
Abstract
The discovery of selective inhibitors of HSP90 two decades ago has enabled both a better understanding of the biology of HSP90 as well as its validation as a pharmacologic target for cancer. A number of HSP90 inhibitors have entered human clinical trials; to date, however, none have been approved for cancer therapy and thus the full potential of this class of agents remains to be realized. In this chapter we review the current status of HSP90 inhibitor development for cancer treatment, with particular emphasis on the second-generation, synthetic classes of compounds. In addition, we highlight various strategies currently being pursued that are designed to exploit specific cancer cell vulnerabilities and provide frameworks for optimization of HSP90 inhibitor-based strategies across a broad spectrum of cancer types.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
By traditional measures, the molecular chaperone heat shock protein 90 (HSP90) represents an unlikely and enigmatic candidate for oncology drug development. Highly conserved and ubiquitously expressed, HSP90 plays an indispensable role in regulating the maturation and functional stability of a vast array of cellular substrates, known as ‘client’ proteins (refer to http://www.picard.ch/downloads for current database). The repertoire of HSP90 clients is diverse, and is particularly enriched for signal transducers such as kinases and transcription factors [1]. In this regard, HSP90 activity impacts a broad range of normal homeostatic and physiological processes, including cell growth and survival, immune modulation, and development. In addition, HSP90 is also an essential component of the cellular heat shock response and may be further transcriptionally induced as a consequence of proteotoxic stress [2]. Not surprisingly, deletion of HSP90 is embryonic lethal and no mutations have been described nor are any polymorphisms known to exist that are suggestive of any association or causal relationship with cancer [3]. For these reasons, HSP90 was not originally considered an intuitively attractive target for the development of antineoplastic therapies.
In 1994, Whitesell and colleagues published a seminal study identifying the first small molecule inhibitors of HSP90 function [4]. This demonstration that natural product benzaquinone ansamycins (such as geldanamycin) were bona fide inhibitors of HSP90 with potent antitumor activity paved the way for a dramatic and exponential increase in our understanding of HSP90 biology and its relationship with malignancy over the past two decades. Most notably, it has emerged that the chaperoning functions of HSP90 can become subverted during tumorigenesis in order to facilitate malignant progression and maintain a transformed cellular phenotype [5]. Indeed, a large number of HSP90 client proteins have been implicated in the pathogenesis of human cancers, and are known to contribute to nearly every aspect of the oncogenic process including immortality, pro-survival/anti-apoptosis, metabolism, genomic instability and dissemination [6]. Often, these oncoproteins are expressed in labile states (e.g. mutant, translocated, or amplified) that are particularly reliant on the HSP90 machinery as a biochemical buffer for their stability and function [7]. Indeed, the buffering capacity of HSP90 against numerous environmental stresses, such as genotoxic, proteotoxic, and/or hypoxic insults, represents an important and broad-based mechanism by which tumor cells may co-opt HSP90 activity for selective advantage [8].
A number of additional considerations arose during the characterization and development of first-generation HSP90 inhibitors that helped establish the feasibility of targeting HSP90 as a viable therapeutic strategy for cancer. First, a unique characteristic of pharmacological HSP90 blockade is that inhibition of the chaperone leads to client protein degradation via the ubiquitin ligase-proteasome machinery. At the cellular level, this results in the simultaneous destabilization of literally hundreds of clients and client-driven signaling pathways. This stands in stark contrast to other molecularly targeted approaches, such as direct kinase inhibition, that selectively ablate only one (or a few) oncoproteins or signaling cascades. Moreover, the concomitant disruption of multiple signaling nodes provides a means to inhibit redundant pathways and feedback loops that can contribute to intrinsic and acquired drug resistance mechanisms [9–11]. Second, a common characteristic displayed by all HSP90 inhibitor compounds to date involves preferential and selective tumor retention characteristics [12]. While the underlying basis of this observation remain to be fully elucidated, HSP90 is frequently overexpressed in tumor tissues and a model has been proposed that the molecule exists in cancer cells as part of a highly active, multi-chaperone complex that exhibits greater affinity for targeted inhibitors than that observed in normal cells [13]. Recently, increased SUMOylation of HSP90, a consequence of cellular transformation, has been shown to sensitize both yeast and mammalian cells to HSP90 inhibitors [14] thus providing mechanism of tumor-selective HSP90 activation. Taken together, the features of tumor selectivity and multimodal impacts on the malignant phenotype combine to provide an exploitable therapeutic index for this collection of compounds (Fig. 15.1).

Summary scheme showing exploitable therapeutic characteristics of next-generation HSP90 inhibitor compounds based on current outcomes from preclinical and clinical evaluations
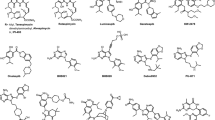
Thus, pharmacological blockade of HSP90 represents an innovative and multifaceted approach for the development of novel antineoplastic agents and small molecule inhibitors of this molecular chaperone rank among the most actively pursued classes of agents in oncology. While evaluation of the prototypical inhibitor class of ansamycin-based compounds (including geldanamycin and its derivatives 17-AAG and 17-DMAG) delivered critical proof-of-concept evidence and validated HSP90 as a druggable target for cancer treatment [7], their clinical application was hampered, and ultimately halted, due to a number of pharmacological and safety limitations [15]. Since then, an increasing number of synthetic small molecule inhibitors of HSP90 have been developed based on a diverse variety of chemical scaffolds, including those presented in Fig. 15.2 [16, 17]. To a large extent these second-generation compounds have overcome many of the original limitations of, and show improved potency relative to, the ansamycin class of HSP90 inhibitors and a number have entered early stage clinical evaluation. The outcomes of these trials have so far resulted in the early termination of clinical development of some candidates based on the emergence of new, potentially confounding safety concerns. For example, SNX-5422 (the prodrug of SNX-2112) has been discontinued based on reports of ocular toxicity and potential irreversible retinal damage [18], with treatment-related cardiac abnormalities also observed with this drug [19]. Others, including the potent resorcinol-based inhibitors NVP-AUY922 and ganetespib, have shown favorable tolerability in studies across a variety of human cancers [20, 21] and, for ganetespib in particular, encouraging signs of clinical activity [22]. At present, however, no HSP90 inhibitors have been approved for any human malignancy. This chapter will highlight recent developments in the therapeutic targeting of HSP90 in human solid tumors. Particular emphasis is placed on next-generation inhibitor compounds that are providing the framework for achieving the full translational potential of this promising group of anticancer agents. Targeting HSP90 is considered an equally promising avenue for intervention in hematological malignancies, including multiple myeloma (where buffering of proteotoxic stress is crucial for cell survival) and those cancers driven by sensitive HSP90 clients e.g., BCR–ABL in chronic myeloid leukemia, NPM–ALK in anaplastic large-cell lymphoma, FLT3 in acute myeloid leukemia, and ZAP70 in chronic lymphocytic leukemia [23].

Chemical structures of second-generation HSP90 inhibitor compounds. Key: Green hexagons currently under clinical evaluation in human solid tumors; red hexagons clinical development discontinued by sponsor; gray hexagons not yet entered clinical trials
1.1 Two Therapeutic Strategies: Client Protein-Driven Tumors Versus Combination Therapy
Many human cancers are known to be ‘oncogenically addicted’ to mutated, overexpressed, and/or chimeric kinases for growth and survival. Importantly, a number of these same oncogenic drivers are established HSP90 clients e.g., mutant EGFR (epidermal growth factor receptor) or ALK (anaplastic lymphoma kinase) translocations in non-small cell lung cancer (NSCLC); HER2 (human epidermal growth factor receptor 2) in breast cancer; mutated BRAF in melanoma; and mutant KIT in gastrointestinal stromal tumors (GIST). Accordingly, a reasonable approach for evaluating the potential of single-agent HSP90 inhibitor therapy involved the selection of tumor types expressing such modifications in order to capitalize on their client protein-driver dependence on HSP90. This premise has been supported by promising clinical activity observed in individuals with defined tumor phenotypes, most notably those exhibiting ALK-driven or HER2-amplified disease. Unexpectedly, similar translational benefit has not been achieved using HSP90 inhibitor monotherapy in other client-protein driven patient populations. This clinical experience has now led to the realization that the pleiotropic effects of HSP90 inhibitors may best be employed in the clinical setting as chemotherapeutic or molecularly-targeted agent sensitizers in order to provide superior antitumor efficacy, overcome resistance mechanisms, and reduce treatment-related toxicities, as summarized in Fig. 15.1 [5]. Here we discuss the potential utility and limitations of both of these strategies, with particular respect to individual cancer types and client protein sensitivity.
2 Non-small Cell Lung Cancer: Highlighting the Relationship Between EML4-ALK Client-Protein Dependence and Clinical Efficacy
NSCLC accounts for 85 % of all cases of lung cancer, which remains the leading cause of cancer-related deaths worldwide [24]. The high mortality is associated, in part, with the fact that a majority of patients present with advanced disease at the time of diagnosis with treatment options limited to systemic therapy. Platinum-based combination chemotherapy provides the foundation for current standard-of-care treatments in the clinical management of advanced NSCLC – a strategy that has largely reached a plateau of effectiveness in improving survival rates for lung cancer patients [25, 26]. Over recent years, an increased understanding of NSCLC biology has transformed lung cancer therapy from such broad-based cytotoxic use towards more tailored treatment approaches for certain patient sub-populations. A prime example involved the introduction of targeted inhibitors of the epidermal growth factor receptor (EGFR), erlotinib and gefitinib, which revolutionized and modified the principles of NSCLC treatment a decade ago [27]. The paradigm shift towards personalized therapies has provided meaningful improvements in overall survival and quality of life for lung cancer patients [28, 29].
NSCLC is characterized by a remarkable degree of genetic diversity and may be classified into distinct molecular subsets based on specific genomic alterations that drive tumorigenesis [29, 30]. This tumor type has long been considered a promising indication for the application of HSP90 inhibitors [6] since many of these oncogenic driver proteins are kinases that are established HSP90 clients, including EGFR, RAF, HER2, and, notably, the EML4-ALK fusion protein [31–36]. Indeed, EML4-ALK provides a compelling example of the relationship between client protein-driver dependence and clinical efficacy of targeted HSP90 inhibition. Approximately 3–7 % of NSCLC tumors are characterized by oncogenic gene rearrangements of ALK, most commonly with echinoderm microtubule-associated protein-like 4 (EML4), resulting in constitutively active kinases with transforming capacity [37, 38]. Crizotinib, a dual MET/ALK small molecule tyrosine kinase inhibitor (TKI), was the first ALK-targeted agent evaluated clinically and was granted accelerated approval in the United States for the treatment of patients with ALK-positive (ALK+) NSCLC [39, 40]. Crizotinib therapy improves overall patient survival compared with crizotinib-naïve controls [41], thereby providing clinical validation for targeting ALK in ‘oncogene-addicted’ lung tumors of this genotype. Since EML4-ALK is a highly sensitive client protein of HSP90 [31, 33], pharmacological blockade of the chaperone has been investigated as an alternative approach to direct kinase inhibition for therapeutic intervention in ALK-driven lung cancer. Confirming preclinical predictions, it was originally shown that ALK+ lung cancer patients could derive therapeutic benefit from targeted degradation of ALK via HSP90 blockade using the ansamycin compound IPI-504 [42]. This finding was supported by the results of a separate phase II study of ganetespib, wherein seven of eight patients harboring EML4-ALK rearrangements had disease control lasting at least 16 weeks, with four of these individuals exhibiting objective responses to inhibitor monotherapy [22].
Recent experimental data with ganetespib suggest that HSP90 inhibition also offers a promising strategy for overcoming acquired TKI resistance in ALK+ lung tumors [43]. Despite its robust clinical success, durable responses to crizotinib therapy are hampered by the invariable development of acquired drug resistance, a common feature of many TKI drugs [44]. Relapses frequently occur due to a spectrum of newly acquired secondary mutations within the ALK kinase domain [45, 46]. To date, a variety of mutations at different amino acid sites have been reported in NSCLC patients who exhibited resistance to crizotinib, and a number of others that can mediate ALK TKI resistance have been identified through in vitro mutagenesis screens [39, 47, 48]. In preclinical models, ganetespib retained robust cytotoxic activity against crizotinib-resistant cell lines irrespective of the mutational site or specific amino acid substitution present [43]. Significantly, this finding was validated by clinically observed tumor responses in a relapsed NSCLC patient who had progressed after 1 year of crizotinib therapy. Despite the presence of a specific kinase domain mutation, a single cycle of ganetespib treatment resulted in a marked shrinkage of lung lesions, underscoring the therapeutic potential of the drug within this refractory population [43].
In addition, systemic resistance to ALK inhibitors may arise in the absence of secondary ALK kinase domain mutations [45]. Although the mechanism(s) remain to be fully elucidated, it has emerged that ligand-mediated activation of secondary and/or separate oncogenic signaling pathways, in particular EGFR and HER2 [49–51], is one process that can bypass the dependency of tumor cells on ALK signaling and contribute to a resistant phenotype. Strategies to counteract these types of acquired resistance in ALK-driven NSCLC have not yet been established and, within this context, the use of ALK-selective inhibitors with increased potency is unlikely to provide any clinical impact. Both EGFR and HER2 are established HSP90 clients and, as such, the broad spectrum of biological activity afforded by HSP90 inhibition represents a potential approach to counteract such compensatory mechanisms. In preclinical models, ganetespib exposure resulted in the simultaneous destabilization of EML4-ALK as well as EGFR in ALK+ NSCLC lines both in vitro and in vivo, with concomitant loss of multiple downstream effector signaling pathways [43]. The effects were distinct to those of crizotinib, with the multimodal activity of the HSP90 inhibitor sufficient to account for its superior relative potency and antitumor efficacy.
Interestingly, chromosomal rearrangements of two additional tyrosine kinases, ROS1 and RET, generate fusion proteins that are sensitive to HSP90 inhibition by ganetespib [43]. Similar to ALK, ROS1 has been reported to define a genomic subset of NSCLC with distinct clinical characteristics [52] and, while the incidence of ROS1 fusions is less than 2 %, preclinical and clinical data suggest that patients whose tumors harbor these rearrangements may benefit from crizotinib therapy [53, 54]. RET kinase fusions have also recently emerged as promising molecular targets in NSCLC [55], where they have been reported to segregate from genetic modifications in EGFR, KRAS, HER2 and ALK [56]. As the clinical significance of these oncogenic drivers in NSCLC becomes realized, it is reasonable to suggest that HSP90 inhibition may warrant evaluation as a potential point of therapeutic intervention.
3 Non-small Cell Lung Cancer: Combining the Modalities of HSP90 and Tyrosine Kinase Inhibition to Improve EGFR therapy
ALK targeting is a prime example of genetics dictating treatment in lung cancer, but what about more frequent genetic alterations that may be targeted through HSP90 blockade? Mutations in EGFR, an established client protein of HSP90, define one of the most prevalent and actionable subgroups of NSCLC. It is estimated that 10–15 % of NSCLCs in Caucasians and up to 30 % in Asian populations harbor activating mutations in EGFR [57]. Aberrant activation of this receptor stimulates a variety of oncogenic signaling cascades, including the JAK/STAT, RAS/RAF/ERK, and PI3K/AKT pathways [58]. EGFR activity may also become dysregulated during tumorigenesis by other mechanisms, including gene amplification and/or receptor overexpression [59]. Importantly, EGFR is a validated therapeutic target in NSCLC, with three small molecule kinase inhibitors (erlotinib, gefitinib, and afatinib) currently approved for the treatment of advanced disease [60]. Each of these TKIs show preferential clinical efficacy in NSCLC patients with EGFR mutant-bearing tumors, although durable responses are rare due to the development of acquired resistance, which typically arises through the acquisition of a second site mutation (T790M) within EGFR [61, 62], or via activation of compensatory signaling pathways that bypass the receptor and restore downstream oncogenic signaling [63]. Mutant EGFR oncoproteins are reliant on the chaperone activity of HSP90 for their conformational stability and function [35, 64], and there are abundant experimental data showing sensitivity of EGFR-driven NSCLC to multiple second-generation HSP90 inhibitors in preclinical studies [65–70]. In addition, it has recently been demonstrated that the mature, wild-type receptor is also a bona fide HSP90 client in cancers that overexpress wild-type EGFR [71], suggesting potential utility for targeted HSP90 inhibition beyond the mutant receptor phenotype. Despite this, the clinical experience with selective HSP90 inhibitors has revealed only modest single-agent activity in molecularly defined subsets of both wild-type and mutant EGFR NSCLC patients [22, 42].
To date, efforts to combine EGFR TKIs with standard chemotherapies have not been associated with survival benefits in clinical trials [29]. Recently, attention has focused on combining the modalities of HSP90 blockade with selective EGFR tyrosine kinase inhibition for optimizing NSCLC tumor responses to EGFR TKIs. Ganetespib can potentiate the efficacy of both erlotinib and afatinib in preclinical models of NSCLC driven by activating EGFR mutations, with combination treatment inducing tumor regressions and a capacity to overcome erlotinib resistance [72]. Similar combinatorial benefit has been reported when erlotinib is combined with SNX-2112 [73] or a new investigational HSP90 inhibitor compound CH5164840 [74]. Moreover, encouraging signs of clinical activity dosing NVP-AUY922 in combination with erlotinib in patients with acquired resistance to EGFR inhibitors have been observed in an ongoing phase II trial (NCT01259089). In the first stage of that study, 2 of 16 patients demonstrated a partial response; both of which had the T790M mutation.Footnote 1 Taken together, these data suggest that HSP90 inhibition alongside EGFR kinase blockade may represent a potential therapeutic pathway for improving patient outcomes and overcoming TKI resistance in EGFR-mutant NSCLC.
4 Non-small Cell Lung Cancer: Targeted HSP90 Inhibition Alongside Taxane Therapy Improves Patient Outcomes
For most individuals with advanced and unresectable NSCLC, treatment options are limited to platinum-based two-drug regimens consisting of either cisplatin or carboplatin in combination with an additional ‘third-generation’ cytotoxic agent (paclitaxel, docetaxel, gemcitabine, vinorelabine, or pemetrexed) [25]. One strategy that has been employed in order to try and improve objective response rates for these patients is through the use of molecularly targeted agents, such as bevacizumab and cetuximab, in combination with front-line chemotherapeutics [28, 76]. Benefit from such therapy combinations is typically observed within subsets of NSCLC patients and correlates with specific tumor histology and/or molecular phenotypes. It is now established that HSP90 blockade can potentiate the activity of a wide variety of chemotherapeutic drugs [77] and, given the pleotropic effects of HSP90 inhibition across a range of NSCLC phenotypes, these considerations provide a strong rationale for evaluating novel HSP90 inhibitors alongside traditional chemotherapy in this malignancy.
An encouraging validation of this is provided by ganetespib improving therapeutic outcomes alongside taxane therapy in NSCLC. Paclitaxel and docetaxel comprise the taxane family of antimitotic agents widely used in the treatment of multiple human cancers. While active in NSCLC therapy, their effectiveness is often hampered by a variety of significant and dose-limiting adverse side effects in clinical practice. Synergistic combinatorial benefit between HSP90 inhibitors and taxanes has previously been described in different cancer models [78–82] suggesting that their non-overlapping but complementary mechanisms of action are conserved across tumor types. Combinations of ganetespib with both taxanes showed synergistic activity and enhanced antitumor efficacy in preclinical models of NSCLC [83]. The mechanistic benefit afforded by the addition of ganetespib to taxane regimens was found to be multifactorial and included loss of pro-survival signaling, direct impacts on the cell cycle machinery, and exacerbation of mitotic catastrophe [83, 84]. In early clinical trials, ganetespib was shown to be well tolerated as monotherapy [20, 22], lacking the severe liver toxicities characteristic of the ansamycin class of inhibitors and also adverse visual disturbances that have emerged as an important clinical concern for some newer HSP90 inhibitor drugs [85]. Together with demonstrated activity in oncogene-driven subsets of NSCLC patients, these findings prompted evaluation of the cytotoxic sensitizing property of ganetespib alongside docetaxel in refractory NSCLC in a recently completed phase II (NCT01348126; GALAXY-1) and ongoing phase III study (NCT01798485; GALAXY-2).
A number of considerations support the selection of docetaxel as the optimal front-line taxane candidate in these trials. Docetaxel is the only agent that is approved for both first- and second-line therapy in advanced NSCLC [26] and was also the first drug to establish superior efficacy and tolerability over other third-generation agents when used in combination with platinum compounds [25]. Meta-analyses of current treatment regimens have shown that docetaxel is associated with better disease control than paclitaxel combinations and tumor histology does not exert any influence over drug activity or efficacy, as opposed to other third-generation cytotoxics [25]. Significantly, the GALAXY-1 trial is the first randomized study to show improvement in efficacy through addition of a targeted HSP90 inhibitor to chemotherapy in cancer patients.Footnote 2 The strongest signals of efficacy were noted improvements in ORR, PFS, and OS observed in a large subset of adenocarcinoma patients with chemosensitive disease. Accordingly, it is this population of NSCLC patients that is presently being enrolled in the ongoing confirmatory GALAXY-2 study. Importantly, the combination of ganetespib with docetaxel was well tolerated and not associated with any excessive toxicity over that seen with docetaxel treatment alone. Moreover, an interesting finding to emerge was that the time to disease progression due to emergence of new metastatic lesions was prolonged in individuals undergoing combination treatment. While this observation warrants confirmation in additional trials, it strongly suggests that HSP90 inhibitor treatment exerted a significant biological impact on the metastatic dissemination and growth in these patients. This provides important validation of a large body of preclinical evidence to support an essential role for HSP90 in regulating multiple facets of the metastatic process [87–89].
5 Breast Cancer: Client Proteins Dictate Sensitivity to HSP90 Inhibitors
The cellular and molecular heterogeneity exhibited by tumors of the breast is such that breast cancer is no longer considered a single disease with variable morphology, but instead a collection of distinct neoplastic disorders each associated with their own pathological features and clinical outcomes [90]. Gene expression profiling has resulted in the classification of human breast cancer into at least five subtypes based on discrete molecular signatures [91–93]. These include normal breast-like, the hormone receptor-positive (estrogen and progesterone receptors; ER and PR) luminal A and luminal B subtypes, HER2-positive, and basal-like. Within this stratification, triple-negative breast cancers (TNBC) are an orphan grouping of tumors characterized by an absence of ER, PR and HER2 expression that primarily fall within the basal-like subtype, however the two definitions are not strictly synonymous [94, 95]. Given this degree of complexity, it is now apparent that pharmacologic targeting of a single pathway or individual component of an oncogenic signal cascade typically fails to translate to long-term efficacy, particularly for metastatic disease. A number of established HSP90 clients have been implicated in the pathogenesis of breast tumors, including the ER and PR steroid hormone receptors, EGFR and HER2 receptor tyrosine kinases, and intermediates of oncogenic signaling cascades (AKT and RAF1) [9]. Accordingly, there is considerable preclinical support for potential therapeutic use of HSP90 inhibitors in breast cancer [96–102].
Similar to ALK-driven lung tumors, HER2-positive breast cancer provides another impressive illustration of HSP90 inhibitor efficacy in client protein-driven disease. HER2 is a highly sensitive HSP90 client protein and overexpression of this receptor defines a clinically relevant subset of breast cancers exquisitely dependent on oncogenic HER2 signaling for growth and survival. In recent years the prognosis for HER2-positive patients has improved following the introduction of selective HER2-targeted agents (such as trastuzumab and lapatinib) as first-line treatments in this disease [103]. Trastuzumab in combination with chemotherapy is the current standard of care for metastatic HER2-positive breast cancer [103]; however the invariable development of resistance presents a significant and unresolved clinical problem. A variety of mechanisms have been proposed to account for the trastuzumab-resistant phenotype, including activation of compensatory growth factor signaling pathways, amplification of the PI3K/AKT cascade, and expression of truncated HER2 receptors that lack the antibody binding epitope [104]. By virtue of its multifaceted mode of action, HSP90 inhibition has been shown to overcome each of these various mechanisms in laboratory models of trastuzumab-resistant breast cancer [98, 101]. Interestingly, recent analyses have suggested a benefit for continued trastuzumab treatment even beyond progression [105] and other selective HER inhibitors such as lapatinib have shown efficacy in patients that have become refractory to trastuzumab therapy [104]. Thus, trastuzumab-resistant tumors appear to remain oncogenically reliant on HER2; raising the possibility that HSP90 inhibition might afford an effective therapeutic approach to abrogate acquired resistance to primary anti-HER2 treatment. In this regard, proof-of-concept was provided in a pivotal phase II trial that demonstrated meaningful clinical efficacy could be achieved by combining 17-AAG with trastuzumab in HER2-positive metastatic breast patients who had previously progressed on trastuzumab [106].
Despite the positive outcome of that study, the clinical development of 17-AAG has been discontinued. Thus a therapeutic opportunity clearly exists within this patient group for an effective and tolerable alternate HSP90 inhibitor. A phase I study evaluating BIIB021, either alone or in combination with trastuzumab (NCT00412412), showed only modest antitumor activity for this purine-based HSP90 inhibitor in a cohort of heavily pre-treated metastatic HER2+ patients [107]. Additional second-generation HSP90 compounds, including clinically advanced agents such as NVP-AUY922 and ganetespib that show superior potency and safety over 17-AAG, also exhibit strong preclinical activity in models of HER2+ breast cancer [99, 108]. NVP-AUY922 has undergone clinical evaluation as monotherapy for locally advanced and metastatic HER2+ breast cancer as part of a completed phase II study expansion arm (NCT00526045) and also as part of combination therapy alongside trastuzumab in trastuzumab-refractory advanced HER2+ breast patients (NCT01271920), essentially reflecting the successful 17-AAG trial. Ganetespib has shown preliminary signs of single-agent activity within the advanced HER2+ breast cancer population in a phase II window-of-opportunity trial (NCT01677455; ENCHANT). While no final data from any of these studies are yet available, the outcomes are likely to be instrumental in guiding the application of HSP90 inhibitor use for this malignancy.
Beyond the HER2+ phenotype, HSP90 blockade may also be considered a logical targeted approach for hormone-responsive, luminal-type breast tumors due to its well-defined role in the chaperoning of steroid receptors, including ER and PR [109]. For patients with estrogen-dependent disease, adjuvant endocrine therapy with ER antagonists such as tamoxifen and aromatase inhibitors are effective first-line treatments [110]. Despite the obvious benefits of these standard-of-care agents for the vast majority of individuals diagnosed with breast cancer, the development of acquired resistance is common, ultimately resulting in disease relapse and death. The mechanisms that underlie hormone refractory phenotypes remain incompletely defined although crosstalk between the ER and growth factor receptor pathways (involving HSP90 clients like EGFR, HER2 and IGF-1R) have been implicated in the resistance process [111]. Interestingly, tamoxifen resistance may arise even as the tumors themselves remain ER positive [112]. Within this context, early reports evaluating ansamycin-based HSP90 inhibitors revealed that chaperone inhibition could overcome endocrine therapy resistance in tamoxifen- and aromatase-resistant breast cancer cell lines [102, 113]. These findings provide a rational framework for investigating the clinical potential of this modality as an alternate, ligand-independent mechanism for sustained degradation of ER/PR in hormone receptor-positive breast tumors. In this regard, the results of a completed phase II trial of BIIB021 in combination with exemestane (NCT01004081) in patients whose hormone receptor-positive cancer had progressed on prior aromatase inhibitor therapy are eagerly awaited. Further, a randomized phase II trial evaluating the addition of ganetespib to the approved ER antagonist fulvestrant in hormone receptor-positive metastatic breast cancer is currently recruiting participants (NCT01560416).
6 Breast Cancer: Will HSP90 Inhibition Be Effective in Triple-Negative Tumors?
In contrast to HER2 or hormone receptor-positive breast cancer, TNBC represents a heterogeneous collection of orphan status tumors that lack a defining molecular vulnerability to serve as a druggable target. TNBC shows a disproportionate mortality amongst breast cancer subtypes and patients with TNBC have a higher likelihood of visceral metastatic disease and early relapse [94, 95]. An absence of reliable predictive biomarkers, combined with the disappointing efficacy of conventional chemotherapy, highlights an urgent need for alternate treatment options for these patients. A variety of potential biological drivers have been incompletely validated in TNBC. Many of these are established client proteins of HSP90, including EGFR, KIT and IGF-1R as well as critical mediators of the RAS/RAF/ERK, PI3K/AKT and mTOR tumorigenic signaling pathways [94]. Indeed, and contrary to an earlier notion that TNBC tumors may not be sufficiently responsive to targeted HSP90 blockade, there is now accumulating preclinical evidence that supports therapeutic sensitivity of triple-negative cancer cell lines to HSP90 inhibition, shown using the second- generation compounds ganetespib and PU-H71, as well as a new investigational agent PF-4942847 [84, 97, 100].
A distinct clinicopathological feature of TNBC is the hematogeneous spread of metastases, showing preferential dissemination to the lungs and brain rather than to bone or soft tissues [94]. Ganetespib treatment negatively impacts orthotopic tumor growth, invasion and distal lung metastasis in experimental metastasis models of TNBC [84, 89]. At a molecular level this has been attributed to, at least in part, potent drug-induced reductions in the expression and activity of HIF-1α (hypoxia-inducible factor 1α) and concomitant target genes that are linked to TNBC progression [89]. Significantly, compelling anecdotal evidence of metastatic tumor responses in TNBC patients undergoing ganetespib therapy has also been obtained in the clinical setting [84]. As shown in the CT scans presented in Fig. 15.3a, four cycles of ganetespib monotherapy resulted in discernible shrinkage of large metastatic lung lesions in a heavily pre-treated TNBC patient as part of a phase I trial (NCT00688116). This cancer was highly aggressive – at the time of enrollment, the patients’ disease had progressed with multiple bilateral pulmonary, liver and bone metastases following six prior, different chemotherapeutic regimens. An overall objective response was not confirmed as the patient was taken off study at the end of cycle 5 due to the detection of brain metastases. Figure 15.3b shows CT scans from another individual currently enrolled in the ongoing ENCHANT monotherapy trial in breast cancer (NCT01677455). In 2011 the patient received six cycles of adjuvant FEC chemotherapy (5-fluorouracil, epirubicin, and cyclophosphamide) but progressed with recurrent disease 2 years later, presenting with multiple axillary and supraclavical lymph node deposits and pulmonary metastases. Within 6 weeks of starting ganetespib treatment, the patient achieved a confirmed partial response, including the marked axillary lymph node tumor shrinkage shown in Fig. 15.3 and the patient remains on therapy at the present time. These robust clinical responses, observed in independent trials, suggest that the TNBC tumors were acutely reliant on the chaperoning function of HSP90, with one or more chaperone-dependent signaling pathways responsible for promoting metastatic growth and survival. Given the remarkable molecular heterogeneity of this collection of breast cancers, an ongoing clinical challenge remains to identify which specific client proteins may ultimately serve as predictive biomarkers for those individuals likely to respond to HSP90 inhibitor treatment.

Clinical activity of ganetespib in TNBC patients. (a) CT scans of a metastatic lung deposit taken prior to ganetespib treatment (left panel) and after 16 weeks on-therapy (right panel). Circles depict location and size of the tumor mass. (b) CT scans taken prior to ganetespib treatment and after 6 weeks on therapy. Circles depict the location and size of multiple axillary lymph node lesions (Images reproduced from [84])
Of note, basal-like breast (including TNBC) and ovarian cancers share striking similarities in terms of genomic and proteomic modifications [114]. The common findings of MYC amplification as well as TP53, RB1 and BRCA1 loss suggest that these represent may represent shared driving events for TNBC and ovarian carcinogenesis, and that common therapeutic approaches might be considered for both these diseases [114]. In addition, bioinformatic meta-analyses recently identified an HSP90-centric hub in ovarian cancer that was susceptible to inhibitor treatment [115]. This finding was validated by the capacity of ganetespib to significantly reduce tumor growth and dissemination in xenograft models and spontaneous ovarian tumors in transgenic mice [115]. Of particular relevance, combining ganetespib with paclitaxel strongly augmented the antitumor efficacy of either agent alone – a result that supports a promising rationale for combination ganetespib and paclitaxel regimens presently being evaluated in two trials in recurrent and metastatic ovarian cancer (NCT01962948; NCT02012192). Further, combination ganetespib plus paclitaxel therapy is undergoing clinical assessment in women with locally advanced breast cancer as part of the ongoing I-SPY2 adaptive phase II study (NCT01042379).
7 Melanoma: Disrupting Oncogene Addiction and Overcoming Resistance
Cutaneous melanoma ranks among the most aggressive and treatment-resistant human cancers, and the worldwide incidence of this disease continues to increase [116]. Mutational activation of BRAF, resulting in dysregulation of the canonical MAPK (RAF/MEK/ERK) signaling cascade, is characteristic of over half of all malignant melanomas [117, 118]. This high frequency underscores a critical role for mutant BRAF activity in melanoma oncogenesis [119]. In this regard, mutated BRAF (most commonly BRAFV600E) also provides an actionable target for molecular therapeutic approaches as evidenced by the recent approval of the first highly selective BRAFV600E inhibitor, vemurafenib, for patients with metastatic melanoma [120]. The conformational stability of mutant BRAF is reliant on the activity of HSP90 [121], the chaperoning function of which thus likely facilitates oncogene addiction in this malignancy. In support of this premise, a number of synthetic small molecule HSP90 inhibitors have shown robust activity in mutant BRAF-driven melanoma cell lines in vitro and in vivo, including NVP-AUY922, SNX-2112, and NVP-BEP800 [122–124]. Extending these observations, ganetespib exhibits superior potency and efficacy compared to vemurafenib in mutant BRAF–driven melanoma models, with drug exposure inducing the simultaneous destabilization of BRAFV600E as well as CRAF, AKT, and RAF/MEK/ERK signaling that is stimulated by mutant BRAF activation [125, 126]. These preclinical findings suggest that targeting the chaperone function of HSP90 represents a rational approach for intervention in mutant BRAF-driven melanoma. Initial clinical studies evaluating the first-generation compound 17-AAG in metastatic melanoma were disappointing, with no objective responses observed [127, 128]. A lack of durable target suppression and pharmacological limitations of the ansamycin inhibitor itself likely contributed to the absence of clinical activity in these trials; however this does not preclude the potential for newer agents with improved potency, durable activity, and favorable tolerability in this malignancy [129].
Indeed, emerging data strongly suggest that combining the modalities of HSP90 inhibition with either selective BRAF or MEK targeting warrants further investigation in melanoma displaying oncogenic addiction to mutated BRAF, particularly as a means to overcome mechanisms of resistance to targeted BRAF agents. Acquired resistance to vemurafenib represents a significant clinical obstacle to its long-term efficacy, and most patients relapse with drug-resistant disease within 6–8 months [130]. Unlike the case for EGFR or EML4-ALK described above, to date there is no evidence of secondary mutations in BRAF to account for a resistant phenotype. Instead, a variety of mechanisms have been identified that allow for either bypass or reactivation of MAPK signaling [119, 131, 132]. Despite the complexity of the signaling intermediates involved that lead to therapeutic escape from BRAF inhibitor treatment, all appear to converge on pathways that are sensitive to HSP90 inhibition. Both XL888, a tropane-derived small molecule HSP90 inhibitor whose clinical development has been halted by the sponsor [133], and ganetespib can overcome a diverse array of intrinsic and acquired vemurafenib resistance mechanisms in relevant melanoma models [125, 134]. NMS-E973, a new investigational inhibitor compound, has also shown activity in models of vemurafenib resistance, including inhibiting the growth of intracranially implanted melanoma xenografts in mice [135].
Given the exquisite dependence on MAPK signaling for both melanoma viability and drug resistance, pharmacological inhibition of MEK has also emerged as an important strategy for therapeutic intervention in mutant BRAF-driven melanoma [136]. While the role of MEK inhibitor monotherapy, given the advent of approved BRAF-targeted agents, remains to be determined [130] a number of combination trials investigating the dual blockade of mutant BRAF and MEK are currently underway, and early evidence suggests that this strategy may represent an effective approach to prevent or delay the onset of resistance due to MAPK reactivation [137, 138]. Within the resistance setting, we have shown that the combination of ganetespib with the allosteric MEK inhibitor TAK-733 provides superior cytotoxic activity and inhibition of MAPK reactivation compared to dual vemurafenib plus TAK733 treatment in melanoma cell lines. Moreover, these effects were recapitulated in vemurafenib-resistant tumors in vivo, where combination treatment induced significant tumor regressions [125]. Such data reinforce the potential utility of HSP90 inhibition as an alternative, and potentially complementary, strategy for treating tumors with acquired resistance to BRAF inhibitors.
8 Gastrointestinal Stromal Tumors and Prostate Cancer: All Clients Are Not Created Equal
ALK-rearranged NSCLC and HER2-positive breast cancer have provided the clearest demonstrations of clinical efficacy in HSP90 client oncoprotein-addicted cancers – however, not all tumor types that appear to meet the same criteria have displayed similar sensitivity to this therapeutic approach. For example, GIST, the most common type of soft-tissue sarcoma, is a malignancy that may be considered particularly amenable to HSP90 inhibitor-directed therapy due to the high frequency (~85 %) of driver KIT mutations [139, 140]. To date, however, the results of a phase I study evaluating IPI-504 in patients with metastatic and/or unresectable GIST and other soft-tissue sarcomas revealed that only 1 of 37 GIST patients exhibited a confirmed partial response; moreover genotyping from this individual failed to identify any KIT mutation present [141]. A subsequent placebo-controlled phase III trial in relapsed/refractory GIST (NCT00688766) was prematurely terminated due an excessive mortality rate in the IPI-504 arm, with treatment-related deaths resulting from liver failure, metabolic acidosis, renal failure, and cardiopulmonary arrest [142]. No results are yet available from an ongoing study of NVP-AUY922 monotherapy in this population (NCT01404650). While its clinical development has now been discontinued, BIIB021 underwent evaluation in a small cohort of refractory GIST patients [143] where metabolic partial responses were observed in 22 % of cases, although no RECIST-defined partial responses were achieved. This study raised the possibility that a lack of durable client protein suppression underscored such modest responses to HSP90 inhibitor monotherapy. Pharmacodynamic assessment showed that significant rebounds in FDG activity were seen in patients between doses, suggesting that although target inhibition was being achieved, it was not sustained. In agreement with this premise, prolonged inhibition of KIT or its downstream pathways has not been observed following ganetespib exposure in either preclinical GIST models or patient biopsies, and consequently only limited efficacy was obtained in a phase II trial evaluating weekly administration of ganetespib to GIST patients (NCT01039519).Footnote 3
A number of considerations have also made prostate cancer an attractive disease indication for HSP90 targeting. Androgen ablation therapy is the foundation of current treatment for patients with locally advanced or metastatic disease and, importantly, it is now clear that advanced and recurrent tumors continue to rely on androgen receptor (AR) signaling, even in the castrate environment [145, 146]. The AR is an established HSP90 client, and the relationship between the chaperoning function of HSP90 with steroid receptor stability, conformation and modulation of ligand binding is well characterized [147]. Accordingly, a number of preclinical studies have provided experimental support for the potential utility of HSP90 inhibitors in prostate cancer [148–152]. Unfortunately, the clinical experience using such compounds in the single-agent setting has not met expectations, with minimal effects on PSA (prostate-specific antigen) levels or tumor burden being observed along with unacceptable toxicities [153, 154]. For these tumors, it appears that combining targeted HSP90 inhibitors alongside other AR antagonists or chemotherapeutics may represent a more feasible path to clinical efficacy. In addition, radiation therapy displays high control rates for low-risk, localized prostatic disease [155] and there is emerging evidence to suggest that the radiosensitizing effects of HSP90 blockade may be exploitable as a valid adjunct to radiotherapy in this disease [156].
9 Colorectal Cancer: A Potential Chemosensitizing Role for HSP90 Inhibitors?
The clinical management of colorectal cancer (CRC) is largely dictated by the stage of the disease. Patients who present with early stage, localized tumors are amenable to curative resection surgery while adjuvant chemotherapy is indicated for patients with Stage III disease or higher, with the aim of improving survival and preventing recurrence [157]. Over the last decade, the addition of targeted biological agents (such as such as the anti-VEGF antibody bevacizumab and the EGFR antagonists cetuximab and panitumumab) to existing chemotherapeutic regimens has effectively doubled the survival outlook for patients with metastatic CRC [158, 159]. This type of approach clearly serves as an encouraging paradigm for providing continuing improvements for the treatment of advanced CRC.
Although there is a considerable literature showing robust activity of HSP90 inhibitors (both first- and second-generation) in preclinical models of CRC [160–163], meaningful responses to HSP90 inhibitor monotherapy within the clinical setting have proven far more modest. As a prime example, the most significant demonstration of efficacy as part of the initial phase I evaluation of ganetespib in solid malignancies involved a patient with metastatic CRC, who achieved a partial response while on-therapy [20]. In a subsequent phase II trial in heavily treated metastatic CRC patients (NCT01111838) no objective responses were observed, although 2/15 evaluable patients achieved durable stable disease. In general terms, this malignancy might not be expected to be highly responsive to targeted HSP90 therapeutic intervention since the proteins that show the highest frequency of alteration as part of colorectal tumorigenesis, APC and KRAS, are themselves not HSP90 clients. However, similar to what is observed following mutation of BRAF in melanoma described above, the canonical MAPK kinase cascade is the primary mitogenic pathway aberrantly stimulated by KRAS under pathological conditions [164] and this signaling axis is particularly sensitive to HSP90 modulation. In addition, mutations in BRAF have also been described in approximately 5–15 % of cases of CRC [165]. An important lesson gleaned from integrating molecularly targeted agents into existing therapies for CRC was that each of the biologics displayed minimal clinical activity as single agents and their full benefit was only realized when they were combined with standard treatment regimens [158]. It is reasonable to suggest then that this profile may be similar for any future application of selective HSP90 inhibitors in CRC.
For five decades, 5-Fluorouracil (5-FU) has played an indispensable role in CRC treatment, both in the curative and palliative settings and constitutes the backbone of the FOLFOX (5-FU/leucovorin (LV) plus oxaliplatin) and FOLFIRI (5-FU/LV plus irinotecan) combination therapies that represent the standard first line cytotoxic regimens for metastatic CRC patients. Due to a short half-life and variations in bioavailability, 5-FU requires intravenous infusion – consequently the first oral prodrug formulation, capecitabine, has more recently received approval for adjuvant monotherapy use [166]. A synergistic interaction between 5-FU and NVP-AUY922 in vitro has been reported in bladder cancer cell lines; no correlative translation into in vivo efficacy was shown in that study [167]. In preclinical CRC models, ganetespib exerts robust antitumor and antiangiogenic activity, through the simultaneous destabilization of multiple key growth and survival pathways, perturbation of cell cycle regulation, and disruption of HIF-1α and STAT3 signaling [168, 169]. Perhaps most importantly, ganetespib co-treatment can significantly improve the efficacy of fluoropyrimidine therapy in CRC xenografts [168]. Using an inherently 5-FU-insensitive HCT116 xenograft model, capecitabine administered on a clinically relevant dosing schedule suppressed tumor growth by over half. Single-agent ganetespib treatment showed a similar, modest degree of tumor growth inhibition, comparable to what has previously been reported for 17-DMAG [163]. However, a combination regimen resulted in over 50 % tumor regression [168]. This represents the first demonstration of combinatorial benefit between a small molecule HSP90 inhibitor and fluoropyrimidine therapy in CRC-derived tumors. Moreover, these data provide a clear rationale for exploiting both the chemosensitizing activity as well as the capacity to overcome intrinsic fluoropyrimidine resistance that was conferred by the addition of a potent HSP90 inhibitor. In light of these considerations, a phase I trial evaluating ganetespib in combination with capecitabine and radiation in rectal cancer [NCT01554969] has been initiated.
10 Future Considerations
Since the initial clinical trials of the natural product-derived HSP90 inhibitors commenced only a decade ago, enormous progress has been made in the discovery and characterization of next-generation synthetic compounds with improved pharmaceutical and tolerability profiles. As outlined in this chapter, continued exploration of biologically informed drug combinations are likely to provide the most direct route to efficacious use of these agents for cancer treatment in the near term. Indeed, it is now possible to envision the eventual application of potent and safe HSP90 inhibitors as adjuncts to both chemotherapy and molecular-targeted strategies for a broad range of human malignancies. However a number of key considerations remain as obstacles on the path to widespread clinical use. Perhaps the most significant factor required for success involves the identification of specific predictive biomarkers within cancer types to stratify those patients most likely to receive benefit from therapeutic HSP90 blockade. In many ways, the pleiotropic effects of HSP90 inhibition represent a double-edged sword – such a wide and diverse range of clients represents an inherent therapeutic advantage, yet at the same time has emerged as a challenge for predicting individual response. Implicit to this endeavor will be a more complete understanding of the relationships that exist between HSP90 and its oncoprotein clients and predicting what makes a client dependent on the chaperoning of HSP90. For example, it was recently discovered that ~30 % of the various EML4-ALK gene rearrangement structures observed in NSCLC patients were insensitive to HSP90 blockade in preclinical studies [170], a finding with clear translational relevance to the application of HSP90 inhibitors in tumors driven by these client proteins. Further, the tumor retention characteristic of targeted HSP90 agents implies that predictive assays of drug sensitivity and pharmacodynamic profiling are likely to require assessment of client protein modulation within tumors themselves, since surrogate tissues are unlikely to reflect true effects of these drugs. Taking into consideration both the multimodal antitumor activity and rapid clearance of small molecule inhibitors from normal tissues and the blood compartment, traditional serum pharmacokinetics may offer only limited guidance for therapeutic dosing of these particular compounds [171]. A number of alternative and non-invasive methods are in development to circumvent this issue, including functional imaging using radiolabeled HSP90 inhibitors such as 124I-PU-H71 [23].
In addition, tumor cells (even within the same cancer type) may display variable degrees of intrinsic ‘resistance’ to HSP90 inhibitory compounds. Pharmacological blockade of HSP90 elicits induction of the heat shock factor-1 (HSF1)-directed heat shock response [172], providing a compensatory mechanism which may mitigate sensitivity to targeted HSP90 inhibition. This cellular stress response is characterized by up-regulation of the inducible molecular chaperone heat shock protein 70 (HSP70). Indeed, elevated HSP70 expression is commonly used in the clinical setting as a pharmacodynamic biomarker for HSP90 blockade [173, 174]. However, in line with its cytoprotective cellular roles, HSP70 possesses strong antiapoptotic activity and experimental evidence has shown that silencing this protein dramatically increases tumor cell sensitivity to selective HSP90 inhibitors [175]. Similarly, knockdown of HSF1 increases tumor cell susceptibility to HSP90 inhibitor treatment [176]. Of interest, in addition to its master regulatory role in orchestrating the heat shock response, HSF1 also drives specific transcriptional programs that maintain the malignant phenotype [177, 178]. Hence, it has become evident that concomitant suppression of the heat shock response represents a potential method for optimizing the full therapeutic potential of HSP90 inhibitors [179].
In an earlier study, Zaarur and colleagues screened a chemical library, in part through evaluation of inducible HSP70 expression, and identified a group of structurally similar benzylisoquinoline alkaloid compounds that could inhibit the stress response and consequently sensitize tumor cells to the cytotoxic effects both HSP90 and proteasome inhibitors [180]. Using a similar experimental approach, we recently uncovered a clinically feasible strategy to overcome this limitation. By performing an immunoassay screen of over 300 late-stage or approved drugs, a number of relevant compounds were identified that effectively blocked HSP70 up-regulation in response to ganetespib treatment [181]. This proof-of-concept study found that targeted inhibitors of the PI3K/mTOR signaling axis could attenuate the HSF1-driven cellular heat shock response at both the genomic and proteomic levels and, importantly, this finding was validated by the capacity of selective mTOR or dual PI3K/mTOR agents to potentiate the antitumor efficacy of ganetespib in multiple in vivo xenograft models [181]. Importantly, these observations provide a molecular framework for novel combinatorial strategies that add PI3K/mTOR inhibitors to exploratory HSP90 inhibitor-based treatment regimens. In this regard, a number of selective mTOR inhibitor drugs are already approved for a variety of solid malignancies and other investigational agents are undergoing late-stage clinical evaluation [182, 183]. The results of two ongoing human trials evaluating combinations of IPI-504 with everolimus in mutant KRAS-driven NSCLC and NVP-AUY922 with the novel PI3K inhibitor BYL719 in metastatic gastric cancer (NCT01427946 and NCT01613950, respectively) are therefore likely to be informative as to clinical applicability of this approach.
Finally, there is now unequivocal evidence to suggest that tumor-specific drug metabolism may also serve as a primary determinant of ‘resistance’ to particular chemical classes of HSP90 inhibitors. It has long been established that the efficacy of the benzoquinone ansamycin inhibitors is related to levels of NAD(P)H dehydrogenase quinone 1 (NQO1), an enzyme required to catalyze the reduction of these compounds to a more active state [184]. For the newer chemical classes of HSP90 inhibitors, however, the possible influence of metabolism underlying diminished cellular activity has thus far received less attention. Of note, two recent studies have uncovered differential sensitivities exhibited by tumor lines to the resorcinol-based inhibitors NVP-AUY922 and ganetespib [185, 186]. Demonstrated for both bladder and CRC-derived lines, cell fate following inhibitor exposure varied according to the chemical class of inhibitor used. While retaining full sensitivity to ansamycin inhibitors, a number of lines of each tumor type were found to be largely resistant to ganetespib treatment or exposure to NVP-AUY922. Both of these resorcinol-containing compounds are primarily metabolized by the UGT1A family of UDP-glucuronosyltransferase enzymes [122, 185], the primary catalysts of glucuronidation reactions in multiple human tissues [187]. Mechanistically, it was determined that rapid metabolism (via glucuronidation) of ganetespib within bladder cells expressing high basal levels of UGT1A enzyme expression was sufficient to account for the lack of HSP90 inhibitory activity [185]. Moreover, targeted knockdown of UGT1A in high-expressing bladder and colorectal lines could sensitize previously resistant cells to HSP90 blockade by ganetespib. This correlation between UGT1A expression and resorcinol-inhibitor resistance suggests that UGT1A detection in tumor biopsy specimens might ultimately allow the development of a specific biomarker with direct translational relevance for the clinical evaluation HSP90-based strategies for this chemical class of inhibitor. Equally, this consideration is likely to be important for the informed application of resorcinol HSP90 drugs for the treatment of neoplasms that arise from tissues known to express the greatest abundance and array of UGT enzymes, including the gastrointestinal (liver, stomach, small intestine, colon) and urinary (kidneys, bladder) tracts [188].
11 Conclusion
By exploiting unique characteristics of HSP90 biology and pharmacology, considerable progress has been made in the development of selective inhibitors of this chaperone for cancer therapy. Indeed, lessons gleaned from characterization of both first- and second-generation classes of targeted HSP90 compounds have now validated inhibitor-based intervention as a promising therapeutic strategy for a wide variety of human malignancies. The clinical experience has identified potential frameworks for realizing the translational potential of this group of anticancer agents, particularly as chemotherapeutic or molecularly targeted agent sensitizers with substantial capacity to promote superior antitumor efficacy, overcome resistance mechanisms, and reduce treatment-related toxicities. Small molecule modulators of HSP90 thus stand at a critical stage of development and, while a number of challenges remain to be overcome, this collection of antineoplastic agents appears poised to achieve their full therapeutic promise in the application of novel cancer management strategies.
Abbreviations
- 5-FU:
-
5-fluorouracil
- 17-AAG:
-
17-allylamino-17-demethoxygeldanamycin
- 17-DMAG:
-
17-dimethylaminoethylamino-17-demethoxygeldanamycin
- ALK:
-
Anaplastic lymphoma kinase
- AR:
-
Androgen receptor
- BCR-ABL:
-
Breakpoint cluster region- Abelson murine leukemia viral oncogene homolog
- BRAF:
-
v-Raf murine sarcoma viral oncogene homolog B
- CRC:
-
Colorectal cancer
- EGFR:
-
Epidermal growth factor receptor
- EML4:
-
Echinoderm microtubule-associated protein-like 4
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular signal-regulated kinase
- FLT3:
-
Fms-related tyrosine kinase 3
- GIST:
-
Gastrointestinal stromal tumor
- HER2:
-
Human epidermal growth factor receptor 2
- HIF-1α:
-
Hypoxia-inducible factor 1 alpha
- HSF1:
-
Heat shock factor-1
- HSP70:
-
Heat shock protein 70
- HSP90:
-
Heat shock protein 90
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- JAK/STAT:
-
Janus kinase/signal transducer and activator of transcription
- KRAS:
-
Kirsten rat sarcoma viral oncogene homolog
- LV:
-
Leucovorin
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
Mitogen/extracellular signal-regulated kinase
- mTOR:
-
Mammalian target of rapamycin
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PI3K/AKT:
-
Phosphoinositide 3-kinase/AKT8 virus oncogene cellular homolog
- PFS:
-
Progression free survival
- PR:
-
Progesterone receptor
- PSA:
-
Prostate-specific antigen
- TKI:
-
Tyrosine kinase inhibitor
- TNBC:
-
Triple-negative breast cancer
- UGT:
-
UDP-glucuronosyltransferase
References
Taipale M, Jarosz DF, Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11:515–528
Morimoto RI (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 12:3788–3796
Barrott JJ, Haystead TA (2013) Hsp90, an unlikely ally in the war on cancer. FEBS J 280:1381–1396
Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM (1994) Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A 91:8324–8328
Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5:761–772
Trepel J, Mollapour M, Giaccone G, Neckers L (2010) Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10:537–549
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18:64–76
Neckers L, Trepel JB (2014) Stressing the development of small molecules targeting HSP90. Clin Cancer Res 20:275–277
Jhaveri K, Modi S (2012) HSP90 inhibitors for cancer therapy and overcoming drug resistance. Adv Pharmacol 65:471–517
Workman P, Burrows F, Neckers L, Rosen N (2007) Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci 1113:202–216
Xu W, Neckers L (2007) Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res 13:1625–1629
Chiosis G, Neckers L (2006) Tumor selectivity of Hsp90 inhibitors: the explanation remains elusive. ACS Chem Biol 1:279–284
Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ (2003) A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425:407–410
Mollapour M, Bourboulia D, Beebe K, Woodford MR, Polier S, Hoang A, Chelluri R, Li Y, Guo A, Lee MJ, Fotooh-Abadi E, Khan S, Prince T, Miyajima N, Yoshida S, Tsutsumi S, Xu W, Panaretou B, Stetler-Stevenson WG, Bratslavsky G, Trepel JB, Prodromou C, Neckers L (2014) Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol Cell 53:317–329
Banerji U, Judson I, Workman P (2003) The clinical applications of heat shock protein inhibitors in cancer – present and future. Curr Cancer Drug Targets 3:385–390
Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee WC (2010) Heat shock protein 90: inhibitors in clinical trials. J Med Chem 53:3–17
Taldone T, Gozman A, Maharaj R, Chiosis G (2008) Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr Opin Pharmacol 8:370–374
Rajan A, Kelly RJ, Trepel JB, Kim YS, Alarcon SV, Kummar S, Gutierrez M, Crandon S, Zein WM, Jain L, Mannargudi B, Figg WD, Houk BE, Shnaidman M, Brega N, Giaccone G (2011) A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin Cancer Res 17:6831–6839
Reddy N, Voorhees PM, Houk BE, Brega N, Hinson JM Jr, Jillela A (2013) Phase I trial of the HSP90 inhibitor PF-04929113 (SNX5422) in adult patients with recurrent, refractory hematologic malignancies. Clin Lymphoma Myeloma Leuk 13:385–391
Goldman JW, Raju RN, Gordon GA, El-Hariry I, Teofilivici F, Vukovic VM, Bradley R, Karol MD, Chen Y, Guo W, Inoue T, Rosen LS (2013) A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer 13:152
Sessa C, Shapiro GI, Bhalla KN, Britten C, Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA, Akimov M, Quadt C, Fernandez-Ibarra C, Lu H, Bailey S, Chica S, Banerji U (2013) First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res 19:3671–3680
Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, Sandler A, Belani CP, Webb T, Harper H, Huberman M, Ramalingam S, Wong KK, Teofilovici F, Guo W, Shapiro GI (2013) A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res 19:3068–3077
Garcia-Carbonero R, Carnero A, Paz-Ares L (2013) Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol 14:e358–e369
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63:11–30
Grossi F, Kubota K, Cappuzzo F, de Marinis F, Gridelli C, Aita M, Douillard JY (2010) Future scenarios for the treatment of advanced non-small cell lung cancer: focus on taxane-containing regimens. Oncologist 15:1102–1112
Ramalingam S, Belani C (2008) Systemic chemotherapy for advanced non-small cell lung cancer: recent advances and future directions. Oncologist 13(Suppl 1):5–13
Stella GM, Luisetti M, Inghilleri S, Cemmi F, Scabini R, Zorzetto M, Pozzi E (2012) Targeting EGFR in non-small-cell lung cancer: lessons, experiences, strategies. Respir Med 106:173–183
Herbst RS, Lynch TJ, Sandler AB (2009) Beyond doublet chemotherapy for advanced non-small-cell lung cancer: combination of targeted agents with first-line chemotherapy. Clin Lung Cancer 10:20–27
Ramalingam SS, Owonikoko TK, Khuri FR (2011) Lung cancer: new biological insights and recent therapeutic advances. CA Cancer J Clin 61:91–112
Gaughan EM, Costa DB (2011) Genotype-driven therapies for non-small cell lung cancer: focus on EGFR, KRAS and ALK gene abnormalities. Ther Adv Med Oncol 3:113–125
Chen Z, Sasaki T, Tan X, Carretero J, Shimamura T, Li D, Xu C, Wang Y, Adelmant GO, Capelletti M, Lee HJ, Rodig SJ, Borgman C, Park SI, Kim HR, Padera R, Marto JA, Gray NS, Kung AL, Shapiro GI, Janne PA, Wong KK (2010) Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res 70:9827–9836
da Rocha Dias S, Friedlos F, Light Y, Springer C, Workman P, Marais R (2005) Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res 65:10686–10691
Normant E, Paez G, West KA, Lim AR, Slocum KL, Tunkey C, McDougall J, Wylie AA, Robison K, Caliri K, Palombella VJ, Fritz CC (2011) The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 30:2581–2586
Schulte TW, Blagosklonny MV, Ingui C, Neckers L (1995) Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem 270:24585–24588
Shimamura T, Lowell AM, Engelman JA, Shapiro GI (2005) Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res 65:6401–6408
Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L (2001) Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem 276:3702–3708
Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Hamada T, Haruta H, Watanabe H, Kurashina K, Hatanaka H, Ueno T, Takada S, Yamashita Y, Sugiyama Y, Ishikawa Y, Mano H (2008) Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res 68:4971–4976
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448:561–566
Camidge DR, Doebele RC (2012) Treating ALK-positive lung cancer-early successes and future challenges. Nat Rev Clin Oncol 9:268–277
Sasaki T, Janne PA (2011) New strategies for treatment of ALK-rearranged non-small cell lung cancers. Clin Cancer Res 17:7213–7218
Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou SH, Butaney M, Salgia R, Maki RG, Varella-Garcia M, Doebele RC, Bang YJ, Kulig K, Selaru P, Tang Y, Wilner KD, Kwak EL, Clark JW, Iafrate AJ, Camidge DR (2011) Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 12:1004–1012
Sequist LV, Gettinger S, Senzer NN, Martins RG, Janne PA, Lilenbaum R, Gray JE, Iafrate AJ, Katayama R, Hafeez N, Sweeney J, Walker JR, Fritz C, Ross RW, Grayzel D, Engelman JA, Borger DR, Paez G, Natale R (2010) Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol 28:4953–4960
Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, Jiang Q, Xue L, Lovly CM, Jimenez JP, Shaw AT, Doebele RC, He S, Bates RC, Camidge DR, Morris SW, El-Hariry I, Proia DA (2013) Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov 3:430–443
Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT (2011) Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A 108:7535–7540
Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 18:1472–1482
Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, Moran L, Mohemmad QK, Anjum R, Wang Y, Narasimhan NI, Dalgarno D, Shakespeare WC, Miret JJ, Clackson T, Rivera VM (2011) Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des 78:999–1005
Ardini E, Galvani A (2012) ALK inhibitors, a pharmaceutical perspective. Front Oncol 2:17
Lovly CM, Pao W (2012) Escaping ALK inhibition: mechanisms of and strategies to overcome resistance. Sci Transl Med 4:120ps122
Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, Capelletti M, Shimamura T, Ercan D, Stumpfova M, Xiao Y, Weremowicz S, Butaney M, Heon S, Wilner K, Christensen JG, Eck MJ, Wong KK, Lindeman N, Gray NS, Rodig SJ, Janne PA (2011) A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 71:6051–6060
Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, Kaneda H, Takezawa K, Kuwata K, Yamaguchi H, Hatashita E, Nishio K, Nakagawa K (2012) Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res 18:6219–6226
Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, Nakamura T, Matsumoto K, Soda M, Mano H, Uenaka T, Yano S (2012) Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res 18:3592–3602
Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang R, Mark EJ, Batten JM, Chen H, Wilner KD, Kwak EL, Clark JW, Carbone DP, Ji H, Engelman JA, Mino-Kenudson M, Pao W, Iafrate AJ (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 30:863–870
Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, Gu TL, Mack JS, Silver MR, Zhou X, Haack H (2012) Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res 18:4449–4457
Stumpfova M, Janne PA (2012) Zeroing in on ROS1 rearrangements in non-small cell lung cancer. Clin Cancer Res 18:4222–4224
Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, Lim Choi Y, Satoh Y, Okumura S, Nakagawa K, Mano H, Ishikawa Y (2012) RET, ROS1 and ALK fusions in lung cancer. Nat Med 18:378–381
Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, Enari M, Schetter AJ, Okayama H, Haugen A, Skaug V, Chiku S, Yamanaka I, Arai Y, Watanabe S, Sekine I, Ogawa S, Harris CC, Tsuda H, Yoshida T, Yokota J, Shibata T (2012) KIF5B-RET fusions in lung adenocarcinoma. Nat Med 18:375–377
Gazdar AF (2009) Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 28(Suppl 1):S24–S31
Scagliotti GV, Selvaggi G, Novello S, Hirsch FR (2004) The biology of epidermal growth factor receptor in lung cancer. Clin Cancer Res 10:4227s–4232s
Ono M, Kuwano M (2006) Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin Cancer Res 12:7242–7251
Reungwetwattana T, Dy GK (2013) Targeted therapies in development for non-small cell lung cancer. J Carcinog 12:22
Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352:786–792
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2:e73
Carrera S, Buque A, Azkona E, Aresti U, Calvo B, Sancho A, Arruti M, Nuno M, Rubio I, de Lobera AR, Lopez C, Vivanco GL (2013) Epidermal growth factor receptor tyrosine-kinase inhibitor treatment resistance in non-small cell lung cancer: biological basis and therapeutic strategies. Clin Transl Oncol 16:339–350
Shimamura T, Li D, Ji H, Haringsma HJ, Liniker E, Borgman CL, Lowell AM, Minami Y, McNamara K, Perera SA, Zaghlul S, Thomas RK, Greulich H, Kobayashi S, Chirieac LR, Padera RF, Kubo S, Takahashi M, Tenen DG, Meyerson M, Wong KK, Shapiro GI (2008) Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res 68:5827–5838
Bao R, Lai CJ, Wang DG, Qu H, Yin L, Zifcak B, Tao X, Wang J, Atoyan R, Samson M, Forrester J, Xu GX, DellaRocca S, Borek M, Zhai HX, Cai X, Qian C (2009) Targeting heat shock protein 90 with CUDC-305 overcomes erlotinib resistance in non-small cell lung cancer. Mol Cancer Ther 8:3296–3306
Garon EB, Finn RS, Hamidi H, Dering J, Pitts S, Kamranpour N, Desai AJ, Hosmer W, Ide S, Avsar E, Jensen MR, Quadt C, Liu M, Dubinett SM, Slamon DJ (2013) The HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growth. Mol Cancer Ther 12:890–900
Graham B, Curry J, Smyth T, Fazal L, Feltell R, Harada I, Coyle J, Williams B, Reule M, Angove H, Cross DM, Lyons J, Wallis NG, Thompson NT (2012) The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci 103:522–527
Shimamura T, Perera SA, Foley KP, Sang J, Rodig SJ, Inoue T, Chen L, Li D, Carretero J, Li YC, Sinha P, Carey CD, Borgman CL, Jimenez JP, Meyerson M, Ying W, Barsoum J, Wong KK, Shapiro GI (2012) Ganetespib (STA-9090), a nongeldanamycin HSP90 inhibitor, has potent antitumor activity in in vitro and in vivo models of non-small cell lung cancer. Clin Cancer Res 18:4973–4985
Ueno T, Tsukuda K, Toyooka S, Ando M, Takaoka M, Soh J, Asano H, Maki Y, Muraoka T, Tanaka N, Shien K, Furukawa M, Yamatsuji T, Kiura K, Naomoto Y, Miyoshi S (2012) Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer 76:26–31
Ying W, Du Z, Sun L, Foley KP, Proia DA, Blackman RK, Zhou D, Inoue T, Tatsuta N, Sang J, Ye S, Acquaviva J, Ogawa LS, Wada Y, Barsoum J, Koya K (2012) Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol Cancer Ther 11:475–484
Ahsan A, Ramanand SG, Whitehead C, Hiniker SM, Rehemtulla A, Pratt WB, Jolly S, Gouveia C, Truong K, Van Waes C, Ray D, Lawrence TS, Nyati MK (2012) Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia 14:670–677
Smith DL, Acquaviva J, Sequeira M, Jimenez J, Zhang C, Sang J, Bates RC, Proia DA (2014) The HSP90 inhibitor ganetespib potentiates the antitumor activity of EGFR tyrosine kinase inhibition in mutant and wild-type non-small cell lung cancer. Target Oncol. doi: 10.1007/s11523-014-0329-6
Rice JW, Veal JM, Barabasz A, Foley B, Fadden P, Scott A, Huang K, Steed P, Hall S (2009) Targeting of multiple signaling pathways by the Hsp90 inhibitor SNX-2112 in EGFR resistance models as a single agent or in combination with erlotinib. Oncol Res 18:229–242
Ono N, Yamazaki T, Tsukaguchi T, Fujii T, Sakata K, Suda A, Tsukuda T, Mio T, Ishii N, Kondoh O, Aoki Y (2013) Enhanced antitumor activity of erlotinib in combination with the Hsp90 inhibitor CH5164840 against non-small-cell lung cancer. Cancer Sci 104:1346–1352
Johnson ML, Hart EM, Rademaker A et al (2013) A phase II study of HSP90 inhibitor AUY922 and erlotinib (E) for patients (pts) with EGFR-mutant lung cancer and acquired resistance (AR) to EGFR tyrosine kinase inhibitors (EGFR TKIs). J Clin Oncol 31 (suppl; abstr 8036)
Dienstmann R, Martinez P, Felip E (2011) Personalizing therapy with targeted agents in non-small cell lung cancer. Oncotarget 2:165–177
Solit DB, Chiosis G (2008) Development and application of Hsp90 inhibitors. Drug Discov Today 13:38–43
Munster PN, Basso A, Solit D, Norton L, Rosen N (2001) Modulation of Hsp90 function by ansamycins sensitizes breast cancer cells to chemotherapy-induced apoptosis in an RB- and schedule-dependent manner. See: E. A. Sausville, Combining cytotoxics and 17-allylamino, 17-demethoxygeldanamycin: sequence and tumor biology matters, Clin. Cancer Res., 7: 2155–2158, 2001. Clin Cancer Res 7:2228–2236
Nguyen DM, Lorang D, Chen GA, Stewart JH, Tabibi E, Schrump DS (2001) Enhancement of paclitaxel-mediated cytotoxicity in lung cancer cells by 17-allylamino geldanamycin: in vitro and in vivo analysis. Ann Thorac Surg 72:371–378, discussion 378–379
Sain N, Krishnan B, Ormerod MG, De Rienzo A, Liu WM, Kaye SB, Workman P, Jackman AL (2006) Potentiation of paclitaxel activity by the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma cell lines with high levels of activated AKT. Mol Cancer Ther 5:1197–1208
Sawai A, Chandarlapaty S, Greulich H, Gonen M, Ye Q, Arteaga CL, Sellers W, Rosen N, Solit DB (2008) Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res 68:589–596
Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N (2003) Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res 63:2139–2144
Proia DA, Sang J, He S, Smith DL, Sequeira M, Zhang C, Liu Y, Ye S, Zhou D, Blackman RK, Foley KP, Koya K, Wada Y (2012) Synergistic activity of the Hsp90 inhibitor ganetespib with taxanes in non-small cell lung cancer models. Invest New Drugs 30:2201–2209
Proia DA, Zhang C, Sequeira M, Jimenez JP, He S, Spector N, Shapiro GI, Tolaney S, Nagai M, Acquaviva J, Smith DL, Sang J, Bates RC, El-Hariry I (2014) Preclinical activity profile and therapeutic efficacy of the HSP90 inhibitor ganetespib in triple-negative breast cancer. Clin Cancer Res 20:413–424
Zhou D, Liu Y, Ye J, Ying W, Ogawa LS, Inoue T, Tatsuta N, Wada Y, Koya K, Huang Q, Bates RC, Sonderfan AJ (2013) A rat retinal damage model predicts for potential clinical visual disturbances induced by Hsp90 inhibitors. Toxicol Appl Pharmacol 273:401–409
Ramalingham SS, Goss GD, Andric ZG et al (2013) A randomized study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel versus docetaxel alone for second-line therapy of lung adenocarcinoma (GALAXY-1). J Clin Oncol 31 (suppl; abstr CRA8007)
Koga F, Kihara K, Neckers L (2009) Inhibition of cancer invasion and metastasis by targeting the molecular chaperone heat-shock protein 90. Anticancer Res 29:797–807
Tsutsumi S, Neckers L (2007) Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci 98:1536–1539
Xiang L, Gilkes DM, Chaturvedi P, Luo W, Hu H, Takano N, Liang H, Semenza GL (2014) Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J Mol Med (Berl) 92:151–164
Tran B, Bedard PL (2011) Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res 13:221
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D (2000) Molecular portraits of human breast tumours. Nature 406:747–752
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 98:10869–10874
Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL, Botstein D (2003) Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 100:8418–8423
Brouckaert O, Wildiers H, Floris G, Neven P (2012) Update on triple-negative breast cancer: prognosis and management strategies. Int J Womens Health 4:511–520
Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N (2009) Triple-negative breast cancer–current status and future directions. Ann Oncol 20:1913–1927
Basso AD, Solit DB, Munster PN, Rosen N (2002) Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene 21:1159–1166
Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A, Guo Y, Wu N, de Stanchina E, White J, Gross SS, Ma Y, Varticovski L, Melnick A, Chiosis G (2009) Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A 106:8368–8373
Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, Fadden P, Partdrige J, Hall S, Steed P, Norton L, Rosen N, Solit DB (2008) SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin Cancer Res 14:240–248
Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, Quadt C, Buckler A, Cozens R, Drysdale MJ, Garcia-Echeverria C, Chene P (2008) NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res 10:R33
Mehta PP, Whalen P, Baxi SM, Kung PP, Yamazaki S, Yin MJ (2011) Effective targeting of triple-negative breast cancer cells by PF-4942847, a novel oral inhibitor of Hsp 90. Clin Cancer Res 17:5432–5442
Scaltriti M, Serra V, Normant E, Guzman M, Rodriguez O, Lim AR, Slocum KL, West KA, Rodriguez V, Prudkin L, Jimenez J, Aura C, Baselga J (2011) Antitumor activity of the Hsp90 inhibitor IPI-504 in HER2-positive trastuzumab-resistant breast cancer. Mol Cancer Ther 10:817–824
Wong C, Chen S (2009) Heat shock protein 90 inhibitors: new mode of therapy to overcome endocrine resistance. Cancer Res 69:8670–8677
De Mattos-Arruda L, Cortes J (2012) Advances in first-line treatment for patients with HER-2+ metastatic breast cancer. Oncologist 17:631–644
Garrett JT, Arteaga CL (2011) Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications. Cancer Biol Ther 11:793–800
Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, Ellis C, Casey M, Vukelja S, Bischoff J, Baselga J, O’Shaughnessy J (2010) Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol 28:1124–1130
Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, D’Andrea G, Dickler M, Moynahan ME, Sugarman S, Ma W, Patil S, Norton L, Hannah AL, Hudis C (2011) HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res 17:5132–5139
Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos MA, Psaltopoulou T (2013) Hsp90 inhibitors in breast cancer: a systematic review. Breast 22:569–578
Friedland JC, Smith DL, Sang J, Acquaviva J, He S, Zhang C, Proia DA (2014) Targeted inhibition of Hsp90 by ganetespib is effective across a broad spectrum of breast cancer subtypes. Invest New Drugs 32:14–24
Pratt WB, Galigniana MD, Morishima Y, Murphy PJ (2004) Role of molecular chaperones in steroid receptor action. Essays Biochem 40:41–58
Rao RD, Cobleigh MA (2012) Adjuvant endocrine therapy for breast cancer. Oncology 26:541–547, 550, 552 passim
Osborne CK, Shou J, Massarweh S, Schiff R (2005) Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 11:865s–870s
Bachleitner-Hofmann T, Pichler-Gebhard B, Rudas M, Gnant M, Taucher S, Kandioler D, Janschek E, Dubsky P, Roka S, Sporn E, Jakesz R (2002) Pattern of hormone receptor status of secondary contralateral breast cancers in patients receiving adjuvant tamoxifen. Clin Cancer Res 8:3427–3432
Beliakoff J, Bagatell R, Paine-Murrieta G, Taylor CW, Lykkesfeldt AE, Whitesell L (2003) Hormone-refractory breast cancer remains sensitive to the antitumor activity of heat shock protein 90 inhibitors. Clin Cancer Res 9:4961–4971
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490:61–70
Liu H, Xiao F, Serebriiskii IG, O’Brien SW, Maglaty MA, Astsaturov I, Litwin S, Martin LP, Proia DA, Golemis EA, Connolly DC (2013) Network analysis identifies an HSP90-central hub susceptible in ovarian cancer. Clin Cancer Res 19:5053–5067
MacKie RM, Hauschild A, Eggermont AM (2009) Epidemiology of invasive cutaneous melanoma. Ann Oncol 20(Suppl 6):vi1–vi7
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF (2011) Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 29:1239–1246
Tsao H, Chin L, Garraway LA, Fisher DE (2012) Melanoma: from mutations to medicine. Genes Dev 26:1131–1155
Finn L, Markovic SN, Joseph RW (2012) Therapy for metastatic melanoma: the past, present, and future. BMC Med 10:23
Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, Rosen N (2006) V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci U S A 103:57–62
Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P (2008) NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res 68:2850–2860
Lemech C, Infante J, Arkenau HT (2012) The potential for BRAF V600 inhibitors in advanced cutaneous melanoma: rationale and latest evidence. Ther Adv Med Oncol 4:61–73
Massey AJ, Schoepfer J, Brough PA, Brueggen J, Chene P, Drysdale MJ, Pfaar U, Radimerski T, Ruetz S, Schweitzer A, Wood M, Garcia-Echeverria C, Jensen MR (2010) Preclinical antitumor activity of the orally available heat shock protein 90 inhibitor NVP-BEP800. Mol Cancer Ther 9:906–919
Acquaviva J, Smith DL, Jimenez JP, Zhang C, Sequeira M, He S, Sang J, Bates RC, Proia DA (2014) Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther 13:353–363
Wu X, Marmarelis ME, Hodi FS (2013) Activity of the heat shock protein 90 inhibitor ganetespib in melanoma. PLoS One 8:e56134
Pacey S, Gore M, Chao D, Banerji U, Larkin J, Sarker S, Owen K, Asad Y, Raynaud F, Walton M, Judson I, Workman P, Eisen T (2012) A phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs 30:341–349
Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N, Chapman PB (2008) Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res 14:8302–8307
Catalanotti F, Solit DB (2012) Will Hsp90 inhibitors prove effective in BRAF-mutant melanomas? Clin Cancer Res 18:2420–2422
Sullivan RJ, Flaherty KT (2013) Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer 49:1297–1304
Liu KS, Liu H, Qi JH, Liu QY, Liu Z, Xia M, Xing GW, Wang SX, Wang YF (2012) SNX-2112, an Hsp90 inhibitor, induces apoptosis and autophagy via degradation of Hsp90 client proteins in human melanoma A-375 cells. Cancer Lett 318:180–188
Corcoran RB, Settleman J, Engelman JA (2011) Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget 2:336–346
Williams R (2013) Discontinued drugs in 2011: oncology drugs. Expert Opin Investig Drugs 22:9–34
Paraiso KH, Haarberg HE, Wood E, Rebecca VW, Chen YA, Xiang Y, Ribas A, Lo RS, Weber JS, Sondak VK, John JK, Sarnaik AA, Koomen JM, Smalley KS (2012) The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res 18:2502–2514
Fogliatto G, Gianellini L, Brasca MG, Casale E, Ballinari D, Ciomei M, Degrassi A, De Ponti A, Germani M, Guanci M, Paolucci M, Polucci P, Russo M, Sola F, Valsasina B, Visco C, Zuccotto F, Donati D, Felder E, Pesenti E, Galvani A, Mantegani S, Isacchi A (2013) NMS-E973, a novel synthetic inhibitor of Hsp90 with activity against multiple models of drug resistance to targeted agents, including intracranial metastases. Clin Cancer Res 19:3520–3532
Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, Rosen N (2006) BRAF mutation predicts sensitivity to MEK inhibition. Nature 439:358–362
Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA (2010) BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal 3:ra84
Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, Messina JL, Flaherty KT, Smalley KS (2010) Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 102:1724–1730
Antonescu CR (2011) The GIST paradigm: lessons for other kinase-driven cancers. J Pathol 223:251–261
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279:577–580
Wagner AJ, Chugh R, Rosen LS, Morgan JA, George S, Gordon M, Dunbar J, Normant E, Grayzel D, Demetri GD (2013) A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin Cancer Res 19:6020–6029
Riedel RF (2011) Targeted agents for sarcoma: is individualized therapy possible in such a diverse tumor type? Semin Oncol 38(Suppl 3):S30–S42
Dickson MA, Okuno SH, Keohan ML, Maki RG, D’Adamo DR, Akhurst TJ, Antonescu CR, Schwartz GK (2013) Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann Oncol 24:252–257
Demetri GD, Heinrich MC, Chmielowski B et al (2011) An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) in patients (pts) with metastatic and/or unresectable GIST. J Clin Oncol 29 (suppl; abstr 10011)
Chen Y, Sawyers CL, Scher HI (2008) Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 8:440–448
Lonergan PE, Tindall DJ (2011) Androgen receptor signaling in prostate cancer development and progression. J Carcinog 10:20
Smith DF, Toft DO (2008) Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol 22:2229–2240
Eskew JD, Sadikot T, Morales P, Duren A, Dunwiddie I, Swink M, Zhang X, Hembruff S, Donnelly A, Rajewski RA, Blagg BS, Manjarrez JR, Matts RL, Holzbeierlein JM, Vielhauer GA (2011) Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 11:468
He S, Zhang C, Shafi AA, Sequeira M, Acquaviva J, Friedland JC, Sang J, Smith DL, Weigel NL, Wada Y, Proia DA (2013) Potent activity of the Hsp90 inhibitor ganetespib in prostate cancer cells irrespective of androgen receptor status or variant receptor expression. Int J Oncol 42:35–43
Lamoureux F, Thomas C, Yin MJ, Kuruma H, Fazli L, Gleave ME, Zoubeidi A (2011) A novel HSP90 inhibitor delays castrate-resistant prostate cancer without altering serum PSA levels and inhibits osteoclastogenesis. Clin Cancer Res 17:2301–2313
O’Malley KJ, Langmann G, Ai J, Ramos-Garcia R, Vessella RL, Wang Z (2011) Hsp90 inhibitor 17-AAG inhibits progression of LuCaP35 xenograft prostate tumors to castration resistance. Prostate 72:1117–1123
Solit DB, Zheng FF, Drobnjak M, Munster PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB, Scher HI, Rosen N (2002) 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res 8:986–993
Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, Harper F, Gaskins M, Pitot HC, Tan W, Ivy SP, Pili R, Carducci MA, Erlichman C, Liu G (2008) A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Cancer Res 14:7940–7946
Oh WK, Galsky MD, Stadler WM, Srinivas S, Chu F, Bubley G, Goddard J, Dunbar J, Ross RW (2011) Multicenter phase II trial of the heat shock protein 90 inhibitor, retaspimycin hydrochloride (IPI-504), in patients with castration-resistant prostate cancer. Urology 78:626–630
Sandler HM, Mirhadi AJ (2009) Radical radiotherapy for prostate cancer is the ‘only way to go’. Oncology (Williston Park) 23:840–843
Gandhi N, Wild AT, Chettiar ST, Aziz K, Kato Y, Gajula RP, Williams RD, Cades JA, Annadanam A, Song D, Zhang Y, Hales RK, Herman JM, Armour E, DeWeese TL, Schaeffer EM, Tran PT (2013) Novel Hsp90 inhibitor NVP-AUY922 radiosensitizes prostate cancer cells. Cancer Biol Ther 14:347–356
Kline CL, El-Deiry WS (2013) Personalizing colon cancer therapeutics: targeting old and new mechanisms of action. Pharmaceuticals (Basel) 6:988–1038
Aparo S, Goel S (2012) Evolvement of the treatment paradigm for metastatic colon cancer. From chemotherapy to targeted therapy. Crit Rev Oncol Hematol 83:47–58
El Zouhairi M, Charabaty A, Pishvaian MJ (2011) Molecularly targeted therapy for metastatic colon cancer: proven treatments and promising new agents. Gastrointest Cancer Res 4:15–21
Azoitei N, Hoffmann CM, Ellegast JM, Ball CR, Obermayer K, Gossele U, Koch B, Faber K, Genze F, Schrader M, Kestler HA, Dohner H, Chiosis G, Glimm H, Frohling S, Scholl C (2012) Targeting of KRAS mutant tumors by HSP90 inhibitors involves degradation of STK33. J Exp Med 209:697–711
Bao R, Lai CJ, Qu H, Wang D, Yin L, Zifcak B, Atoyan R, Wang J, Samson M, Forrester J, DellaRocca S, Xu GX, Tao X, Zhai HX, Cai X, Qian C (2009) CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy. Clin Cancer Res 15:4046–4057
Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA (2001) Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res 61:4003–4009
Moser C, Lang SA, Kainz S, Gaumann A, Fichtner-Feigl S, Koehl GE, Schlitt HJ, Geissler EK, Stoeltzing O (2007) Blocking heat shock protein-90 inhibits the invasive properties and hepatic growth of human colon cancer cells and improves the efficacy of oxaliplatin in p53-deficient colon cancer tumors in vivo. Mol Cancer Ther 6:2868–2878
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D (2011) RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11:761–774
Pakneshan S, Salajegheh A, Smith RA, Lam AK (2013) Clinicopathological relevance of BRAF mutations in human cancer. Pathology 45:346–356
Hirsch BR, Zafar SY (2011) Capecitabine in the management of colorectal cancer. Cancer Manag Res 3:79–89
Lee KH, Lee JH, Han SW, Im SA, Kim TY, Oh DY, Bang YJ (2011) Antitumor activity of NVP-AUY922, a novel heat shock protein 90 inhibitor, in human gastric cancer cells is mediated through proteasomal degradation of client proteins. Cancer Sci 102:1388–1395
He S, Smith DL, Sequeira M, Sang J, Bates RC, Proia DA (2014) The HSP90 inhibitor ganetespib has chemosensitizer and radiosensitizer activity in colorectal cancer. Invest New Drugs 32:577–586
Nagaraju GP, Park W, Wen J, Mahaseth H, Landry J, Farris AB, Willingham F, Sullivan PS, Proia DA, El-Hariry I, Taliaferro-Smith L, Diaz R, El-Rayes BF (2013) Antiangiogenic effects of ganetespib in colorectal cancer mediated through inhibition of HIF-1alpha and STAT-3. Angiogenesis 16:903–917
Richards MW, Law EW, Rennalls LP, Busacca S, O‘Regan L, Fry AM, Fennell DA, Bayliss R (2014) Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical β-propeller domain. Proc Natl Acad Sci U S A 111:5195–5200
Jhaveri K, Taldone T, Modi S, Chiosis G (2012) Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta 1823:742–755
Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ, Whitesell L (2000) Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res 6:3312–3318
Dakappagari N, Neely L, Tangri S, Lundgren K, Hipolito L, Estrellado A, Burrows F, Zhang H (2010) An investigation into the potential use of serum Hsp70 as a novel tumour biomarker for Hsp90 inhibitors. Biomarkers 15:31–38
Whitesell L, Bagatell R, Falsey R (2003) The stress response: implications for the clinical development of hsp90 inhibitors. Curr Cancer Drug Targets 3:349–358
Powers MV, Clarke PA, Workman P (2008) Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14:250–262
Chen Y, Chen J, Loo A, Jaeger S, Bagdasarian L, Yu J, Chung F, Korn J, Ruddy D, Guo R, McLaughlin ME, Feng F, Zhu P, Stegmeier F, Pagliarini R, Porter D, Zhou W (2013) Targeting HSF1 sensitizes cancer cells to HSP90 inhibition. Oncotarget 4:816–829
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1018
Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–562
Piper PW, Millson SH (2011) Mechanisms of resistance to Hsp90 inhibitor drugs: a complex mosaic emerges. Pharmaceuticals 4:1400–1422
Zaarur N, Gabai VL, Porco JA Jr, Calderwood S, Sherman MY (2006) Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res 66:1783–1791
Acquaviva J, He S, Sang J, Smith DL, Sequeira M, Zhang C, Bates RC, Proia DA (2014) mTOR inhibition potentiates HSP90 inhibitor activity via cessation of HSP synthesis. Mol Cancer Res 12:703–713
Martini M, Ciraolo E, Gulluni F, Hirsch E (2013) Targeting PI3K in cancer: any good news? Front Oncol 3:108
Owonikoko TK, Khuri FR (2013) Targeting the PI3K/AKT/mTOR Pathway. Am Soc Clin Oncol Educ Book:395–401
Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, Ross D (2006) The bioreduction of a series of benzoquinone ansamycins by NAD(P)H:quinone oxidoreductase 1 to more potent heat shock protein 90 inhibitors, the hydroquinone ansamycins. Mol Pharmacol 70:1194–1203
Acquaviva J, He S, Zhang C, Jimenez JP, Nagai M, Sang J, Sequeira M, Smith DL, Shin Ogawa L, Inoue T, Tatsuta N, Knowles MA, Bates RC, Proia DA (2014) FGFR3 translocations in bladder cancer: differential sensitivity to HSP90 inhibition based on drug metabolism. Mol Cancer Res 12:1042–1054
Landmann H, Proia DA, He S, Ogawa LS, Kramer F, Breissbarth T, Grade M, Gaedcke J, Ghadami MB, Moll UM, Dobblestein M (2014) UDP glucuronosyltransferase 1A expression levels determine the response of colorectal cancer cells to the heat shock protein 90 inhibitor ganetespib. Cell Death Dis 5:e1411
Strassburg CP, Kalthoff S, Ehmer U (2008) Variability and function of family 1 uridine-5′-diphosphate glucuronosyltransferases (UGT1A). Crit Rev Clin Lab Sci 45:485–530
Rowland A, Miners JO, Mackenzie PI (2013) The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 45:1121–1132
Acknowledgements
The authors wish to thank members of Synta Pharmaceuticals, past and present, who contributed to the discovery of ganetespib and to the continued effort of developing the drug for patients. In addition, we would like to acknowledge our many collaborators and colleagues across academia, industry, and medical practice for their outstanding contributions to this constantly evolving field, including Cheryl London, Len Neckers, Erica Golemis, Denise Connolly, Susan Lindquist, Luke Whitesell, Mikko Taipale, Ute Moll, Suzanne Conzen, Geoffrey Shapiro, Roberto Diaz, Bassel El-Rayes, Dean Fennell, and members of their laboratories. Lastly, Dr. Proia would like to express gratitude to Theresa Proia, Trudy Morrison, Nancy Weigel, Patricia McCoon, Dennis Huszar, Ronald Blackman, Kevin Foley, Yumiko Wada, Safi Bahcall, Lan Bo Chen, Weiwen Ying, and Dinesh Chimmanamada for their support and guidance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Proia, D.A., Bates, R.C. (2015). HSP90 Inhibitor-Based Strategies for Cancer Therapy: Advancing Toward Clinical Impact. In: Asea, A., Almasoud, N., Krishnan, S., Kaur, P. (eds) Heat Shock Protein-Based Therapies. Heat Shock Proteins, vol 9. Springer, Cham. https://doi.org/10.1007/978-3-319-17211-8_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-17211-8_15
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17210-1
Online ISBN: 978-3-319-17211-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)