
Abstract
Lung cancer remains the leading cause of cancer-related death. Non-small cell lung cancer (NSCLC) represents 85 % of all lung cancer cases and it is classified into three major subtypes: adenocarcinoma, squamous cell carcinoma and large-cell carcinoma. In the past years, molecular-targeted therapies have been developed in order to improve response, survival and quality of life in patients with advanced NSCLC. Lung cancers harboring mutations in the epidermal growth factor receptor (EGFR) respond to EGFR tyrosine-kinase inhibitors (TKIs). However, virtually all patients with initial response relapse due to acquired resistance. Better understanding the biology of these tumors and mechanisms of EGFR TKIs resistance could shed some light on research of new therapeutic options in this setting. This review aims to emphasize on EGFR involved lung cancer pathway, primary and acquired mechanisms of TKIs resistance, and discuss agents currently used in clinical development in this emerging scenario.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: understanding the biological basis
EGFR family encloses a group of 4 members: ERBB1 (EGFR-HER1), ERBB2 (Neu or HER2), ERBB3 (HER3) and ERBB4 (HER4). EGFR belongs to tyrosine-kinase receptor family (RTKs); each of these proteins possesses three domains: the extracellular domain, involved in recognizing and binding the ligands; the transmembrane domain, involved in interactions between receptors; and the intracellular domain, with intrinsic tyrosine-kinase activity [1].
This receptor family recognizes different related growth factors [2], such as epidermal growth factor (EGF), transforming growth factor alpha (TGFα), heparin-binding EGF-like growth factor (HBEGF), amphiregulin, betacellulin, epiregulin and neuregulins [3]. Receptor-ligand interaction induces a conformational change which produces a receptor homo or heterodimerization, with a subsequent phosphorylation of tyrosine residues on different intracellular coupled proteins and of the receptor itself. This process leads to activation of intracellular signal transduction pathways, such as phosphatidylinositol 3-kinase PI3K/AKT/mTOR and the RAS/RAF/Mitogen-activated protein kinase (MAPK)/ERK kinase (MEK)/extracellular-signal-regulated kinase (ERK) pathways [4].
These pathways are involved in cell transformation and tumor progression:
-
(a)
PI3K is a family of proteins involved in the regulation of cell growth, metabolism, proliferation, glucose homeostasis and vesicle trafficking. A direct antagonist of PI3K is the phosphatase and tensin homologue (PTEN) which directly reverses the activity PI3K by dephosphorylating phosphatidylinositol 3,4,5-triphosphate (PIP3) into phosphatidylinositol 4,5-bisphosphate (PIP2), and therefore plays an important role as a negative controlling element of incoming signals. The loss and/or mutation of PTEN in various cancers lead to hyperactive PI3K pathway. Under phosphorylation of mediated proteins, the prosurvival AKT kinase is recruited. Likewise, AKT represents one of the main regulators of mTORc1 (mammalian target of rapamycin complex 1), a complex involved in protein translation, ribosome construction and autophagy [5].
-
(b)
The RAS/RAF/MEK/ERK pathway plays an essential role in cell proliferation, differentiation and survival. Activated RAS recruits RAF family members (BRAF). RAF stimulates MEK, a kinase that phosphorylates tyrosine/threonine residues of MAPK, allowing its activation and the modulation of different cellular process, such as growth, proliferation, differentiation, survival, motility and angiogenesis [6].
In physiological conditions, ligand binding to extracellular portion of RTKs leads to receptor dimerization and phosphorylation of tyrosine residues located at the intracytoplasmatic enzymatic domain; this activates downstream pathways involved in many functions of the cell [7]. But inappropriate mechanism of RTKs activation might lead to activation of cellular transduction, even in absence of ligand. Mutation in TK domain of this receptor generates hyperactivation of cellular downstream pathways signaling, a key role for cell proliferation and tumorigenesis [8] (Fig. 1).

EGFR pathway and TKI mechanism of action in cell with EGFR-sensitizing mutation. 1 Ligand binding to extracellular portion of RTKs leads to receptor dimerization and phosphorylation of tyrosine residues located at the intracytoplasmatic enzymatic domain. This activates downstream pathways involved in many functions of the cell. 2 EGFR mutations in ATP cleft of the tyrosine-kinase domain generate stabilization in the interaction with ATP, stimulating phosphorylation of tyrosine residues with a hyperactivation of cellular downstream pathways, even in absence of ligands. 3 ERLOTINIB and GEFITINIB inhibit the phosphorylation and tyrosine-kinase activity of the intracellular ATP-binding domain of EGFR through a competitive mechanism
EGFR and lung cancer: the paradigm of targeted therapy
Different mechanisms activate EGFR signaling in lung cancers. EGFR is overexpressed, assessed by immunohistochemistry (IHC), in more than 60 % of lung cancers [9]. In some cases, genomic analyses show the amplification of chromosomal region 7p12, where the EGFR gene is located [10]. NSCLC cells can release EGF ligands. These ligands may induce juxtacrine, autocrine, paracrine, and/or endocrine signaling [11]. Another important mechanism of RTK activity disruption is a mutation that affects the activity of tyrosine-kinase domain [12]. Thus, NSCLC cells that depend on EGFR for survival constitutively activate the receptor through a combination of genetic mutations, overexpression of EGFR and their ligands.
In virtue of the above, pharmacological selective block of EGFR has evolved as a treatment paradigm in NSCLCs patients with mutations in EGFR TK domain. Actually, gefitinib [13] and erlotinib [14] represent the two major first-generation TKIs approved for advanced NSCLC.
Mutational analysis of the entire EGFR coding sequence of gefitinib-responsive tumors was performed in 2004 [15]. The identification of specific somatic-sensitizing mutations within the tyrosine-kinase domain of EGFR and its correlation with encouraging responses in this subgroup of patients marked a turning point in lung cancer targeted therapy. These mutations are allocated near the ATP cleft of the tyrosine-kinase domain, generating stabilization in the interaction with ATP, stimulating phosphorylation of tyrosine residues and causing intracellular transduction activation in an aberrant manner [12].
Erlotinib and gefitinib inhibit the phosphorylation and tyrosine-kinase activity of the intracellular ATP-binding domain of EGFR through a competitive mechanism. Thus, the inhibition of the receptor achieves a down-regulation of its related intracellular pathways [16]. From a chemical perspective, gefitinib and erlotinib are effective in EGFR-mutant NSCLC because they are more potent inhibitors of EGFR mutants than of the wild-type (WT) EGFR kinase. In fact, inhibition of WT EGFR in the normal tissues contributes to the dose-limiting toxicity of EGFR TKIs.
In the ISEL phase III trial [17], gefitinib was compared to placebo in second-line setting, and it did not demonstrate any overall survival (OS) gain. Subgroups of never smokers and Asians achieved better median OS with gefitinib and some patients developed amazing tumor responses with gefitinib [18]. BR21 trial [19] showed an OS improvement with erlotinib vs. placebo in second- and third-line setting. Response rate (RR) was 8.9 % for erlotinib arm and <1 % for placebo arm. OS for the erlotinib group was 6.7 months compared with 4.7 months for placebo arm (HR 0.7, p < 0.001). Exploratory multivariate analyses showed that Asian origin, adenocarcinomas histology and no smoking history were significantly independent predictors of survival.
The identification of somatic mutations of EGFR has led to the development of numerous trials. IPASS (Iressa Pan-Asia Study) phase III trial [20] compared for the very first time EGFR TKIs (gefitinib) with a chemotherapy doublet (carboplatin/paclitaxel) in a first-line setting of advanced NSCLC. Superiority of gefitinib in terms of progression-free survival (PFS) was reported (HR 0.74, 95 % CI 0.65–0.85; p < 0.001). Subgroups’ analysis showed that patients with EGFR mutations had higher overall response rate (ORR) with gefitinib compared with chemotherapy (71.2 vs. 47.3 %, p = 0.0001); a significant difference in PFS favoring gefitinib in this subgroup was reported (9.5 vs. 6.3 months, HR 0.48; p < 0.001). In the subgroup of patients with EGFR mutation negative tumors, ORR with gefitinib was 1.1 %, and PFS was significantly longer with chemotherapy (HR 2.85, 95 % CI 2.05–3.98; p < 0.001). Higher ORR in EGFR mutation-positive patients, who received chemotherapy (47.3 vs. 23 %) than in wild-type EGFR patients, raises the question about enhanced chemosensitivity of EGFR-mutated tumors, and is object of further researches.
EURTAC (Erlotinib vs. standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive NSCLC) phase III trial [21] included NSCLC patients with EGFR mutations in metastatic first-line setting. Patients (n = 173) were randomized in a 1:1 ratio to receive erlotinib or standard chemotherapy (platinum plus docetaxel or gemcitabine). Median PFS was 9.7 months for patients treated with erlotinib and 5.2 months for those treated with chemotherapy (HR 0.37, 95 % CI 0.25–0.54; p < 0.0001). ORR with erlotinib was 64 and 18 % for chemotherapy. Median OS did not differ significantly between the two arms.
As a result of these TKIs studies, erlotinib and gefitinib were approved in a first-line setting of advanced NSCLC with EGFR somatic-sensitizing mutations.
EGFR mutations: drug sensitive and drug resistant
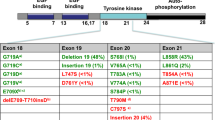
In lung cancer, EGFR mutations occur in exons encoding the ATP-binding pocket of the kinase domain (exon 18 to 21). The most relevant drug-sensitive mutations are deletions in exon 19 and point mutations in exon 21 (L858R). Taken together, they account for approximately 85 % of EGFR mutations. These mutations are oncogenic and intrinsically active, and they provide receptor-increased affinity from gefitinib and erlotinib over ATP [22]. Other drug-sensitive mutations are reported at much lower prevalence such as G719X (3 %), L861X (2 %), and exon 19 insertions (1 %) [23]. On the other hand, drug-resistant mutations have been defined; these resistance mutations can appear simultaneously with a drug-sensitive mutation or as an acquired event during TKIs treatment. The most noteworthy are: L747S and D761Y in exon 19, T790M and insertions in exon 20 [24], and T854A in exon 21 [25] (Fig. 2).

EGFR kinase domain major mutations are represented; sensitizing mutations to reversible EGFR TKI (at the top) and resistant mutations to reversible EGFR TKI (below)
Prevalence of EGFR mutations varies among ethnicities: approximately 50 % of adenocarcinomas of East Asia harbor EGFR mutations; about 7–16 % of patients from an unselected population of North America and Europe have these somatic mutations [26]. EGFR mutations can be found in all histological subtypes. In a Spanish study [27], lung cancers from 2,105 patients were screened for EGFR mutations and they were found in 350 patients (16.6 %); mutations were more frequent in women (69.7 %), in patients who had never smoked (66.6 %) and in adenocarcinomas (80.9 %). EGFR mutations have also been described in squamous cell carcinoma, although its prevalence is about 3.5 % in different series [28, 29].
TKIs have demonstrated an increase in ORR and PFS compared with chemotherapy in patients with NSCLC and EGFR-sensitizing mutations in a first-line setting [14, 30]. No randomized prospective studies have yet shown that EGFR TKIs prolong OS compared with chemotherapy (mainly because once the patients in the chemotherapy arm suffer disease progression, they can benefit from switching to a TKI therapy). But there is a growing body of evidence that patients with EGFR-sensitizing mutations treated with TKIs have higher survival rates, longer than 2 years, with a median PFS among 9.2–13.1 months and an ORR ranging from 58 to 83 % [31]. Unfortunately, about 30 % of these patients do not respond to TKIs therapy and the responders will inevitably develop acquired treatment resistance [32, 33].
Conversely, a small proportion of patients whose tumors respond to TKIs have no evidence of EGFR mutations [34]. As mentioned before, NSCLC cells can release EGF ligands, establishing EGFR autocrine loops. If this loop is dependent on continued EGFR signaling and inhibited by TKIs, it could be an explanation for why some WT EGFR tumors can respond to these therapies. Taken together these considerations, we must hypothesize that more factors than EGFR mutations confer sensitivity to EGFR inhibition, such as other Erb receptors and ligands [35].
EGFR mutations and primary or acquired resistance
Primary resistance
The three major mechanisms involved in primary EGFR TKIs resistance are: KRAS mutations, PTEN losses and concurrent T790M mutation (Table 1; Fig. 3). KRAS mutations and PTEN losses are, almost always, mutually exclusive with EGFR mutations, which could confer TKI resistance in EGFR wild-type tumors.

EGFR primary and acquired resistance. 1 EGFR drug-resistant mutations increase tyrosine kinase affinity for ATP, which competitively displaces erlotinib–gefitinib from receptor. Afatinib covalently and irreversibly binds a cysteine residue in EGFR to the amino acid position 797, leading to EGFR kinase activity inhibition even in presence of an EGFR T790M mutation. 2 MET amplification can stimulate HER3 dependent activation of PI3K and also RAS downstream pathway. 3 PTEN downregulates PI3K signaling by dephosphorylating PIP3 into PIP2. The loss and/or mutation of PTEN lead to hyperactive PI3K pathway. Mutations in PI3K protein can also stimulate this molecular pathway. 4 RAS-RAF-MEK1 mutations can lead to persistent activation of downstream pathways. NF1 RasGAP increases KRAS persistent activation
KRAS: the Kirsten rat sarcoma viral oncogene homolog
Mutations in KRAS, NRAS, BRAF and MEK1 rarely occur in EGFR-mutant tumors. Oncogenic driver mutations of this pathway downstream of EGFR in lung cancer appear with the following frequencies: KRAS 15–30 %, NRAS 1 %, BRAF 3–5 % [36] and MEK1 1 % [37].
KRAS mutations are found in approximately 30 % of lung adenocarcinomas and 5 % of squamous carcinomas. These mutations occur primarily at codon 12 or 13 of exon 2, and they are associated with a history of tobacco use. A metaanalysis of 28 studies evaluating NSCLC found that mutant KRAS was a negative prognostic indicator for OS with a HR of 1.30 for all studies, and of 1.52 in adenocarcinomas studies [38]. KRAS was initially thought to be a poor prognostic marker of survival, but data are contradictory [39].
In addition, NF1 RasGAP disabling mutations [40] increase the occurrence of persistent RAS activation in NSCLC to approximately 40 %. Retrospective analyses suggest the association between KRAS mutations and a lack of response to EGFR TKI therapy, but it remains unclear whether there is a relationship between KRAS mutations and EGFR TKI PFS and OS [41].
In an analysis of approximately 200 lung cancer sample tumors with acquired resistance to TKIs, no RAS or MEK1 mutations were identified, but two BRAF mutations (V600E/G469A) were described. This opens a possible way towards BRAF inhibition as a strategy to overcome resistance to EGFR TKIs [42].
PTEN
Loss of function mutations in PTEN leads to a hyperactive PI3K pathway [43] which is hypothesized to cause de novo TKIs resistance. Loss of PTEN permits high level of AKT activity independent of TK receptor status, causing stimulation of downstream pathways [44]. Indeed, inhibition of AKT should lead to overcome TKIs resistance in this subset of patients. Several PI3K-AKT inhibitors are in clinical development for NSCLC [45].
Concurrent T790M mutation
EGFR mutation at T790M accounts for more than 50 % of acquired TKI resistance [46]. The coexistence of a drug-sensitive and a drug-resistant EGFR mutation has become a trending topic in Oncology. Are T790M mutations second events which appear after a long exposure to EGFR TKIs, or do they pre-exist before TKI therapy? Are both statements true? Is there a T790M cell population selection after prolonged exposure to TKI drugs which can emerge as the dominant tumor clone, conferring EGFR TKIs resistance? [47].
A retrospective study included 73 lung cancer samples before TKI treatment, and they were analyzed for correlation with TKI response [48]. In this sample, 31.5 % of patients had pretreatment T790M mutation using Matrix-Associated Laser Desorption Ionization- Time of Flight Mass Spectrometry (MALDI-TOF MS) detection method, whereas only 2.7 % of patients had T790M by direct sequencing. All T790M mutations detected by direct sequencing were also positive by MALDI-TOF MS. All T790M mutations coexisted with EGFR-sensitizing mutations; of the 56 patients with EGFR mutations, 23 had also de novo T790M and showed significantly shorter PFS compared with 33 patients without T790M mutation (median PFS, 6.7 vs. 10.2, 95 % CI 1.044–3.292, p < 0.05). However, the TKI RR of patients with T790M was not different from those without this mutation, in concordance with other reports.
In a retrospective subgroup analysis of EURTAC [49], 123 patients with available pretreatment tumor tissue were reanalyzed for the concomitant presence of T790M and sensitizing mutation with Taqman assay. The T790M mutation was detected in 21/64 (32.8 %) patients in the erlotinib arm and 26/59 (44.1 %) in the chemotherapy arm. PFS was 12.1 months for patients with mutant T790M in the erlotinib arm, 8.8 months for patients without T790M in the erlotinib arm, 6.3 months for patients with mutant T790M in the chemotherapy arm, and 4.5 months for patients without T790M in the chemotherapy arm (p < 0.0001). Excitement about maximum clinical benefit in patients with double mutation treated with erlotinib was expressed by the authors.
The variability observed between studies when comparing different mutation testing methods opens the question about the ideal test that should be done to detect mutations in NSCLC. Using high-sensitive detection methods, the T790M mutation is detected in up to 68 % of rebiopsied patients [50]. Clinical EGFR mutation test should be able to include T790M mutation not only after TKI progression.
Acquired resistance
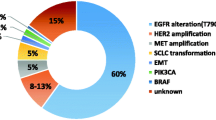
For the last several years, many have been the potential mechanisms related to the acquisition of TKIs treatment resistance (Table 1). Most outstanding mechanisms are: secondary mutations in exons 19 and 20 of the TK domain of EGFR, MET amplification, PI3K pathway mutations and phenotypic transformation. Unknown mechanisms underlying TKIs resistance constitute about 30 % [51] (Fig. 3).
Secondary mutations
Different secondary mutations in the TK domain of the EGFR have been related to acquisition of resistance to reversible EGFR TKIs. The most relevant is T790M of exon 20, which is detected in about 50 % of NSCLC tumors harboring resistance to erlotinib–gefitinib. This point mutation at the gatekeeper position T790 of exon 20 generates a substitution of threonine with a bulkier residue, methionine, which sterically hinders drug binding. T790M also increases the EGFR kinase affinity for ATP, which competitively displaces erlotinib–gefitinib from receptor [52].
Those patients who harbor T790M acquired resistance mutations have longer survival than patients with another acquired resistance mechanism [53]. EGFR-mutated NSCLC cell lines, with or without T790M mutation, exhibited more sensitivity to irradiation in contrast to WT EGFR [54]. Data derived from retrospective studies and preclinical researches show that T790M mutation does not appear as a ubiquitous mutation and that possesses a dynamic behavior [55]. The first observation means that, many times, T790M emerges as an acquired resistance mutation in some tumor lesions whereas other lesions continue responding to TKIs treatment; the second observation refers to a curious phenomenon named oncogene addiction [56].
It has been widely known that human cancers usually evolve through a multistage process with accumulation of mutations and epigenetic changes that affect different genes. But in many occasions, only one or few of these disturbances can promote tumor cell growth and survival. The concept of oncogene addiction refers to the dependence of a cancer cell on one overactive gene or pathway for its development. The apparition of secondary mutations as T790M in EGFR TK domain in patients treated with gefitinib–erlotinib is an example of cancer growth and survival dependence on “one and only” genetic event.
This cancer growth selection under one well-defined genetic way can be interpreted as the tumor Achilles heel, becoming a molecular target that may be candidate to specific drug development.
Met amplification
Mesenchymal–epithelial transition factor (MET) receptor tyrosine kinase can be mutated or overexpressed in lung cancer. The gene for MET is located on chromosome 7q and its ligand is the hepatocyte growth factor (HGF). Downstream molecules involved in the regulation of MET induce motility, migration and regulation of tumor angiogenesis [57].
MET amplification has been reported in 20 % of EGFR TKI-naïve patients, but its primary role seems to be related to acquired TKIs resistance. It has been shown that amplification of MET generates erlotinib–gefitinib resistance by stimulating HER3 dependent activation of PI3K, even in presence of activating EGFR mutations or by secondary amplification of RAS downstream pathway [58].
Concomitant presence of T790M mutation and MET amplification has been described in patients under treatment with TKIs, so concurrent inhibition of both mechanisms could be critical for overcoming resistance [59].
Mutations in PI3K protein family have been also described as an acquired resistance mechanism to EGFR TKIs in about 4 % of NSCLC [60].
Phenotypic transformation
Acquired resistance to EGFR TKIs may also appear through tumor transformation to other histological types:
-
Epithelial to mesenchymal transition (EMT): mesenchymal status is related with intrinsic resistance to TKIs. EMT is a process in which epithelial cells acquire phenotypic characteristics of mesenchymal cells, such as down-regulation of E-cadherin and up-regulation of vimentin, fibronectin and n-cadherin [61]. E-cadherin, a cell surface transmembrane molecule, is involved with EGFR in activating downstream signaling pathways and its repression renders cell insensitive to TKIs therapy [62].
-
Transformation into other lung cancer histological type: most recently reports [51] publish transformation of EGFR-mutant adenocarcinoma to small cell lung carcinoma (SCLC); another case report of transformation to a high-grade neuroendocrine carcinoma with combined features of SCLC and NSCLC with neuroendocrine morphology has been published [63]. Most patients with SCLC transformation after TKIs progression can respond to platinum-etoposide therapy [64].
Role of rebiopsy in resistant EGFR tumors
Emerging data support the value of tumor rebiopsy in NSCLC patients after TKI progression. A retrospective analysis [65] with NSCLC patients whose tumors became resistant to treatment with TKIs, included biopsies taken before and after TKI treatment in patients with either EGFR mutation or who had demonstrated a duration of response to TKIs of more than 24 weeks. Comparison of pre- and post-TKI biopsies showed that the frequency of T790M mutation was 47.6 % following TKI treatment; two patients lost T790M and exon 21 mutations that were recorded prior to treatment. A total of 13 patients developed EGFR exon 19 mutations. Furthermore, 17 patients following TKI treatment showed both T790M and exon 19 mutations. Finally, one patient with an exon 19 deletion in the pre-TKI treatment biopsy exhibited transformation to SCLC.
Taken together these data and described resistance mechanisms, rebiopsy can be a powerful arm and must be considered especially in patients who become resistant to TKI, since it can add information on tumor characteristics that may directly affect treatment decision and define mechanisms under the development of resistance.
Other resistance mechanisms under evaluation
EML4-ALK translocation confers resistance to EGFR TKI therapy, although this could be explained by the absence of EGFR-sensitizing mutations [66]. EGFR mutations and EML4-ALK translocation were initially thought to be mutually exclusive, but coexistence of these two alterations has been reported. In fact, concomitant EML4-ALK translocation has been detected in 15.8 % of patients of Eurtac trial [67]. Crizotinib is an ALK kinase inhibitor which also targets c-MET. A potential role of crizotinib in MET amplification mediated EGFR TKI resistance is being explored [68].
HER2/neu kinase domain mutations are found in approximately 1–4 % of lung adenocarcinomas. They appear typically in women and never smokers with no concurrent EGFR mutations. This subset of patients is also object of research with targeted therapies [69].
Overcoming resistance to TKIs
In order to define more specifically TKIs acquired resistance concept, selection criteria have been proposed [70]. All patients should have the following:
-
Previously received treatment with a single-agent EGFR TKI (gefitinib–erlotinib).
-
Either or both the following: a tumor that harbors an EGFR mutation known to be associated with drug sensitivity or objective clinical benefit from treatment with an EGFR TKI as defined by either documented partial or complete response (RECIST or WHO) or significant and durable (≥6 months) clinical benefit (SD as defined by RECIST or WHO) after initiation of erlotinib or gefitinib.
-
Systemic progression of disease (RECIST or WHO) while on continuous treatment with gefitinib or erlotinib within the last 30 days.
-
No systemic therapy between cessation of gefitinib–erlotinib and initiation of new therapy.
EGFR-mutated tumors tend to grow slowly despite evidence of RECIST progression suggesting that some tumor cells remain sensitive to TKI. Patients with EGFR-mutant tumors can display a disease flare with symptomatic and radiographic progression after stopping TKIs, while improvement is noted after restarting the treatment. Moreover, clinical observations in patients with acquired clinical resistance to EGFR TKIs have shown a symptomatic disease flare attributable to disease progression after treatment discontinuation. Using TKI therapies, NSCLC tends not only to decrease in size but also to undergo morphologic changes on computed tomography (CT), such as ground glass opacity, cavitations and attenuation changes within target lesions [71]. Therefore, the response to TKIs may be inadequately assessed by RECIST criteria.
These clinical and radiographic observations call for the development of additional response criteria which complements RECIST [72]. Combination with positron emission tomography (PET) showing the tumor metabolic behavior could be useful in defining response criteria.
Second-generation irreversible EGFR TKIs
Erlotinib and gefitinib reversibly block tyrosine-kinase receptor; theoretically, an irreversible block should be more effective. Activity of reversible EGFR TKIs in T790M mutant samples is limited or even non-existent [73]. In order to get higher affinity for the tyrosine-kinase domain, second-generation irreversible EGFR TKIs are emerging.
-
AFATINIB: a highly selective and irreversible ErbB family inhibitor of both EGFR and HER2 kinases. In cell assays, afatinib has a similar potency than gefitinib for inhibiting L858R EGFR and comparable to lapatinib inhibiting HER2; however, afatinib has shown 100-fold greater activity in preclinical models against L858R-T790M EGFR double mutants than gefitinib. Afatinib covalently and irreversibly binds a cysteine residue in EGFR to the amino acid position 797, leading to EGFR kinase activity inhibition even in presence of an EGFR T790M mutation [74]. Afatinib has also shown preliminary clinical activity in patients NSCLC patients harboring a HER2 mutation, present in approximately 2–4 % of adenocarcinomas [75].
-
LUX-LUNG3 phase III trial [76] results have been recently reported. This is a randomized study of afatinib vs. pemetrexed–cisplatin as first-line treatment for patients with advanced lung adenocarcinoma harboring EGFR-sensitizing mutations; 345 patients were randomized to afatinib 40 mg or chemotherapy. Significant improvement in median PFS (11.1 months in afatinib group vs. 6.9 months in chemotherapy arm, HR 0.58, 95 % CI 0.43–0.78; p = 0.0004) was observed. In 308 patients with common mutations (del19/L858R), median PFS was 13.6 vs. 6.9 months, respectively (HR 0.47, 95 % CI 0.34–0.65, p < 0.0001). ORR was significantly higher with afatinib (56 vs. 23 %; p < 0.0001).
LUX-LUNG3 represents the largest trial in EGFR mutation-positive lung cancer patients. A potential weakness of these afatinib trials is that no data about concomitant or acquired T790M mutations have been reported; this should provide an opportunity to dispel some doubts about the initial promising afatinib activity in T790M-mutated cells.
Combined EGFR targeting with afatinib and cetuximab has induced near complete responses in T790M murine models, with no responses with erlotinib and cetuximab combination. This observation has lead into further research in afatinib and cetuximab combination in patients with acquired resistance to erlotinib or gefitinib, with a theoretical benefit of double-inhibition blockade of EGFR and HER2. In a phase II dose trial [77] with 26 patients with TKI acquired resistance, disease control was observed in all of them including 36 % of confirmed partial responses.
-
DACOMITINIB: a pan-HER inhibitor that irreversibly and covalently binds to the ATP domain of each of three kinase-active member of the HER family: EGFR, HER2 and HER4. In patients with progressive NSCLC after treatment with an EGFR TKI and one or more chemotherapy regimens, dacomitinib showed antitumor activity in phase I and II trials [78]. Phase III trials are ongoing.
Met-acquired resistance
Several agents are being developed to specifically target MET, including monoclonal antibodies (onartuzumab) and TKIs (tivantinib). Dual inhibition of MET and EGFR has also demonstrated activity in preclinical models of EGFR-resistant NSCLC [79].
In a phase II trial [80] in previously treated patients with EGFR TKI naïve advanced NSCLC, 167 patients were randomized to erlotinib plus tivantinib or erlotinib plus placebo. PFS and OS significant improvement in planned subset analysis were reported in patients with non-squamous histology who were treated with erlotinib plus tivantinib. Biomarker studies showed that among non-squamous tumors 75 % were MET-positive by IHC, compared with only 12 % of squamous tumors. Based on these data, a randomized, double-blind, placebo controlled phase III study [81] of tivantinib plus erlotinib vs. placebo plus erlotinib, in patients who have received 1–2 prior lines of chemotherapy but TKI naïve, was designed. Unfortunately, the trial has been discontinued since no OS benefit has been found after an interim analysis [82].
Onartuzumab (MetMab) is a humanized monoclonal antibody that binds to MET preventing HGF ligand binding and blocking downstream signaling. In a phase II study [83], patients with MET-positive tumors (IHC) who received erlotinib plus onartuzumab had significant reduction in the risk of death and disease progression compared with erlotinib alone. A phase III trial in MET-positive NSCLC patients previously treated with at least one but no more than two prior lines of chemotherapy, but TKI naïve, is open for accrual [84].
Other therapeutic targets in overcoming resistance
Resistance to anti-EGFR therapy has been also supposed to be secondary to increased vascular endothelial growth factor (VEGF) expression. These two signaling pathways are independent but are closely interlinked [85]. EGF and TGFα both induce VEGF expression via activation of EGFR in cell culture models. It has been postulated that overactivation of pathways driving VEGF expression independently of EGFR might result in resistance to EGFR inhibitors due to the inability of these agents to downregulate VEGF to a non-angiogenic “point of no return”, because EGFR inhibition does not substantially inhibit angiogenesis.
BELIEF, a phase II prospective trial of erlotinib and bevacizumab in patients with advanced NSCLC and sensitizing EGFR mutations with or without T790M mutations at diagnosis, represents an ongoing trial with VEGFR and EGFR inhibition approach [86].
Vandetanib, an inhibitor of VEGF receptor, EGFR and RET signaling, was compared with placebo [87] in patients with advanced NSCLC who had received no more than two prior chemotherapy regimens and had experienced treatment failure with an EGFR TKI. The trial did not demonstrate any OS benefit and serious adverse events rate was high.
The insulin-like growth factor-1 receptor (IGF-1R) is also interconnected with EGFR pathway. Preclinical models [88] with acquired EGFR TKIs treatment resistance have shown up-regulation of IGF-1. Mammalian target of rapamycin (mTOR) [89] and heat shock protein 90 (HSP90) chaperone [90] are also other targets under evaluation in this setting.
Alternation of reversible TKIs and combination of chemotherapy plus TKI
Based on the premise that erlotinib is administered at maximum tolerated dose (MTD) whereas gefitinib is approximately given at one-third of this, there is some evidence derived from small clinical series that erlotinib could be used after progression on gefitinib [91–93]. Pooled analysis of the reports of erlotinib after failure of gefitinib for NSCLC showed that erlotinib might produce clinical benefits in patients who had shown long SD on prior gefitinib therapy [94].
EGFR-mutant patients treated with EGFR TKIs often present progression of one or more tumor lesions while others remain unchanged or continue responding. Taking into account that acquired resistance mutations have a dynamic behavior and having in mind the oncogene addiction model, it could be hypothesized that a TKI treatment-free interval might result in restoration of gefitinib–erlotinib sensitivity. As a consequence of the foregoing considerations, another explored approach is the association with a chemotherapeutic agent at the time of TKI resistance acquisition. A retrospective study [95] of gefitinib plus paclitaxel in patients with gefitinib progression disease provided a response rate of 13 %, a PFS of 4.2 months and OS of 8.1 months. LUX-LUNG5 is an ongoing randomized, open-label, active-controlled trial of afatinib plus weekly paclitaxel vs. investigator’s choice of chemotherapy following afatinib monotherapy in NSCLC patients failing erlotinib or gefitinib [96].
Conclusions
Understanding the biological basis of lung cancer development is essential in order to design therapeutical approaches. EGFR TKIs primary and acquired resistance mechanisms are complex and heterogeneous, but definition of EGFR onco-addicted lung cancer into molecular subgroups could better clarify researches in targeted specific drugs (Fig. 4).

Some noteworthy EGFR TKI resistance mechanisms and potential therapeutical approaches
TKIs constitute the best choice of treatment in NSCLC patients with sensitizing EGFR mutations; once their disease is under progression a critical scenario opens: a clinical trial or chemotherapy seem the most reasonable options in daily clinical practice. Many are the open issues in the setting of TKIs resistance: which is the best treatment option? What “kind of progression disease” is the patient suffering? Which are the mechanisms under resistance acquisition? Is a rebiopsy worthy? Are RECIST/WHO criteria adequate in EGFR-mutant patients? Is PET with CT scan a better evaluation instrument than CT scan alone?
EGFR second-generation TKIs are called to play a role in NSCLC treatment options but their best place in the therapeutic arsenal is an object of discussion, since their results are similar to those of erlotinib–gefitinib. Clinical evaluation of potential drugs in patients with EGFR TKI-resistant disease is warranted.
Abbreviations
- NSCLC:
-
Non-small cell lung cancer
- SCLC:
-
Small cell lung carcinoma
- EGFR:
-
Epidermal growth factor receptor
- VEGF:
-
Vascular endothelial growth factor
- TKI:
-
Tyrosine-kinase inhibitor
- RTKs:
-
Tyrosine-kinase receptor family
- PI3K:
-
Phosphatidylinositol 3-kinase
- WT:
-
Wild-type
- KRAS:
-
Kirsten rat sarcoma viral oncogene homolog
- PTEN:
-
Phosphatase and tensin homologue
- MET:
-
Mesenchymal–epithelial transition factor
- EMT:
-
Epithelial to mesenchymal transition
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- WHO:
-
World Health Organization
References
Gullick WJ, Downward J, Parker PJ, Whittle N, Kris R, Schlessinger J, et al. The structure and function of the epidermal growth factor receptor studied by using antisynthetic peptide antibodies. Proc R Soc Lond B Biol Sci. 1985;226(1242):127–34.
Lo HW. Nuclear mode of the EGFR signaling network: biology, prognostic value, and therapeutic implications. Discov Med. 2010;10(50):44–51.
Zaczek A, Brandt B, Bielawski KP. The diverse signaling of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol Histopathol. 2005;20(3):1005–15.
Normanno N, Maiello MR, De Luca A. Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs): simple drugs with a complex mechanism of action? J Cell Physiol. 2003;194(1):13–9.
Burris HA 3rd. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013. doi:10.1007/s00280-012-2043-3.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3(9):954–87.
Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, Wemmer DE, et al. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152(3):557–69.
Jiang J, Greulich H, Jänne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65(19):8968–74.
Zhang Z, Stiegler AL, Boggon TJ, Kobayashi S, Halmos B. EGFR-mutated lung cancer: a paradigm of molecular oncology. Oncotarget. 2010;1(7):497–514.
Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–81.
Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. 2009;218(3):460–6. doi:10.1002/jcp.21635.
Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–31.
Tiseo M, Bartolotti M, Gelsomino F, Bordi P. Emerging role of gefitinib in the treatment of non-small-cell lung cancer (NSCLC). Drug Des Devel Ther. 2010;4:81–98.
Nguyen KS, Neal JW. First-line treatment of EGFR-mutant non-small-cell lung cancer: the role of erlotinib and other tyrosine kinase inhibitors. Biologics. 2012;6:337–45. doi:10.2147/BTT.S26558.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39.
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39.
Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet. 2005;366(9496):1527–37.
Chang A, Parikh P, Thongprasert S, Tan EH, Perng RP, Ganzon D, et al. Gefitinib (IRESSA) in patients of Asian origin with refractory advanced non-small cell lung cancer: subset analysis from the ISEL study. J Thorac Oncol. 2006;1(8):847–55.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–32.
Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol. 2011;29(21):2866–74.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46.
Soria JC, Mok TS, Cappuzzo F, Jänne PA. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev. 2012;38(5):416–30.
Otto C, Csanadi A, Fisch P, Werner M, Kayser G. Molecular modeling and description of a newly characterized activating mutation of the EGFR gene in non-small cell lung cancer. Diagn Pathol. 2012;22(7):146.
Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012;13(1):e23–31.
Gotoh N. Somatic mutations of the EGF receptor and their signal transducers affect the efficacy of EGF receptor-specific tyrosine kinase inhibitors. Int J Clin Exp Pathol. 2011;4(4):403–9.
Calvo E, Baselga J. Ethnic differences in response to epidermal growth factor receptor tyrosine kinase inhibitors. J Clin Oncol. 2006;24(14):2158–63.
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–67.
Miyamae Y, Shimizu K, Hirato J, Araki T, Tanaka K, Ogawa H, et al. Significance of epidermal growth factor receptor gene mutations in squamous cell lung carcinoma. Oncol Rep. 2011;25(4):921–8.
Park SH, Ha SY, Lee JI, Lee H, Sim H, Kim YS, et al. Epidermal growth factor receptor mutations and the clinical outcome in male smokers with squamous cell carcinoma of lung. J Korean Med Sci. 2009;24(3):448–52.
Stella GM, Luisetti M, Inghilleri S, Cemmi F, Scabini R, Zorzetto M, et al. Targeting EGFR in non-small-cell lung cancer: lessons, experiences, strategies. Respir Med. 2012;106(2):173–83.
Pallis AG, Syrigos KN. Epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of NSCLC. Lung Cancer. 2013. doi:10.1016/j.lungcan.2012.12.025.
Ou SH. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence. Crit Rev Oncol Hematol. 2012;83(3):407–21.
Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013. doi:10.1200/JCO.2012.43.3912.
Wu W, O'Reilly MS, Langley RR, Tsan RZ, Baker CH, Bekele N, et al. Expression of epidermal growth factor/transforming growth factor alpha by human lung cancer determines their response to EGF receptor tyrosine kinase inhibition in the lungs of mice. Mol Cancer Ther. 2007;6(10):2652–63.
Gazdar AF, Minna JD. Deregulated EGFR signaling during lung cancer progression: mutations, amplicons and autocrine loops. Cancer Prev Res (Phila). 2008;1(3):156–60. doi:10.1158/1940-6207.
Sequist LV, Heist RS, Shaw AT, Fidias P, Rosovsky R, Temel JS, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22(12):2616–24.
Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68(14):5524–8.
Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92(1):131.
Tsao MS, Aviel-Ronen S, Ding K, Lau D, Liu N, Sakurada A, et al. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol. 2007;25(33):5240.
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–75.
Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26(26):4268–75.
Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–33.
Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6(4):707–15.
Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, et al. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22(18):2812–22.
Wojtalla A, Arcaro A. Targeting phosphoinositide 3-kinase signaling in lung cancer. Crit Rev Oncol Hematol. 2011;80(2):278–90.
Bar J, Onn A. Overcoming molecular mechanisms of resistance to first-generation epidermal growth factor receptor tyrosine kinase inhibitors. Clin Lung Cancer. 2012;13(4):267–79.
Ghosh G, Lian X, Kron SJ, Palecek SP. Properties of resistant cells generated from lung cancer cell lines treated with EGFR inhibitors. BMC Cancer. 2012;20(12):95.
Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30(4):433–40.
Rosell R, Molina-Vila MA, Taron M, Bertran-Alamillo, Mayo C, Vergnenegre A, et al. EGFR compound mutants and survival on erlotinib in non-small cell lung cancer (NSCLC) patients (p) in the EURTAC study. J Clin Oncol. 2012 (suppl; abstr 7522).
Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiobsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17(5):1169–80.
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26.
Kosaka T, Yamaki E, Mogi A, Kuwano H. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. J Biomed Biotechnol. 2011;2011:165214. doi:10.1155/2011/165214.
Oxnard GR, Arcila ME, Sima CS, Riely GJ, Chmielecki J, Kris MG, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17(6):1616–22.
Rosell R, Molina MA, Costa C, Simonetti S, Gimenez-Capitan A, Beltran-Alamillo J. Pretreatment EGFR T790M mutation and BRCA-1 mRNA expression in erlotinib treated advanced non-small cell lung cancer patients with EGFR mutations. Clin Cancer Res. 2011;17(5):1160–8.
Politi K, Pao W. How genetically engineered mouse tumor models provide insights into human cancers. J Clin Oncol. 2011;29(16):2273–81.
Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–80 (discussion 3080).
Kim ES, Salgia R. MET pathway as a therapeutic target. J Thorac Oncol. 2009;4(4):444–7.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43.
Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88.
Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borrás AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116(10):2695–706.
Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6(7):1152–61.
Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66(2):944–50.
Popat S, Wotherspoon A, Nutting CM, Gonzalez D, Nicholson AG, O’Brien M. Transformation to “high grade” neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer. 2013. doi:10.1016/j.lungcan.2012.12.019.
Hata A, Katakami N, Yoshioka H, Takeshita J, Tanaka K, Nanjo S, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to EGFR-TKI: Comparison between T790M mutation-positive and -negative populations. J Clin Oncol. 2012 (suppl; abstr 7528).
Kuyper J. Rebiopsy results in EGFR-mutated NSCLC patients with TKI resistance. http://www.esmo.org/Conferences/Past-Conferences/EMCTO-2013-Lung-Cancer/News/Findings-support-the-value-of-tumour-rebiopsy-in-NSCLC-patients.
Shaw AT, Engelman JA. ALK in lung cancer: past, present and future. J Clin Oncol. 2013;. doi:10.1200/JCO.2012.44.5353.
Rosell R, Massuti B, Costa C, Molina MA, Gimenez-Capitan A, Karachaliou N, et al. Concomitant actionable mutations and overall survival in EGFR mutant non small cell lung cancer patients included in the EURTAC trial: EGFR L858R, EFR T790M, TP53 R273H and EML4-ALK (v3). 37th ESMO congress. Abstract 929. 2012.
Tanizaki J, Okamoto I, Okamoto K, Takezawa K, Kuwata K, Yamaguchi H, et al. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J Thorac Oncol. 2011;6(10):1624–31.
Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–33.
Jackman D, Pao W, Riely JG, Engelman JA, Kris MK, Janne PA, et al. Clinical definition of acquired resistance to epidermal Growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. J Clin Oncol. 2010;28(2):357–60.
Nishino M, Cardarella S, Dahlberg SE, Jackman DM, Ramaiya NH, Hatabu H, et al. Radiographic assessment and therapeutic decisions at RECIST progression in EGFR mutant NSCLC treated with EGFR tyrosine kinase inhibitors. Lung Cancer. 2013;79(3):283–8.
Lee HY, Lee KS, Ahn MJ, Hwang HS, Lee JW, Park K, et al. New CT response criteria in non small cell lung cancer: proposal and application in EGFR tyrosine kinase inhibitor therapy. Lung Cancer. 2011;73(1):63–9.
Heuckmann JM, Rauh D, Thomas RK. Epidermal growth factor receptor (EGFR) signaling and covalent EGFR inhibition in lung cancer. J Clin Oncol. 2012;30(27):3417–20.
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–11.
De Grève J, Teugels E, Geers C, Decoster L, Galdermans D, De Mey J, et al. Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer. 2012;76(1):123–7.
Yang JC, Schuler MH, Yamamoto N, O'Byrne KJ, Hirsch V, Mok T, et al. LUX-Lung 3: a randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol. 2012 (suppl; abstr LBA7500).
Janjigian YY, Groen HJ, Horn L, Smit EF, Fu Y, Wang F, et al. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol. 2011 (suppl; abstr 7525).
Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012;30(27):3337–44.
Huang MH, Lee JH, Chang YJ, Tsai HH, Lin YL, Lin AM, et al. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol Oncol. 2013;7(1):112–20.
Sequist LV, von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29(24):3307–15.
Scagliotti GV, Novello S, Schiller JH, Hirsh V, Sequist LV, Soria JC, et al. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13(5):391–5.
GEN: Genetic Engineering and Biotechnology news (Internet). http://www.genengnews.com/gen-news-highlights/lack-of-efficacy-halts-pivotal-tivantinib-lung-cancer-trial/81247420. Accessed 19 Mar 2013.
Spigel DR, Ervin TJ, Ramlau R, Daniel DB, Goldschmidt JH, Blumenschein GR, et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J Clin Oncol. 2011 (suppl; abstr 7505).
Spigel DR, Edelman MJ, Mok T, O′Byrne KJ, Paz-Ares L, Yu W, et al. The MetLUNG study: a randomized, double-blind, phase III study of onartuzumab (MetMAb) plus erlotinib versus placebo plus erlotinib in patients with advanced, MET-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2012 (suppl; abstr TPS7616).
Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res. 2007;5(3):203–20.
ETOP 2-11 BELIEF. http://www.etop-eu.org/index.php?option=com_content&view=category&layout=blog&id=180&Itemid=220.
Lee JS, Hirsh V, Park K, Qin S, Blajman CR, Perng RP, et al. Vandetanib Versus placebo in patients with advanced non-small-cell lung cancer after prior therapy with an epidermal growth factor receptor tyrosine kinase inhibitor: a randomized, double-blind phase III trial (ZEPHYR). J Clin Oncol. 2012;30(10):1114–21.
Kato Y, Mascaux M, Wynes MW, Reyna Asuncion B, Tran C, Yoshida K et al. The role of IGF-1R in EGFR TKI resistance in NSCLC using IHC and AQUA technology. J Clin Oncol. 2011 (suppl; abstr 10556).
Sano T, Takeuchi S, Nakagawa T, Ishikawa D, Nanjo S, Yamada T, et al. The novel phosphoinositide 3-kinase-mammalian target of rapamycin inhibitor, BEZ235, circumvents erlotinib resistance of epidermal growth factor receptor mutant lung cancer cells triggered by hepatocyte growth factor. Int J Cancer. 2013. doi:10.1002/ijc.28034.
Koizumi H, Yamada T, Takeuchi S, Nakagawa T, Kita K, Nakamura T, et al. Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs in EGFR-mutant lung cancer by decreasing client protein expression and angiogenesis. J Thorac Oncol. 2012;7(7):1078–85.
Grossi F, Rijavec E, Dal Bello MG, Defferrari C, Brianti A, Barletta G, et al. The administration of gefitinib in patients with advanced non-small-cell lung cancer after the failure of erlotinib. Cancer Chemother Pharmacol. 2012;69(6):1407–12.
Maruyama R, Wataya H, Seto T, Ichinose Y. Treatment after the failure of gefitinib in patients with advanced or recurrent non-small cell lung cancer. Anticancer Res. 2009;29(10):4217–21.
Hata A, Katakami N, Yoshioka H, Fujita S, Kunimasa K, Nanjo S, et al. Erlotinib after gefitinib failure in relapsed non-small cell lung cancer: clinical benefit with optimal patient selection. Lung Cancer. 2011;74(2):268–73.
Kaira K, Naito T, Takahashi T, Ayabe E, Shimoyama R, Kaira R, et al. Pooled analysis of the reports of erlotinib after failure of gefitinib for non-small cell lung cancer. Lung Cancer. 2010;68(1):99–104.
Shukuya T, Takahashi T, Tamiya A, Ono A, Igawa S, Nakamura Y, et al. Gefitinib plus paclitaxel after failure of gefitinib in non-small cell lung cancer initially responding to gefitinib. Anticancer Res. 2009;29(7):2747–51.
Hirsch V. Afatinib (BIBW 2992) development in non-small-cell lung cancer. Future Oncol. 2011;7(7):817–25. doi:10.2217/fon.11.62.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Carrera, S., Buque, A., Azkona, E. et al. Epidermal growth factor receptor tyrosine-kinase inhibitor treatment resistance in non-small cell lung cancer: biological basis and therapeutic strategies. Clin Transl Oncol 16, 339–350 (2014). https://doi.org/10.1007/s12094-013-1143-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-013-1143-9