
Abstract
Whole genome amplification (WGA) have been applied to single cell copy number variations (CNVs) analysis, which is a common genomic mutation associated with various diseases and provides new insight for the fields of biology and medicine. However, the WGA-induced bias based on multiple displacement amplification (MDA) significantly limits sensitivity and specificity for CNVs detection. To address the limitations, an empirical algorithm for CNVs detection at single cell level was developed. This proposed method consists of base call amplification, alig- nment and analysis to remove the MDA-induced bias. We generated and analyzed about 50G short read data sets based on MDAsim, a software to amplify the chromosome 21 into various coverage. Simulation experiments have shown that the coverage tended to be less than average in genomic GC-enriched (>45 %) regions, implying a significant amplification bias within these regions. Base substitution error frequencies with G > A transversion is being among the most frequent and C > T, G > T transversions are among the least frequent substitution errors. The estimated substitution was employed to compensate errors to correct bias readings.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Whole genome amplification (WGA), a widespread approach to amplify inadequate amounts of DNA samples for sequencing in Single-cell genomics analysis [1–3] have been extensively used to single-cell copy number variations (CNVs) analysis, at the cost of introducing biases [4–8]. As a key factor in cancer mutation [1, 9], CNVs is a common genomic variation closely associated with assorted diseases, the detection and analysis of which contributes to the research of biology and medicine.
Limited by the number of the specimens, WGA methods are widely used to facilitate the CNVs detection and analysis at single-cell level, such as Polymerase chain reaction (PCR) and multiple displacement amplification (MDA). Multiple displacement amplification (MDA), a DNA amplification method widely used in Single-cell genomics studies, uses Φ29 DNA polymerase and random primers to generate large amount of DNA template for genome samples [10]. Compared with PCR-based amplification method, MDA can be amplifid to the output with high quality and low error rates while not limited by the target length [10].
The introduction of WGA method insures the accuracy of CNVs’ detection, nevertheless, it at the same time gives rise to amplification biases [4–8]. Although the mechanism of how the DNA polymerase function is influenced by GC content remains unsettled, it has been suggested that the amplification quality of template is closely associated with GC content [5]. The over-amplification or under-amplification of specific region of template can result from the rich or poor GC content [5], causing misrepresentation of that region. Thus, the WGA-induced bias significantly limits sensitivity and specificity for CNVs detection.
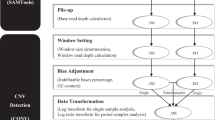
To investigate the limitation, an empirical algorithm was developed for CNVs detection at single cell level. The proposed method consists of base call amplification, alignment and analysis for MDA-induced bias removal, with the aid of Multiple Displacement Amplification Simulator (MDAsim). MDAsim is a software developed to simulate MDA process, which generates simulated reads well approximated to the experimental ones [11]. By comparing the simulated outputs with the input chromosome 21, corrective measures were carried out to remove and compensate the MDA-induced biases. The proposed algorithm is expected to optimize the MDAsim analysis and improve the accuracy of the simulation process.
In the proposed algorithm, chromosome 21 from human genome was selected as reference template and was amplified into various coverage based on MDAsim, thus generating about 50G short read data sets. Each read has been trimmed to 50 bases and aligned to chromosome 21 by BWA [12]. Extensive statistical analysis has been conducted to investigate the correlation between genomic GC content and corresponding read coverage, per-positon error numbers considering the wrong base calls only, per-base error rate considering all base calls.Finally, we conclude the base substitution error frequencies.
2 Methods
A systematical pipeline was designed to analyze the simulated data set. The pipeline consists of three steps: amplification, alignment and analysis. The chromosome 21 was selected to amplify its base calls by MDAsim [11].
Step 1: Amplification. Since the whole chromosome 21 is too large to analysis by the amplification software. The 48 M reference was splitted into 45 subgroups, each of those is 3 M in length with the index repeating 2 M each time (1–3, 2–4……). The resulted 3 M fasta file was then used to amplify the chromosome 21. With the help of MDAsim [11], chromosome 21 is amplified into different coverage range under various parameter settings to simulate the reads with different GC contents.
Step 2: Alignment. BWA [12] is used to map the amplified reads in different coverage against the reference template. Its alignment process generates the intermediate binary sai file and final sam file. In the sam file, BWA outputs the sam file in the SAM format [14], each line of which consists of the alignment information of each read.
Step 3: Analysis. To extract the classified errors from the BWA outputs and analyze the MDA-induces biases, an extensive statistical analysis has been developed to analyze the correlation between the read coverage and GC content, base substitution errors in reads, per-postion error numbers considering the wrong base calls and per-base error rate considering all the base calls.
3 Results
The chromosome 21 was amplified into different coverage, extending from 40 to 60. The BWA analysis was then conducted on the resulted data sets.
Because only in that coverage can we find the output with U1 (match with exactly one error(insertion or replacement).Finally, we acquired 90923032 50mer reads from the process that the perl scripts reported to be uniquely matched against the chr21 reference sequence which were labeled U0, U1 or U2 respectively (Fig. 1).

Pie chart of the read analysis. The four categories are NM, no match found; U0, exact match found without any error; U1, match with exactly one error (insertion or replacement); U2, match with exactly two errors (insertion or replacement); U0’, exact match found without any error, but its length is less than 50.
-
Correlation between the read coverage and GC content
The amplification was amplified and aligned with the coverage 50 from the chromosome 21 to analyze the GC biases in WGA. The number of reads starting in a sliding window of length in 1kbp is estimated firstly. The analysis of the correlation between the statistic and the characteristic of the sequence of chromosome 21 shows a positive correlation between the read coverage and GC content. The coverage increases as well as GC content. However, when GC content is larger than 45 %, the coverage decreases with the GC content.increasing.
We defined the quotient between the reads number of each observation window and the average reads number as relative read number (RRN) [13], which ideally would be equal to one. By comparing the GC content and RRN, we discovered that the RRN tended to be less than average in genomic GC-rich (>45 %) (shown in Fig. 2), implying the amplification bias within these regions. Futhermore, the base substitutional analysis was done in these regions to correct the biases.
Fig. 2. 
Correlation of the read coverage and GC content: 50mer reads acquired from the chromosome 21. Each bar corresponds to the number of reads recorded for a 1-kbp window.
-
Analysis of base substitution errors in reads
The overall substitution error is calculated and summarized in Table 1. There are twelve possible substitution errors (8 transversions and 4 transitions) when a base call happens. The transition error of G > A happens most frequently, which accounts for almost half of the substitution errors, and the least frequent substitution error is G > T and C > T. The most frequent base to happen substitution error is G, and the least is A. However, A is the most frequent base to be changed into while T is the least.
Table 1. Base substitution frequencies in the read data sets Futher experiment is also done to analyze the GC-enriched (>45 %, Fig. 2) region’s substantial error, through which we can compensate the biased region’ (>45 %, Fig. 2) substitutional base call. The transition error of T > C happens most frequently, which accounts for almost half of the substitution errors, and the least frequent substitution error is T > A and G > C. The most frequent base to happen substitution error is T, and the least is A. However, A is the most frequent base to be changed into while G is the least. With these estimated substitutional information, we compensate errors to correct bias readings in the GC-enriched (>45 %, Fig. 2) regions.
-
Numbers of wrong base calls in reads verses the position along the read
All the U1 U2 and U3 reads are selected for analysis, i.e. 3817 read (cf. Fig. 1), on the occurrence of errors per position. Two types of measurements are provided to quantify the errors. The first measurement calculated per-positon error numbers considering all the wrong base calls. The second measurement calculated per-base error rate among all the base calls. The results are shown in Fig. 3. The figure (a) shows that the high fraction of the wrong base calls occurs at the first and last position of the read. 8.2 % of the errors in the data sets are found at read position 1, and 6.7 % of errors are found at the last read position (position 50 in the data set Fig. 3a). The rate of the wrong base calls (Fig. 3b) has shown similar tendency. The rate is the highest at the first position along the read and the second highest at the last position of the read.
Fig. 3. 
Numbers of wrong base calls in reads depending on the position along the read. (a) Per-positon error numbers considering all the wrong base calls. (b) Per-base error rate among all the base calls.


4 Conclusion
In this study, an algorithm was developed to detect the bias of multiple displacement amplification and the relation between GC content and coverage at the single cell level. The proposed method consists of base call amplification, alignment, analysis and base call substitutional compensate. The chromosome 21 was selected and amplified into 50 coverage. The defined RRN shows that the coverage tends to be less than average within GC-rich regions (Fig. 2). The GC-rich regions’ substitution error and overall substitution error were extensively analyzed and estimated to compensate the base substitution error. For the overall reads, wrong base calls are frequently preceded by base G. Base substitution error frequencies vary with G > A transversion being among the most frequent and C > T, G > T transversions among the least frequent substitution errors. With these estimated substitutional information, we compensate errors to correct bias readings in the GC-enriched (>45 %, Fig. 2) regions. For the biased region (GC-rich regions), the transition error of T > C happens most frequently, and the least frequent substitution error is T > A and G > C. With these estimated substitutional information, we compensate the errors to correct the MDA-induced bias.
References
Navin, N., Kendall, J., Troge, J., Andrews, P., Rodgers, L., McIndoo, J., Cook, K., Stepansky, A., Levy, D., Esposito, D.: Tumour evolution inferred by single-cell sequencing. Nature 472(7341), 90–94 (2011)
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R.: The sequence alignment/map format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009)
Benjamini, Y., Speed, T.P.: Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 40(10), e72 (2012)
Dohm, J.C., Lottaz, C., Borodina, T., Himmelbauer, H.: Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 36(16), e105 (2008)
Voet, T., Kumar, P., Van Loo, P., Cooke, S.L., Marshall, J., Lin, M., Esteki, M.Z., Van der Aa, N., Mateiu, L., McBride, D.J.: Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res (2013)
Hou, Y., Song, L., Zhu, P., Zhang, B., Tao, Y., Xu, X., Li, F., Wu, K., Liang, J., Shao, D.: Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148(5), 873–885 (2012)
Talkowski, M.E., Rosenfeld, J.A., Blumenthal, I., Pillalamarri, V., Chiang, C., Heilbut, A., Ernst, C., Hanscom, C., Rossin, E., Lindgren, A.M.: Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149(3), 525–537 (2012)
Dean, F.B., Nelson, J.R., Giesler, T.L., Lasken, R.S.: Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 11(6), 1095–1099 (2001)
Tagliavi, Z., Draghici, S.: MDAsim: A multiple displacement amplification simulator. In: 2012 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pp. 1–4. IEEE (2012)
Paez, J.G., Lin, M., Beroukhim, R., Lee, J.C., Zhao, X., Richter, D.J., Gabriel, S., Herman, P., Sasaki, H., Altshuler, D.: Genome coverage and sequence fidelity of Φ29 polymerasee-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 32(9), e71 (2004)
Li, H., Durbin, R.: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14), 1754–1760 (2009)
Arriola, E., Lambros, M.B., Jones, C., Dexter, T., Mackay, A., Tan, D.S., Tamber, N., Fenwick, K., Ashworth, A., Dowsett, M.: Evaluation of Phi29-based whole-genome amplification for microarray-based comparative genomic hybridisation. Lab. Invest. 87(1), 75–83 (2007)
Bredel, M., Bredel, C., Juric, D., Kim, Y., Vogel, H., Harsh, G.R., Recht, L.D., Pollack, J.R., Sikic, B.I.: Amplification of whole tumor genomes and gene-by-gene mapping of genomic aberrations from limited sources of fresh-frozen and paraffin-embedded DNA. J. Mol. Diagn. 7(2), 171–182 (2005)
Zhang, C., Zhang, C., Chen, S., Yin, X., Pan, X., Lin, G., Tan, Y., Tan, K., Xu, Z., Hu, P.: A single cell level based method for copy number variation analysis by low coverage massively parallel sequencing. PLoS ONE 8(1), e54236 (2013)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this paper
Cite this paper
Xu, B., Li, T., Luo, Y., Xu, R., Cai, H. (2014). An Empirical Algorithm for Bias Correction Based on GC Estimation for Single Cell Sequencing. In: Peng, WC., et al. Trends and Applications in Knowledge Discovery and Data Mining. PAKDD 2014. Lecture Notes in Computer Science(), vol 8643. Springer, Cham. https://doi.org/10.1007/978-3-319-13186-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-13186-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13185-6
Online ISBN: 978-3-319-13186-3
eBook Packages: Computer ScienceComputer Science (R0)