
Abstract
Maternal obesity increases the risk of health complications in both the mother and the infant. In turn, the offspring of obese mothers are inclined to develop obesity, cardiovascular disease, and type 2 diabetes as adults, establishing a vicious cycle of metabolic disease. Obese mothers exhibit dyslipidemia, inflammation, and insulin resistance, factors which can negatively influence fetal development via effects on placental function. Optimal nutrient delivery to the fetus is a critical function of the placenta, and impairments in this process may modulate fetal growth and/or compromise fetal tissue development. Additionally, nutrients, in particular fatty acids, function as bioactive molecules to alter placental inflammatory status. Recent findings demonstrate increased posttranslational processing of IL-1β by an inflammatory process known as the inflammasome, in placentas of obese mothers. Inflammasome activation results in caspase-1-mediated maturation of IL-1β prior to cellular release. High circulating levels of saturated fatty acids activate and polyunsaturated fatty acids inhibit the inflammasome. In addition to its role as an immunogenic molecule, IL-1β impairs placental insulin signaling and function, thereby altering the metabolic function of the placenta. Hence placental inflammasomes may represent one of the key inflammatory mechanisms linking maternal nutrient excess to aberrant placental function and fetal development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Gestational Diabetes
- Fetal Growth Restriction
- Inflammasome Activation
- Maternal Obesity
- Placental Function
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
In the United States, over two-thirds of women of reproductive age have a high body mass index (BMI >25 kg/m2), and more than one-third are obese (BMI >30 kg/m2) [1]. Maternal obesity represents significant health risks for both mother and child. Pregnant mothers who are obese have an increased risk of developing hypertension, preeclampsia, and gestational diabetes. Infants born to obese and gestational diabetic mothers are likely to have greater adiposity, elevated levels of proinflammatory cytokines, and insulin resistance [2]. Children of obese mothers are susceptible to childhood obesity and to develop cardiovascular disease, type 2 diabetes, and obesity as adults [3]. Animal models and clinical studies suggest that the intrauterine environment plays a critical role in mediating the adverse effects of maternal obesity on the offspring and therefore offers a unique window of opportunity for intervention. However, few strategies are currently available for the prevention of obesity or metabolic dysfunction in children of obese mothers. Recent studies demonstrate that children born to women who had undergone bariatric surgery and weight loss had a lower prevalence of obesity compared to their siblings born before surgical weight loss [4]. Although bariatric surgery is limited to morbidly obese patients, this finding establishes the importance of the maternal metabolic environment on the in utero transmission of obesity to the next generation.
In the past decade, the search for potentially unifying mechanisms underlying the pathogenesis of obesity-associated diseases has revealed a surprisingly close relationship between signaling pathways regulating cellular immune response and metabolic homeostasis. This has given rise to the concept of “metaflammation” (metabolically induced inflammation) which refers to a distinct set of inflammatory responses principally triggered by nutrients and metabolites [5]. In contrast to the acute innate immune response induced by microbial stimuli, metaflammation is characterized by multisystemic chronic low-grade inflammation leading to insulin resistance in several tissues including the adipose, liver, and muscle [6]. While pregnancy itself represents a physiologic inflammatory state, women entering pregnancy with preexisting obesity exhibit enhanced systemic inflammation associated with peripheral insulin resistance [7], which may lead to the development of gestational diabetes. In recent years, placental inflammation has emerged as a common observation in these pregnancy disorders [8–12], although the mechanisms regulating placental inflammatory processes in maternal obesity have been a matter for debate [8, 10, 11].
The human placenta is the direct interface between maternal and fetal circulations. The placenta performs many indispensable tasks including hormone production, nutrient transfer, and gas exchange. Optimal placental function is thus paramount to support the growth and development of a healthy infant. Maternal diet and disease can influence placental development and function, leading to changes in the supply of nutrients, hormones, and oxygen to the fetus. The placenta therefore plays a central role in determining the impact of maternal obesity on fetal development and the long-term health of the infant. Clinical studies now indicate that maternal obesity is associated with changes in placental function [13–17]. In particular, placental circulation [17, 18], nutrient transporters [13, 19, 20], and metabolic function [14, 15] have been reported to be influenced by maternal adiposity. Whether these changes are associated with or caused by placental inflammatory processes is currently unclear.
2 Inflammatory Mechanisms Involved in Metabolic Disorders
All tissues possess inflammatory mechanisms which mediate a highly coordinated homeostatic response to harmful stimuli. The short-term adaptive inflammatory response is a crucial component of tissue repair involving the integration of multiple signals in distinct cells and tissues. However, if left unresolved, long-term inflammation may result in permanent organ damage. The chronic nature of obesity produces low-grade inflammation that disrupts metabolic homeostasis over time. An adverse in utero environment may place individuals at risk of lifelong metaflammation predisposing the infant to the development of metabolic syndrome in later life.
Insulin resistance is a hallmark of the metabolic syndrome, occurring as a result of decreased insulin sensitivity in the adipose, liver, and muscle. To maintain homeostasis, insulin secretion from pancreatic β-cells is increased. Over time, β-cells may fail to meet the increasing demand for insulin, resulting in hyperglycemia and diabetes. A link between inflammation and insulin resistance was first established by the demonstration that TNFα knockout mice were protected against obesity-induced insulin resistance [21]. Compared to wild-type obese mice, TNFα-null obese mice exhibited increased insulin signaling in adipose and muscle tissues, improved glucose metabolism, and reduced circulating levels of free fatty acids. Indeed, TNFα levels during pregnancy predicted maternal insulin resistance even in euglycemic women [22], and a proinflammatory maternal milieu is associated with a number of pregnancy disorders.
Inflammation is initiated by cytokines and pathogen-associated molecular patterns (PAMPs), such as carbohydrates, lipoproteins, and lipopolysaccharide components of bacterial and fungal cell walls, that stimulate plasma membrane-bound cytokine receptors or toll-like receptors (TLR) to initiate an inflammatory signaling cascade. These signaling pathways in turn activate transcription factors which drive the expression of genes which collectively assist in the recruitment and activation of immune cells and removal of pathogens and accelerate tissue repair. In recent years, TLR4 has emerged as a sensor for both microbial products and nutrients. The archetypal TLR4 agonist is lipopolysaccharide (LPS), a component of Gram-negative bacteria cell wall. Interestingly, saturated fatty acids (SFAs) acylated in the lipid A moiety of LPS were sufficient to invoke TLR4-mediated biological effects [23], implicating a role for circulating fatty acids in the inflammatory response. The effect of fatty acids on TLR4 activity depends on chain length and saturation [24]. Saturated fatty acids such as palmitic (C16:0) and stearic (C18:0) acid promote TLR4-mediated inflammatory response, whereas the omega-3 polyunsaturated fatty acid (n-3 PUFA) docosahexaenoic acid (C22:6) attenuates TLR4 signaling. Additionally, TLR4 is activated by other endogenous molecules especially in response to stress. Otherwise known as danger-associated molecular patterns (DAMPs), these endogenous TLR4 agonists include heat shock proteins, oxidized lipids and sterols, and breakdown products of the extracellular matrix [25–27].
TLR4 activates two major signaling pathways, the mitogen-associated protein kinase (MAPK) pathways (p38 MAPK and c-Jun N-terminal kinases (JNK)) and nuclear factor kappa B (NF-κB). Activation of JNK results in nuclear translocation where it regulates the activity of activating protein-1 (AP-1), a heterodimeric protein composed of multiple transcription factors belonging to c-Fos, c-Jun, ATF, and JDP families that regulate a vast array of genes. Conversely, NF-κB activation results in nuclear translocation of one of its five subunits which directly controls gene transcription. Moreover, reports of AP-1 regulation of NF-κB and vice versa suggest significant crosstalk between these signaling pathways [28, 29]. Collectively, activation of these signaling pathways results in increased transcription of cytokines, such as IL-6, IL-1β, and TNFα, which inhibit insulin signaling in many tissues. Research in the last decade provides significant evidence for the involvement of these pathways in the initiation, propagation, and development of metabolic diseases [30, 31]. Figure 6.1 illustrates an overview of the major inflammatory mechanisms implicated in metabolic disorders.

Regulation of placental inflammatory response by dietary fatty acids. Saturated and monounsaturated fatty acids activate TLR4 leading to downstream inflammatory pathways JNK and NF-κB. JNK activates AP-1 transcription factors, while NF-κB subunits directly translocate to the nucleus and bind promoter regions of proinflammatory cytokines. Omega-3 fatty acids bind to the GPR120 plasma membrane receptor to inhibit inflammatory pathways. The intracellular NLRP3 inflammasome, which is responsible for IL-1β maturation, is also activated by dietary fatty acids and inhibited by omega-3 fatty acids. AP-1 activating protein-1, GPR120 G protein-coupled receptor 120, IKK-β inhibitor of nuclear factor kappa B kinase, JNK c-jun-N-terminal kinase, MUFA monounsaturated fatty acid, NF-κB nuclear factor kappa B, NLRP3/Inflammasome nod-like receptor 3 inflammasome, SFA saturated fatty acid
3 Placental Inflammation in Maternal Obesity
While there have been numerous animal models of maternal obesity in pregnancy demonstrating placental inflammation [18, 32–34], we have limited our discussion to human studies. Several studies show increased expression of proinflammatory cytokines IL-6, IL-1β, and TNFα in placental tissues of obese mothers [8, 10–12]. The mechanism(s) of placental inflammation in maternal obesity, however, has been a source of contention. Analogous to adipose tissues, increased accumulation of maternal macrophages in placentas of obese mothers has been reported by some investigators [9, 10], while others were unable to show any differences [11]. Similarly, Saben et al. have implicated placental JNK and NF-κB with maternal obesity [12, 35], while we could not find any changes in these pathways, but discovered increased STAT3 and p38 MAPK activity in placentas of obese mothers [8]. One possible explanation for these differences may be related to subject selection, since maternal obesity is also associated with several comorbidities including hypertension and gestational diabetes, conditions potentially more likely to invoke a greater multisystemic inflammatory response than obesity per se.
While activation of placental inflammatory processes in maternal obesity has been described [8, 12, 35, 36], the physiological significance of placental inflammation has not been addressed in adequate detail. Inflammation of the fetal membranes (chorioamnionitis) is a major risk factor for preterm birth, which may result in premature rupture of the membranes (PROM). Recent epidemiological studies demonstrate increased risk of preterm deliveries in obese mothers [37, 38]. However, it is currently unclear if placental inflammation associated with maternal obesity increases the risk of PROM. Systemic or placental inflammation may also result in endothelial dysfunction. Pre-gravid obesity increases maternal levels of markers of endothelial dysfunction [39] and is associated with impaired vascular function in placental arteries [17]. Consequently, this may impact upon fetal heart development, because uteroplacental circulation has been linked with fetal vascular function [40].
Previous work from our lab demonstrated that proinflammatory cytokines influence placental nutrient transport functions [41, 42]. Using concentrations similar to circulating cytokine concentrations in obese women, IL-6 and TNFα were shown to stimulate amino acid transport activity [41], while higher concentrations were required to stimulate fatty acid uptake into primary trophoblasts [42]. STAT3 activity was required for IL-6 (and leptin)-stimulated amino acid uptake [41, 43], while our preliminary data suggests a role for p38 MAPK in regulating the TNFα response (Aye et al, 2015 unpublished observations). In addition to cytokines, insulin and leptin (hormones elevated in maternal obesity) also significantly increase placental amino acid transport [44, 45]. Interestingly, IL-1β which is also upregulated in the placentas of obese mothers [8, 10, 11] was shown to inhibit insulin-dependent [46] and insulin-independent [47, 48] amino acid transport in placental primary trophoblasts and trophoblast cell lines. These findings suggest complex interactions between proinflammatory cytokines and maternal hormones in the regulation of placental nutrient transfer to the fetus.
4 The Emerging Role of Placental Inflammasomes in Pregnancy Disorders
Inflammasomes are multi-protein complexes composed of a cytoplasmic receptor, an apoptosis-associated speck-like protein (ASC) containing a caspase activation and recruitment domain (CARD) adaptor, and pro-caspase-1, which associate upon cellular exposure to microbial or endogenous danger signals. Upon activation, the inflammasome complex formation results in caspase-1 maturation leading to proteolytic cleavage of pro-IL-1β into mature IL-1β (Fig. 6.2). This is then followed by either secretion of IL-1β into the circulation which may lead to systemic inflammatory effects or more locally induced pyroptosis. Currently, six members of the nod-like receptor (NLR) family are known to function as “pathogen” or “danger” sensors to initiate inflammasome complex formation. Of these, NLRP3 is best characterized for its role in both immune and metabolic disorders.

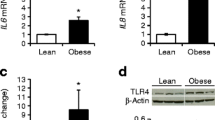
Increased placental inflammasomes in maternal obesity – regulation by dietary fatty acids. (a) Representative immunoblot showing caspase-1 activation and increased IL-1β maturation with maternal adiposity. (b) Immunoblot demonstrating increased inflammasome activation by oleic and palmitic acid (100 μM) in primary human trophoblasts. (c) Immunoblot of decreased inflammasome activation by 50 μM DHA treatment in primary human trophoblasts. BMI body mass index, DHA docosahexaenoic acid, N normal, OW overweight, OB obese, Cnt BSA-control, OA oleic acid, PA palmitic acid
The placenta expresses the cytosolic NLRP1, NLRP3, and NLRC4 [49], which respond to multiple factors associated with both pathogenic and sterile inflammation. Activation of inflammasomes in gestational tissues has been reported in a number of pregnancy complications including preterm birth [50–52], preeclampsia [53–55], and microbial infections [56]. The resulting release of mature IL-1β mediates a myriad of effects in gestational tissues including induction of IGFBP-1 expression in decidual tissues thereby decreasing the bioavailability of IGF-I in the feto-maternal interface and in the maternal circulation [57] and stimulating placental production of progesterone [58], hCG [59], and activin-A [60]. Furthermore, IL-1β is a highly apoptotic agent in human gestational tissues [61]. Hence knowledge of the mechanisms governing IL-1β production in the placenta plays a critical role in understanding the pathogenesis of several pregnancy disorders.
We recently identified increased caspase-1 activation in placentas of women with high BMI ([62] and Fig. 6.2), despite no changes in placental NF-κB DNA-binding activity. Likewise, expression of the mature IL-1β protein (17 kDa) in placental tissues was also positively correlated with maternal BMI, whereas pro-IL-1β (32 kDa) was not significantly altered by maternal BMI. These findings indicate a novel mechanism in which IL-1β is regulated at the posttranslational level by maternal obesity. We further determined the impact of IL-1β on placental function and demonstrated that IL-1β inhibits insulin signaling and function (as measured by insulin-mediated amino acid transport) in primary human trophoblasts [46]. Inhibition of insulin signaling was mediated at the level of the insulin receptor substrate-1 (IRS-1), where IL-1β increased the inhibitory serine phosphorylation of IRS-1 (Ser307), decreased tyrosine phosphorylation-mediated activation of IRS-1 (Tyr612), and reduced total IRS-1 levels. Decreased IRS-1 activity led to inactivation of both the PI3K and GRB2 signaling pathways downstream of IRS-1. Taken together, these findings suggest that the increased inflammasome activation in placentas of high BMI mothers may lead to decreased placental insulin sensitivity. However, the signals activating inflammasomes in these placentas are currently unclear.
Because the classical inflammatory pathways associated with the microbial response (namely, NF-κB and JNK) were not altered in the placenta in pregnancies complicated by maternal obesity, we hypothesized that placental inflammasome activation is a result of DAMPs or nutritional and/or oxidative stress. Robust activation of the inflammasomes requires two signals: a first “priming” signal, which promotes NF-κB activity leading to pro-IL-1β expression, and a second “activating” signal that activates caspase-1. In the placenta, constitutive NF-κB activity is likely to contribute to pro-IL-1β expression, whereas caspase-1 activation requires a specific event. DAMPs including cholesterol crystals, ATP, potassium efflux, and ceramide initiate caspase-1 maturation in many cell types [63]. However, it is currently unclear if DAMPs provide the mechanistic link to placental inflammasome activation because these factors have not been implicated in maternal obesity. On the other hand, maternal obesity is associated with oxidative stress, which may provide the necessary signal required for caspase-1 activation [12, 14, 64]. Dietary SFAs activate inflammasomes in a cell-specific manner [65, 66]. In immune cells palmitic acid, but not oleic acid, triggered inflammasome activation [67]. In contrast, both these saturated fatty acids increased caspase-1 activity and IL-1β maturation in cultured primary human trophoblast cells (Fig. 6.2). This discrepancy is particularly relevant in pregnancy because obese mothers typically have high circulating levels of SFA, especially palmitic acid and monounsaturated fatty acid (MUFA) such as oleic acid [68]. Interestingly, both palmitic and oleic acids are capable of activating TLR4 [35, 69], suggesting that these fatty acids stimulate both the necessary pathways required for robust inflammasome activation. Moreover, most Western diets have low levels of long-chain n-3 PUFA – in particular docosahexaenoic acid (DHA) [70], which has previously been shown to prevent inflammasome activation in macrophages [71–73]. Consistent with these reports, our preliminary data demonstrates that DHA decreases (pro-)caspase-1 expression thereby attenuating IL-1β maturation (Fig. 6.2). The high dietary SFA and MUFA combined with low n-3 PUFA may therefore favor inflammasome activation in the placentas of high BMI mothers (Fig. 6.3).

Mechanisms linking dietary inflammasome activation to placental insulin resistance. Inflammasome-mediated IL-1β maturation requires a “priming” signal involving TLR4-NF-κB-mediated pro-IL-1β expression. A secondary “activation” signal in the form of DAMPs or dietary fatty acids promotes NLRP3-inflammasome complex formation resulting in caspase-1-mediated proteolytic cleavage of IL-1β. Mature IL-1β may function in an autocrine manner to degrade IRS-1 protein leading to decreased insulin signaling and insulin-mediated amino acid transport. AAT amino acid transport, ASC apoptosis-associated speck-like protein containing a CARD adaptor, IR insulin receptor, IRS-1 insulin receptor substrate-1, MUFA monounsaturated fatty acid, NF-κB nuclear factor kappa B, NLRP3 nod-like receptor 3, PAMP pathogen-associated molecular pattern, ROS reactive oxygen species, SFA saturated fatty acid, TLR4 toll-like receptor 4
The clinical significance of increased inflammasome activation leading to IL-1β-mediated insulin resistance in the placenta may be substantial. Although, increased placental amino acid transport activity has been reported in obese [13] or diabetic mothers [74], other studies demonstrate either no changes [75] or reduced transport activity [76, 77]. The discrepancy between these reports may be due to the inflammatory status of these placentas. It is possible that an increased placental inflammatory response associated with maternal obesity [8, 10–12, 35] or diabetes [78–81] may limit placental insulin-mediated transport, featuring an adaptive mechanism to limit excessive fetal growth in the presence of maternal hyperinsulinemia. Providing circumstantial support for this concept are reports demonstrating that the inflammatory processes associated with fetal growth restriction also inhibit placental transport activity in pregnancies complicated by malaria, a condition which increases the risk of fetal growth restriction [48].
Glyburide is a sulfonylurea agent which stimulates insulin release in the pancreas. The use of glyburide in the management of gestational diabetes has been actively explored, due to its glucose-lowering effects. While generally considered a safe alternative to insulin treatment in gestational diabetes pregnancies [82], a recent meta-analysis demonstrated that glyburide use in pregnancy is associated with increased fetal growth [83]. Although it is currently unclear if the association represents a cause and effect, it is interesting to note that glyburide is a potent inhibitor of inflammasome activation [84]. Hence inhibition of inflammasomes by glyburide may reduce IL-1β maturation, resulting in unrestricted insulin signaling in the placenta. Because insulin stimulates placental amino acid transport [45, 85], this may result in increased nutrient transfer to the fetus. In vitro studies testing this hypothesis will establish a mechanistic link between glyburide use in pregnancy and fetal growth.
5 Conclusions
The incidence of maternal obesity and its comorbidities (diabetes and cardiovascular disease) continues to surge, with major public health ramifications. Maternal obesity not only affects newborn health, but it also impacts the long-term health of the child leading to increased risk of childhood obesity and diabetes. Given all the evidence for the critical role of the in utero environment for lifelong health, understanding the impact of obesity in pregnancy represents a significant challenge, but also an intriguing opportunity to improve the health of future generations. Obesity is associated with the triad of metabolic complications: insulin resistance, dyslipidemia, and inflammation. Studies in recent years have demonstrated intimate links between these metabolic pathways, for example, inflammation causes insulin resistance and dyslipidemia, and dyslipidemia causes inflammation and insulin resistance. Excess nutrients, in particular dietary fatty acids, have emerged as a common instigator of these metabolic complications influencing insulin signaling and inflammatory pathways. As an organ of exchange, the placenta is a critical mediator of fetal health and is highly responsive to maternal health and diet. We recently reported inflammasome activation in the placentas of high BMI mothers, which inhibits placental insulin signaling and nutrient transport function. In vitro data suggests that placental inflammasomes are highly responsive to dietary fatty acids, with SFA and MUFA promoting inflammasome activation and n-3 PUFA inhibiting its activity. Future clinical studies are warranted to determine whether dietary interventions during pregnancy can impact upon infant and placental health by altering placental inflammatory pathways.
References
Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–7.
Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32:1076–80.
Drake AJ, Reynolds RM. Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction. 2010;140:387–98.
Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, Marceau P. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics. 2006;118:e1644–9.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7.
Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–7.
Basu S, Haghiac M, Surace P, Challier JC, Guerre-Millo M, Singh K, Waters T, Minium J, Presley L, Catalano PM, Hauguel-de Mouzon S. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring). 2011;19:476–82.
Aye IL, Lager S, Ramirez VI, Gaccioli F, Dudley DJ, Jansson T, Powell TL. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod. 2014;90:129.
Basu S, Leahy P, Challier JC, Minium J, Catalano P, Hauguel-de Mouzon S. Molecular phenotype of monocytes at the maternal-fetal interface. Am J Obstet Gynecol. 2011;205(265):e261–8.
Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29:274–81.
Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, Denison FC. Placental structure and inflammation in pregnancies associated with obesity. Placenta. 2011;32:247–54.
Saben J, Lindsey F, Zhong Y, Thakali K, Badger TM, Andres A, Gomez-Acevedo H, Shankar K. Maternal obesity is associated with a lipotoxic placental environment. Placenta. 2014;35:171–7.
Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, Jansson T, Powell TL. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2013;98:105–13.
Mele J, Muralimanoharan S, Maloyan A, Myatt L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am J Physiol Endocrinol Metab. 2014;307(5):E419–25.
DuBois BN, O’Tierney-Ginn P, Pearson J, Friedman JE, Thornburg K, Cherala G. Maternal obesity alters feto-placental cytochrome P4501A1 activity. Placenta. 2012;33:1045–51.
Higgins L, Mills TA, Greenwood SL, Cowley EJ, Sibley CP, Jones RL. Maternal obesity and its effect on placental cell turnover. J Matern Fetal Neonatal Med. 2013;26:783–8.
Hayward CE, Higgins L, Cowley EJ, Greenwood SL, Mills TA, Sibley CP, Wareing M. Chorionic plate arterial function is altered in maternal obesity. Placenta. 2013;34:281–7.
Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, Grove KL. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology. 2011;152:2456–64.
Brass E, Hanson E, O’Tierney-Ginn PF. Placental oleic acid uptake is lower in male offspring of obese women. Placenta. 2013;34:503–9.
Desforges M, Ditchfield A, Hirst CR, Pegorie C, Martyn-Smith K, Sibley CP, Greenwood SL. Reduced placental taurine transporter (TauT) activity in pregnancies complicated by pre-eclampsia and maternal obesity. Adv Exp Med Biol. 2013;776:81–91.
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4.
Walsh JM, McGowan CA, Byrne JA, Rath A, McAuliffe FM. The association between TNF-alpha and insulin resistance in euglycemic women. Cytokine. 2013;64:208–12.
Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–9.
Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, Adams SH, Hwang DH. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. 2012;53:2002–13.
Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23:509–12.
Erridge C, Kennedy S, Spickett CM, Webb DJ. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. 2008;283:24748–59.
Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–9.
Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol. 2004;24:7806–19.
Udalova IA, Kwiatkowski D. Interaction of AP-1 with a cluster of NF-kappa B binding elements in the human TNF promoter region. Biochem Biophys Res Commun. 2001;289:25–33.
Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22.
Vallerie SN, Hotamisligil GS. The role of JNK proteins in metabolism. Sci Transl Med. 2010;2:60rv65.
Rebholz SL, Jones T, Burke KT, Jaeschke A, Tso P, D’Alessio DA, Woollett LA. Multiparity leads to obesity and inflammation in mothers and obesity in male offspring. Am J Physiol Endocrinol Metab. 2012;302:E449–57.
Li HP, Chen X, Li MQ. Gestational diabetes induces chronic hypoxia stress and excessive inflammatory response in murine placenta. Int J Clin Exp Pathol. 2013;6:650–9.
Heerwagen MJ, Stewart MS, de la Houssaye BA, Janssen RC, Friedman JE. Transgenic increase in N-3/n-6 Fatty Acid ratio reduces maternal obesity-associated inflammation and limits adverse developmental programming in mice. PLoS One. 2013;8:e67791.
Saben J, Zhong Y, Gomez-Acevedo H, Thakali KM, Borengasser SJ, Andres A, Shankar K. Early growth response protein-1 mediates lipotoxicity-associated placental inflammation: role in maternal obesity. Am J Physiol Endocrinol Metab. 2013;305:E1–14.
Lappas M. Cellular inhibitors of apoptosis (cIAP) 1 and 2 are increased in placenta from obese pregnant women. Placenta. 2014;35:831–8.
Cnattingius S, Villamor E, Johansson S, Edstedt Bonamy AK, Persson M, Wikstrom AK, Granath F. Maternal obesity and risk of preterm delivery. JAMA. 2013;309:2362–70.
Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, Coustan DR, Hadden DR, McCance DR, Hod M, McIntyre HD, Oats JJ, Persson B, Rogers MS, Sacks DA. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358:1991–2002.
Stewart FM, Freeman DJ, Ramsay JE, Greer IA, Caslake M, Ferrell WR. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J Clin Endocrinol Metab. 2007;92:969–75.
Thornburg KL, Louey S. Uteroplacental circulation and fetal vascular function and development. Curr Vasc Pharmacol. 2013;11:748–57.
Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol. 2009;297:C1228–35.
Lager S, Jansson N, Olsson AL, Wennergren M, Jansson T, Powell TL. Effect of IL-6 and TNF-alpha on fatty acid uptake in cultured human primary trophoblast cells. Placenta. 2011;32(2):121–7.
von Versen-Hoynck F, Rajakumar A, Parrott MS, Powers RW. Leptin affects system A amino acid transport activity in the human placenta: evidence for STAT3 dependent mechanisms. Placenta. 2009;30:361–7.
Jansson N, Greenwood SL, Johansson BR, Powell TL, Jansson T. Leptin stimulates the activity of the system A amino acid transporter in human placental villous fragments. J Clin Endocrinol Metab. 2003;88:1205–11.
Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino Acid transport in human primary trophoblast cells. Diabetes. 2010;59:1161–70.
Aye IL, Jansson T, Powell TL. Interleukin-1beta inhibits insulin signaling and prevents insulin-stimulated system A amino acid transport in primary human trophoblasts. Mol Cell Endocrinol. 2013;381:46–55.
Thongsong B, Subramanian RK, Ganapathy V, Prasad PD. Inhibition of amino acid transport system a by interleukin-1beta in trophoblasts. J Soc Gynecol Investig. 2005;12:495–503.
Boeuf P, Aitken EH, Chandrasiri U, Chua CL, McInerney B, McQuade L, Duffy M, Molyneux M, Brown G, Glazier J, Rogerson SJ. Plasmodium falciparum malaria elicits inflammatory responses that dysregulate placental amino acid transport. PLoS Pathog. 2013;9:e1003153.
Pontillo A, Girardelli M, Agostinis C, Masat E, Bulla R, Crovella S. Bacterial LPS differently modulates inflammasome gene expression and IL-1beta secretion in trophoblast cells, decidual stromal cells, and decidual endothelial cells. Reprod Sci. 2013;20:563–6.
Ammala M, Nyman T, Salmi A, Rutanen EM. The interleukin-1 system in gestational tissues at term: effect of labour. Placenta. 1997;18:717–23.
Gotsch F, Romero R, Chaiworapongsa T, Erez O, Vaisbuch E, Espinoza J, Kusanovic JP, Mittal P, Mazaki-Tovi S, Kim CJ, Kim JS, Edwin S, Nhan-Chang CL, Hamill N, Friel L, Than NG, Mazor M, Yoon BH, Hassan SS. Evidence of the involvement of caspase-1 under physiologic and pathologic cellular stress during human pregnancy: a link between the inflammasome and parturition. J Maternal Fetal Neonatal Med. 2008;21:605–16.
Jaiswal MK, Agrawal V, Mallers T, Gilman-Sachs A, Hirsch E, Beaman KD. Regulation of apoptosis and innate immune stimuli in inflammation-induced preterm labor. J Immunol. 2013;191:5702–13.
Mulla MJ, Myrtolli K, Potter J, Boeras C, Kavathas PB, Sfakianaki AK, Tadesse S, Norwitz ER, Guller S, Abrahams VM. Uric acid induces trophoblast IL-1beta production via the inflammasome: implications for the pathogenesis of preeclampsia. Am J Reprod Immunol. 2011;65:542–8.
Mulla MJ, Salmon JE, Chamley LW, Brosens JJ, Boeras CM, Kavathas PB, Abrahams VM. A role for uric acid and the Nalp3 inflammasome in antiphospholipid antibody-induced IL-1beta production by human first trimester trophoblast. PLoS One. 2013;8:e65237.
Shen F, Wei J, Snowise S, DeSousa J, Stone P, Viall C, Chen Q, Chamley L. Trophoblast debris extruded from preeclamptic placentae activates endothelial cells: a mechanism by which the placenta communicates with the maternal endothelium. Placenta. 2014;35(10):839–47.
Kavathas PB, Boeras CM, Mulla MJ, Abrahams VM. Nod1, but not the ASC inflammasome, contributes to induction of IL-1beta secretion in human trophoblasts after sensing of Chlamydia trachomatis. Mucosal Immunol. 2013;6:235–43.
Strakova Z, Srisuparp S, Fazleabas AT. Interleukin-1beta induces the expression of insulin-like growth factor binding protein-1 during decidualization in the primate. Endocrinology. 2000;141:4664–70.
Seki H, Zosmer A, Elder MG, Sullivan MH. The regulation of progesterone and hCG production from placental cells by interleukin-1beta. Biochim Biophys Acta. 1997;1336:342–8.
Tsukihara S, Harada T, Deura I, Mitsunari M, Yoshida S, Iwabe T, Terakawa N. Interleukin-1beta-induced expression of IL-6 and production of human chorionic gonadotropin in human trophoblast cells via nuclear factor-kappaB activation. Am J Reprod Immunol. 2004;52:218–23.
Keelan JA, Groome NP, Mitchell MD. Regulation of activin-A production by human amnion, decidua and placenta in vitro by pro-inflammatory cytokines. Placenta. 1998;19:429–34.
Fortunato SJ, Menon R. IL-1 beta is a better inducer of apoptosis in human fetal membranes than IL-6. Placenta. 2003;24:922–8.
Aye IL, Ramirez VI, Gaccioli F, Lager S, Jansson T, Powell T. Activation of placental inflammasomes in pregnant women with high BMI. Reprod Sci. 2013;20(S3):73A–73A.
Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. 2009;119:3502–11.
Hastie R, Lappas M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta. 2014;35(9):673–83.
Luo X, Yang Y, Shen T, Tang X, Xiao Y, Zou T, Xia M, Ling W. Docosahexaenoic acid ameliorates palmitate-induced lipid accumulation and inflammation through repressing NLRC4 inflammasome activation in HepG2 cells. Nutr Metab. 2012;9:34.
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15.
L’Homme L, Esser N, Riva L, Scheen A, Paquot N, Piette J, Legrand-Poels S. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J Lipid Res. 2013;54:2998–3008.
Chen X, Scholl TO, Leskiw M, Savaille J, Stein TP. Differences in maternal circulating fatty acid composition and dietary fat intake in women with gestational diabetes mellitus or mild gestational hyperglycemia. Diabetes Care. 2010;33:2049–54.
Lager S, Gaccioli F, Ramirez VI, Jones HN, Jansson T, Powell TL. Oleic acid stimulates system A amino acid transport in primary human trophoblast cells mediated by toll-like receptor 4. J Lipid Res. 2013;54:725–33.
Grant WF, Gillingham MB, Batra AK, Fewkes NM, Comstock SM, Takahashi D, Braun TP, Grove KL, Friedman JE, Marks DL. Maternal high fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PLoS One. 2011;6:e17261.
Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. 2013;191:4337–47.
Williams-Bey Y, Boularan C, Vural A, Huang NN, Hwang IY, Shan-Shi C, Kehrl JH. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-kappaB activation and enhancing autophagy. PLoS One. 2014;9:e97957.
Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, Guarda G, Tian Z, Tschopp J, Zhou R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154–63.
Jansson T, Ekstrand Y, Bjorn C, Wennergren M, Powell TL. Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes. 2002;51:2214–9.
Dicke JM, Henderson GI. Placental amino acid uptake in normal and complicated pregnancies. Am J Med Sci. 1988;295:223–7.
Farley DM, Choi J, Dudley DJ, Li C, Jenkins SL, Myatt L, Nathanielsz PW. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta. 2010;31:718–24.
Kuruvilla AG, D’Souza SW, Glazier JD, Mahendran D, Maresh MJ, Sibley CP. Altered activity of the system A amino acid transporter in microvillous membrane vesicles from placentas of macrosomic babies born to diabetic women. J Clin Invest. 1994;94:689–95.
Mrizak I, Grissa O, Henault B, Fekih M, Bouslema A, Boumaiza I, Zaouali M, Tabka Z, Khan NA. Placental infiltration of inflammatory markers in gestational diabetic women. Gen Physiol Biophys. 2014;33:169–76.
Radaelli T, Varastehpour A, Catalano P, Hauguel-de Mouzon S. Gestational diabetes induces placental genes for chronic stress and inflammatory pathways. Diabetes. 2003;52:2951–8.
Sisino G, Bouckenooghe T, Aurientis S, Fontaine P, Storme L, Vambergue A. Diabetes during pregnancy influences Hofbauer cells, a subtype of placental macrophages, to acquire a pro-inflammatory phenotype. Biochim Biophys Acta. 2013;1832:1959–68.
Yu J, Zhou Y, Gui J, Li AZ, Su XL, Feng L. Assessment of the number and function of macrophages in the placenta of gestational diabetes mellitus patients. J Huazhong Univ Sci Technolog Med Sci. 2013;33:725–9.
Langer O, Conway DL, Berkus MD, Xenakis EM, Gonzales O. A comparison of glyburide and insulin in women with gestational diabetes mellitus. N Engl J Med. 2000;343:1134–8.
Zeng YC, Li MJ, Chen Y, Jiang L, Wang SM, Mo XL, Li BY. The use of glyburide in the management of gestational diabetes mellitus: a meta-analysis. Adv Med Sci. 2014;59:95–101.
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187:61–70.
Aye IL, Gao X, Weintraub ST, Jansson T, Powell TL. Adiponectin inhibits insulin function in primary trophoblasts by PPARalpha-mediated ceramide synthesis. Mol Endocrinol. 2014;28:512–24.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Aye, I.L.M.H., Lager, S., Powell, T.L. (2015). The Role of Placental Inflammasomes in Linking the Adverse Effects of Maternal Obesity on Fetal Development. In: Ferrazzi, E., Sears, B. (eds) Metabolic Syndrome and Complications of Pregnancy. Springer, Cham. https://doi.org/10.1007/978-3-319-16853-1_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-16853-1_6
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16852-4
Online ISBN: 978-3-319-16853-1
eBook Packages: MedicineMedicine (R0)