targeted delivery of calicheamicin . IO is the calicheamicin conjugated to a humanized IgG4 anti-CD22 mAb, G544, with the linker containing an acid-labile hydrazone. Therefore, the action mechanisms of IO are similar to GO, except that these conjugates recognize distinct molecular targets. Clinical efficacies have been reported in several B cell malignancies [90–92].
7.3.2 Drug Resistance of IO
The reports about the resistant mechanism of IO have not be more frequently found than those of GO. However, the similar resistant mechanisms observed in the studies of GO can be found in IO. The effect of IO was analyzed in relation to CD22 and P-gp in B-cell chronic lymphocytic leukaemia (CLL) and non-Hodgkin lymphoma (NHL) in vitro [47]. The cell lines used were the CD22-positive parental Daudi and Raji and their P-gp positive sublines, Daudi/MDR and Raji/MDR. The effect of IO was analyzed by morphology, annexin-V staining, and cell cycle distribution. A dose-dependent, selective cytotoxic effect of IO was observed in cell lines that expressed CD22. CMC-544 was not effective on Daudi/MDR and Raji/MDR cells compared with their parental cells. The MDR modifiers, PSC833 and MS209, restored the cytotoxic effect of CMC-544 in P-gp-expressing sublines. In clinical samples, the cytotoxic effect of CMC-544 was inversely related to the amount of P-gp, and to intracellular rhodamine-123 accumulation. The effect positively correlated with the amount of CD22.
7.4 Conclusion
Antibody-targeted chemotherapy using immunoconjugates of calicheamicin is theoretically an effective therapeutic method in the treatment of cancers. They have improved the specificity and therapeutic effects. They have been used as a single agent or in combination with conventional chemotherapies or other molecular target therapies, and several successes have been reported. However, the immunoconjugates of calicheamicin also acquire drug resistance and, hence, it should be used with understanding of their characteristic features.
GO has introduced a new perspective into the treatment of AML. However, the second evaluation of this treatment did not yield positive results mainly due to MDR. Recent studies have shown the efficacy of GO in AML, with a favorable risk in APL as well. Subsequent evaluations should focus on the efficacy of GO in the core binding factor (CBF) leukemia and its mechanism of action, which may lead to the re-approval of GO. IO is a very potent agent against B cell malignancies . IO action and resistant mechanisms will be similar to GO. Combination therapies with other agents will be promising.
Conflict of Interest
No potential conflicts of interest were disclosed.
Abbreviations
- AML:
-
Acute myeloid leukemia
- mAb:
-
Monoclonal antibody
- CR:
-
Complete remission
- GO:
-
Gemtuzumab ozogamicin
- MDR:
-
Multidrug resistant
- P-gp:
-
P-glycoprotein
- CyA:
-
Cyclosporine A
- APL:
-
Acute promyelocytic leukemia
- IO:
-
Inotuzumab ozogamicin
- OS:
-
Overall survival
- DFS:
-
Disease-free survival
- MRP1:
-
MDR-associated protein 1
- CRp:
-
Complete remission with thrombocytopenia
- RFS:
-
Relapse-free survival
- OR:
-
Overall response
- SOS:
-
Sinusoid obstructed syndrome
- HSCT:
-
Hematopoietic stem cell transplantation
- Ara-C:
-
Cytosine arabinoside
- DNR:
-
Daunorubicin
- RAEB:
-
Refractory anemia with excess blasts
- EFS:
-
Event-free survival
- pBzRs:
-
Peripheral benzodiazepine receptors
- HiDAC:
-
High dose AraC
- ETP:
-
Etoposide
- WHO:
-
World Health Organization
- ATRA:
-
All-trans retinoic acid
- Am80:
-
Tamibarotene
- ATO:
-
Arsenic trioxide
- MR:
-
Molecular remission
- ITAM:
-
Immunoreceptor tyrosine activation motifs
- ITIM:
-
Immunoreceptor tyrosine inhibitory motifs
- BCR:
-
B-cell antigen receptor
- CLL:
-
Chronic lymphocytic leukaemia
- NHL:
-
Non-Hodgkin lymphoma
- CBF:
-
Core binding factor
References
Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106:1154–1163.
Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013;88:318–27.
Stein EM, Tallman MS. Remission induction in acute myeloid leukemia. Int J Hematol. 2012;96:164–70.
Kuhnl A, Grimwade D. Molecular markers in acute myeloid leukaemia. Int J Hematol. 2012;96:153–63.
Naoe T, Kiyoi H. Gene mutations of acute myeloid leukemia in the genome era. Int J Hematol. 2013;97:165–74.
Freeman SD, Kelm S, Barber EK, Crocker PR. Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood. 1995;85:2005–12.
Lee MD, Dunne TS, Siegel MM, Chang CC, Morton GO. Borders DB. Calicheamicins, a novel family of antitumor antibiotics. 1: Chemistry and partial structure of calicheamicinγ1. J Am Chem Soc. 1987;109:3464–6.
Lee MD, Dunne TS, Chang CC. Calicheamicins, a novel family of antitumor antibiotics. 2: Chemistry and structure of calicheamicinγ1I. J Am Chem Soc. 1987; 109:3466–8.
Zein, N, Poncin M, Nilakantan, R, Ellestad, GA. Calicheamicin gamma 1I and DNA: molecular recognition process responsible for site-specificity. Science. 1989;244:697–9.
Hangeland JJ, De Voss JJ, Heath JA, Townsend CA, Ding WD, Ashcroft JS, Ellestad GA. Specific abstraction of the 5’(S)- and 4’-deoxyribosyl hydrogen atoms from DNA by calicheamicinγ1 I. J Am Chem Soc. 1992;114:9200–2.
Tchilian EZ, Beverley PC, Young BD, Watt SM. Molecular cloning of two isoforms of the murine homolog of the myeloid CD33 antigen. Blood.1994; 83: 3188–98.
Gao, Z, McAlister VC, Williams GM. Repopulation of liver endothelium by bone-marrow-derived cells. Lancet. 2001;357:932–3.
Crocker PR. Siglecs: sialic-acid-binding immunoglobulin-like lectins in cell-cell interactions and signalling. Curr Opin Struct Biol. 2002;12:609–15.
Taylor VC, Buckley CD, Douglas M, Cody AJ, Simmons DL, Freeman SD. The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. J Biol Chem. 1999;274: 11505–12.
Andrews RG, Torok-Storb B, Bernstein ID. Myeloid-associated differentiation antigens on stem cells and their progeny identified by monoclonal antibodies. Blood. 1983;62:124–32.
Andrews RG, Takahashi M, Segal GM, Powell JS, Bernstein ID, Singer JW. The L4F3 antigen is expressed by unipotent and multipotent colony-forming cells but not by their precursors. Blood. 1986;68:1030–5.
Griffin, J. D, Linch, D, Sabbath, K, Larcom, P, and Schlossman, S. F. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. 1984;8:521–34.
Robertson MJ, Soiffer RJ, Freedman AS, Rabinowe SL, Anderson KC, Ervin TJ, Murray C, Dear K, Griffin JD, Nadler LM, Human bone marrow depleted of CD33-positive cells mediates delayed but durable reconstitution of hematopoiesis: clinical trial of MY9 monoclonal antibody-purged autografts for the treatment of acute myeloid leukemia. Blood. 1992;79:2229–36.
Wagner JE, Collins D, Fuller S, Schain LR, Berson AE, Almici C, Hall MA, Chen KE, Okarma TB, Lebkowski JS. Isolation of small, primitive human hematopoietic stem cells: distribution of cell surface cytokine receptors and growth in SCID-Hu mice. Blood. 1995;86:512–23.
Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood. 1986;67:1048–53.
Terstappen, LW, Safford, M, Konemann, S, Loken, MR, Zurlutter, K, Buchner, T, Hiddemann, W, and Wormann, B. Flow cytometric characterization of acute myeloid leukemia. Part II. Phenotypic heterogeneity at diagnosis. Leukemia. 1992;6:70–80.
Putti MC, Rondelli R, Cocito MG, Arico M, Sainati L, Conter V, Guglielmi C, Cantu-Rajnoldi A, Consolini R, Pession A, Zanesco L, Masera G, Biondi A, Basso G. Expression of myeloid markers lacks prognostic impact in children treated for acute lymphoblastic leukemia: Italian experience in AIEOP-ALL 88–91 studies. Blood. 1998;92:795–801.
Jilani I, Estey E, Huh Y, Joe Y, Manshouri T, Yared M, Giles F, Kantarjian H, Cortes J, Thomas D, Keating M, Freireich E, Albitar M. Differences in CD33 intensity between various myeloid neoplasms. Am J Clin Pathol. 2002;118: 560–6.
Iwamoto S, Deguchi T, Ohta H, Kiyokawa N, Tsurusawa M, Yamada T, Takase K, Fujimoto J, Hanada R, Hori H, Horibe K, Komada Y. Flow cytometric analysis of de novo acute lymphoblastic leukemia in childhood: report from the Japanese Pediatric Leukemia/Lymphoma Study Group. Int J Hematol. 2011;94: 185–92.
McGrath MS, Rosenblum MG, Philips MR, Scheinberg DA. Immunotoxin resistance in multidrug resistant cells. Cancer Res. 2003;63:72–9.
Scheinberg DA, Lovett D, Divgi CR, Graham MC, Berman E, Pentlow K, Feirt N, Finn RD, Clarkson BD, Gee TS, et al. A phase I trial of monoclonal antibody M195 in acute myelogenous leukemia: specific bone marrow targeting and internalization of radionuclide. J Clin Oncol. 1991;9:478–90.
Caron PC, Co MS, Bull MK, Avdalovic NM, Queen C, Scheinberg DA. Biological and immunological features of humanized M195 (anti-CD33) monoclonal antibodies. Cancer Res. 1992;52:6761–7.
Borthakur G, Rosenblum MG, Talpaz M, Daver N, Ravandi F, Faderl S, Freireich EJ, Kadia T, Garcia-Manero G, Kantarjian H, Cortes JE. Phase 1 study of an anti-CD33 immunotoxin, humanized monoclonal antibody M195 conjugated to recombinant gelonin (HUM-195/rGEL), in patients with advanced myeloid malignancies. Haematologica. 2013;98:217–21.
La Russa VF, Griffin JD, Kessler SW, Cutting MA, Knight RD, Blattler WA, Lambert JM, Wright DG. Effects of anti-CD33 blocked ricin immunotoxin on the capacity of CD34+ human marrow cells to establish in vitro hematopoiesis in long-term marrow cultures. Exp Hematol. 1992;20:442–8.
Matsui H, Takeshita A, Naito K, Shinjo K, Shigeno K, Maekawa M, Yamakawa Y, Tanimoto M, Kobayashi M, Ohnishi K, Ohno R. Reduced effect of gemtuzumab ozogamicin (CMA-676) on P-glycoprotein and/or CD34-positive leukemia cells and its restoration by multidrug resistance modifiers. Leukemia. 2002;16:813–9.
Sievers EL, Larson RA, Stadtmauer EA, Estey E, Lowenberg B, Dombret H, Karanes C, Theobald M, Bennett JM, Sherman ML, Berger MS, Eten CB, Loken MR, van Dongen JJ, Bernstein ID, Appelbaum FR. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244–54.
Hamann PR, Hinman LM, Hollander I, Beyer CF, Lindh D, Holcomb R, Hallett W, Tsou HR, Upeslacis J, Shochat D, Mountain A, Flowers DA, Bernstein I. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13:47–58.
Zhao B, Konno S, Wu JM, Oronsky AL. Modulation of nicotinamide adenine dinucleotide and poly(adenosine diphosphoribose) metabolism by calicheamicin gamma 1 in human HL-60 cells. Cancer Lett. 1990;50:141–7.
Nicolaou KC, Pitsinos EN, Theodorakis EA, Saimoto H, Wrasidlo W. Synthetic calicheamicin mimics with novel initiation mechanisms: DNA cleavage, cytotoxicity, and apoptosis. Chem Biol. 1994;1:57–66.
Lode HN, Reisfeld RA, Handgretinger R, Nicolaou KC, Gaedicke G, Wrasidlo W. Targeted therapy with a novel enediyene antibiotic calicheamicin theta(I)1 effectively suppresses growth and dissemination of liver metastases in a syngeneic model of murine neuroblastoma. Cancer Res. 1998;58:2925–8.
Naito K, Takeshita A, Shigeno K, Nakamura S, Fujisawa S, Shinjo K, Yoshida H, Ohnishi K, Mori M, Terakawa S, Ohno R. Calicheamicin-conjugated humanized anti-CD33 monoclonal antibody (gemtuzumab zogamicin, CMA-676) shows cytocidal effect on CD33-positive leukemia cell lines, but is inactive on P-glycoprotein-expressing sublines. Leukemia. 2000;14:1436–43.
Amico D, Barbui AM, Erba E, Rambaldi A, Introna M, Golay J. Differential response of human acute myeloid leukemia cells to gemtuzumab ozogamicin in vitro: role of Chk1 and Chk2 phosphorylation and caspase 3. Blood. 2003;101: 4589–97.
Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood. 2005;105:1295–302.
Jedema I, Barge RM, van der Velden VH, Nijmeijer BA, van Dongen JJ, Willemze R, Falkenburg JH. Internalization and cell cycle-dependent killing of leukemic cells by Gemtuzumab Ozogamicin: rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia. 2004;18:316–25.
Linenberger ML. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia. 2005;19:176–82.
Sievers EL, Appelbaum FR, Spielberger RT, Forman SJ, Flowers D, Smith FO, Shannon-Dorcy K, Berger MS, Bernstein ID. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: a phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood. 1999;93:3678–84.
Larson RA, Sievers EL, Stadtmauer EA, Lowenberg B, Estey EH, Dombret H, Theobald M, Voliotis D, Bennett JM, Richie M, Leopold LH, Berger MS, Sherman ML, Loken MR, van Dongen JJ, Bernstein ID, Appelbaum FR. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer. 2005;104:1442–52.
Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, Roy S, Sridhara R, Rahman A, Williams G, Pazdur R. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7: 1490–6.
Kartner N, Evernden-Porelle D, Bradley G, Ling V. Detection of P-glycoprotein in multidrug-resistant cell lines by monoclonal antibodies. Nature. 1985;316:820–3.
Gottesman MM, Pastan I, Ambudkar SV. P-glycoprotein and multidrug resistance. Curr Opin Genet Dev. 1996;6:610–7.
Takeshita A, Yamakage N, Shinjo K, Ono T, Hirano I, Nakamura S, Shigeno K, Tobita T, Maekawa M, Kiyoi H, Naoe T, Ohnishi K, Sugimoto Y, Ohno R. CMC-544 (inotuzumab ozogamicin), an anti-CD22 immuno-conjugate of calicheamicin, alters the levels of target molecules of malignant B-cells. Leukemia. 2009;23:1329–36.
Takeshita A, Shinjo K, Yamakage N, Ono T, Hirano I, Matsui H, Shigeno K, Nakamura S, Tobita T, Maekawa M, Ohnishi K, Sugimoto Y, Kiyoi H, Naoe T, Ohno R. CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma. Br J Haematol. 2009;146:34–43.
Matsumoto T, Jimi S, Hara S, Takamatsu Y, Suzumiya J, Tamura K. Importance of inducible multidrug resistance 1 expression in HL-60 cells resistant to gemtuzumab ozogamicin. Leuk Lymphoma. 2012;53:1399–405.
Apostolidou E, Cortes J, Tsimberidou A, Estey E, Kantarjian H, Giles FJ. Pilot study of gemtuzumab ozogamicin, liposomal daunorubicin, cytarabine and cyclosporine regimen in patients with refractory acute myelogenous leukemia. Leuk Res. 2003;27:887–91.
Tsimberidou A, Estey E, Cortes J, Thomas D, Faderl S, Verstovsek S, Garcia-Manero G, Keating M, Albitar M, O’Brien S, Kantarjian H, Giles F. Gemtuzumab, fludarabine, cytarabine, and cyclosporine in patients with newly diagnosed acute myelogenous leukemia or high-risk myelodysplastic syndromes. Cancer. 2003;97:1481–7.
Tsimberidou A, Cortes J, Thomas D, Garcia-Manero G, Verstovsek S, Faderl S, Albitar M, Kantarjian H, Estey E, Giles FJ. Gemtuzumab ozogamicin, fludarabine, cytarabine and cyclosporine combination regimen in patients with CD33+primary resistant or relapsed acute myeloid leukemia. Leuk Res. 2003;27: 893–7.
Takeshita A. Efficacy and resistance of gemtuzumab ozogamicin for acute myeloid leukemia. Int J Hematol. 2013;97:703–16.
Legrand O, Zittoun R, Marie JP. Role of MRP1 in multidrug resistance in acute myeloid leukemia. Leukemia. 1999;13:578–84.
Cianfriglia M, Mallano A, Ascione A, Dupuis ML. Multidrug transporter proteins and cellular factors involved in free and mAb linked calicheamicin-gamma1 (gentuzumab ozogamicin, GO) resistance and in the selection of GO resistant variants of the HL60 AML cell line. Int J Oncol. 2010;36:1513–20.
Moore, J, Seiter, K, Kolitz, J, Stock, W, Giles, F, Kalaycio, M, Zenk, D, and Marcucci, GA Phase II study of Bcl-2 antisense (oblimersen sodium) combined with gemtuzumab ozogamicin in older patients with acute myeloid leukemia in first relapse. Leuk Res. 2006;30:777–83.
Walter RB, Raden BW, Cronk MR, Bernstein ID, Appelbaum FR, Banker DE. The peripheral benzodiazepine receptor ligand PK11195 overcomes different resistance mechanisms to sensitize AML cells to gemtuzumab ozogamicin. Blood. 2004;103:4276–84.
Haag P, Viktorsson K, Lindberg ML, Kanter L, Lewensohn R, Stenke L. Deficient activation of Bak and Bax confers resistance to gemtuzumab ozogamicin-induced apoptotic cell death in AML. Exp Hematol. 2009;37:755–66.
Rosen DB, Harrington KH, Cordeiro JA, Leung LY, Putta S, Lacayo N, Laszlo GS, Gudgeon CJ, Hogge DE, Hawtin RE, Cesano A, Walter RB. AKT signaling as a novel factor associated with in vitro resistance of human AML to gemtuzumab ozogamicin. PLoS One. 2013;8: e53518.
van der Velden VH, Boeckx N, Jedema I, te Marvelde JG, Hoogeveen PG, Boogaerts M, van Dongen JJ. High CD33-antigen loads in peripheral blood limit the efficacy of gemtuzumab ozogamicin (Mylotarg) treatment in acute myeloid leukemia patients. Leukemia. 2004;18:983–8.
Kell WJ, Burnett AK, Chopra R, Yin JA, Clark RE, Rohatiner A, Culligan D, Hunter A, Prentice AG, Milligan DW. A feasibility study of simultaneous administration of gemtuzumab ozogamicin with intensive chemotherapy in induction and consolidation in younger patients with acute myeloid leukemia. Blood. 2003;102:4277–83.
Fianchi L, Pagano L, Leoni F, Storti S, Voso MT, Valentini CG, Rutella S, Scardocci A, Caira M, Gianfaldoni G, Leone G. Gemtuzumab ozogamicin, cytosine arabinoside, G-CSF combination (G-AraMy) in the treatment of elderly patients with poor-prognosis acute myeloid leukemia. Ann Oncol. 2008;19:128–34.
ten Cate B, Samplonius DF, Bijma T, de Leij LF, Helfrich W, Bremer E. The histone deacetylase inhibitor valproic acid potently augments gemtuzumab ozogamicin-induced apoptosis in acute myeloid leukemic cells. Leukemia. 2007; 21:248–52.
Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, Larson RA, Erba HP, Stiff PJ, Stuart RK, Walter RB, Tallman MS, Stenke L, Appelbaum FR. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121:4854–60.
Delaunay J, Recher C, Pigneux A. Addition of gemtuzumab ozogamycin to chemotherapy improves event-free survival but not overall survival of AML patients with intermediate cytogenetics not eligible for allogeneic transplantation. Results of the GOELAMS AML 2006 IR study (Abstract#79). Blood. 2011;118:37–8.
Burnett AK, Hills RK, Milligan D, Kjeldsen L, Kell J, Russell NH, Yin JA, Hunter A, Goldstone AH, Wheatley K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29:369–77.
Burnett AK, Russell NH, Hills RK, Kell J, Freeman S, Kjeldsen L, Hunter AE, Yin J, Craddock CF, Dufva IH, Wheatley K, Milligan D. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012;30:3924–31.
Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Reman O, de Revel T, Gastaud L, de Gunzburg N, Contentin N, Henry E, Marolleau JP, Aljijakli A, Rousselot P, Fenaux P, Preudhomme C, Chevret S, Dombret H. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379:1508–16.
Swerdlow SH, Campo E, Harris NL. Acute myeloid leukaemia with recurrent genetic abnormalities: WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC; 2008. pp. 110–23
Warrell RP, Jr, de The H, Wang ZY, Degos L. Acute promyelocytic leukemia. N Engl J Med. 1993;329:177–89.
Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–15.
Fenaux P, Le Deley MC, Castaigne S, Archimbaud E, Chomienne C, Link H, Guerci A, Duarte M, Daniel MT, Bowen D et al. Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. European APL 91 Group. Blood. 1993;82:3241–9.
Ohno R, Yoshida H, Fukutani H, Naoe T, Ohshima T, Kyo T, Endoh N, Fujimoto T, Kobayashi T, Hiraoka A et al. Multi-institutional study of all-trans-retinoic acid as a differentiation therapy of refractory acute promyelocytic leukemia. Leukaemia Study Group of the Ministry of Health and Welfare. Leukemia. 1993;7:1722–7.
Asou N, Adachi K, Tamura U, Kanamaru A, Kageyama S, Hiraoka A, Omoto E, Akiyama H, Tsubaki K, Saito K, Kuriyama K, Oh H, Kitano K, Miyawaki S, Takeyama U, Yamada O, Nishikawa K, Takahashi M, Matsuda S, Ohtake H, Ohno R. Analysis of prognostic factors in newly diagnosed patients with acute promyelocytic leukemia: the APL92 study of the Japan Adult Leukemia Study Group (JALSG). Cancer Chemother Pharmacol. 2001;48(Suppl. 1):S65–71.
Ohno R, Asou N, Ohnishi K. Treatment of acute promyelocytic leukemia: strategy toward further increase of cure rate. Leukemia. 2003;17:1454–63.
Douer D. New advances in the treatment of acute promyelocytic leukemia. Int J Hematol. 2002;76(Suppl. 2):179–87.
Tobita T, Takeshita A, Kitamura K, Ohnishi K, Yanagi M, Hiraoka A, Karasuno T, Takeuchi M, Miyawaki S, Ueda R, Naoe T, Ohno R. Treatment with a new synthetic retinoid, Am80, of acute promyelocytic leukemia relapsed from complete remission induced by all-trans retinoic acid. Blood. 1997;90:967–73.
Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, Pandolfi PP, Warrell RP Jr. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339:1341–8.
Estey EH, Giles FJ, Beran M, O’Brien S, Pierce SA, Faderl SH, Cortes JE, Kantarjian HM. Experience with gemtuzumab ozogamycin ("mylotarg") and all-trans retinoic acid in untreated acute promyelocytic leukemia. Blood. 2002;99:4222–4.
Lo-Coco F, Cimino G, Breccia M, Noguera NI, Diverio D, Finolezzi E, Pogliani EM, Di Bona E, Micalizzi C, Kropp M, Venditti A, Tafuri A, Mandelli F. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood. 2004;104:1995–9.
Takeshita A, Shinjo K, Naito K, Matsui H, Sahara N, Shigeno K, Suzumura T, Horii T, Shirai N, Maekawa M, Yada Y, Teshima H, Takeuchi J, Ohnishi K, Ohno R. Two with all-trans retinoic acid-resistant acute promyelocytic leukemia treated successfully with gemtuzumab ozogamicin as a single agent. Int J Hematol. 2005;82:445–8.
Takeshita A, Shinjo K, Naito K, Matsui H, Sahara N, Shigeno K, Horii T, Shirai N, Maekawa M, Ohnishi K, Naoe T, Ohno R. Efficacy of gemtuzumab ozogamicin on ATRA- and arsenic-resistant acute promyelocytic leukemia (APL) cells. Leukemia. 2005;19:1306–11.
Aribi A, Kantarjian HM, Estey EH, Koller CA, Thomas DA, Kornblau SM, Faderl SH, Laddie NM, Garcia-Manero G, Cortes JE. Combination therapy with arsenic trioxide, all-trans retinoic acid, and gemtuzumab ozogamicin in recurrent acute promyelocytic leukemia. Cancer. 2007;109:1355–9.
Collins BE, Blixt O, Han S, Duong B, Li H, Nathan JK, Bovin N, Paulson JC. High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J Immunol. 2006;177:2994–3003.
Engel P, Wagner N, Miller AS, Tedder TF. Identification of the ligand-binding domains of CD22, a member of the immunoglobulin superfamily that uniquely binds a sialic acid-dependent ligand. J Exp Med. 1995; 181:1581–6.
Haas KM, Sen S, Sanford IG, Miller AS, Poe JC, Tedder TF. CD22 ligand binding regulates normal and malignant B lymphocyte survival in vivo. J Immunol. 2006;177:3063–73.
Tedder TF, Poe JC, Haas KM. CD22: a multifunctional receptor that regulates B lymphocyte survival and signal transduction. Adv Immunol. 2005; 88:1–50.
Tedder TF, Tuscano J, Sato S, Kehrl JH. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu Rev Immunol. 1997;15:481–504.
Tuscano J, Engel P, Tedder TF, Kehrl JH. Engagement of the adhesion receptor CD22 triggers a potent stimulatory signal for B cells and blocking CD22/CD22 L interactions impairs T-cell proliferation. Blood. 1996;87:4723–30.
Tuscano JM, Riva A, Toscano SN, Tedder TF, Kehrl JH. CD22 cross-linking generates B-cell antigen receptor-independent signals that activate the JNK/SAPK signaling cascade. Blood. 1999;94:1382–92.
Kantarjian H, Thomas D, Jorgensen J, Jabbour E, Kebriaei P, Rytting M, York S, Ravandi F, Kwari M, Faderl S, Rios MB, Cortes J, Fayad L, Tarnai R, Wang SA, Champlin R, Advani A, O’Brien S. Inotuzumab ozogamicin, anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13:403–11.
Advani A, Coiffier B, Czuczman MS, Dreyling M, Foran J, Gine E, Gisselbrecht C, Ketterer N, Nasta S, Rohatiner A, Schmidt-Wolf IG, Schuler M, Sierra J, Smith MR, Verhoef G, Winter JN, Boni J, Vandendries E, Shapiro M, Fayad L. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: results of a phase I study. J Clin Oncol. 2010;28:2085–93.
Ogura M, Tobinai K, Hatake K, Uchida T, Kasai M, Oyama T, Suzuki T, Kobayashi Y, Watanabe T, Azuma T, Mori M, Terui Y, Yokoyama M, Mishima Y, Takahashi S, Ono C, Ohata J. Phase I study of inotuzumab ozogamicin (CMC-544) in Japanese patients with follicular lymphoma pretreated with rituximab-based therapy. Cancer Sci. 2010;101:1840–5.
Acknowledgements
We would like to express sincere gratitude Dr. Ryuzo Ohno (Aichi Cancer Center, Nagoya, Japan) and Tomoki Naoe (National Hospital Organization Nagoya Medical Center, Nagoya, Japan) for their continuous advises of this work.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Adachi, M., Takeshita, A. (2015). Drug Resistance to Calicheamicin Conjugated Monoclonal Antibody Therapy. In: Verma, R., Bonavida, B. (eds) Resistance to Immunotoxins in Cancer Therapy. Resistance to Targeted Anti-Cancer Therapeutics, vol 6. Springer, Cham. https://doi.org/10.1007/978-3-319-17275-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-17275-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17274-3
Online ISBN: 978-3-319-17275-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)