
Abstract
The complex multistep mechanism of oxygen activation in P450 is reviewed as a sequence of the following reactions: Substrate binding, reduction of the heme iron from ferric to the ferrous state, binding of dioxygen, second electron transfer and formation of peroxo-ferric intermediate, two sequential protonation events to give hydroperoxo-ferric intermediate, and finally, after O–O bond scission, the ferryl-oxo intermediate, termed compound I. Details of these processes and the role of interactions with redox partners, as well as substrate variability in the overall efficiency of P450 catalysis, are discussed. In addition, common points and variations between the soluble prokaryotic and membrane-bound eukaryotic cytochromes P450 with respect to the oxygen activation mechanisms are briefly compared.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Substrate binding
- Water ligand
- Oxygen binding
- Spin shift
- Redox potential
- Redox partner
- Ferrous dioxygen complex (oxy-complex)
- Peroxo intermediate
- Hydroperoxo intermediate
- O–O bond
- Compound I
- Compound II
- Peroxide dissociation
- Superoxide
- Uncoupling
- Mössbauer spectroscopy
- Resonance Raman (rR)
- Nanodiscs
1 A Brief History of “Oxygen Activation”
The cytochrome P450s have been the focus of attention for legions of investigators. For the basic scientists, the unique spectral properties of this heme protein provided fascinating challenges for the bioinorganic chemist. The difficult chemistry of adding an oxygen atom to an unactivated alkane intrigued the bioorganic chemist, and the need for electron transfer with proton involvement brought the physical biochemists to the table. With the known processes of the archetypical heme proteins myoglobin and hemoglobin, as well as the reductive chemistry operating in the cytochrome oxidases, it was no surprise that investigators from these fields were amongst the first to focus their attention on cytochrome P450 and its redox partner s. The concept of “oxygen activation” thus comes from two directions. First, although atmospheric dioxygen can be reactive at room temperature, e.g., in the formation of rust, typical hydrocarbons are stable until combustion at elevated temperatures. Thus, facile hydrocarbon hydroxylation or epoxidation near 37 °C requires enzymatic “activation.” From the protein standpoint, the reversible binding and release of atmospheric dioxygen by hemoglobin led to documentation of an intermediate state—the ferrous heme—O2 complex. Nature evolved the protoporphyrin IX prosthetic group within the globins to protect this oxy-ferrous complex, resulting in a relatively stable species, although after long time intervals this intermediate would “auto-oxidize” releasing superoxide and converting the heme iron to the ferric state. Since it was realized early on [1] that the cytochrome P450s also contained protoporphyrin IX heme as a prosthetic group, and hence could bind atmospheric dioxygen, the protein must be doing something to “activate” the bound dioxygen for catalysis.
The canonical overall reaction of cytochrome P450 involves the reductive scission of the O–O bond of atmospheric dioxygen to release a single molecule of water with the transfer of a single oxygen atom to the substrate:
Cytochrome P450s are thus “oxygenases” as one or more oxygen atoms from O2 are incorporated into a substrate molecule, following the discovery of this class of enzymes by Hayaishi and Mason in 1955 [2, 3]. Very soon thereafter the first experimental proof of steroid hydroxylation by a mammalian oxygenase was identified by using 18O2 for the reaction [4], although the enzyme responsible for this, CYP11B1, was not identified until 1965 [5]. A beautiful review of the early P450 history was provided by Estabrook [6]. Since a single oxygen atom is inserted into a substrate, the P450s are “monoxygenases” and require additional redox transfer partners to provide the two electrons (and potentially the two protons) necessary to reduce the other oxygen atom from O2 to water. Historically, this led to the cytochrome P450s also being called “mixed function oxidases” as they operated like a half-way point of the cytochrome oxidase stoichiometry in which four electrons and protons are used to fully convert O2 to two molecules of water. As we now know, the cytochromes P450 can carry out a variety of additional organic transformations, including carbon–carbon bond scission and formation, dealkylation, heteroatom oxygenation, and halogenation/dehalogenation. We will discuss these other reactivities of the cytochromes P450 later in the chapter in the context of what they teach us about the various states of “oxygen activation.” Since this chapter is devoted to the mechanisms of oxygen activation, it is useful to briefly mention early ideas of how the relatively inert O2 molecule could be “activated.” Again we can organize the discussion along two lines of focus: the enzymology of the P450 hemoprotein and the dioxygen molecule itself.
Debates as to the mechanisms of oxygen activation heated up in the early 1970s. From the standpoint of O2, this was also the era of intense arguments as to the chemical reactivity of superoxide , a one electron reduced O2. Early discussion by Fridovich and others [7] suggested O2− itself could attack unactivated carbon centers, while others, led by Fee et al. [8], argued that superoxide was at best a mild reductant and could not by itself institute carbon oxidation. At the same time in history, enzymologists documented the existence of the ferrous dioxygen complex of P450 isolated from Pseudomonas putida (P450 CYP101A1). Since this protein could be obtained in large quantities, it could be investigated by a plethora of spectroscopies. Using Mössbauer spectroscopy it was shown that in the ferrous-dioxygen intermediate of CYP101A1, stabilized at cryogenic temperatures, the iron was in the ferric state, analogous to the Weiss model proposed for hemoglobin and myoglobin. If the iron looks ferric and there is an extra electron in the Fe–O2 system, then the electron density must favor the superoxide resonance form. Hence the thought: Was the active form of O2 in P450 catalysis the superoxide anion? Reality set in, however, when it was noted that the Fe–O2 complex of heme proteins could not carry out even simple oxygenation reactions—a second electron was required for “activation.”
The early 1970s was also the time when interesting chemistries of the second-row nonmetals carbon and nitrogen were revealed when they were missing two electrons from their valence shell. These so-called carbene and nitrene species were shown to be able to directly insert into C–C, C–H and other organic bonds. What about oxygen? Could a six-electron oxygen atom provide the observed reactivity of the P450 enzymes? The term “oxene transferase” was proposed by Ullrich and coworkers to describe this form of activated oxygen [9]. Simple electron counting from a ferrous–dioxygen complex after a second electron input and the release of water indicated the presence of a six-electron oxygen atom somehow bound to a ferric heme. On the other hand, it was difficult to see how such an electron-deficient species could dissociate from the heme and react with a nearby substrate.
The solution to the identification of the “active oxygen species” in P450 catalysis came in 1976 through the efforts of Groves in collaboration with the Coon laboratory [10, 11]. Understanding the nature of an “oxene” bound to ferric heme, Groves realized that there would be two open orbitals on the oxygen that could initiate radical chemistry. He proposed an “oxygen rebound” mechanism wherein this species, formally at the redox state of compound I as observed in the peroxidase class of enzymes, could abstract a hydrogen from a substrate C–H bond, formally generating a hydroxyl radical bound to heme that could then undergo radical recombination with the substrate carbon radical to generate the hydroxylated product. Showing the transient existence of a substrate carbon radical intermediate was strong evidence for this being the intermediate in oxygen activation [11, 12].
2 The Plethora of Chemical Reactivities of Cytochrome P450
The initial focus on the ability of P450 to catalyze the oxidation of an unactivated carbon center was a driver for the chemical community, while parallel interests that focused on metabolic transformations in humans expanded the spectrum of activities associated with P450 metabolism. In addition to hydroxylation of unactivated alkanes , this includes epoxidation of olefinic substrates, the addition of oxygen to heteroatoms such as sulfur, the dealkylation of amines, and the formation and breakage of carbon–carbon bonds. These include reactions involved in human health and disease, such as the epoxidation of aromatics as part of carcinogen activation (e.g., benzo(a)pyrene) and facile heteroatom dealkylation as exemplified by the O-demethylation that converts codeine into morphine. These human relevancies brought the large body of pharmacologists and toxicologists into the community studying the cytochromes P450. With the growing involvement of P450 in multiple biotransformations, a natural question emerged as to the number of isozymes that might be present. Initially, several variants were found in animal liver, a key site for first pass metabolism. They were first isolated as pure proteins though enormous efforts by the Coon laboratory and others, with the isozymes labeled LM1, LM2, LM3, LM4, etc., for liver microsomal fraction 1, etc. The general feeling at the time was that there could be a dozen or even 20 different isozymes of P450 in animals and perhaps a few more in bacteria and plants. As is beautifully described elsewhere in this volume, there are now over 20,000 P450 genes identified [13]! The functions of all these P450s can be artificially separated into two classes: Those involved in the synthesis of intermediary metabolites, such as prostaglandins and hormones in humans, and those involved in catabolic reactions often associated with xenobiotic breakdown—most prevalent in the human liver, kidney, and epithelial tissues. This classification also applies to plants, insects, etc., as described by Schuler et al. in this volume (Chap. 7).
3 The Three-Dimensional Structure of Cytochrome P450
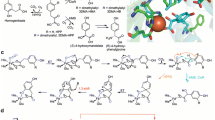
By the mid-1970s enzymologists were comforted by the availability of a three-dimensional structure of their enzyme, although the technology was primitive by today’s standards. One requirement to obtain an X-ray structure in this era was for a substantial amount of highly purified protein. The only P450 available in the needed quantity and quality was P450cam or CYP101A1. The crystallization and solution of the structure is told by Poulos and Johnson in this volume together with those of several other soluble P450s. The vast majority of the P450s in nature, however, are anchored to a membrane and the solution of membrane protein structure remains a significant hurdle today. Indeed, one entire session of an international P450 meeting was devoted to the debate as to how good a structural model CYP101A1 would be for the membrane-bound P450s [14]. Johnson and Poulos (Chap. 1 in this volume) summarize the amazing progress in solving the structure of the membrane-bound P450s. We now recognize that all members of this large super family of P450s possess basically the same fold, with subtle differences being present that reflect specificity for substrates and redox partner s. Additionally, the past four decades of work have unambiguously shown that all P450s operate by basically the same reaction cycle (Fig. 3.1), including the stoichiometry of oxygen and reducing equivalents. However, the degree of coupling, or efficiency of converting atmospheric O2 and electrons to substrate-derived products can vary widely.

Reaction cycle of cytochrome P450, reproduced with permission from American Chemical Society from [15]
Most P450s operate with a single substrate-binding site, often with the high degree of specificity needed, for example, in hormone biosynthesis. However, some P450s can bind more than one substrate molecule, either in an enlarged active site or in a distant effector or allosteric site. This can lead to a profound effect on metabolic throughput as will be discussed in detail subsequently.
The remainder of this chapter, as well as contributions from other authors, will address the spectroscopic characterization of the intermediate states that lead to the ultimate oxygenating species operating in the cytochromes P450. We will also address the aspects of the protein structure that allow control of electron and proton input into the catalytic cycle to control the stability and reactivity of these intermediate states. Appropriate results that define the side uncoupling pathways, as well as other forms of the reduced oxygen-bound P450 heme that have the potential for substrate metabolism, will conclude this review and also provide the critical link to other forms of “active oxygen.”
4 Substrate Binding , Spin Shift , and Redox Potential s
Substrate binding to cytochrome P450 is an important step in the overall mechanism of P450 catalysis, not only because it is necessary to position the substrate in the proper orientation in the immediate vicinity of the heme bound catalytically competent “active oxygen.” Equally important, it serves as the trigger activating the electron transfer from the redox partner to the heme iron resulting in reduction of the iron from the ferric Fe3 + to the ferrous Fe2 + state. This, in turn, is necessary for binding of oxygen to the ferrous cytochrome P450 and formation of the oxygenated intermediate. In general, the regulatory role of substrate binding as the trigger initiating the reduction is used in the cytochromes P450 [16, 17], as well as in some nonheme enzymes [18], to minimize production of reactive oxygen species and unproductive waste of nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH).
Usually much tighter substrate binding is observed for P450s involved in specific biosynthesis of hormones and other regulatory compounds. Examples include the high affinity of cytochromes P450 involved in steroid hormone biosynthesis towards their natural substrates and other synthetic steroid compounds [19–21]. Interestingly, many P450s that formally belong to this class can also bind and metabolize compounds not related to their native substrates, albeit with lower affinity and efficiency. Such examples are described for CYP101A1 [22–24], CYP102 [25–27], and CYP46 [28]. For xenobiotic metabolizing cytochromes P450, which can bind and catalyze oxidative transformations of various organic molecules with a very broad distribution of chemical structures and molecular masses, lower substrate affinities with dissociation constants in the range of 10− 5 to 10− 3 M are more typical. Apparently, weaker substrate binding is the price for their broad substrate specificity , a requirement for the first line of chemical defense of the organism against myriads of alien, potentially toxic, and dangerous compounds. Multiple examples are described in comprehensive reviews [29–31].
The binding of hydrophobic substrates usually leads to displacement of water from the substrate-binding pocket, including the water molecule coordinated to the heme iron as the sixth (axial) ligand, as shown in Fig. 3.2 for CYP101A1. The transition of the ferric iron atom Fe3 + from the hexacoordinated to the pentacoordinated state results in a spin-state transition from low spin (S = 1/2) to high spin (S = 5/2). This change in the coordination state of the heme iron gives rise to an upshift of redox potential , which is essential for efficient reduction of the enzyme from the ferric to the ferrous state.

The difference in redox potential s between pentacoordinated and hexacoordinated porphyrins can be illustrated using the thermodynamic cycle shown in Fig. 3.3:

Coupling of the ligand L binding equilibria to the ferric and ferrous heme protein with binding constants K 1 and K 3 and of redox equilibria in the substrate free or substrate bound protein with equilibrium constants K 2 and K 4
Here the ligand L can bind to the heme iron with binding constants K 1 and K 3, which are different for the ferric and ferrous states, while K 2 and K 4 define the redox equilibria for the five-coordinated high-spin and six-coordinated low-spin heme iron respectively [16]. The overall redox equilibrium between Fe3 + and Fe2 +, i.e., the midpoint potential, can be shifted towards the strong binder, if the ligand is present [34]. In the aqueous solution, water or a hydroxide always favors the ferric state as compared to ferrous, so that K 1 > 1 and K 3 < 1, and Fe3 + is typically six-coordinated in cytochromes P450 in the absence of a substrate, while Fe2 + is five-coordinated. As a result, the thermodynamic (redox) equilibrium between the ferric and ferrous states of the heme iron is shifted to the former in the absence of substrates, while substrate binding displaces the water molecule from the sixth coordination position, thus destabilizing the ferric state and lifting the midpoint potential. Experimentally measured shifts of the redox potential s in cytochromes P450 caused by substrate binding are in the range 80–170 mV [16, 35–37]. In most cases, cytochromes P450 saturated with substrates are reduced much faster [37–41]. Acceleration of the first electron transfer to cytochromes P450 in the presence of a bound substrate represents an important thermodynamic regulatory mechanism, preventing futile consumption of redox equivalents and formation of toxic superoxide and peroxide, as will be discussed in a subsequent section of this chapter. In addition, Marcus theory analysis suggests a faster electron transfer in the presence of substrate due to a lower reorganization energy [42]. The spin-state equilibrium in cytochromes P450 is temperature dependent and can be probed by temperature jump studies. Direct kinetic measurements show that the typical rates of spin-state relaxation after temperature jump are in the range of 400–2000 s- 1 for CYP101A1 [43] and 800–2500 s- 1 for CYP102A1 [44]. The same thermodynamic coupling is responsible for the higher affinity of cytochromes P450 with respect to hydrophobic substrates at higher temperatures that is observed experimentally [44].
Substrate binding is usually fast for the soluble P450s, with apparent rates of 102 to 103 s- 1 [45] and second-order rates ~ 106 to 107 M− 1s− 1 [45, 46]. This fast binding and simple 1:1 stoichiometry is usually observed for the efficient bacterial P450s with their natural substrates, i.e., CYP101A1 with camphor [46, 47]. Comparison of camphor binding and dissociation kinetics with mutants generated to perturb the equilibrium-binding constant demonstrated fast binding in all cases, with the affinity exclusively dependent on the dissociation rate [24]. For instance, the T101M mutant had the same camphor-binding rate as the wild-type enzyme, k on = 3´107 M− 1s− 1, but an almost tenfold higher dissociation rate k off= 192 s− 1. Fast substrate binding was also reported for many other cytochromes P450, such as CYP102A1 [48] and other soluble bacterial enzymes. In many cases purified and solubilized eukaryotic cytochromes also show fast substrate binding [49]. However, in some cases, very slow substrate-binding kinetics have been observed, such as those reported for cholesterol derivatives binding to P450scc in lipid vesicles, where type I spectral changes were monitored on the scale of 15 min and apparent first-order rates obtained in the range of (4–9) 10– 4 s− 1 [50]. Such results are probably due to the extremely low solubility of cholesterol and its derivatives and slow redistribution between the aqueous phase and lipid bilayers [19]. The kinetics of NAD(P)H-dependent reduction of cytochromes P450 in the presence of their redox partner s almost always strongly depends on the presence of their substrates. Exceptions from this general rule are reported for several cytochromes P450 that are predominantly in the high-spin ferric state even before addition of a substrate, such as CYP1A2 [51–53]. These observations are in line with the redox thermodynamics modulated by substrate binding described above (Fig. 3.3). Typically, reduction of substrate-free P450 enzymes is very slow with apparent rates in the range of 10− 4–10− 2 s− 1 [54], and is much faster (sometimes by several orders of magnitude) in the substrate-bound state [17]. Sometimes the first electron-transfer step is identified as the rate-limiting step, as shown for CYP7A1 [55]. The significant acceleration of P450 reduction in the presence of substrates is easily seen in the steady-state kinetics of NAD(P)H consumption, as reported for both bacterial and eukaryotic cytochromes [56]. The acceleration of NAD(P)H oxidation can be used as an empirical test for the screening of new compounds as potential substrates for a given cytochrome P450 [57, 58] or as a rough measure of P450 activity [59].
Interactions with redox partner s are not only necessary to bring the electron donor close to the heme for efficient electron transfer . Recent structural studies of the complex of CYP101A1 with its natural redox partner, the iron–sulfur protein putidaredoxin (Pdx) [60, 61], confirmed the important allosteric regulatory role of these interactions that was first suggested in 1974 [62]. Perturbations of the CYP101A1 heme environment when complexed with its redox partner Pdx have been detected using various spectroscopic methods [63–67]. Early work by Davies and Sligar demonstrated the redox-dependent affinities of Pdx and P450 and the critical residues involved [68]. What was missing, however, was a linkage between structure and the functional implications caused by Pdx binding. It was the X-ray structure of the complex [60, 61] that clearly demonstrated that Pdx binding results in opening of the cleft in the I-helix that is necessary for directed proton delivery to the coordinated dioxygen. Thus, interactions with oxidized Pdx favor the open conformational state of CYP101A1 [60, 61]. However, Goodin et al. demonstrated an opposite effect of reduced Pdx binding on the conformational state of CYP101A1 [69]. Taken together, these results reveal a sophisticated pattern of allosteric regulatory effects of Pdx on the structure, dynamics, and functional properties of CYP101A1. In the first step, binding of reduced Pdx stabilizes the substrate-bound closed state of ferric CYP101A1 and provides optimal conditions for the first electron transfer. After reduction of the heme, oxidized Pdx dissociates and oxygen binds to the heme iron atom. Binding of reduced Pdx to the oxy-complex stabilizes the latter against autoxidation, preventing the heme from autoxidation and resulting in transfer of the second electron and formation of the peroxo-ferric intermediate. Finally, bound oxidized Pdx favors the open conformational state of CYP101A1, with the functionally important rearrangement of residues Asp251 and Thr252 in the I-helix that are necessary for efficient proton delivery to the dioxygen moiety of the peroxo-intermediate and formation of compound I (see Chap. 1 by Poulos and Johnson). Allosteric effects of interactions with redox partners have also been suggested in other systems. Fusion with different redox partners changed the regiospecificity of catalysis and also the range of chemical transformations catalyzed by the multipurpose cytochrome P450 MycG [70].
5 Oxygen Binding and the Structure of the Ferrous Dioxygen Complex
The binding of dioxygen to ferrous cytochrome P450 leads to formation of the oxy-complex, which is the last relatively stable intermediate in the catalytic cycle. This complex has dioxygen coordinated end-on to the heme iron with partial transfer of electron density from the iron to the dioxygen moiety. Based on spectroscopic and structural data [71–75], the latter can be described as partially superoxide . In general, most properties of the oxy-complexes in cytochromes P450 are similar to those of other heme proteins, including the myoglobins, hemoglobins, and heme oxygenases. An overview of the structural studies of oxy-complexes in various heme enzymes was published in 2007 [76]. Oxygen binding to cytochromes P450 is usually fast and not rate limiting for P450 catalysis at ambient conditions. Kinetic studies show second-order binding rates for CYP101A1 in the range of (0.8–1.7) 106 M− 1 s− 1 at 4–25 °C in the presence of camphor [77, 78]. These rates correspond to apparent first-order binding rates 200–300 s− 1 in aerated solutions. Similar rates have been reported for CYP1A2 [45], CYP2A6 [79], and CYP158A1 [80]. The presence of substrates can significantly impede the access of O2 and other diatomic ligands to the heme iron, and the scale of this effect can vary to a great extent with various substrates. For instance, different oxygen binding rates have been reported for CYP158A1 saturated with flaviolin (120 s− 1) or 2-hydroxy-1,4-naphthoquinone (15 s− 1) [80]. The effect of substrates on oxygen binding and autoxidation have been systematically studied for monomeric CYP3A4 incorporated into 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) Nanodiscs [81, 82]. The experimentally observed rate of O2 binding in the presence of testosterone TST and bromocryptine (BC) varies by more than an order of magnitude (350–400 s− 1 with TST and 24 s− 1 with BC at 279 K). The effect of TST is even more dramatic with respect to the binding of cyanide to CYP3A4, which is 60 times slower in the presence of substrate than in its absence [82], as is also observed for the association of small ligands like cyanide and imidazole to the ferric enzyme [83, 84] and of carbon monoxide to ferrous P450. [85]. The rate of CO binding to CYP101A1 in the absence of camphor (5´106 M− 1 s− 1) is two orders of magnitude faster than in its presence (4 · 104 M− 1 s− 1) [86]. A similar slowing of CO binding by substrate was observed for CYP108, but not for CYP102 [87]. This reduction in the binding rates of diatomic ligands in the presence of substrates is commonly observed with other heme enzymes, including indoleamine 2,3-dioxygenase (IDO) [88] and nitric oxide synthase (NOS) [89].
An interesting aspect of the kinetics of CO binding to CYP102A1 has been described by Munro et al. [90] using laser photooxidation of NAD(P)H to reduce the heme iron in the microsecond timescale ( k obs = 14,000 s− 1), a much faster rate than the typical dead time in stopped-flow studies (~ 1 ms). The surprisingly fast CO binding observed in this work, with apparent rates of 1700–3000 s− 1, was attributed to the presence of CO molecules inside the protein in the immediate vicinity of the heme iron. In this case, there is no need for penetration of the diatomic ligand from the solution into the substrate-binding pocket and diffusion towards the heme iron. It is reasonable to expect that the same may be true for other diatomic neutral gases, such as O2, and as a result the oxygen-binding step may happen in aerobic solution with apparent rates significantly higher than those measured in stopped-flow experiments, where the reduced protein is equilibrated with deoxygenated buffer before mixing with oxygenated solvent and oxygen must access the heme from outside. The same effects have also been described for CYP121 and CYP51B1 from Mycobacterium tuberculosis [91].
The first X-ray structure of the ferrous dioxygen (or ferrous-oxy complex) of a cytochrome P450 was solved by Schlichting and coworkers in 2000 using the bacterial CYP101A1 [92]. The oxygen molecule was found to fit tightly between the substrate camphor and the small cleft in the I-helix as suggested by the structure of the ferric protein [33]. Importantly, the binding of dioxygen resulted in a change in the active site hydrogen-bonding structure through the addition of two new water molecules not observed in the ferric structures. These are illustrated in Fig. 3.4. The appearance of these water molecules in the oxygenated form of CYP101A1 strongly suggested a likely path for the delivery of protons to the distal oxygen atom of the heme bound O2. This provided the first structure-based suggestion of a mechanism of oxygen activation in the cytochromes P450 [74] through site-specific proton delivery to the distal atom of the dioxygen ligand [93–95]. A first proton transfer would lead to the hydroperoxide intermediate and a second proton delivery would then lead to cleavage of the O–O bond , releasing a molecule of water and generating the higher valent compound I oxidizing species. Oxy-complex structures of wild type and mutant CYP101A1 [74] and P450eryF [73] were subsequently solved by Poulos and coworkers.

a X-ray structure of the oxy-complex of CYP101A1 (1DZ8.pdb [92]) with two new water molecules appearing in the cleft opening in the I-helix next to the coordinated dioxygen molecule. b A tentative proton delivery pathway with two new water molecules is shown in yellow
Because the resolution of the structures of oxygenated cytochromes P450 is not high enough for precise evaluation of the geometric parameters of the heme ligands, information about bond lengths and angles can be best obtained from the structures of closely related model complexes. A comprehensive review published in 1994 [96] provides an extensive description of the physical inorganic chemistry of heme oxygen complexes. For many years, the classical reference for the geometric parameters of the iron–porphyrin oxy-complex was the X-ray crystallographic study by the Collman group [97] of two picket-fence iron porphyrins with imidazole and dioxygen as axial ligands. These structures provided a clear picture of the end-on coordinated dioxygen molecule with a Fe–O–O angle of 135°–137° and O–O bond lengths of 1.23 and 1.26 Å. These structures also revealed significant mobility and multiple orientations of the coordinated dioxygen, both in plane and out of plane together with the axial histidine ligand [97, 98]. Recently, a new high-resolution structure of the oxy-complex of an iron picket fence porphyrin has been determined and the oxidation state of the iron atom was characterized by temperature dependent Mössbauer spectroscopy [99] and provided the geometric parameters of a heme iron end-on coordinated dioxygen with the highest precision. The values determined are Fe−O = 1.811 Å, Fe−O−O = 118.2°, and O−O = 1.281 Å, and an off-axis tilt of 6.2° in the complex with 2-methyl imidazole as the axial ligand. This O–O distance is in good agreement with the range expected from the Fourier transform infrared spectroscopy (FTIR) experimental frequencies of the O–O stretch mode observed for such model complexes (1150–1163 cm− 1) [100] and with the general dioxygen—superoxide —peroxide formal assignment [96, 101]. Similar bond lengths for Fe–O (1.81–1.83) Å and O–O (1.24–1.25) Å are reported in two high-resolution X-ray structures of the oxy-complex of sperm whale myoglobin [102, 103]. For reference, in various models the O–O bond length increases from 1.21 Å in dioxygen to 1.33 Å in the superoxide anion [104], and to 1.49 Å in the peroxide anion [96], concomitant with reduction of the O–O bond order from 2 to 1.5 to 1.
Another recent and important study of the oxy-complex of the picket-fence iron-porphyrin model combined the L edge extended X-ray absorption fine structure (EXAFS) and density functional theory calculations with a goal of characterizing the electronic structure of iron in this complex [105]. Comparison of X-ray absorption spectra (XAS) results obtained for the oxy-complex of several other hexa-coordinated ferrous and ferric low-spin complexes revealed strong σ-donation and strong π-interaction of the dioxygen moiety with iron, indicating a highly covalent Fe–O bond. This fact restricts the formal application of the oxidation state formalism and explains the absence of the hole in the dπ orbital of the iron, which is characteristic of all low-spin ferric complexes. XAS spectra of the oxy-complex are similar to the spectra of the bis-imidazole ferrous porphyrin, (Fig. 12 in Ref. [105]) and do not look like the spectra of ferric complexes [105]. However, the electronic configuration in the oxy-complexes strongly depends on the presence or absence of hydrogen bonds to the coordinated oxygen [103]. In the model porphyrin complexes there is no hydrogen bonding [105], while in most heme proteins there are proton-donating amino acid side chains or water molecules that can form one or two hydrogen bonds and shift the electron density towards the ferric-superoxide configuration [103].
Local interactions in the immediate vicinity of the heme and axial ligands can strongly affect the electronic structure of the oxy-complex. These can be detected by comparison of the ultraviolet–visible (UV–vis) spectra of various oxygenated cytochromes P450, nitric oxide synthase (NOS), and chloroperoxidase (CPO) , which all have identical iron coordination spheres and the same heme prosthetic group. While all display a split Soret band [106, 107], the position of the main band changes from 418 nm in CYP101A1 [77] to 430 nm in CPO [108]. Even for the same cytochrome P450 the position of the main Soret band may vary significantly in the presence of various substrates, as documented for CYP102A1 (422–425 nm) [36, 109] and for CYP3A4 (420–425 nm) [81, 110]. In CYP2B4 the UV–vis and magnetic circular dichroism (MCD) spectra of the oxy-complex with and without substrate are very similar, with the Soret maximum at 423 nm [111]. However, a red shift of the Soret band to 426–427 nm is observed in the CYP2B4 E301Q and T302A mutants, respectively, indicating slightly different configurations of the hydrogen-bonding network caused by these mutations. This is in contrast to the same mutations (D251N and T252A) in CYP101A1, where no changes in the UV–vis and MCD spectra were observed relative to the wild-type protein [112].
The most detailed and site-specific information on the bond strength and hydrogen-bonding environment of the coordinated dioxygen, as well as on the main heme vibrational modes, can be obtained using resonance Raman (rR) spectroscopy [113]. Because of the limited stability of the oxy-complex at ambient conditions, most Raman measurements are performed under cryogenic conditions using frozen solutions. The first successful rR characterization of the oxy-complex of CYP101A1 in the presence of camphor was published in 1986 [71]. A strong O–O mode at 1140 cm− 1 was identified based on isotopic shift of this band using 16O2 and 18O2 of 1121–1131 cm− 1, near that reported for isolated superoxide ions in solid matrices [104]. Note that the O–O stretching mode is usually not active in rR spectra of heme proteins that have histidine as a proximal iron ligand, although it was identified in infrared (IR) spectra of the oxy-complexes of hemoglobin and myoglobin at 1135 cm− 1 [114]. However, in some cases the O–O stretch mode was experimentally observed, i.e., in oxyhemoglobins from Chlamydomonas (1136 cm− 1) and from Sinechocystis (1133 cm− 1) [115], and also in indoleamine dioxygenase (IDO) (1138 cm− 1) [116]. Subsequent rR spectra of oxy-complexes in CYP101A1 provided new information on perturbation of the Fe–OO moiety by various substrates [72] and by Pdx [117].
The rR spectra of the oxy-complexes of several human cytochromes P450 have also been measured recently for recombinant purified CYP11A1 [118] and for purified CYP17A1 [119] and CYP19A1 incorporated in Nanodisc bilayers [120]. In general, all features of these spectra are similar to those previously reported for CYP101A1. The position of the O–O mode varies from 1147 to 1124 cm− 1 and the range of the Fe–OO mode frequencies is between 540 and 529 cm− 1, with the expected linear correlation observed [118, 121–123]. The positions of these modes are not significantly different in the thiolate -ligated cytochromes P450 and nitric oxide synthase s, but can be substantially perturbed by hydrogen bonding to the dioxygen ligand [120, 124–126] and by steric effects caused by size and positioning of substrates [72]. In addition, detailed analysis of the spectra of oxy-complexes in the presence of various substrates revealed a striking difference in the configuration of the hydrogen-bonding network that includes the hydroxyl group of the substrate, the coordinated dioxygen moiety, and possibly other amino acid side chains and active site waters. For example, based on the different pattern of perturbations of the O–O and Fe–OO modes by 17-hydroxypregnenolone and 17-hydroxyprogesterone, hydrogen bonding to the proximal oxygen atom for the former and the distal oxygen atom for the latter, has been observed in CYP17A1 [119]. This difference correlates with the efficiency of the lyase reaction catalyzed by CYP17A1 and speaks directly to the intermediate states involved in the catalytic cycle and the identity of the “active oxygen” involved. More information about the Fe–O vibrational modes, as well as detection of new modes not seen in rR spectra, was provided by nuclear resonance vibrational spectroscopy (NRVS) [127, 128]. Using this method, Sage and collaborators demonstrated the strongly mixed character of two Fe–O modes observed in Raman spectra and claimed that the unambiguous assignment of these modes to either bending or stretching vibrations is not always valid.
The ferrous dioxygen complexes of heme proteins are not stable species, with the overall lifetime of this state in the cytochromes P450 ranging from milliseconds to minutes (Table 3.1). Autoxidation of the Fe–O2 complex proceeds through spontaneous dissociation of superoxide , which in turn quickly dismutates into hydrogen peroxide and dioxygen in aqueous solution. The heme is returned to the resting ferric state. The rates of autoxidation strongly depend on the presence of substrate, which sometimes can extend the half-life of the oxy-complex by a factor of 100 [81]. Another common property of the oxy-complexes in cytochromes P450 is a strong temperature dependence of autoxidation, with high activation energies implying substantial conformational changes involved in the release of superoxide [50, 129–132]. For this reason the oxy-complexes of substrate-free cytochromes P450 are prepared at low temperatures, often with the help of cryosolvents to suppress the freezing point and extend the temperature range for solutions down to 250–240 K [109, 111, 133–138]. The observed stabilization of the oxy-complexes in the presence of substrate is a general property of cytochromes P450 and is usually attributed to steric restrictions for superoxide escape from the active site . The concept of conformational gating is also supported by a similar slowing of the dissociation rates of CO, CN− and other diatomic ligands in the presence of a substrate. The same mechanism can be observed even when the substrate is present far from the catalytic site, as evidenced in human CYP3A4 when steroids bind at a peripheral allosteric site [82]. For CYP3A4, which can bind up to three TST molecules, the substrate dependence of the autoxidation rate is not trivial, with the major stabilization of the oxy-complex caused by the first binding event. Although the first TST molecule is likely bound at the same peripheral binding site as progesterone in the crystal structure described by Williams et al. [139], with no spin shift and no product formed at this stage [140], both autoxidation and geminate rebinding of CO undergo substantial changes and almost reach saturation with no changes caused by the second and third substrate binding [82]. Together with the high activation energies observed for autoxidation (15–18 kcal/mol in CYP3A4 with and without substrates) [81], these results suggest the existence of “conformational gating” in the binding and dissociation of diatomic ligands in various cytochromes P450. The presence of open and closed forms in equilibrium is now considered as a common property of the cytochrome P450 fold [95, 141] (see also Chap. 1 by Johnson and Poulos) and substrate binding is known to strongly affect the position of this equilibrium [142–145] as well as the likely rates of transitions between these states. Apparently, substrate binding to the peripheral binding site in CYP3A4 can play an effector role by stabilizing the closed form and thereby significantly decreasing the dissociation rate of diatomic ligands, as well as possibly other substrate or product molecules, from the active site. Manifestations of such effects of substrate or effector binding at the peripheral sites were observed as substrate or product inhibition at high substrate concentrations in other P450s such as CYP3A4 [146] and CYP2E1 [147].
Autoxidation together with direct peroxide dissociation from cytochromes P450 is responsible for the formation of reactive oxygen species and their formation is suggested to be an important source of toxic and potentially carcinogenic compounds [148, 149]. In some cases autoxidation is the main uncoupling pathway, as in CYP3A4 with poorly coupled substrates, for which autoxidation is faster than the second electron transfer . This is suggested based on the very fast autoxidation rates, for example 20 s− 1 with TST bound at 37 °C, as compared with the relatively slow overall steady-state NADPH consumption rate (about 4 s−1 under the same conditions) [81, 82]. Substrate binding significantly stabilizes the oxy-complex by both kinetic and thermodynamic mechanisms. Kinetic stabilization due to steric restriction of the escape pathway for superoxide in the presence of substrate was mentioned in the previous section. The thermodynamic stabilization is due to the changes in redox potential of the heme iron as described [36, 132]. The oxy-complex can decompose via dissociation of dioxygen from the ferrous heme, or by dissociation of superoxide anion from the ferric heme, as shown in Fig. 3.5. The overall process can be represented by two steps, fast equilibration in the immediate vicinity of the heme inside the active site , and slower escape of diatomic ligand into the solvent.

Decomposition pathways of the Fe–O2 complex via reversible dissociation of dioxygen from the ferrous heme ( top) or quasi-irreversible dissociation of superoxide from the ferric heme
Here the first reversible steps, breakage of the coordination bond, and geminate rebinding of the neutral dioxygen (top pathway) or superoxide (bottom pathway), are equilibrated on the ~ 10 ns timescale [150, 151]. The relative probability of superoxide dissociation is determined by the partitioning constant K part, which can be calculated by combining the two redox equilibria in Fig. 3.6:

Redox equilibria between ferrous and ferric states in the heme iron and between dioxygen and superoxide
The fraction of dioxygen dissociating from the protein via the bottom pathway as superoxide can be calculated as shown in the equation below:
Here the midpoint potential of the dioxygen–superoxide pair is − 0.33 V for unprotonated superoxide [101], so the first term is constant, and partitioning between the autoxidation pathway and reversible dissociation of dioxygen depends exponentially on the midpoint redox potential of the heme iron. As a result, the observed apparent autoxidation rate k autox also increases exponentially when the redox potential of the heme iron decreases:
This exponential dependence of the apparent autoxidation rates on the midpoint redox potential of the heme iron in cytochromes P450 explains the higher stability of the oxy-ferrous intermediates in the presence of substrates [36, 37, 132]. In addition, the presence of substrate at the active site of the cytochrome P450 creates steric restrictions on the mobility of diatomic ligands and furthermore increases the lifetime of oxy-complexes and thus improves the efficiency of the overall catalytic cycle by reducing unproductive dissociation of superoxide . [81, 82]. Overall, this regulatory role of substrate on the efficiency of oxygen activation is a critical factor in the mechanism of cytochrome P450.
6 Second Electron Transfer and the Peroxo- and Hydroperoxo-Intermediates
The rate of the second electron transfer , [4] ( [5] in Fig. 3.1 is difficult to measure. The marginal stability of the oxy-complex in many cases is a serious obstacle, made more difficult by the rate-limiting formation of a productive complex with the protein redox partner , either an iron–sulfur ferredoxin (e.g., Pdx ), or the flavoprotein cytochrome P450 reductase. In several experimental studies the reduction rates of oxy-complexes in cytochromes P450 were measured using stopped-flow absorption spectroscopy by monitoring the decay of the oxy-complex after rapid mixing with the reduced redox partner [152–156]. The measured rates varied greatly, from > 100 s− 1 for the fast CYP101A1 reduction by Pdx, to 8.4 and 0.37 s− 1 for the slower and multiphasic reduction of CYP2B4 by cytochrome P450 reductase (CPR) . Steady-state kinetic studies, conducted over many years, did not reveal any detectable spectral intermediate following the second electron transfer to the oxy-complex before the appearance of the ferric resting state. From the first investigations using the soluble CYP101A1, it was apparent that this second electron transfer is at least partially rate limiting, as the ferrous-oxy complex accumulates to some degree during turnover. The same observations were made by monitoring the steady-state turnover of microsomal cytochromes P450 [157] and in stopped-flow spectroscopic studies of oxygen binding to purified microsomal cytochromes P450 [158]. Thus, despite numerous attempts, no success has been achieved in cleanly observing a peroxo- or hydroperoxo-ferric intermediate in wild-type P450 at room temperature with the normal redox partners and atmospheric dioxygen. Early stopped-flow studies did claim the observation of such an intermediate state [159, 160], and with the D251N mutant of CYP101A1, where protonation is impaired, some level of a peroxo- or hydroperoxo-ferric species could be observed [161]. As will be discussed, the spectral and structural characterization of these intermediates in the cytochromes P450, as well as in other oxygen reactive heme proteins, requires the use of cryoradiolytic reduction. A shunt pathway exists, however, wherein two oxygen atoms and two reducing equivalents can be brought to the ferric heme together in the form of a peroxide or peroxy acid. With this approach, a transient species with red-shifted Soret band has been observed in horseradish peroxidase [162, 163].
Since the early 1970s, it was understood that one-electron reduction of the ferrous-oxy complex would generate a state with two redox equivalents and dioxygen—a ferric–“peroxo” state, with the electron going somewhere in the liganded prosthetic group. As the oxygenated intermediate has a dominant “ferric-superoxo” resonance form, as evidenced by Mossbauer measurements [75], addition of the second electron was thought to form a ferric iron with the oxygen reduced to the level of peroxide in resonance with a ferrous-superoxo configuration. However, neither the peroxo- [5a] nor hydroperoxo-ferric [5b] complexes has ever been cleanly observed at ambient temperatures. This intermediate, termed “compound 0” by analogy to similar states in the peroxidases, undergoes further transformation and disappears faster than it is formed. A pioneering breakthrough was realized though the work of Davydov, wherein the oxygen intermediate was trapped in a frozen matrix and the second electron was added by radiolysis. Although low-temperature matrix-isolation techniques were well established in the 1950s and 1960s [164–168], the first applications of this method to heme protein solutions were those of Davydov [169–174] and Symons [175–179]. Cryoradiolysis uses ionizing radiation to produce hydrated electrons, which can either interact directly with the protein molecule or the solvent matrix. For dilute protein solutions, the volume fraction of the mixed solvent is much larger and hence the predominant effect is to produce hydrated electrons that are highly mobile even at cryogenic temperatures. The radiation chemistry of aqueous solutions has been well studied, with reviews focused on frozen aqueous solutions of proteins also appearing in the literature [134, 174, 180, 181]. It is worth noting that the solvent itself also plays a crucial role as a selective quencher of undesired radiolysis products. For example, glycerol or ethylene glycol efficiently trap and quench hydroxyl radicals in the cryogenic radiolytic reduction of metalloproteins, with the result that a higher net yield of solvated electrons is available to reduce the proteins of interest [182].
A pioneering publication in the P450 literature was that of Davydov, Huttermann, and Peterson, who demonstrated that radiolytic reduction of the ferrous dioxygen complex of CYP101A1 at liquid nitrogen temperature yielded an electron paramagnetic resonance (EPR) signal identified as a peroxo intermediate [183]. Although referenced in several reviews of the P450 mechanism, it was not until Davydov moved to the Hoffman laboratory that electron nuclear double resonance (ENDOR), and additional magnetic resonance investigations at variable frequencies, spectroscopically defined the peroxoanion and hydroperoxo forms of ligated heme. Since that time cryoradiolytic reduction of oxy-complexes has been the method of choice for stabilization of the fleeting intermediates in the P450 catalytic cycle with the goal of obtaining the detailed structural and spectroscopic information necessary for evaluation of the mechanism of oxygen activation and metalloenzyme catalysis. Several reviews on experimental applications of these methods and the results obtained using the cryoradiolytic approach have been published recently [134, 135, 180, 184]. For CYP101A1, radiolysis at 77 K trapped the hydroperoxo intermediate [185]. In the D251N mutant of CYP101A1, which altered the occupancy of active site waters as observed in the crystal structure of the ferrous dioxygen complex [92, 186], the species observed upon 77 K radiolysis was the peroxoanion. Thermal annealing of this trapped state, with monitoring by EPR and ENDOR spectroscopy [185], allowed direct observation of the protonation event and quantitative conversion of the peroxoanion to the hydroperoxo and product.
These first detailed characterizations of the peroxo- and hydroperoxo-ferric intermediates in CYP101A1 [185, 187] provided several important results and enabled further experimental studies with other heme proteins. Clear EPR signatures for the unprotonated peroxo-ferric ( g 1 < 2.27) and protonated hydroperoxo-ferric ( g 1 > 2.27) intermediates in cytochromes P450 were thus defined. These parameters are very similar to those observed in other heme proteins, such as myoglobin, hemoglobin, and horseradish peroxidase (see Table 3.2). Thus, the first immediate product of cryoradiolytic reduction of the oxy-complex is the peroxo anion, as proton transfer events are prevented at low temperature. In some cases, such as with wild-type CYP10A1, protonation can occur even at 77 K and one needs to do the cryoreduction at helium temperatures to trap the peroxo anion. This temperature dependence of the proton transfer events provides a recipe for stepwise annealing of the trapped peroxo anion to follow the transformation of metastable intermediates along the reaction coordinate through to product formation. The catalytic competence of the cryoradiolytic reduction of CYP101A1 can be directly demonstrated by analysis of product formation, with the overall yield proportional to the irradiation dose [185].
Subsequent work with CYP101A1 demonstrated that various substrates significantly modulate proton delivery to the coordinated dioxygen, as monitored by EPR and ENDOR of the cryoreduced oxy-complexes in the presence of different substrates [188]. Controlled annealing at elevated temperatures (170–180 K) demonstrated that the presence of any substrate dramatically increases the stability of the hydroperoxo-ferric complex, with lifetimes at least 20 times longer than in the absence of a substrate. With all substrates, alternate and multiple conformational substates have been detected in the heme-iron center by changes in 14N,1H hyperfine couplings in the ENDOR spectra. Unusual EPR and ENDOR spectra and reactivity have been observed with CYP101A1 bound with (1R)-methylenyl camphor . One well-defined conformational substate of the heme was observed, but the decay rates of the hydroperoxo-ferric complexes of wild-type CYP101A1 and its T252A mutant at 180 K were much lower than with all other substrates. Although the T252A mutant does not yield a product with normal substrates, in this case the epoxide of (1R)-methylenyl camphor was generated. Dawson, Hoffman, and colleagues thus suggested that there may be a direct involvement of compound 0 in the epoxidation reaction, rather than a reaction involving proton transfer, O–O bond scission and compound I formation. The results of this work suggest a potential for the involvement of substrates in modulating the chemical properties of peroxo- and hydroperoxo-ferric intermediates and selection of the appropriate “active oxygen” for catalysis. Such a role may help explain the numerous proposals for the involvement of multiple oxidants and catalytic mechanisms in P450 function [56].
In addition to the critical acid–alcohol pair that is directly involved in the protonation of peroxo-ferric complexes in cytochromes P450, other amino acids in the immediate vicinity also can perturb the proton delivery and significantly change the functional properties of the enzyme. The CYP101A1 single G248 mutants [189], which retain the native acid–alcohol pair of D251 and T252, show significant perturbation of the proton delivery, although both mutant proteins still catalyze camphor hydroxylation in a reconstituted system. Functional studies suggest that the second protonation of the hydroperoxo-anion is inhibited by mutations at the 248 position. EPR of the cryoreduced oxy-complex shows that the first protonation is also impeded, since the immediate product of the cryoreduction at 77 K is almost completely the unprotonated peroxo-anion, in contrast to the wild-type and the T252A mutant, for which cryoreduction at 77 K produces the hydroperoxo state [185, 187, 190].
With the low-temperature oxygenation protocols developed for the preparation of unstable oxy-complexes in cytochromes P450 and NOS [108, 109, 135, 180, 191–193], cryoradiolytic reduction and characterization of the peroxo- and hydroperoxo-ferric intermediates have been realized for the mammalian CYP2B4 [133] and the steroid metabolizing P450s CYP17A1, CYP19A1 [194], and CYP11A1 [195]. In addition to the substrate free protein, samples of CYP2B4 have been prepared in the presence of two substrates, benzphetamine (BP) and 3-hydroxy-tert-butyl toluene (BHT). Because no high-spin signal was detected by EPR in the frozen solution of CYP2B4 with BP, dissociation of the substrate at low temperature in the cryosolvent (60 % glycerol with Tris buffer, pH 8.0) was suggested, in contrast to CYP2B4 bound with BHT, which revealed a mostly high-spin EPR signal. Oxygenation of the reduced protein was realized at − 40 °C in order to minimize autoxidation [109, 111]. The yield of hydroperoxo-ferric complex was been estimated at ~ 40 % by comparison of the EPR signal with the calibrated standard [133]. As with CYP101A1 and heme oxygenase [196], the immediate product of cryoradiolytic reduction in CYP2B4 with or without substrate was the already protonated hydroperoxo-ferric complex characterized by g 1 > 2.27.
The peroxo- and hydroperoxo-ferric intermediates in the mammalian cholesterol side-chain cleaving cytochrome P450 (CYP11A1 ) has been recently documented [195]. The oxy-complex of CYP11A1 with cholesterol bound was radiolytically reduced at 77 K in 33 % glycerol/phosphate buffer at pH 7.5. After irradiation the main cryoreduced intermediate had an EPR signal with g 1 = 2.34 characteristic of a protonated hydroperoxo-ferric complex. However, two minor signals with g 1 = 2.214 and g 1 = 2.28 indicated the presence of some unprotonated peroxo-ferric intermediates. These latter intermediates both converted to the hydroperoxo-ferric intermediate after annealing at 145 K, with a considerable protium/deuterium (H/D) solvent isotope effect for the conversion of the 2.214 signal, but no isotope effect for the reaction of the 2.28 intermediate. Taken together, these observations indicate the presence of multiple conformers of coordinated dioxygen and one or more water molecules in the immediate vicinity that may serve as proton donors to the peroxo-anion in CYP11A1. Annealing at 185 K and further to 220 K, resulted in decay of the hydroperoxo-ferric intermediate and formation of the 22R-hydroxycholesterol product. This step also featured a substantial solvent H/D isotope effect consistent with the expected partially rate-limiting second proton-transfer step, which is necessary for formation of the catalytically active compound I . This suggests that the C–C bond scission of a vicinal diol, as is the case in the generation of pregnenolone from cholesterol by CYP11A1, uses compound I as the “active oxygen” for catalysis.
Generation and decay of peroxo-states can also be monitored by optical absorption spectroscopy [137, 180, 197, 198], although with UV–vis methods it is not possible to differentiate between the peroxo and hydroperoxo intermediate s [198]. The main spectral feature of these intermediates in cytochromes P450 and in other thiolate -ligated proteins is a significant red-shift of the Soret band from 420–430 to 440–450 nm, and the appearance of a second minor band at ~ 375 nm. These properties are consistent with the split Soret band characteristic of the optical spectra of the ferrous O2 and CO complexes and the ferric-cyanide adduct of cytochrome P450 [106, 107, 199]. Interestingly, only a minor red-shift of the Soret band (3–8 nm) is observed for the peroxo-complexes in heme proteins with histidine as the proximal iron ligand [134, 192, 200, 201].
As noted in the previous discussion of the earlier intermediates in the P450 reaction cycle, rR spectroscopy is a powerful tool to reveal critical information regarding P450 structure and function, including mechanistic details of P450 catalytic oxygen activation and substrate metabolism and their linkage to the delivery of protons to the reduced heme-dioxygen complex. rR spectroscopy probes the vibrational modes associated with the active site , is operational in all states of the reaction wheel, and hence is uniquely positioned to provide key information on the mechanism of “oxygen activation” in cytochromes P450.
Low-temperature rR investigations have been extensively conducted by the Kincaid laboratory on radiolytically reduced oxy-ferrous cytochromes P450 [183, 185, 187, 188, 197, 198, 201–205]. Using the D251N mutant of CYP101A1 and experiments analogous to the EPR investigations already discussed, the peroxoanion intermediate and the formation of the protonated hydroperoxo state following thermal annealing was characterized. These studies demonstrated that the hydroperoxo-anion retains the end-on structure of the oxy-ferrous precursor and forms a relatively strong bond with the heme iron that is characterized by ν(Fe–O) ~ 617 cm− 1 in the hydroperoxo-ferric complex in myoglobin [201] and 564 cm− 1 in CYP101A1 [198, 205], while the unprotonated ferric-peroxo complex [5a] in the D251N mutant of CYP101A1 displays a slightly weaker Fe–O bond with ν(Fe–O) 553 cm− 1 [198]. These complexes reflected the typical features of low-spin heme-thiolate complexes with a narrow span of g values in the EPR spectra and a red-shifted split Soret band with maxima at 436–440 and 370–375 nm [185, 197, 204].
Spectroscopic and theoretical studies reveal that the length and strength of the O–O bond in the peroxo states (termed Compound 0 by analogy to the peroxidase literature) are similar to those observed in the low-spin oxygen activating nonheme (hydro)peroxo-ferric complexes. Particularly interesting is the direct observation of the downshift of ν(O–O) from 792 cm− 1 in the peroxo anion state ([5a] in Fig. 3.1) to 774 cm− 1 in the hydroperoxo [5b], indicating a weakening of the O–O bond as a result of protonation of the peroxo-anion coordinated to iron [198]. Notably, the ν(O–O) in the P450 peroxo-ferric intermediates is significantly lower than in myoglobin with cobalt-substituted heme, where this mode was observed at 851 cm− 1 [206]. This difference is attributed to the strong electron donating capabilities of the thiolate proximal ligand in cytochrome P450 as compared to the imidazole nitrogen of the proximal histidine in myoglobin. The thiolate trans-effect weakens the O–O bond and promotes its heterolytic cleavage, with concomitant formation of the high-valent catalytically active ferryl-oxo intermediate ([6] in Fig. 3.1). However, the presence of the distinct hydroperoxo-ferric heme intermediate in the frozen solutions and in crystals of cytochromes P450 and other heme proteins suggests that there is no spontaneous breakage of the O–O bond, but rather the enzyme/substrate provides a catalytically important function. Thus, efficient formation of the main active intermediate Compound I requires catalytic delivery of the second proton to the distal oxygen atom (Fig. 3.1 [5b] ( [6]). The application of cryoreduction and annealing of native and mutant proteins, with concerted spectroscopic characterization by EPR/ENDOR and Raman spectroscopy, offers a means for revealing these critical steps in oxygen activation by the cytochromes P450.
Additional information on the structure and reactivity of peroxo-ferric heme intermediates can be obtained from the recent porphyrin models developed by Naruta and coworkers [207–209]. High-quality rR spectra of oxy-complexes and both low-spin end-on and high-spin side-on peroxo-ferric complexes have been measured in acetonitrile and in methanol at low temperatures (208 K) or in frozen solutions at 77 K. However, the proximal ligand to the iron in these model complexes is imidazole, and hence they can be considered as appropriate models for the oxygen activation intermediates in peroxidases, rather than the P450 enzymes. Interestingly, both Fe–OO and O–O modes have been observed in these complexes, contrary to the peroxo- and hydroperoxo-ferric complexes in myoglobin, where the O–O stretch mode was not detected in rR spectra [201, 210].
While early X-ray crystallographic investigations did not fully appreciate the in citu reduction of the prosthetic groups of metalloproteins, it is now clear that the X-ray beam, particularly from intense synchrotron sources, can efficiently add electrons to the system. An important advance in protein X-ray crystallography was achieved when cryoradiolytic reduction of the oxy-complex in CYP101A1 was intentionally used [92]. The unavoidable reduction of the heme complexes during data collection at cryogenic temperatures was carefully monitored and controlled by combining data obtained on multiple crystals [181, 211]. Following this approach, the first well-characterized structures of the unstable Compound 0 in horseradish peroxidase [211] and in CPO [212] were realized, and a high-resolution structure of the Compound 0 (peroxo-intermediate) in myoglobin was obtained [213]. The latter structures provide good experimental data on the O–O and Fe–O bond lengths in the protein hydroperoxo-ferric complexes.
7 Reactivities of the Peroxo States: O–O Bond Scission Versus Peroxide Dissociation
A second protonation of Compound 0 at the distal oxygen atom reduces the O–O bond order to zero and results in immediate scission and departure of a water molecule [214]. In cryoradiolytic experiments, Compound 0 is stable below the glass transition temperature, typically 180–190 K. This suggests that the second proton delivery requires sufficient mobility and diffusion of solvent molecules, with the potential relaxation of the protein matrix to a new conformation . Experiments with native CYP101A1 and the D251N mutant proved that at higher temperatures, Compound 0 disappears with formation of Compound I [6] (Fig. 3.1) and concomitant product formation [185]. For the CYP101A1 T252A mutant, where the native proton transfer mechanism is perturbed, the dissociation of peroxide with no product formation is the dominant path of Compound 0 decomposition. The latter reaction is considered as the main source of reactive oxygen species in the poorly coupled P450 systems. In general, the coupling efficiency measured by the ratio of the product molecules formed per NADPH molecule consumed can be very different for the same cytochrome P450 with different substrates. Efficient proton delivery requires specific positioning and stabilization of water molecules in the vicinity of the dioxygen moiety, which can be significantly perturbed by variations in the structure of the substrate.
The chemical mechanisms describing the hydroxylation of unactivated substrates most assuredly involves the Compound I intermediate state generated after O–O heterolysis following second proton transfer as described above. This is not necessarily the case for reactions involving carbon–carbon bond scission. For instance, in the case of CYP19 (aromatase) -catalyzed androstenedione (AD) metabolism, it has been a long-standing question as to whether the conversion of 19-oxo-AD to estrone by CYP19A1 occurs via the classic higher valence Compound I intermediate that operates in the normal hydroxylation cycle, or via the precursor peroxo-anion (Compound 0) intermediate. This is shown schematically in Fig. 3.7.

Two alternative mechanisms of C–C bond scission in CYP19A1
Evidence supporting both hypotheses is present in the literature [215–218], as the availability of a nearby proton for abstraction makes both a radical and nucleophilic mechanism plausible. In the first experiments with human CYP19A1 self-assembled into Nanodiscs, we discovered that when the ferrous-oxy complex was radiolytically reduced in the presence of AD, the peroxo state formed and stabilized at 77 K was the anionic form rather than the protonated hydroperoxo that had been seen in all previous P450s investigated [194]. This suggested that there was perhaps a different hydrogen-bonding configuration provided by active site water molecules in this P450. However, experiments monitoring the conversion of AD to 19-hydroxy-AD in an EPR-annealing experiment revealed a kinetic solvent isotope effect of greater than 3.5, suggesting one or more protons were involved in product formation from AD [219]. More recent EPR results demonstrated that when the substrate is 19-oxo AD, the immediate precursor to the carbon–carbon lyase reaction, the species stabilized at 77 K after radiolysis, and before product formation by CYP19A1, is the protonated (hydroperoxo) intermediate, as one would expect for a normal Compound I- mediated reaction. There are thus subtle differences in the active site structure that dictate a key variability in distal pocket hydrogen bonding and proton transfer, but it appears that Compound I is the “active oxygen” leading to C–C bond cleavage and aromatization of the A-ring. Exactly the opposite is true in the case of CYP17A1, where a nucleophilic reactivity of Compound 0 appears to be operating. This will be discussed further in the following section.
A carbon–carbon bond cleavage required for conversion of the pro-drug nabumetone to the active form is also catalyzed by the peroxo-ferric intermediate of human CYP1A2, as reported based on a thorough study comparing the activities of several human cytochromes P450 [220]. Only CYP1A2 and CYP3A4 (CYP2B6 with significantly lower efficiency) supported the C–C cleavage reaction with nabumetone and 3-hydroxy-nabumetone as substrates. In addition, C–C cleavage did not proceed when the peroxide shunt pathway with cumene hydroperoxide was used instead of NADPH supported catalysis. However, the NADPH-supported hydroxylation of nabumetone in reconstituted systems and in commercial Supersome® preparations was observed with almost all the isozymes, the most efficient being CYP2C19, CYP2B6, and CYP3A4. These observations suggest that the unprotonated peroxo-ferric intermediate is the main catalytic species for C–C bond cleavage in this system.
8 Compound I as the “Active Oxygen” in Alkane Hydroxylations
Despite the great variety of chemical transformations catalyzed by cytochromes P450, the vast majority of them are undoubtedly driven by Compound I . This ferryl-oxo intermediate with a π-cation radical delocalized on the porphyrin is a very reactive species. All attempts to observe this species in a P450 system using atmospheric dioxygen have so far failed. However, important spectroscopic characterization and reactivity measurements have been obtained by using the peroxide shunt pathway [3] ([6] in Fig. 3.1. In this approach, which bypasses the dioxygen reduction process, rapid mixing of the ferric heme enzyme with peroxides or peroxy acids such as meta-chloroperoxybenzoic ( m-CPBA) can generate the Compound I intermediate directly [221–223]. Unlike the usual P450 pathway of oxygen activation, where two electrons and two protons have to be channeled to the dioxygen via coordination to the heme iron and proton delivery pathways, the peroxide pathway benefits from the fact that peroxides or peroxyacids already have the two electrons and protons on the dioxygen moiety. The role of the enzyme in this case is the efficient rearrangement of the proton from the proximal oxygen atom, which forms the transient coordination bond with the heme iron, to the distal oxygen to facilitate heterolytic scission of the O–O bond and thus create the same Compound I, as happens in the normal catalytic pathway of horseradish peroxidase [224–226]. However, in general, the cytochromes P450 are inefficient peroxidases or peroxygenases, and the yield of Compound I by this pathway is low. Thus the first experiments devoted to revealing this intermediate via stopped flow realized a yield of ~ 10 % or less [221–223]. This low level of protein made it all but impossible to obtain detailed structural and spectroscopic characterization of the Compound I in cytochromes P450. Until recently, the only way to address experimentally the physico-chemical and functional properties of this intermediate was via model porphyrin systems [10, 227–230] or by analogy to other closely related thiolate- ligated heme enzymes such as CPO [231–233] and peroxygenases [234, 235], for which Comopund I is much more stable.
This situation changed with the work of Rittle and Green who achieved a breakthrough on the peroxide pathway by radically improving the purification protocol for thermostable CYP119 from the extremophile archae Sulfolobus acidocaldarius [236–238]. Careful multistep removal of endogenous substrate analogs from the purified, heterologously expressed protein, which hampered earlier studies [222], allowed them to dramatically increase the yield of Compound I in a stopped-flow reaction with m-CPBA, reaching a conversion of greater than 75 % [236]. This made possible high-precision UV–vis spectra to quantitate the reaction kinetics, which in turn provided the necessary information for the preparation of highly concentrated samples for EPR and Mössbauer spectroscopy . The UV–vis spectra of Compound I confirmed the main features of the ferryl-oxo π-cation radical known from the earlier experiments: a broad Soret band at 367 nm and a pronounced charge-transfer band at 690 nm. The EPR spectrum of CYP119 Compound I [236] had a different shape as compared to that previously reported for CPO, another thiolate- ligated heme protein [239]. Fitting of both spectra to the S = 1 Fe(IV)-oxo unit coupled with S = 1/2 porphyrin radical resulted in a higher ratio of the exchange coupling ( J) to zero-field splitting ( D) for CYP119 ( J/D = 1.3) than in CPO ( J/D = 1.02) [236]. The higher J value in CYP119 was tentatively attributed to either a higher spin density on the thiolate sulfur atom or a shortened Fe–S bond. The Mössbauer parameters measured for the CYP119 Compound I were more similar to those of CPO [239], with the isomer shift δ = 0.11 mm/s (0.13 mm/s for CPO) and quadrupole splitting ΔE Q = 0.96 mm/s (0.90 mm/s for CPO). These parameters also correspond to the ferryl-oxo S = 1 unit exchange coupled to the porphyrin radical (S = 1/2).
The functional competence of this Compound I intermediate was confirmed in fatty acid hydroxylation assays using a double-mixing stopped-flow technique. After premixing CYP119 with m-CPBA and incubating for 100 ms, the reaction mixture containing 35–40 % of Compound I was rapidly mixed with solutions of the substrates at various concentrations at 4 °C. The kinetics of the reactions were monitored spectroscopically and the product yield was verified by gas chromatography ([236] and supporting online material). The observed apparent rates were very high, up to 220 s− 1 for lauric acid, with the rate constants varying from 4.4´104 to 1.1´107 M− 1s− 1 for hexanoic and dodecanoic (lauric) acids, respectively. In addition, the kinetic isotope effects (KIE) for these reactions measured experimentally with protonated and perdeuterated substrates strongly depended on the chain lengths of the fatty acids , varying from 12.5 for hexanoic acid to 1.0 for lauric acid. This disappearance of the KIE for the fast-reacting substrate is explained by strong masking of the isotope effect by tight substrate binding and rate-limiting unproductive substrate dissociation for lauric acid. The true isotope effect value can be measured only when substrate binding is at rapid equilibrium, as demonstrated in [236] and supporting material. Thus, the high-unmasked KIE strongly confirms the catalytic competence of the Compound I obtained in CYP119 by rapid mixing with m-CPBA and the kinetic parameters expected for the hydrocarbon hydroxylation via a hydrogen-abstraction mechanism [11, 227].
Recently, the same improved multistep purification approach proved to be critically important for the generation of high populations of Compound I in another cytochrome P450, P450ST [238]. Following similar experimental protocols, Green and his group were able to trap Compound I in a high concentration and to measure its EPR and Mössbauer spectra. The results were similar to those measured for CYP119 [236]. Mössbauer spectra could be fitted well with an isomer shift δ = 0.12 mm/s and a quadrupole splitting ΔE Q = 0.85 mm/s, and J/D = 1.3 obtained from EPR spectra that were the same as for CYP119 [238].
The oxygen-rebound mechanism of hydrocarbon hydroxylation catalyzed by Compound I presumes formation of the transient heme intermediate equivalent to Compound II following hydrogen abstraction from the substrate. In this case, the iron-oxo unit is protonated, and the electron fills the π-cation radical of the porphyrin [236, 240]. The critical importance of thiolate ligation in P450 catalysis was evaluated by recent work from the Green group [241]. By direct measurements of pK a of the Compound II of CYP158, they estimated and compared the relative contributions of redox potential and proton affinity to the thermodynamics of hydrogen atom abstraction by Compound I in cytochrome P450, CPO, and nitric oxide synthase . The key difference between histidine-ligated peroxidases and thiolate-ligated P450 enzymes is the large shift of the pK a of Compound II from ~ 3.5 in the former to ~ 12 in the latter, due to the much stronger electron-donating abilities of a thiolate than a histidine. At the same time, the large contribution from the strong proton affinity term makes the redox potential term low enough to prevent fast inactivation of this catalytically active intermediate by intra-protein electron transfer and reduction to Compound II [242, 243]. The same effect of the thiolate proximal ligand was observed by Hoffrichter and Groves in a thiolate-ligated peroxygenase [235]. Thus we have for the first time a clear mechanistic rationale as to why the cytochromes P450 utilize cysteine as the axial ligand to the iron [235, 242, 243].
9 Bleed Points of Inefficiency: Uncoupling Pathways in the Cytochromes P450
The key characteristics of enzymatic catalysis are the maximum rate of product formation given by V max or k cat, and the substrate-binding constant, or Michaelis constant K m. For comparison of different enzymes and/or substrates, the efficiency of the enzyme is characterized by the ratio of these two parameters. For cytochromes P450, these parameters also can be used as the essential quantitative measures of their ability to metabolize xenobiotic compounds or to synthesize their specific products. The case of P450 catalysis, however, is complicated by the consumption of redox equivalents and the nature of atmospheric dioxygen as a reactant. The ideal stoichiometry of P450 catalysis requires one NAD(P)H and one O2 molecule to make one molecule of product. This rarely happens in reality. In addition to product formation, a fraction of oxygen is released in the form of superoxide after one redox transfer event, as peroxide after two-electron reduction, or as water after four-electron reduction, as shown in Fig. 3.1. Superoxide and hydrogen peroxide belong to a class of compounds termed “reactive oxygen species” or ROS. A comprehensive review on ROS production by P450 summarizes the main mechanisms as well as the implications of the release of these potentially toxic products [149]. Other side reactions, such as formation of protein radicals, covalent coupling of the heme to the protein or to active radical products, heme loss, or accumulation of the inactive P420 form, can also be considered as the consequences of uncoupling and have been reviewed elsewhere [244].
Oxygen activation in cytochromes P450 is a multistep process with several branching points. As seen from Fig. 3.1, there are at least three steps where the reaction flow can partition between productive and unproductive pathways. The first one is the oxy-complex, which can decompose with dissociation of superoxide if the second electron transfer is not efficient or simply not fast enough. The second branching point is dissociation of the hydroperoxo-anion, if the second protonation is not accomplished. The third one is unproductive reduction of compound I with consumption of a second molecule of NAD(P)H, which can occur if a productive catalytic reaction with the substrate does not happen. All these branching points are essentially kinetic, and the result at each step is determined by the corresponding rate constants. Under steady-state conditions, the overall degree of uncoupling is determined by the ratio of the reaction flux along the productive pathway [3]→[4]→[5]→[6] →[7] in Fig. 3.1 and the sum of fluxes along the unproductive pathways [4]→[2], [5]→[2], and [6]→[2]. Partitioning at each branch point is proportional to the absolute microscopic rate constants leading out of the intermediate. For progress from the oxy-complex, the fraction of the overall reaction flux that follows the uncoupling pathway is proportional to the autoxidation rate k 42, while the fraction of oxy-complex reduced to the peroxo-ferric state is proportional to k 45. Thus, the fraction of the reaction flux following the productive pathway, or the coupling ratio at the level of the oxy-complex, is k 45/( k 45 + k 42). The second branch point lies with the peroxo states, where it is possible to release a twice-reduced dioxygen to regenerate the ferric prosthetic group. Similarly, a coupling ratio taking together the peroxo- and hydroperoxo intermediate s as [5] is determined by the ratio of protonation rates and the rate of peroxide dissociation , k 56/( k 56 + k 52). The third uncoupling point in the P450 reaction cycle centers on the Compound I intermediate [6], which can be reduced by two additional electrons to form water. Since the overall stoichiometry is then four electrons and four protons added to one dioxygen yielding two water molecules, this has been termed the oxidase pathway. From the scheme in Fig. 3.1, the overall partition coefficient for the oxidase branch point is given by k 67/( k 67 + k 62). Overall, the experimentally observed uncoupling is proportional to the product of these three fractions, while the absolute rates of substrate conversion to product and of NAD(P)H and O2 consumption, in most cases, depends on the rates of the first and second electron transfers, protonation of the dioxygen moiety, and the catalytic step, although in some cases substrate binding and product release may also be rate limiting.
In reality, many of these individual rate constants are not known and difficult to measure. In most cases, there is no single and well-defined rate-limiting step in the overall catalytic cycle of the cytochromes P450 and thus several intermediates are present at any one time. Early attempts to monitor the steady state of P450 catalysis usually focused on the oxy-ferrous intermediate, which was observed experimentally during turnover using optical absorption spectroscopy [157, 158, 245]. The rate of autoxidation k 42 can be measured separately with high precision, as described earlier in this review and in previous publications and review articles [36, 82, 87, 246–248]. The rate of the second electron transfer to the oxy-complex, k 45, which competes with autoxidation, is more difficult to probe at ambient conditions due to autoxidation and possible rate-limiting interactions with redox partner s. Successful examples are represented by stopped-flow studies with various concentrations of Pdx and CYP101A1 [154, 155]. Measurements of the peroxide dissociation rate, k 52, and oxidase uncoupling rate, k 62, are even more difficult because the steady-state concentrations of the key intermediates [5] and [6] are exceedingly small. To our knowledge, no independent reports of these rate constants at ambient conditions are available. However, the steady-state rates of peroxide production and water production via the oxidase uncoupling channel are known for many cytochromes P450 in reconstituted systems, so the fractional partition coefficients at the peroxide [5] and Compound I [6] branching points can be estimated.
Partitioning at these three uncoupling branch points and the corresponding rates determine the overall efficiency of substrate turnover. This depends on various factors, which include the substrate structure and its positioning inside the substrate-binding pocket, the efficiency of proton delivery to the coordinated dioxygen via the hydrogen-bonded network of several protein groups together with strategically placed and conserved water molecules, and the efficiency of electron transfer from the protein redox partner . Clearly, changes of each of these factors may significantly affect the result of oxygen activation and change the partitioning between productive and unproductive pathways. These two pathways may be described as oxygen activation for either the oxidative transformation of organic substrates or the production of peroxide and water. For the most efficient cytochromes P450, such as CYP101A1 and CYP102A1, where catalysis via the productive pathway with optimal substrates is realized with almost 100 % efficiency, even small variations in the substrate structure or single-point mutations at the active center result in significant uncoupling and redistribution of the reaction flow towards peroxide production. For those cytochromes P450 that are significantly uncoupled (either in vitro or in vivo), the same mutations and/or substrate variations may be favorable for the increase or the productive consumption of NAD(P)H and O2. The same is true for inefficient metabolism of nonnative substrates by the wild-type enzymes, where mutations may significantly improve the rate of oxygen activation and coupling to the productive pathways. Multiple mutations at the substrate-binding pocket not only can drastically change the regio- and stereo-specificity of substrate binding but also can be engineered to alter uncoupling and extend the range of chemical transformations of nonnative substrates catalyzed by cytochrome P450. These results have been extensively reviewed for the self-sufficient CYP102A1, which is considered as the most promising cytochrome P450 for bioengineering and synthetic biology purposes [27, 249, 250].
Uncoupling at the oxidase branch point has been observed in microsomes by comparing the rates of NADPH and O2 consumption with a natural substrate with those of a non-metabolized analog, as in the case for CYP21 in bovine adrenocortical microsomes [38]. Here, the 2:1 NADPH/O2 stoichiometry was correctly assigned to the oxidase uncoupling pathway. Similar observations were reported by Ullrich [39, 251] using perfluorinated substrate analogs to prevent productive reactions of Compound I . They also observed a 2:1 NADPH/O2 consumption stoichiometry. Later Coon and coworkers documented both productive and unproductive pathways, including peroxide and oxidase uncoupling, using purified liver microsomal cytochrome P450 and demonstrated that the results strongly depend on the substrate [252]. A review of the overall stoichiometry of coupled and uncoupled P450 reactions has been provided by Zhukov and Archakov [253, 254].
The availability of X-ray structures for CYP101A1 [32, 33, 255–259] made possible a detailed analysis of uncoupling in P450 catalysis. The hydroxylation of the natural substrate 1R-camphor by wild-type CYP101A1 is very fast ( k cat up to 35 s− 1) and almost 100 % coupled, with an NADH/product ratio of ~ 1.02–1.03. This provides an excellent reference system for systematic study of the relative importance of essential features of the enzyme active site as well as substrate structural variations for overall catalysis. A series of CYP101A1 mutants has been generated based on the available X-ray structures with the goal of deciphering the structural determinants of efficient substrate hydroxylation [23, 24, 260–268]. Various substrate analogs were also employed to explore the regio- and stereo-specificity of chemical transformations catalyzed by CYP101A1 together with the rate and efficiency of steady-state turnover [23, 261–263, 265]. Taken together, these works revealed a great variability of both rates and coupling efficiencies that depend on both on single mutations and variations of the substrate structure.
The discovery of the critically important acid–alcohol pair D251-T252 in CYP101A1 was very important from a mechanistic point of view. The dramatic effect of a D251N mutation, which resulted in a 50–100-fold slowing of the product-formation rate, without loss of the efficiency of NADH consumption, and the same ~ 95 % coupling as in the wild-type enzyme [267], clearly indicated the gate-keeping role of this residue in proton delivery. Its role was later confirmed by kinetic solvent isotope effect (KSIE) and proton inventory measurements [269]. Impaired proton delivery in the D251N mutant as the main cause for slow turnover was also confirmed by directly measuring the rate of the first electron transfer from Pdx , which was even faster than in the wild-type enzyme [270]. In addition, significant acceleration (5–10 times) of NADH consumption and camphor hydroxylation observed at moderately acidic pH (5.5–5.0) also supported protonation as the strongly rate-limiting step in the CYP101A1 D251N mutant [267]. The effect of the salt link between D251 and K178 was tested separately by mutating this residue to glutamine, K178Q [267]. This mutant was highly coupled and only moderately slower than the wild-type enzyme, implying that the position of the side-chain of D251 is not the main factor determining the overall turnover efficiency of CYP101A1. In contrast, the T252A mutation of the neighboring residue uncoupled hydroxylation catalysis by ~ 95 % with no inhibition in NADH consumption, efficiently channeling redox equivalents into peroxide production [271, 272]. The essential role played by the alcohol side-chain of T252 in oxygen activation was confirmed by the high activity and 81 % coupling of the T252S mutant [272]. The importance of these two residues in the CYP101A1 mechanism has been analyzed in great detail using X-ray structures of the oxy-complexes of these mutants [74, 92]. The structures of oxy-complexes reveal important conformational rearrangements of the I-helix, with reorientation of the T252 side-chain opening the cleft between T252 and G248, and appearance of two new well-resolved water molecules that most likely represent the main proton delivery channel [74, 92, 93, 95]. The same opening in the I-helix and same water molecules were also observed in the structure of the cyanide complex of CYP101A1 [273] and the highly similar CYP101D1 [274], possibly due to very similar geometries of the Fe–O2 and Fe–CN− complexes with an angle of ~ 125° between the ligand axis and heme plane, and similar H-bonding properties.
The recent discovery and crystallization of other members of the CYP101 family, CYP101D1 [274, 275] and CYP101D2 [276], opened additional means to probe the finely tuned and highly efficient mechanism of oxygen activation. Both CYP101D1 and CYP101D2 bind camphor in the same orientation as CYP101A1 and catalyze the same hydroxylation with similar high rates (1000–2000 min− 1) and almost 100 % efficiency [274, 277]. Despite the same activity towards the same substrate, there are structural and functional differences between these three isozymes that provide a better understanding of the essential (and not essential) features for optimal P450 catalysis. Mutations of the acid–alcohol pair residues D259N and T260A in CYP101D1, analogous to D251N and T252A in CYP101A1, had the same effect: Little or no activity in the Asp/Asn mutant and highly uncoupled NADH consumption in the Thr/Ala mutant in both proteins [274]. Critical variations in CYP101D1, as compared to CYP101A1 (where G180 replaces the homologous K178, D182 is used instead of N184 and A366 in CYP101D1 replaces L358 in CYP101A1) may have changed the functional properties with respect to interactions with the redox partner Pdx and/or protonation/substrate binding. When these mutants were introduced into CYP101A1 to check for the functional implications of these residues using the native redox partner Pdx [275], the single mutants L358A and K178G had little effect on the activity or structure of CYP101A1. However, the double mutant L358A/K178G had a tenfold slower rate of NADPH consumption than the wild type due to the mostly low-spin state even in the presence of camphor. The addition of 400 mM K+ converted the double mutant protein to the high-spin form and diminished the difference in steady-state NADPH turnover. The crystal structure of the cyanide complex of the mutant CYP101A1 shows the same structural changes as in the wild type, including the key water molecules in the I-helix cleft, indicating that the proton delivery pathway is not perturbed by these mutations [275].
Homology analysis revealed that the acid–alcohol pair in the I-helix is a common feature in the great majority of cytochromes P450, although some deviations are evident. In the CYP51 class the semi-conserved acid side-chain (D251 in CYP101A1) is replaced by a histidine [278]. In the rat CYP51 enzyme, the mutation H314D resulted in a sevenfold lower 14-demethylase activity [279]. In some cytochromes P450 the alcohol side-chain from threonine or serine is replaced by alanine, as in P450eryF (CYP107A1) and CYP158A2. Based on the X-ray structural studies of these two enzymes, the concept of substrate-assisted catalysis was proposed as an alternative to the missing side-chain of Thr/Ser [80, 280]. The functionally important water molecules forming the proton delivery pathway are stabilized at the proper position close to the coordinated dioxygen by hydrogen bonding to the substrate hydroxyl group instead of the alcohol side-chain [73, 80, 280]. In CYP107A1 the mutants A245S and A245T have lower activity and higher uncoupling to produce peroxide, which was attributed to perturbations in positioning of the functionally important waters [73, 280–282]. However, the A245T mutant gained the ability to catalyze the hydroxylation of alternative substrates such as TST [283] and 7-benzyloxyquinoline [284], providing further evidence in support of the general importance of this alcohol side-chain for the P450 catalytic cycle. Other examples of naturally occurring variations in the acid–alcohol pair include the replacement of threonine by asparagine N242 in CYP176A1 [285] and N240 in P450 OxyB [286], glutamine Q230 in CYP1 65D3 [287], and proline P237 in CYP134A1 [288]. Still, these enzymes perform the usual P450 chemistry with oxygen activation. In contrast, in P450 peroxygenases, which do not follow the regular P450 oxygen activation cycle, but rather react via the peroxide shunt mechanism, both acid and alcohol residues are replaced by other amino acids, such as V245-A246 in CYP152A1 [289] and I248-A249 in CYP152L1 [290].
Both the semi-conserved acid and alcohol residues were mutated in other cytochromes P450 in order to understand the importance of these features for P450 catalysis. In CYP102A1 the T268A mutation similar to T252A in CYP101A1 resulted in a decrease in both NADPH consumption and product turnover rates with several substrates [291, 292], but not with pentadecanoic acid, for which coupling was maintained at the same level as in the wild-type enzyme [293]. The high coupling with some substrates and strong dependence on the chain length of the fatty acid (34 vs. 10 % for C12, 88 vs. 74 % for C14, 88 vs. 89 % for C15, and 93 vs. 21 % for C16, in the wild type vs. T268A mutant correspondingly) clearly demonstrates variations due to the packing of the substrate in the overall efficiency of P450 turnover, as well as the difference caused by the Thr268 mutation. These results again stress the critical role of the substrate in the modulating water access to the active site and thus the overall catalytic efficiency defined as the ratio between productive and non-productive pathways. At the same time, the absolute rates of NADPH consumption also strongly depend on the T268A mutation in CYP102A1 [292]. The general conclusion based on the comparison of the wild-type CYP102A1 and T268A mutant is that the presence of this threonine is not absolutely essential for hydroxylation, but it is certainly important in providing an efficient proton delivery pathway with most substrates [293].
CYP176A1 (P450cin) is a close analog of CYP101A1, with its natural substrate cineol also being similar to camphor . However, in wild-type CYP176A1 the conserved threonine residue is replaced by asparagine N242 [294]. The fact that this enzyme is nevertheless catalytically competent in cineole hydroxylation with almost the same efficiency as CYP101A1 in camphor hydroxylation (rate of NADH consumption 950 min− 1 and coupling ~ 80 %) [295] attracted the attention of several research groups and inspired a detailed analysis of its structure and mechanistic issues [285, 295–300]. Mutation of the unusual N242 to a threonine (N242T) [295] or alanine (N242A) [298] resulted in a moderate decrease in coupling, 54 and 72 % respectively, as compared to 80 % in the wild-type enzyme. At the same time significant (sixfold and fourfold) decreases in the absolute rates of NADH consumption were observed with these mutants. Both results are very different from the very strong uncoupling with fast NADH consumption in the CYP101A1 T252A mutant. Attempts to test in CYP176A1 the substrate-assisted mechanism of oxygen activation found for CYP107A1 (P450eryF ), where the hydroxyl group of the substrate replaces the threonine side-chain in stabilizing the proton delivery pathway, proved inconclusive [298]. Unlike in CYP107A1, where replacement of the key hydroxyl group of the substrate by a ketone inhibited hydroxylation by more than 100-fold [301], similar modifications of the native substrate cineol produced only a moderate decrease in activity and coupling [298]. Therefore, no clear understanding of the predominant structural features affecting proton delivery and efficiency of oxygen activation came out of these mutation studies. Alternatively, a comparison of CYP176A1 and CYP101A1 suggests the presence of multiple pathways for protonation of the dioxygen moiety, which compensate to a certain extent for the loss of an important functional group in the active site .
The same conclusion may be made based on the effect caused by the T252N mutation in CYP101A1, which mimics the N242 residue in CYP176A1. This was done to test the ability of asparagine to replace the key T252 [302]. As in CYP176A1, the Asn252 mutant in CYP101A1 demonstrated efficient camphor hydroxylation with an overall 42 % coupling determined as the ratio between the rates of product formation and NADH consumption. The main difference between the wild-type and mutant protein was an almost 20-fold lower affinity for camphor binding in the T252N mutant. This work provides one more example of the great flexibility and robust design of active centers in cytochromes P450, which remain functional despite various mutations.
Both the acid and alcohol residues, Glu318 and Thr319, have been mutated in CYP1A2 in order to evaluate the mechanism of oxygen activation in this cytochrome P450 [303, 304]. Surprisingly, in some cases the mutations improved the overall coupling of methanol oxidation from 9 % in the wild-type protein to 16 % in the E318A mutant, and even to 40 % in the T319A mutant [303], despite the slower product-formation rates, 25 % and 39 % of the rate of 4.4 min− 1 observed in the wild-type enzyme. In addition, no H2O2 was detected with the E318A and T319A mutants, with most of the uncoupling attributed to the oxidase (water) channel. In contrast, the same E318A mutant was almost inactive when 7-ethoxycoumarin was used as a substrate by the same authors, whereas the T319A mutant was even more active than wild-type [304]. These observations led the authors to suggest that the role of the conserved threonine in oxygen activation may be different, or at least not as critical, in CYP1A2. However, later studies demonstrated that even changes in the buffer composition, pH, and temperature could significantly change uncoupling by a factor of three [305].
In CYP2B4 the T302A mutation also significantly inhibits N-demethylation of benzphetamine and hydroxylation of cyclohexane with the rates decreasing 20-fold [56]. The steady-state NADPH consumption rates are also significantly slower when the T302A mutant is used, but H2O2 production is sometimes even higher. This fact can be interpreted as a slower proton delivery in the mutant enzyme and a longer lifetime of the peroxo- and hydroperoxo-ferric intermediates, with a predominantly dissociative unproductive pathway favored over productive protonation and Compound I formation. This hypothesis is also consistent with the tenfold increase in the rate of cyclohexane carboxaldehyde deformylation, apparently catalyzed by the peroxo-ferric intermediate, and not by Compound I [56]. The peroxo-anion reaction with aldehydes to give a peroxyhemiacetal intermediate, followed by homolytic scission of the O–O bond , was also invoked to explain the mechanism of substrate-assisted heme destruction and the faster rate of heme loss in the T302A mutant than in the wild-type protein [306]. The activation of the conversion of a p-hydroxybenzene derivative to a hydroquinone caused by the T303A mutation in CYP2E1 was attributed to a more efficient catalysis of the ipso-substitution reaction by the hydroperoxo-ferric intermediate in the mutant enzyme due to perturbed protonation and loss of the Compound I pathway [307]. Interestingly, the effects observed in these works strongly depended on the substrates, with the k cat increase caused by T303A in CYP2E1 varying from 1.1 with 4-fluoro phenol to 31 with 4-bromo phenol [307], indicating the importance of the electron-withdrawing halogen substituents. At the same time, the authors noted that this variation may indicate a difference in the predominant mechanism in the wild-type and CYP2E1 T303A mutant, consistent with the concept of multiple “active oxygen” intermediates in P450 catalysis as recently reviewed [308, 309].
Substrate dependent uncoupling is clearly manifested in CYP3A4, which can bind up to three substrate molecules such as TST [82, 140, 310]. Using global analysis of multiple experimental data sets measured under identical conditions, it was possible to resolve the fractional contributions of intermediates with one, two, or three TST molecules bound to CYP3A4 in the overall NADPH consumption and product formation rates. The first binding of steroid substrate to the remote binding site did not result in formation of product, but increased the NADPH consumption rate by a factor of four, likely due to stabilization of the oxy-complex [82] and more efficient second electron transfer . Binding of the second substrate molecule caused almost complete shift to the high-spin state and resulted in product formation at almost the maximal rate. At the same time, the NADPH consumption rate also increased, so that the coupling in this case was only 5 %. Binding of the third substrate did not change the rate of product formation, but improved coupling to ~ 13 % [140]. This nontrivial dependence of the rate and efficiency of CYP3A4 catalysis on the substrate concentration demonstrates the complexity of the mechanism of oxygen activation with many parameters determining the overall outcome.
The substrate dependence of the oxidase uncoupling channel in CYP3A4 provided more information about productive and non-productive pathways and the role of the lipid bilayer in the overall efficiency of TST hydroxylation [310]. Even in the absence of substrate the oxidase uncoupling channel accounted for almost 20 % of the total oxygen consumption, indicating possible formation of Compound I in substrate-free CYP3A4. At saturating TST concentrations, the absolute rate of water production increased from 8 to 52 min− 1, or ~ 25 % of the total oxygen consumption, with total peroxide production decreasing from 80 to 65 %, and the product-formation rate to 25 min− 1. These results provide a rough estimate of the partitioning between productive (TST hydroxylation, [6] - > [7] Fig. 3.1) and unproductive (water production, [6] - > [2] Fig. 3.1) pathways at the Compound I level. In this scheme, the ratio k 67/k 62= 0.5 gives an estimate of the relative probability of a successful catalytic event as Compared to the unproductive decay of Compound I with TST as a substrate. The same rate of oxidase uncoupling was observed with bromocriptine as a substrate [310], although the product formation rate was slower. The presence of 30 % anionic lipid 1-palmitoyl-2-oleoyl-phosphatidylserine improved overall coupling and facilitated product formation for TST and bromocriptine by a factor of 1.5–2.
Important results on the specific mechanisms of uncoupling in CYP101A1 have been obtained by Makris et al. [189] by comparison of the G248T and G248V mutants with the wild-type protein. The second proton delivery was significantly inhibited in both mutants, so that the overall NADH consumption rate decreased by factors of 4 and 13, respectively. In addition, the coupling efficiency (ratio of the product formation rate to the NADH consumption rate) fell from 98 % to 74 % and 28 %. Additional information has been provided by comparison of the steady-state kinetic parameters measured in H2O and D2O. Increased uncoupling in D2O was observed in all cases, but to a different extent, with the ratio of the product formation rate constants ranging from 1.1 in the wild-type protein to 1.75 in the G248V mutant. This variation indicates that the second proton transfer is at least in part rate limiting in the CYP101A1 catalytic cycle; otherwise, there would be no apparent difference in the steady-state kinetic parameters in H2O and D2O. In both the G248T and G248V mutants, the steady-state rate of NADH consumption was slightly higher in D2O, despite the slower rate of product formation in deuterated solvent. This is the result of impaired protonation of the peroxo- or hydroperoxo-ferric intermediate in D2O and redistribution of the reaction flux towards H2O2 production.
Because the productive pathway of substrate metabolism includes protonation steps, it depends on the solvent H/D composition and is slower in D2O than in H2O. In contrast, the autoxidation and peroxide dissociation rates are not as strongly proton-dependent and hence are less affected by solvent composition. Therefore, P450 catalysis in D2O is usually slower and less efficient (more uncoupled) than in H2O. The observation of an inverse isotope effect is usually interpreted as an indication of some mechanistic change or different catalytic pathway. This is the case in the C–C cleavage reaction catalyzed by CYP17A1 [311]. The hydroxylation of pregnenolone at C17 proceeds through the common P450 pathway with Compound I as the catalytic intermediate and thus is proton dependent. In agreement with this mechanism, the KSIE measured for this step is small (~ 1.3), as is seen in other cytochromes P450. However, a large inverse isotope effect with k H/k D = 0.39 for the second lyase step with the 17-hydroxypregnenolone as a substrate, cannot be rationalized using the same catalytic pathway. Analysis of the protonation-dependent and protonation-independent pathways (Fig. 3.1) supports the alternative mechanism of lyase catalysis via the unprotonated peroxo-ferric intermediate, first proposed by Akhtar [216] and still debated in the literature [312]. Unlike the regular P450 pathway via Compound I, this reaction goes directly from [5] to product and does not require proton delivery. A productive pathway in such mechanism is not expected to show any KSIE. Alternatively, the peroxo-ferric intermediate can be protonated and form a hydroperoxo-ferric intermediate, which can then dissociate without product formation and contribute to proton-dependent uncoupling . Unlike normal P450 catalysis via proton-dependent formation of Compound I, for the peroxo-driven pathway product formation is not proton-dependent, while uncoupling involves protonation of peroxide and dissociation of H2O2. As a result, for peroxo-ferric driven catalysis of the lyase reaction in CYP17A1 an inverse KSIE is expected, exactly as observed experimentally [311].
10 Summary
The complex multistep mechanism of oxygen activation in P450 represents a finely orchestrated process in which contributions from multiple players have to be delivered timely and in a proper order. Oxygen activation is necessary for accelerating reactions in which dioxygen as a free molecule in gas or solution would never engage because of very high activation barriers. Considering dioxygen as a reagent, the P450 cycle (Fig. 3.1) can be viewed as an oxygen activation process with multiple possible outcomes. The most important for living organisms are the two pathways that result in oxidative transformations of organic molecules catalyzed by heme-oxygen intermediates, Compound I or in some cases peroxo-ferric complexes. In an ideal system (as in CYP101A1 with camphor ) there is almost no peroxide production in this pathway, and all dioxygen consumed in the process of P450 catalysis is evenly distributed between organic products and water. However, other pathways, which do not involve product formation, also can be termed “oxygen activation,” because ROS are released as a result of formation of superoxide and hydrogen peroxide using electrons from NAD(P)H. Taken together, all these pathways result in oxygen consumption and represent the process of dioxygen activation catalyzed by P450. Product and peroxide are produced with 1:1 stoichiometric consumption of NAD(P)H and O2, while the oxidase unproductive pathway has 2:1 stoichiometry of NAD(P)H/O2.
In order to start the activation of atmospheric dioxygen, the heme iron must be reduced to the Fe2 + state, to enable oxygen binding and formation of the oxy-complex of P450. Reduction is performed by electron transfer from a protein redox partner . The rate of reduction [2] → [3] (Fig. 3.1) in most cases strongly depends on the presence of a substrate and on the ability of this substrate to shift the spin state of ferric cytochrome P450 from low-spin (S = 1/2) to high-spin (S = 5/2). Therefore, substrates and their analogs significantly facilitate the NAD(P)H consumption and concomitantly the first step of oxygen consumption [3] → [4]. In case of fast autoxidation [4] → [2] the efficiency of the productive pathway is not high, so the overall effect of the presence of substrate may be predominantly acceleration of superoxide production. This is “the bad side” of the same coin, which is suggested to be especially important for functioning of xenobiotic metabolizing cytochromes P450 in liver, because it may cause oxidative damage by ROS production in the presence of “poor” substrates or substrate analogs that are metabolized with high uncoupling . The effect of ROS production is more pronounced if both the absolute rates of the first electron transfer and autoxidation are high, and the efficiency of the productive pathway is determined by the ratio of the rates of the second electron transfer and uncoupling.
The same logic holds for the second uncoupling branching point between protonation of Compound 0 and O–O bond heterolysis to give Compound I [5] ( [6], versus dissociation of peroxide [5] ( [2]. Again, the higher these rates are, the faster the overall O2 consumption and peroxide production if the enzyme does not provide timely delivery of protons to the distal oxygen of the peroxo-ferric complex. Protonation pathways are formed by side-chains of functionally important residues in the active site , which also help to stabilize several water molecules strategically positioned to form the hydrogen-bonded network essential for proton transfer towards the (hydro)peroxo-anion coordinated to the heme iron. The configuration and continuity of this proton-delivery network, and hence the rate and efficiency of protonation, strongly depend on the structure of substrate and it’s positioning and dynamics in the vicinity of the heme. Even minor variations in the substrate structure can significantly perturb the optimal protonation network and result in highly uncoupled oxygen consumption with high absolute rates. The same is true for mutations of critically important residues, such as T252A in CYP101A1. On the other hand, the D251N mutant in CYP101A1 highlights the role of the absolute rate of the first proton delivery in determining the overall absolute rate of catalysis with no loss of coupling.
This update, in defining the state of our understanding of P450 “oxygen activation,” has encompassed many aspects of the catalytic wheel. On the nature of the species responsible for the critical transformative event of substrate into product, it is clear that there can be more than one oxidant operating. For functionalization of unactivated carbon centers, the mechanism most certainly involves radical chemistry initiated by the iron-oxo, Compound I , intermediate. This state is generated from the dioxygen bound ferrous heme by the input of a second electron and two protons that result in the cleavage of the O–O bond of atmospheric dioxygen. However, the precursor peroxo state can also be reactive in some special cases. The transformation of the initial reactants, O2, and substrate with redox input is dependent on all the steps in the reaction cycle—from substrate binding through product release. The overall efficiency of catalysis is dependent on the protein's ability to control the critical electron and proton input and the position of the substrate near the heme active site . With > 104 isozymes of P450 present throughout living organisms, this enzyme superfamily has clearly learned how to control the utilization of atmospheric oxygen and the “hot” oxidants generated upon its reduction.
References
Omura T, Sato R (1964) The carbon monoxide-binding pigment of liver microsomes. I evidence for its hemoprotein nature. J Biol Chem 239:2370–2378
Hayaishi O (2005) An odyssey with oxygen. Biochem Biophys Res Commun 338:2–6
Waterman MR (2005) Professor Howard Mason and oxygen activation. Biochem Biophys Res Commun 338:7–11
Hayano M, Lindberg MC, Dorfman RI, Hancock JE, Von Doering WE (1955) On the mechanism of the c-11beta-hydroxylation of steroids; a study with H2O18 and O218. Arch Biochem Biophys 59:529–532
Cooper DY, Narasimhulu S, Slade A, Raich W, Foroff O, Rosenthal O (1965) Hemoprotein content and activity of solubilized steroid 11 beta-hydroxylase preparations from adrenocortical mitochondria. Life Sci 4:2109–2114
Estabrook RW (2005) Steroid hydroxylations: a paradigm for cytochrome P450 catalyzed mammalian monooxygenation reactions. Biochem Biophys Res Commun 338:290–298
McCord JM, Keele BB Jr, Fridovich I (1971) An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci U S A 68:1024–1027
Fee JA (1982) Is superoxide important in oxygen poisoning? Trends Biochem Sci 7:84–84
Lichtenberger F, Nastainczyk W, Ullrich V (1976) Cytochrome P450 as an oxene transferase. Biochem Biophys Res Commun 70:939–946
Groves JT, McClusky GA (1976) Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J Am Chem Soc 98:859–861
Groves JT, McClusky GA, White RE, Coon MJ (1978) Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem Biophys Res Commun 81:154–160
Gelb MH, Heimbrook DC, Malkonen P, Sligar SG (1982) Stereochemistry and deuterium isotope effects in camphor hydroxylation by the cytochrome P450cam monoxygenase system. Biochemistry 21:370–377
Nelson DR (2013) http://drnelson.uthsc.edu/P450.stats.Aug2013.png. Accessed 28 Dec. 2014
Coon MJ, Usanov SA (1989) Cytochrome P450 structure and function: summary of the discussion. In: Schuster I (ed) Cytochrome P450: biochemistry and biophysics. Taylor and Francis Press, London, pp 235–241
Denisov IG, Makris TM, Sligar SG, Schlichting I (2005) Structure and chemistry of cytochrome P450 Chem Rev 105:2253–2277
Sligar SG (1976) Coupling of spin, substrate, and redox equilibria in cytochrome P450. Biochemistry 15:5399–5406
Daff SN, Chapman SK, Turner KL, Holt RA, Govindaraj S, Poulos TL, Munro AW (1997) Redox control of the catalytic cycle of flavocytochrome P-450 BM3. Biochemistry 36:13816–13823
Liu LV, Hong S, Cho J, Nam W, Solomon EI (2013) Comparison of high-spin and low-spin nonheme Fe(III)-OOH complexes in O-O bond homolysis and H-atom abstraction reactivities. J Am Chem Soc 135:3286–3299
Ebel RE, O’Keeffe DH, Peterson JA (1978) Substrate binding to hepatic microsomal cytochrome P-450. Influence of the microsomal membrane. J Biol Chem 253:3888–3897
Mast N, Pikuleva IA (2005) A simple and rapid method to measure cholesterol binding to P450s and other proteins. J Lipid Res 46:1561–1568
Murtazina DA, Andersson U, Hahn IS, Bjorkhem I, Ansari GA, Pikuleva IA (2004) Phospholipids modify substrate binding and enzyme activity of human cytochrome P450 27A1. J Lipid Res 45:2345–2353
Atkins WM, Sligar SG (1990) Tyrosine-96 as a natural spectroscopic probe of the cytochrome P-450cam active site. Biochemistry 29:1271–1275
Filipovic D, Paulsen MD, Loida PJ, Sligar SG, Ornstein RL (1992) Ethylbenzene hydroxylation by cytochrome P450cam. Biochem Biophys Res Comm 189:488–495
Mueller EJ, Loida PJ, Sligar SG (1995) Twenty five years of P450cam research. Mechanistic insights into oxygenase catalysis. In: Ortiz de Montellano PR (ed) Cytochrome P450. Structure, mechanism, and biochemistry. Plenum Press, New York, pp 83–124
Rea V, Kolkman AJ, Vottero E, Stronks EJ, Ampt KA, Honing M, Vermeulen NP, Wijmenga SS, Commandeur JN (2012) Active site substitution A82W improves the regioselectivity of steroid hydroxylation by cytochrome P450 BM3 mutants as rationalized by spin relaxation nuclear magnetic resonance studies. Biochemistry 51:750–60
Venkataraman H, Beer B, Bergen LA, Essen N, Geerke DP., Vermeulen NP, Commandeur JN (2012) A single active site mutation inverts stereoselectivity of 16-hydroxylation of testosterone catalyzed by engineered cytochrome P450 BM3. Chembiochem 13:520–523
Whitehouse CJ, Bell SG, Wong LL (2012) P450(BM3) (CYP102A1): connecting the dots. Chem Soc Rev 41:1218–1260
Mast N, Zheng W, Stout CD, Pikuleva IA (2013) Binding of a cyano-and fluoro-containing drug bicalutamide to cytochrome P450 46A1: unusual features and spectral response. J Biol Chem 288:4613–4624
Niwa T, Murayama N, Yamazaki H (2009) Oxidation of endobiotics mediated by xenobiotic-metabolizing forms of human cytochrome P450. Curr Drug Metab 10:700–712
Rendic S, Guengerich FP (2010) Update information on drug metabolism systems–2009, part II: summary of information on the effects of diseases and environmental factors on human cytochrome P450 (CYP) enzymes and transporters. Curr Drug Metab 11:4–84
Rendic S, Guengerich FP (2012) Contributions of human enzymes in carcinogen metabolism. Chem Res Toxicol 25:1316–1383
Poulos TL, Finzel BC, Howard AJ (1986) Crystal structure of substrate-free pseudomonas putida cytochrome P-450. Biochemistry 25:5314–5322
Poulos TL, Finzel BC, Howard AJ (1987) High-resolution crystal structure of cytochrome P450cam. J Mol Biol 195:687–700
Nesset MJM, Shokhirev NV, Enemark PD, Jacobson SE, Walker FA (1996) Models of the cytochromes. Redox properties and thermodynamic stabilities of complexes of\“hindered\” iron(III) and iron(II) tetraphenylporphyrinates with substituted pyridines and imidazoles. Inorg Chem 35:5188–5200
Das A, Grinkova YV, Sligar SG (2007) Redox potential control by drug binding to cytochrome P 450 3A4. J Am Chem Soc 129:13778–13779
Ost TWB, Clark J, Mowat CG, Miles CS, Walkinshaw MD, Reid GA, Chapman SK, Daff S (2003) Oxygen activation and electron transfer in flavocytochrome P 450 BM3. J Am Chem Soc 125:15010–15020
Clark JP, Miles CS, Mowat CG, Walkinshaw MD, Reid GA, Daff SN, Chapman SK (2006) The role of Thr268 and Phe393 in cytochrome P450 BM3. J Inorg Biochem 100:1075–1090
Narasimhulu S (1971) Uncoupling of oxygen activation from hydroxylation in the steroid C-21 hydroxylase of bovine adrenocortical microsomes. Arch Biochem Biophys 147:384–390
Ullrich V, Diehl H (1971) Uncoupling of monooxygenation and electron transport by fluorocarbons in liver microsomes. Eur J Biochem 20:509–512
Blanck J, Smettan G, Jaenig GR, Ruckpaul K (1977) Substrate binding kinetics and its role in the cytochrome P450 hydroxylation sequence. Croat Chem Acta 49:271–277
Backes WL, Tamburini PP, Jansson I, Gibson GG, Sligar SG, Schenkman JB (1985) Kinetics of cytochrome P-450 reduction: evidence for faster reduction of the high-spin ferric state. Biochemistry 24:5130–5136
Honeychurch MJ, Hill AO, Wong LL (1999) The thermodynamics and kinetics of electron transfer in the cytochrome P450cam enzyme system. FEBS Lett 451:351–353
Fisher MT, Sligar SG (1987) Temperature jump relaxation kinetics of the P-450cam spin equilibrium. Biochemistry 26:4797–4803
Brenner S, Hay S, Girvan HM, Munro AW, Scrutton NS (2007) Conformational dynamics of the cytochrome P450 BM3/N-palmitoylglycine complex: the proposed “proximal-distal” transition probed by temperature-jump spectroscopy. J Phys Chem B 111:7879–7886
Guengerich FP, Krauser JA, Johnson WW (2004) Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry 43:10775–10788
Griffin BW, Peterson JA (1972) Camphor binding of Pseudomonas putida cytochrome P450. Kinetics and thermodynamics of the reaction. Biochemistry 11:4740–4746
Behera RK, Mazumdar S (2008) Roles of two surface residues near the access channel in the substrate recognition by cytochrome P450cam. Biophys Chem 135:1–6
Munro AW, Daff S, Coggins JR, Lindsay JG, Chapman SK (1996) Probing electron transfer in flavocytochrome P-450 BM3 and its component domains. Eur J Biochem 239:403–409
Isin EM, Guengerich FP (2008) Substrate binding to cytochromes P450. Anal Bioanal Chem 392:1019–1030
Hume R, Kelly RW, Taylor PL, Boyd GS (1984) The catalytic cycle of cytochrome P-450scc and intermediates in the conversion of cholesterol to pregnenolone. Eur J Biochem 140:583–591
Isin EM, Sohl CD, Eoff RL., Guengerich FP (2008) Cooperativity of cytochrome P450 1A2: interactions of 1,4-phenylene diisocyanide and 1-isopropoxy-4-nitrobenzene. Arch Biochem Biophys 473:69–75
Guengerich FP (2002) Rate-limiting steps in cytochrome P450 catalysis. Biol Chem 383:1553–1564
Yun CH, Miller GP, Guengerich FP (2000) Rate-determining steps in phenacetin oxidations by human cytochrome P450 1A2 and selected mutants. Biochemistry 39:11319–11329
McLean KJ, Carroll P, Lewis DG, Dunford AJ, Seward HE, Neeli R, Cheesman MR, Marsollier L, Douglas P, Smith WE, Rosenkrands I, Cole ST, Leys D, Parish T, Munro AW (2008) Characterization of active site structure in CYP121. A cytochrome P450 essential for viability of Mycobacterium tuberculosis H37Rv. J Biol Chem 283:33406–33416
Shinkyo R, Guengerich FP (2011) Cytochrome P450 7A1 cholesterol 7alpha-hydroxylation: individual reaction steps in the catalytic cycle and rate-limiting ferric iron reduction. J Biol Chem 286:4632–4643
Vaz ADN, Pernecky SJ, Raner GM, Coon MJ (1996) Peroxo-iron and oxenoid-iron species as alternative oxygenating agents in cytochrome P450-catalyzed reactions: switching by threonine-302 to alanine mutagenesis of cytochrome P450 2B4. Proc Natl Acad Sci U S A 93:4644–4648
Johnson BM, Bolton JL, van Breemen RB (2001) Screening botanical extracts for quinoid metabolites. Chem Res Toxicol 14:1546–1551
Rabe KS, Gandubert VJ, Spengler M, Erkelenz M, Niemeyer CM (2008) Engineering and assaying of cytochrome P450 biocatalysts. Anal Bioanal Chem 392:1059–1073
Butler CF, Peet C, McLean KJ, Baynham MT, Blankley RT, Fisher K, Rigby SE, Leys D, Voice MW, Munro AW (2014) Human P450-like oxidation of diverse proton pump inhibitor drugs by “gatekeeper” mutants of flavocytochrome P450 BM3. Biochem J 460:247–259
Hiruma Y, Hass MA, Kikui Y, Liu WM, Olmez B, Skinner SP, Blok A, Kloosterman A, Koteishi H, Lohr F, Schwalbe H, Nojiri M, Ubbink M (2013) The structure of the cytochrome p450cam-putidaredoxin complex determined by paramagnetic NMR spectroscopy and crystallography. J Mol Biol 425:4353–4365
Tripathi S, Li H, Poulos TL (2013) Structural basis for effector control and redox partner recognition in cytochrome P450. Science 340:1227–1230
Sligar SG, Debrunner PG, Lipscomb JD, Namtvedt MJ, Gunsalus IC (1974) A role of the putidaredoxin COOH-terminus in P-450cam (cytochrome m) hydroxylations. Proc Natl Acad Sci U S A 71:3906–3910
Shimada H, Nagano S, Ariga Y, Unno M, Egawa T, Hishiki T, Ishimura Y, Masuya F, Obata T, Hori H (1999) Putidaredoxin-cytochrome p450cam interaction. Spin state of the heme iron modulates putidaredoxin structure. J Biol Chem 274:9363–9369
Tosha T, Yoshioka S, Ishimori K, Morishima I (2004) L358P mutation on cytochrome P 450cam simulates structural changes upon putidaredoxin binding: the structural changes trigger electron transfer to oxy-P450cam from electron donors. J Biol Chem 279:42836–42843
Tosha T, Yoshioka S, Takahashi S, Ishimori K, Shimada H, Morishima I (2003) NMR study on the structural changes of cytochrome P 450cam upon the complex formation with putidaredoxin: functional significance of the putidaredoxin-induced structural changes. J Biol Chem 278:39809–39821
Unno M, Christian JF, Benson DE, Gerber NC, Sligar SG, Champion PM (1997) Resonance Raman investigations of cytochrome P450cam complexed with putidaredoxin. J Am Chem Soc 119:6614–6620
Unno M, Christian JF, Sjodin T, Benson DE, Macdonald IDG, Sligar SG, Champion PM (2002) Complex formation of cytochrome P450cam with putidaredoxin: evidence for protein-specific interactions involving the proximal thiolate ligand. J Biol Chem 277:2547–2553
Davies MD., Sligar SG (1992) Genetic variants in the putidaredoxin-cytochrome P-450cam electron-transfer complex: identification of the residue responsible for redox-state-dependent conformers. Biochemistry 31:11383–11389
Myers WK, Lee YT, Britt RD, Goodin DB (2013) The conformation of P450cam in complex with putidaredoxin is dependent on oxidation state. J Am Chem Soc 135:11732–11735
Zhang W, Liu Y, Yan J, Cao S, Bai F, Yang Y, Huang S, Yao L, Anzai Y, Kato F, Podust LM, Sherman DH, Li S (2014) New reactions and products resulting from alternative interactions between the P450 enzyme and redox partners. J Am Chem Soc 136:3640–3646
Bangcharoenpaurpong O, Rizos AK, Champion M, Jllie D, Sligar SG (1986) Resonance Raman detection of bound dioxygen in cytochrome P-450cam. J Biol Chem 261:8089–8092
Hu S, Schneider AJ, Kincaid JR (1991) Resonance Raman studies of oxycytochrome P450cam: effect of substrate structure on n(O–O) and n(Fe–O2). J Am Chem Soc 113:4815–4822
Nagano S, Cupp-Vickery JR, PoulosTL (2005) Crystal structures of the ferrous dioxygen complex of wild-type cytochrome P450eryF and its mutants, A245S and A245T: investigation of the proton transfer system in P450eryF. J Biol Chem 280:22102–22107
Nagano S, Poulos TL (2005) Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J Biol Chem 280:31659–31663
Sharrock M, Debrunner PG, Schulz C, Lipscomb JD, Marshall V, Gunsalus IC (1976) Cytochrome P450cam and its complexes. Moessbauer parameters of the heme iron. Biochim Biophys Acta 420:8–26
Carrondo MA, Bento I, Matias PM, Lindley PF (2007) Crystallographic evidence for dioxygen interactions with iron proteins. J Biol Inorg Chem 12:429–442
Peterson JA, Ishimura Y, Griffin BW (1972) Pseudomonas putida cytochrome P-450: characterization of an oxygenated form of the hemoprotein. Arch Biochem Biophys 149:197–208
Tyson CA, Lipscomb JD, Gunsalus IC (1972) The role of putidaredoxin and P450 cam in methylene hydroxylation. J Biol Chem 247:5777–5784
Yun CH, Kim KH, Calcutt MW, Guengerich FP (2005) Kinetic analysis of oxidation of coumarins by human cytochrome P450 2A6. J Biol Chem 280:12279–12291
Zhao B, Guengerich FP, Voehler M, Waterman MR (2005) Role of active site water molecules and substrate hydroxyl groups in oxygen activation by cytochrome P450 158A2: a new mechanism of proton transfer. J Biol Chem 280:42188–42197
Denisov IG, Grinkova YV, Baas BJ, Sligar SG (2006) The ferrous-dioxygen intermediate in human cytochrome P450 3A4: substrate dependence of formation of decay kinetics. J Biol Chem 281:23313–23318
Denisov IG, Grinkova YV, McLean MA, Sligar SG (2007) The one-electron autoxidation of human cytochrome P450 3A4. J Biol Chem 282:26865–26873
Das A, Varma SS, Mularczyk C, Meling DD (2014) Functional investigations of thromboxane synthase (CYP5A1) in lipid bilayers of nanodiscs. Chembiochem 15:892–899
Totani K, Iizuka T, Shimada H, Makino R, Ishimura Y (1983) Proton coupling in the ligand-binding reaction of ferric cytochrome P-450 from Pseudomonas putida. Arch Biochem Biophys 222:207–215
Greschner S, Davidov RM, Stepanov SV, Ruckpaul K (1980) Comparative studies on carbon monoxide binding to cytochrome P-450 analyzed by flash-photolysis and stopped-flow-techniques. Stud Biophys 81:133–134
Peterson JA, Griffin BW (1972) Carbon monoxide binding by Pseudomonas putida cytochrome P-450. Arch Biochem Biophys 151:427–433
Sevrioukova IF, Peterson JA (1995) Reaction of carbon monoxide and molecular oxygen with P450terp (CYP108) and P450BM-3 (CYP102). Arch Biochem Biophys 317:397–404
Weber B, Nickel E, Horn M, Nienhaus K, Nienhaus GU (2014) Substrate inhibition in human indoleamine 2,3-dioxygenase. J Phys Chem Lett 5:756–761
Berka V, Tsai AL (2000) Characterization of interactions among the heme center, tetrahydrobiopterin, and L-arginine binding sites of ferric eNOS using imidazole, cyanide, and nitric oxide as probes. Biochemistry 39:9373–9383
Girvan HM, Heyes DJ, Scrutton NS, Munro AW (2007) Laser photoexcitation of NAD(P)H induces reduction of P450 BM3 heme domain on the microsecond time scale. J Am Chem Soc 129: 6647–6653
Dunford AJ, McLean KJ, Sabri M, Seward HE, Heyes DJ, Scrutton NS, Munro AW (2007) Rapid P450 heme iron reduction by laser photoexcitation of Mycobacterium tuberculosis CYP121 and CYP51B1. Analysis of CO complexation reactions and reversibility of the P450/P420 equilibrium. J Biol Chem 282:24816–24824
Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG (2000) The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287:1615–1622
Poulos TL (2005) Intermediates in P450 catalysis. Philos Transact A 363:793–806; discussion 1035–1040
Poulos TL, Madrona Y (2013) Oxygen activation and redox partner binding in cytochromes P450. Biotechnol Appl Biochem 60:128–133
Poulos TL (2014) Heme enzyme structure and function. Chem Rev 114:3919–3962
Momenteau M, Reed CA (1994) Synthetic heme dioxygen complexes. Chem Rev 94:659–698
Collman JP, Gagne RR, Reed CA, Robinson WT, Rodley GA (1974) Structure of an iron(II) dioxygen complex; a model for oxygen carrying hemeproteins. Proc Natl Acad Sci U S A 71:1326–1329
Collman JP (1977) Synthetic models for the oxygen-binding hemoproteins. Acc Chem Res 10:265–272
Li J, Noll BC, Oliver AG, Schulz CE, Scheidt WR (2013) Correlated ligand dynamics in oxyiron picket fence porphyrins: structural and Mossbauer investigations. J Am Chem Soc 135:15627–15641
Collman JP, Brauman JI, Halbert TR, Suslick KS (1976) Nature of O2 and CO binding to metalloporphyrins and heme proteins. Proc Natl Acad Sci U S A 73:3333–3337
Friesner RA, Baik M-H, Gherman BF, Guallar V, Wirstam M, Murphy RB, Lippard SJ (2003) How iron-containing proteins control dioxygen chemistry: a detailed atomic level description via accurate quantum chemical and mixed quantum mechanics/molecular mechanics calculations. Coord Chem Rev 238–239:267–290
Vojtechovsky J, Chu K, Berendzen J, Sweet RM, Schlichting I (1999) Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys J 77:2153–2174
Chen H, Ikeda-Saito M, Shaik S (2008) Nature of the Fe-O2 bonding in oxy-myoglobin: effect of the protein. J Am Chem Soc 130:14778–14790
Dietzel PD, Kremer RK, Jansen M (2004) Tetraorganylammonium superoxide compounds: close to unperturbed superoxide ions in the solid state. J Am Chem Soc 126:4689–4696
Wilson SA, Kroll T, Decreau RA, Hocking RK, Lundberg M, Hedman B, Hodgson KO, Solomon EI (2013) Iron L-edge X-ray absorption spectroscopy of oxy-picket fence porphyrin: experimental insight into Fe-O2 bonding. J Am Chem Soc 135:1124–1136
Hanson LK, Eaton WA, Sligar SG, Gunsalus IC, Gouterman M, Connell CR (1976) Origin of the anomalous Soret spectra of carboxycytochrome P-450. J Am Chem Soc 98:2672–2674
Hanson LK, Sligar SG, Gunsalus IC (1977) Electronic structure of cytochrome P450. Croat Chem Acta 49:237–250
Denisov IG, Dawson JH, Hager LP, Sligar SG (2007) The ferric-hydroperoxo complex of chloroperoxidase. Biochem Biophys Res Commun 363:954–958
Perera R, Sono M, Raner M Dawson JH (2005) Subzero-temperature stabilization and spectroscopic characterization of homogeneous oxyferrous complexes of the cytochrome P450 BM3 (CYP102) oxygenase domain and holoenzyme. Biochem Biophys Res Commun 338:365–371
Denisov IG, Sligar SG (2010) Cytochromes P450 in nanodiscs. Biochim Biophys Acta 1814:223–229
Perera R, Sono M, Kinloch R, Zhang H, Tarasev M, Im SC, Waskell L, Dawson JH (2011) Stabilization and spectroscopic characterization of the dioxygen complex of wild-type cytochrome P4502B4 (CYP2B4) and its distal side E301Q, T302A and proximal side F429H mutants at subzero temperatures. Biochim Biophys Acta 1814:69–75
Sono M, Perera R, Jin S, Makris TM, Sligar SG, Bryson TA, Dawson JH (2005) The influence of substrate on the spectral properties of oxyferrous wild-type and T252A cytochrome P450-CAM. Arch Biochem Biophys 436:40–49
Kincaid JR (2000) Resonance Raman spectra of heme proteins and model compounds. In: Kadish KM, Smith KM, Guilard R (eds) Porphyrin handbook. Academic, New York, pp 225–291
Bruha A, Kincaid JR (1988) Resonance Raman studies of dioxygen adducts of cobalt-substituted heme proteins and model compounds. Vibrationally coupled dioxygen and the issues of multiple structures and distal side hydrogen bonding. J Am Chem Soc 110:6006–6014
Das TK, Couture M, Ouellet Y, Guertin M, Rousseau DL (2001) Simultaneous observation of the O–O and Fe–O2 stretching modes in oxyhemoglobins. Proc Natl Acad Sci U S A 98:479–484
Yanagisawa S, Horitani M, Sugimoto H, Shiro Y, Okada N, Ogura T (2011) Resonance Raman study on the oxygenated and the ferryl-oxo species of indoleamine 2,3-dioxygenase during catalytic turnover. Faraday Disc 148:239–247; discussion 299–314
Sjodin T, Christian JF, Macdonald IDG Davydov R, Unno M, Sligar SG, Hoffman BM, Champion PM (2001) Resonance Raman and EPR investigations of the D251N oxycytochrome P450cam/putidaredoxin complex. Biochemistry 40:6852–6859
Tosha T, Kagawa N, Arase M, Waterman MR, Kitagawa T (2008) Interaction between substrate and oxygen ligand responsible for effective O–O bond cleavage in bovine cytochrome P450 steroid 21-hydroxylase proved by Raman spectroscopy. J Biol Chem 283:3708–3717
Gregory M, Mak PJ, Sligar SG, Kincaid JR (2013) Differential hydrogen bonding in human CYP17 dictates hydroxylation versus lyase chemistry. Angew Chem Int Ed Engl 52:5342–5345
Mak PJ, Luthra A, Sligar SG, Kincaid JR (2014) Resonance Raman spectroscopy of the oxygenated intermediates of human CYP19A1 implicates a compound I intermediate in the final lyase step. J Am Chem Soc 136:4825–4828
Macdonald IDG, Sligar SG, Christian JF, Unno M, Champion PM (1999) Identification of the Fe–O–O bending mode in oxycytochrome P450cam by resonance Raman spectroscopy. J Am Chem Soc 121:376–380
Soldatova AV, Ibrahim M, Spiro TG (2013) Electronic structure and ligand vibrations in FeNO, CoNO, and FeOO porphyrin adducts. Inorg Chem 52:7478–7486
Spiro TG, Soldatova AV, Balakrishnan G (2013) CO, NO and O2 as vibrational probes of heme protein interactions. Coord Chem Rev 257:511–527
Li D, Kabir M, Stuehr DJ, Rousseau DL, Yeh SR (2007) Substrate- and isoform-specific dioxygen complexes of nitric oxide synthase. J Am Chem Soc 129:6943–6951
Chartier FJ, Couture M (2007) Substrate-specific interactions with the heme-bound oxygen molecule of nitric-oxide synthase. J Biol Chem 282:20877–20886
Mak PJ, Gregory MC, Sligar SG, Kincaid JR (2014) Resonance Raman spectroscopy reveals that substrate structure selectively impacts the heme-bound diatomic ligands of CYP17. Biochemistry 53:90–100
Li J, Peng Q, Barabanschikov A, Pavlik JW, Alp EE, Sturhahn W, Zhao J, Schulz CE, Sage JT, Scheidt WR (2011) New perspectives on iron-ligand vibrations of oxyheme complexes. Chemistry 17:11178–11185
Zeng W, Barabanschikov A, Wang N, Lu Y, Zhao J, Sturhahn W, Alp EE., Sage JT (2012) Vibrational dynamics of oxygenated heme proteins. Chem Commun 48:6340–6342
Lambeth JD, Kitchen SE, Farooqui AA, Tuckey R, Kamin H (1982) Cytochrome P-450scc-substrate interactions. Studies of binding and catalytic activity using hydroxycholesterols. J Biol Chem 257:1876–1884
Tuckey RC, Kamin H (1982) The oxyferro complex of adrenal cytochrome P-450scc. Effect of cholesterol and intermediates on its stability and optical characteristics. J Biol Chem 257:9309–9314
Lipscomb JD, Sligar SG, Namtvedt MJ, Gunsalus IC (1976) Autooxidation and hydroxylation reactions of oxygenated ctohrome P-450cam. J Biol Chem 251:1116–1124
Denisov IG, Sligar SG (2010) Cytochrome P450 enzymes. In: Kadish KM, Smith KM, Guilard R (eds) Handbook of porphyrin science, v.5. World Scientific, Singapore, pp 165–201
Davydov R, Razeghifard R, Im SC, Waskell L, Hoffman BM (2008) Characterization of the microsomal cytochrome P450 2B4 O2 activation intermediates by cryoreduction and electron paramagnetic resonance. Biochemistry 47:9661–9666
Denisov IG (2010) Cryoradiolysis as a method for mechanistic studies in inorganic biochemistry. In: Bakac A (ed) Physical inorganic chemistry. Principles, methods, models. Wiley, New York, pp 109–142
Denisov IG, Grinkova YV, Sligar SG (2012) Cryoradiolysis and cryospectroscopy for studies of heme-oxygen intermediates in cytochromes p450. Meth Mol Biol 875:375–391
Eisenstein L, Debey P, Douzou P (1977) P 450cam: oxygenated complexes stabilized at low temperature. Biochem Biophys Res Comm 77:1377–1383
Luthra A, Denisov IG, Sligar SG (2011) Spectroscopic features of cytochrome P450 reaction intermediates. Arch Biochem Biophys 507:26–35
Luthra A, Denisov IG, Sligar SG (2011) Temperature derivative spectroscopy to monitor the autoxidation decay of cytochromes P450. Anal Chem 83:5394–5399
Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305:683–686
Denisov IG, Baas BJ, Grinkova YV, Sligar SG (2007) Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation. J Biol Chem 282:7066–7076
Johnson EF, Stout CD (2013) Structural diversity of eukaryotic membrane cytochrome P450s. J Biol Chem 288:17082–17090
Lee YT, Wilson RF, Rupniewski I, Goodin DB (2010) P450cam visits an open conformation in the absence of substrate. Biochemistry 49:3412–3419
Lee YT, Glazer EC, Wilson RF, Stout CD, Goodin DB (2011) Three clusters of conformational states in p450cam reveal a multistep pathway for closing of the substrate access channel. Biochemistry 50:693–703
Markwick PR, Pierce LC, Goodin DB, McCammon JA (2011) Adaptive accelerated molecular dynamics (Ad-AMD) revealing the molecular plasticity of P450cam. J Phys Chem Lett 2:158–164
Stoll S, Lee YT, Zhang M, Wilson RF, Britt RD, Goodin DB (2012) Double electron-electron resonance shows cytochrome P450cam undergoes a conformational change in solution upon binding substrate. Proc Natl Acad Sci U S A 109:12888–12893
Nath A, Koo PK, Rhoades E, Atkins WM (2008) Allosteric effects on substrate dissociation from cytochrome P450 3A4 in nanodiscs observed by ensemble and single-molecule fluorescence spectroscopy. J Am Chem Soc 130:15746–15747
Collom SL, Laddusaw RM, Burch AM, Kuzmic P, Perry MD Jr, Miller GP (2008) CYP2E1 substrate inhibition. Mechanistic interpretation through an effector site for monocyclic compounds. J Biol Chem 283:3487–3496
Lu Y, Cederbaum AI (2008) CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med 44:723–738
Zangar RC, Davydov DR, Verma S (2004) Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol 199:316–331
McLean MA, Yeom H, Sligar SG (1996) Carbon monoxide binding to cytochrome P450BM-3: evidence for a substrate-dependent conformational change. Biochimie 78:700–705
Tian WD, Wells AV, Champion PM, Di Primo C, Gerber N, Sligar SG (1995) Measurements of CO geminate recombination in cytochromes P450 and P420. J Biol Chem 270:8673–8679
Brewer CB, Peterson JA (1986) Single turnover studies with oxy-cytochrome P-450cam. Arch Biochem Biophys 249:515–521
Brewer CB, Peterson JA (1988) Single turnover kinetics of the reaction between oxycytochrome P-450cam and reduced putidaredoxin. J Biol Chem 263:791–798
Kuznetsov VY, Poulos TL, Sevrioukova IF (2006) Putidaredoxin-to-cytochrome P450cam electron transfer: differences between the two reductive steps required for catalysis. Biochemistry 45:11934–11944
Tosha T, Yoshioka S, Hori H, Takahashi S, Ishimori K, Morishima I (2002) Molecular mechanism of the electron transfer reaction in cytochrome P450cam-putidaredoxin: roles of glutamine 360 at the heme proximal site. Biochemistry 41:13883–13893
Zhang H, Gruenke L, Arscott D, Shen A, Kasper C, Harris Danni L, Glavanovich M, Johnson R, Waskell L (2003) Determination of the rate of reduction of oxyferrous cytochrome P450 2B4 by 5-deazariboflavin adenine dinucleotide T491V cytochrome P450 reductase. Biochemistry 42:11594–11603
Baron J, Hildebrandt AG, Peterson JA, Estabrook RW (1973) The role of oxygenated cytochrome P-450 and of cytochrome b5 in hepatic microsomal drug oxidations. Drug Metab Dispos 1:129–138
Guengerich FP, Ballou DP, Coon MJ (1976) Spectral intermediates in the reaction of oxygen with purified liver microsomal cytochrome P-450. Biochem Biophys Res Comm 70:951–956
Blake RC 2nd, Coon MJ (1980) On the mechanism of action of cytochrome P-450. Spectral intermediates in the reaction of P-450LM2 with peroxy compounds. J Biol Chem 255:4100–4111
Blake RC 2nd, Coon MJ (1989) On the mechanism of action of cytochrome P-450. Spectral intermediates in the reaction with iodosobenzene and its derivatives. J Biol Chem 264:3694–3701
Benson DE, Suslick KS, Sligar SG (1997) Reduced oxy Intermediate observed in D251N cytochrome P450cam. Biochemistry 36:5104–5107
Baek HK, Van Wart HE (1989) Elementary steps in the formation of horseradish peroxidase compound I: direct observation of compound 0, a new intermediate with a hyperporphyrin spectrum. Biochemistry 28:5714–5719
Tanaka M, Matsuura K, Yoshioka S, Takahashi S, Ishimori K, Hori H, Morishima I (2003) Activation of hydrogen peroxide in horseradish peroxidase occurs within approximately 200 micro s observed by a new freeze-quench device. Biophys J 84:1998–2004
Norman I, Porter G (1954) Trapped atoms and radicals in a glass ‘cage’. Nature 174:508–509
Whittle E, Dows DA, Pimentel GC (1954) Matrix isolation method for the experimental study of unstable species. J Chem Phys 22:1943
Pimentel GC (1958) Reaction kinetics by the matrix isolation method: diffusion on Argon; cis-trans isomerization of nitrous acid. J Am Chem Soc 80:62–64
Symons MCR, Townsend MG (1959) Unstable intermediates. I. Photochemical reactions in rigid glasses. J Chem Soc :263–269
Pimentel GC (1962) Matrix technique and its application in the field of chemical physics. Pure Appl Chem 4:61–70
Greschner S, Davydov RM, Jaenig GR, Ruckpaul K, Blumenfeld LA (1979) Spectral properties of nonequilibrium states in cytochrome P-450 formed by reduction at subzero temperatures. Acta Biol Med Germ 38:443–448
Davydov RM, Greschner S, Ruckpaul K (1979) Study of absorption and magnetic circular dichroism spectra of hemoproteins in conformationally nonequilibrium states. V. Cytochrome P 450 and its complexes with substrates. Molek Biol 13:1397–1406
Davydov RM, Greshner S, Magonov SN, Ruckpaul K, Blyumenfel’d LA (1978) Absorption spectra of cytochrome P-450 nonequilibrium states formed during the low-temperature reduction of protein. Doklady Akad Nauk SSSR 241:707–709
Blyumenfel’d LA, Davydov RM, Fel NS, Magonov SN, Vilu R (1974) Studies on the conformational changes of metalloproteins induced by electrons in water-ethylene glycol solutions at low temperatures. Cytochrome c. FEBS Lett 45:256–258
Blumenfeld LA, Davydov RM, Magonov SN, Vilu R (1974) Conformational changes of metalloproteins induced by electrons in water-ethyleneglycol solutions at low temperatures. Hemoglobin. II. FEBS Lett 49:246–248
Blyumenfel’d LA, Davydov RM (1979) Chemical reactivity of metalloproteins in conformationally out-of-equilibrium states. Biochim Biophys Acta 549:255–280
Kappl R, Hoehn-Berlage M, Huettermann J, Bartlett N, Symons MCR (1985) Electron spin and electron nuclear double resonance of the [FeO2]− [ferrite] center from irradiated oxyhemo- and oxymyoglobin. Biochim Biophys Acta 827:327–343
Bartlett N, Symons MC (1983) Electron addition to the (FeO2) unit of oxyhaemoglobin glycera. Biochim Biophys Acta 744:110–114
Symons MCR, Petersen RL (1978) Electron capture at the iron-oxygen center in single crystals of oxymyoglobin studied by electron spin resonance spectroscopy. Biochim Biophys Acta 535:241–246
Symons MCR, Petersen RL (1978) Electron capture by oxyhaemoglobin: an e.s.r. study. Proc R Soc London B 201:285–300
Symons MCR, Petersen RL (1978) The relative electron affinities of the alpha and beta chains of oxyhaemoglobin as a function of pH and added inositol hexaphosphate. An electron spin resonance study. Biochim Biophys Acta 537:70–76
Denisov IG, Makris TM, Sligar SG (2002) Cryoradiolysis for the study of P450 reaction intermediates. Meth Enzymol 357:103–115
Beitlich T, Kuehnel K, Schulze-Briese C, Shoeman RL, Schlichting I (2007) Cryoradiolytic reduction of crystalline heme proteins: analysis by UV-vis spectroscopy and X-ray crystallography. J Synchrotron Radiat 14:11–23
Davydov R, Kuprin S, Graeslund A, Ehrenberg A (1994) Electron paramagnetic resonance study of the mixed-valent diiron center in Escherichia coli ribonucleotide reductase produced by reduction of radical-free protein R2 at 77 K. J Am Chem Soc 116:11120–11128
Davydov R, Kappl R, Huettermann J, Peterson JA (1991) EPR-spectroscopy of reduced oxyferrous-P450cam. FEBS Lett 295:113–115
Davydov R, Hoffman BM (2011) Active intermediates in heme monooxygenase reactions as revealed by cryoreduction/annealing, EPR/ENDOR studies. Arch Biochem Biophys 507:36–43
Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM (2001) Hydroxylation of camphor by reduced oxy-cytochrome P450cam: mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J Am Chem Soc 123:1403–1415
Nagano S, Tosha T, Ishimori K, Morishima I, Poulos TL (2004) Crystal structure of the cytochrome p450cam mutant that exhibits the same spectral perturbations induced by putidaredoxin binding. J Biol Chem 279:42844–9
Davydov R, Macdonald IDG, Makris TM, Sligar SG, Hoffman BM (1999) EPR and ENDOR of catalytic intermediates in cryoreduced native and mutant oxy-cytochromes P450cam: mutation-Induced changes in the proton delivery system. J Am Chem Soc 121:10654–10655
Davydov R, Perera R, Jin S, Yang T-C, Bryson TA, Sono M, Dawson JH, Hoffman BM (2005) Substrate modulation of the properties and reactivity of the oxy-ferrous and hydroperoxo-ferric intermediates of cytochrome P450cam as shown by cryoreduction-EPR/ENDOR spectroscopy. J Am Chem Soc 127:1403–1413
Makris TM, von Koenig K, Schlichting I, Sligar SG (2007) Alteration of P450 distal pocket solvent leads to impaired proton delivery and changes in heme geometry. Biochemistry 46:14129–14140
Kim SH, Yang T-C, Perera R, Jin S, Bryson TA, Sono M, Davydov R, Dawson JH, Hoffman BM (2005) Cryoreduction EPR and 13C, 19F ENDOR study of substrate-bound substates and solvent kinetic isotope effects in the catalytic cycle of cytochrome P450cam and its T252A mutant. Dalton Trans :3464–3469
Davydov R, Ledbetter-Rogers A, Martasek P, Larukhin M, Sono M, Dawson JH, Siler Masters BS, Hoffman BM (2002) EPR and ENDOR characterization of intermediates in the cryoreduced oxy-nitric oxide synthase heme domain with bound L-arginine or NG-hydroxyarginine. Biochemistry 41:10375–10381
Denisov IG, Ikeda-Saito M, Yoshida T, Sligar SG (2002) Cryogenic absorption spectra of hydroperoxo-ferric heme oxygenase, the active intermediate of enzymatic heme oxygenation. FEBS Lett 532:203–206
Ledbetter AP, McMillan K, Roman LJ, Masters BS, Dawson JH, Sono M (1999) Low-temperature stabilization and spectroscopic characterization of the dioxygen complex of the ferrous neuronal nitric oxide synthase oxygenase domain. Biochemistry 38:8014–8021
Gantt SL, Denisov IG, Grinkova YV, Sligar SG (2009) The critical iron-oxygen intermediate in human aromatase. Biochem Biophys Res Commun 387:169–173
Davydov R, Gilep AA, Strushkevich NV, Usanov SA, Hoffman BM (2012) Compound I is the reactive intermediate in the first monooxygenation step during conversion of cholesterol to pregnenolone by cytochrome P450scc: EPR/ENDOR/cryoreduction/annealing studies. J Am Chem Soc 134:17149–17156
Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman BM (2002) Catalytic mechanism of heme oxygenase through EPR and ENDOR of cryoreduced oxy-heme oxygenase and its Asp 140 mutants. J Am Chem Soc 124:1798–1808
Denisov IG, Makris TM, Sligar SG (2001) Cryotrapped reaction intermediates of cytochrome P450 studied by radiolytic reduction with phosphorus-32. J Biol Chem 276:11648–11652
Denisov IG, Mak PJ, Makris TM, Sligar SG, Kincaid JR (2008) Resonance Raman characterization of the peroxo and hydroperoxo intermediates in cytochrome P450. J Phys Chem A 112:13172–13179
Dawson JH, Andersson LA, Sono M, Hager LP (1992) Systematic trends in the spectroscopic properties of low-spin ferric ligand adducts of cytochrome P450 and chloroperoxidase: the transition from normal to hyper spectra. New J Chem 16:577–582
Denisov IG, Makris TM, Sligar SG (2002) Formation and decay of hydroperoxo-ferric heme complex in horseradish peroxidase studied by cryoradiolysis. J Biol Chem 277:42706–42710
Ibrahim M, Denisov IG, Makris TM, Kincaid JR, Sligar SG (2003) Resonance Raman spectroscopic studies of hydroperoxo-myoglobin at cryogenic temperatures. J Am Chem Soc 125:13714–13718
Makris TM, Davydov R, Denisov IG, Hoffman B, Sligar SG (2002) Mechanistic enzymology of oxygen activation by the cytochromes P450. Drug Metab Rev 34:691–708
Makris TM, Denisov IG, Sligar SG (2003) Heme-oxygen reactive intermediates: catalysis by the two-step. Biochem Soc Trans 31:516–519
Sligar SG, Makris TM, Denisov IG (2005) Thirty years of microbial P450 monooxygenase research: peroxo-heme intermediates–the central bus station in heme oxygenase catalysis. Biochem Biophys Res Commun 338:346–354
Mak PJ, Denisov IG, Victoria D, Makris TM, Deng T, Sligar SG, Kincaid JR (2007) Resonance Raman detection of the hydroperoxo intermediate in the cytochrome P450 enzymatic cycle. J Am Chem Soc 129:6382–6383
Mak PJ, Kincaid JR (2008) Resonance Raman spectroscopic studies of hydroperoxo derivatives of Cobalt-substituted myoglobin. J Inorg Biochem 102:1952–1957
Liu JG, Ohta T, Yamaguchi S, Ogura T, Sakamoto S, Maeda Y, Naruta Y (2009) Spectroscopic characterization of a hydroperoxo-heme intermediate: conversion of a side-on peroxo to an end-on hydroperoxo complex. Angew Chem Int Ed Engl 48:9262–9267
Liu JG, Shimizu Y, Ohta T, Naruta Y (2010) Formation of an end-on ferric peroxo intermediate upon one-electron reduction of a ferric superoxo heme. J Am Chem Soc 132:3672–3673
Ohta T, Liu J-G, Naruta Y (2013) Resonance Raman characterization of mononuclear heme-peroxo intermediate models. Coord Chem Rev 257:407–413
Ibrahim M, Kincaid JR (2004) Spectroscopic studies of peroxo/hydroperoxo derivatives of heme proteins and model compounds. J Porphyrins Phthalocyanines 8:215–225
Berglund GI, Carlsson GH, Smith AT, Szoeke H, Henriksen A, Hajdu J (2002) The catalytic pathway of horseradish peroxidase at high resolution. Nature 417:463–468
Kuhnel K, Derat E, Terner J, Shaik S, Schlichting I (2007) Structure and quantum chemical characterization of chloroperoxidase compound 0, a common reaction intermediate of diverse heme enzymes. Proc Natl Acad Sci U S A 104:99–104
Unno M, Chen H, Kusama S, Shaik S, Ikeda-Saito M (2007) Structural characterization of the fleeting ferric peroxo species in myoglobin: experiment and theory. J Am Chem Soc 129:13394–13395
Harris DL, Loew GH (1998) Theoretical investigation of the proton assisted pathway to formation of cytochrome P450 compound I. J Am Chem Soc 120:8941–8948
Akhtar M, Corina D, Miller S, Shyadehi AZ, Wright JN (1994) Mechanism of the acyl-carbon cleavage and related reactions catalyzed by multifunctional P-450s: studies on cytochrome P-450(17)alpha. Biochemistry 33:4410–4418
Akhtar M, Njar VC, Wright JN (1993) Mechanistic studies on aromatase and related C-C bond cleaving P-450 enzymes. J Steroid Biochem Mol Biol 44:375–387
Hackett JC, Brueggemeier RW, Hadad CM (2005) The final catalytic step of cytochrome p450 aromatase: a density functional theory study. J Am Chem Soc 127:5224–5237
Sen K, Hackett JC (2012) Coupled electron transfer and proton hopping in the final step of CYP19-catalyzed androgen aromatization. Biochemistry 51:3039–3049
Sligar SG, Luthra A, Denisov I, Davydov R, Hoffman B, Mak P, Kincaid J (2010) Oxygen intermediates in heme monoxygenases. Pacifichem 2010, international chemical congress of pacific basin societies, Honolulu, HI, United States, 15–20 Dec 2010:BIOL–102
Varfaj F, Zulkifli SN, Park HG, Challinor VL, De Voss JJ, Ortiz de Montellano PR (2014) Carbon–carbon bond cleavage in activation of the prodrug nabumetone. Drug Metab Dispos 42:828–838
Egawa T, Shimada H, Ishimura Y (1994) Evidence for compound I formation in the reaction of cytochrome-P450cam with m-chloroperbenzoic acid. Biochem Biophys Res Comm 201:1464–1469
Kellner DG, Hung S-C, Weiss KE, Sligar SG (2002) Kinetic characterization of compound I formation in the thermostable cytochrome P450 CYP119. J Biol Chem 277:9641–9644
Spolitak T, Dawson John H, Ballou David P (2006) Rapid kinetics investigations of peracid oxidation of ferric cytochrome P450cam: nature and possible function of compound ES. J Inorg Biochem 100:2034–2044
Dolphin D, Felton RH (1974) The biochemical significance of porphyrin p cation radicals. Acc Chem Res 7:26–32
Dunford HB (1999) Heme peroxidases. Wiley, New York, p 507
George P (1952) Intermediate compound formation with peroxidase and strong oxidizing agents. J Biol Chem 201:413–426
Groves JT (1985) Key elements of the chemistry of cytochrome P-450. The oxygen rebound mechanism. J Chem Educ 62:928–931
Groves JT (2003) The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. Proc Natl Acad Sci U S A 100:3569–3574
Groves JT (2006) High-valent iron in chemical and biological oxidations. J Inorg Biochem 100:434–447
Bell SR, Groves JT (2009) A highly reactive P450 model compound I. J Am Chem Soc 131:9640–9641
Palcic MM, Rutter R, Araiso T, Hager LP, Dunford HB (1980) Spectrum of chloroperoxidase compound I. Biochem Biophys Res Commun 94:1123–1127
Kim SH, Perera R, Hager LP, Dawson JH, Hoffman BM (2006) Rapid freeze-quench ENDOR study of chloroperoxidase compound I: the site of the radical. J Am Chem Soc 128:5598–5599
Egawa T, Proshlyakov DA, Miki H, Makino R, Ogura T, Kitagawa T, Ishimura Y (2001) Effects of a thiolate axial ligand on the pi -> pi* electronic states of oxoferryl porphyrins: a study of the optical and resonance Raman spectra of compounds I and II of chloroperoxidase. J Biol Inorg Chem 6:46–54
Wang X, Peter S, Kinne M, Hofrichter M, Groves JT (2012) Detection and kinetic characterization of a highly reactive heme-thiolate peroxygenase compound I. J Am Chem Soc 134:12897–12900
Wang X, Peter S, Ullrich R, Hofrichter M, Groves JT (2013) Driving force for oxygen-atom transfer by heme-thiolate enzymes. Angew Chem Int Ed Engl 52:9238–9241
Rittle J, Green MT (2010) Cytochrome P450 compound I: capture, characterization, and C–H bond activation kinetics. Science 330:933–937
Rittle J, Younker JM, Green MT (2010) Cytochrome P450: the active oxidant and its spectrum. Inorg Chem 49:3610–3617
Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT (2013) Reactive intermediates in cytochrome p450 catalysis. J Biol Chem 288:17074–17081
Rutter R, Hager LP, Dhonau H, Hendrich M, Valentine M, Debrunner P (1984) Chloroperoxidase compound I: electron paramagnetic resonance and Mossbauer studies. Biochemistry 23:6809–6816
Green MT, Dawson JH, Gray HB (2004) Oxoiron(IV) in chloroperoxidase compound II is basic: implications for P450 chemistry. Science 304:1653–1656
Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov A, Calixto JC, Behan RK, Green MT (2013) Iron(IV)hydroxide pK(a) and the role of thiolate ligation in C–H bond activation by cytochrome P450. Science 342:825–829
Groves JT (2014) Enzymatic C–H bond activation. Using push to get pull. Nat Chem 6:89–91
McQuarters AB, Wolf MW, Hunt AP, Lehnert N (2014) 1958–2014: after 56 years of research, cytochrome P450 reactivity is finally explained. Angew Chem Int Ed Engl 53:4750–4752
Jung C (2007) Leakage in cytochrome P450 reactions in relation to protein structural properties. In: Sigel A, Sigel H, Sigel RKO (eds) Ubiquitous roles of cytochrome P450 proteins. Wiley, New York, pp 187–234
Estabrook RW, Hildebrandt AG, Baron J, Netter KJ, Leibman K (1971) New spectral intermediate associated with cytochrome P-450 function in liver microsomes. Biochemic Biophys Res Comm 42:132–139
Ishimura Y, Ullrich V, Peterson JA (1971) Oxygenated cytochrome P-450 and its possible role in enzymic hydroxylation. Biochem Biophys Res Commun 42:140–146
Sligar SG, Lipscomb JD, Debrunner PG, Gunsalus IC (1974) Superoxide anion production by the autoxidation of cytochrome P450cam. Biochem Biophys Res Commun 61:290–296
Ullrich V, Kuthan H (1980) Autoxidation and uncoupling in microsomal monooxygenations. Developm Biochem 13:267–272
Lewis JC, Bastian S, Bennett CS, Fu Y, Mitsuda Y, Chen MM, Greenberg WA, Wong CH, Arnold FH (2009) Chemoenzymatic elaboration of monosaccharides using engineered cytochrome P450BM3 demethylases. Proc Natl Acad Sci U S A 106:16550–16555
Lewis JC, Mantovani SM, Fu Y, Snow CD, Komor RS, Wong CH, Arnold FH (2010) Combinatorial alanine substitution enables rapid optimization of cytochrome P450BM3 for selective hydroxylation of large substrates. Chembiochem 11:2502–2505
Staudt H, Lichtenberger F, Ullrich V (1974) The role of NADH in uncoupled microsomal monoxygenations. Eur J Biochem 46:99–106
Gorsky LD, Koop DR, Coon MJ (1984) On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450. Products of oxygen reduction. J Biol Chem 259:6812–6817
Zhukov AA, Archakov AI (1982) Complete stoichiometry of free NADPH oxidation in liver microsomes. Biochem Biophys Res Commun 109:813–818
Archakov AI, Bachmanova GI (1990) Cytochrome P450 and active oxygen. Taylor & Francis, London, 339 pp
Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J (1985) The 2.6- ANG. Crystal structure of Pseudomonas putida cytochrome P-450. J Biol Chem 260:16122–16130
Poulos TL, Howard AJ (1987) Crystal structures of metyrapone- and phenylimidazole-inhibited complexes of cytochrome P-450cam. Biochemistry 26:8165–8174
Raag R, Swanson BA, Poulos TL, Ortiz de Montellano PR (1990) Formation, crystal structure, and rearrangement of a cytochrome P-450cam iron-phenyl complex. Biochemistry 29:8119–8126
Raag R, Martinis SA, Sligar SG, Poulos TL (1991) Crystal structure of the cytochrome P-450CAM active site mutant Thr252Ala. Biochemistry 30:11420–11429
Raag R, Poulos TL (1991) Crystal structures of cytochrome P-450CAM complexed with camphane, thiocamphor, and adamantane: factors controlling P-450 substrate hydroxylation. Biochemistry 30:2674–2684
Atkins WM, Sligar SG (1988) Deuterium isotope effects in norcamphor metabolism by cytochrome P-450cam: kinetic evidence for the two-electron reduction of a high-valent iron-oxo intermediate. Biochemistry 27:1610–1616
Atkins WM, Sligar SG (1988) The roles of active site hydrogen bonding in cytochrome P-450cam as revealed by site-directed mutagenesis. J Biol Chem 263:18842–18849
Atkins WM, Sligar SG (1989) Molecular recognition in cytochrome P-450: alteration of regioselective alkane hydroxylation via protein engineering. J Am Chem Soc 111:2715–2717
Loida J, Sligar SG (1993) Molecular recognition in cytochrome P-450: mechanism for the control of uncoupling reactions. Biochemistry 32:11530–11538
Paulsen MD, Filipovic D, Sligar SG, Ornstein RL (1993) Controlling the regiospecificity and coupling of cytochrome P450cam: T185F mutant increases coupling and abolishes 3-hydroxynorcamphor product. Protein Sci 2:357–365
Loida PJ, Sligar SG (1994) Molecular recognition in cytochrome P450: control of uncoupling reactions via site-directed mutagenesis. 8th international conference on cytochrome P450, pp 463–466
Loida PJ, Sligar SG, Paulsen MD, Arnold GE, Ornstein RL (1995) Stereoselective hydroxylation of norcamphor by cytochrome P450cam. Experimental verification of molecular dynamics simulations. J Biol Chem 270:5326–5330
Gerber NC, Sligar SG (1994) A role for Asp251 in the activation of oxygen by cytochrome P450cam. 8th international conference on cytochrome P450, pp 753–756
Kimata Y, Shimada H, Hirose T, Ishimura Y (1995) Role of Thr-252 in cytochrome P450(Cam)—a study with unnatural amino-acid mutagenesis. Biochem Biophys Res Commun 208:96–102
Vidakovic M, Sligar SG, Li H, Poulos TL (1998) Understanding the role of the essential Asp251 in cytochrome P450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry 37:9211–9219
Gerber NC., Sligar SG (1992) Catalytic mechanism of cytochrome P-450: evidence for a distal charge relay. J Am Chem Soc 114:8742–8743
Martinis SA, Atkins WM, Stayton PS, Sligar SG (1989) A conserved residue of cytochrome-P-450 is involved in heme- oxygen stability and activation. J Am Chem Soc 111:9252–9253
Imai M, Shimada H, Watanabe Y, Matsushima-Hibiya Y, Makino R, Koga H, Horiuchi T, Ishimura Y (1989) Uncoupling of the cytochrome P-450cam monooxygenase reaction by a single mutation, threonine-252 to alanine or valine: possible role of the hydroxy amino acid in oxygen activation. Proc Natl Acad Sci U S A 86:7823–7827
Fedorov R, Ghosh DK, Schlichting I (2003) Crystal structures of cyanide complexes of P450cam and the oxygenase domain of inducible nitric oxide synthase—structural models of the short-lived oxygen complexes. Arch Biochem Biophys 409:25–31
Batabyal D, Poulos TL (2013) Crystal structures and functional characterization of wild-type CYP101D1 and its active site mutants. Biochemistry 52:8998–8906
Batabyal D, Li H, Poulos TL (2013) Synergistic effects of mutations in cytochrome P450cam designed to mimic CYP101D1. Biochemistry 52:5396–5402
Yang W, Bell SG, Wang H, Zhou W, Bartlam M, Wong LL, Rao Z (2011) The structure of CYP101D2 unveils a potential path for substrate entry into the active site. Biochem J 433:85–93
Bell SG, Yang W, Dale A, Zhou W, Wong LL (2013) Improving the affinity and activity of CYP101D2 for hydrophobic substrates. Appl Microbiol Biotechnol 97:3979–3990
Lepesheva GI, Waterman MR (2011) Structural basis for conservation in the CYP51 family. Biochim Biophys Acta 1814:88–93
Nitahara Y, Kishimoto K, Yabusaki Y, Gotoh O, Yoshida Y, Horiuchi T, Aoyama Y (2001) The amino acid residues affecting the activity and azole susceptibility of rat CYP51 (sterol 14-demethylase P450). J Biochem 129:761–768
Cupp-Vickery JR, Han O, Hutchinson CR, Poulos TL (1996) Substrate-assisted catalysis in cytochrome P450eryF. Nat Struct Biol 3:632–637
Cupp-Vickery JR, Poulos TL (1997) Structure of cytochrome P450eryF: substrate, inhibitors, and model compounds bound in the active site. Steroids 62:112–116
Kim C, Kim H, Han O (2000) The role of serine-246 in cytochrome P450eryF-catalyzed hydroxylation of 6-deoxyerythronolide B. Bioorg Chem 28:306–314
Xiang H, Tschirret-Guth RA, Ortiz De Montellano PR (2000) An A245T mutation conveys on cytochrome P450eryF the ability to oxidize alternative substrates. J Biol Chem 275:35999–36006
Khan KK, Halpert JR (2002) 7-Benzyloxyquinoline oxidation by P450eryF A245T: finding of a new fluorescent substrate probe. Chem Res Toxicol 15:806–814
Stok JE, Yamada S, Farlow AJ, Slessor KE, De Voss JJ (2013) Cytochrome P450(cin) (CYP176A1) D241N: investigating the role of the conserved acid in the active site of cytochrome P450s. Biochim Biophys Acta 1834:688–696
Zerbe K, Pylypenko O, Vitali F, Zhang W, Rouset S, Heck M, Vrijbloed JW, Bischoff D, Bister B, Sussmuth RD, Pelzer S, Wohlleben W, Robinson JA, Schlichting I (2002) Crystal structure of OxyB, a cytochrome P450 implicated in an oxidative phenol coupling reaction during vancomycin biosynthesis. J Biol Chem 277:47476–47485
Cryle MJ, Staaden J, Schlichting I (2011) Structural characterization of CYP165D3, a cytochrome P450 involved in phenolic coupling in teicoplanin biosynthesis. Arch Biochem Biophys 507:163–173
Cryle MJ, Bell SG, Schlichting I (2010) Structural and biochemical characterization of the cytochrome P450 CypX (CYP134A1) from bacillus subtilis: a cyclo-L-leucyl-L-leucyl dipeptide oxidase. Biochemistry 49:7282–7296
Shoji O, Fujishiro T, Nagano S, Tanaka S, Hirose T, Shiro Y, Watanabe Y (2010) Understanding substrate misrecognition of hydrogen peroxide dependent cytochrome P450 from bacillus subtilis. J Biol Inorg Chem 15:1331–1339
Belcher J, McLean KJ, Matthews S, Woodward LS, Fisher K, Rigby SEJ, Nelson DR, Potts D, Baynham MT, Parker DA, Leys D, Munro AW (2014) Structure and biochemical properties of the alkene producing cytochrome P450 oleTJE (CYP152L1) from the jeotgalicoccus sp. 8456 bacterium. J Biol Chem. 289:6535–6550
Truan G, Peterson JA (1998) Thr268 in substrate binding and catalysis in P450BM-3. Arch Biochem Biophys 349:53–64
Yeom H, Sligar SG, Li H, Poulos TL, Fulco AJ (1995) The role of Thr268 in oxygen activation of cytochrome P450BM-3. Biochemistry 34:14733–14740
Cryle MJ, De Voss JJ (2008) The role of the conserved threonine in P450 BM3 oxygen activation: substrate-determined hydroxylation activity of the Thr268Ala mutant. Chembiochem 9:261–266
Hawkes DB, Adams GW, Burlingame AL, Ortiz de Montellano PR, De Voss JJ (2002) Cytochrome P450cin (CYP176A): isolation, expression and characterisation. J Biol Chem 277:27725–27732
Meharenna YT, Slessor KE, Cavaignac SM, Poulos TL, De Voss JJ (2008) The critical role of substrate-protein hydrogen bonding in the control of regioselective hydroxylation in P450cin. J Biol Chem 283:10804–10812
Meharenna YT, Li H, Hawkes DB, Pearson AG, De Voss J, Poulos TL (2004) Crystal structure of P450cin in a complex with Its substrate, 1,8-Cineole, a close structural homologue to d-camphor, the substrate for P450cam. Biochemistry 43:9487–9494
Kimmich N, Das A, Sevrioukova I, Meharenna Y, Sligar SG, Poulos TL (2007) Electron transfer between cytochrome P450cin and its FMN-containing redox partner, cindoxin. J Biol Chem 282:27006–27011
Slessor KE, Farlow AJ, Cavaignac SM, Stok JE, De Voss JJ (2011) Oxygen activation by P450(cin): protein and substrate mutagenesis. Arch Biochem Biophys 507:154–162
Madrona Y, Tripathi S, Li H, Poulos TL (2012) Crystal structures of substrate-free and nitrosyl cytochrome P450cin: implications for O2 activation. Biochemistry 51:6623–6631
Madrona Y, Hollingsworth SA, Khan B, Poulos TL (2013) P450cin active site water: implications for substrate binding and solvent accessibility. Biochemistry 52:5039–5050
Andersen JF, Tatsuta K, Gunji H, Ishiyama T, Hutchinson CR (1993) Substrate specificity of 6-deoxyerythronolide B hydroxylase, a bacterial cytochrome P450 of erythromycin a biosynthesis. Biochemistry 32:1905–1913
Kim D, Heo YS, Ortiz de Montellano PR (2008) Efficient catalytic turnover of cytochrome P450(cam) is supported by a T252N mutation. Arch Biochem Biophys 474:150–156
Hiroya K, Ishigooka M, Shimizu T, Hatano M (1992) Role of Glu318 and Thr319 in the catalytic function of cytochrome P450d (P4501A2): effects of mutations on the methanol hydroxylation. FASEB J 6:749–751
Ishigooka M, Shimizu T, Hiroya K, Hatano M (1992) Role of Glu318 at the putative distal site in the catalytic function of cytochrome P450d. Biochemistry 31:1528–1531
Traylor MJ, Chai J, Clark DS (2011) Simultaneous measurement of CYP1A2 activity, regioselectivity, and coupling: Implications for environmental sensitivity of enzyme-substrate binding. Arch Biochem Biophys 505:186–193
Raner GM, Chiang EW, Vaz AD, Coon MJ (1997) Mechanism-based inactivation of cytochrome P450 2B4 by aldehydes: relationship to aldehyde deformylation via a peroxyhemiacetal intermediate. Biochemistry 36:4895–4902
Vatsis KP, Coon MJ (2002) Ipso-substitution by cytochrome P450 with conversion of p-hydroxybenzene derivatives to hydroquinone: evidence for hydroperoxo-iron as the active oxygen species. Arch Biochem Biophys 397:119–129
Ortiz De Montellano PR, De Voss JJ (2002) Oxidizing species in the mechanism of cytochrome P450. Nat Prod Rep 19:477–493
Ortiz de Montellano PR, De Voss JJ (2005) Substrate oxidation by cytochrome P450 enzymes. In: Ortiz de Montellano PR (Ed) Cytochrome P450: structure, mechanism, and biochemistry. Kluwer Academic/Plenum Publishers, New York, pp 183–245
Grinkova YV, Denisov IG, McLean MA, Sligar SG (2013) Oxidase uncoupling in heme monooxygenases: human cytochrome P450 CYP3A4 in nanodiscs. Biochem Biophys Res Commun 430:1223–1227
Gregory MC, Denisov IG, Grinkova YV, Khatri Y, Sligar SG (2013) Kinetic solvent isotope effect in human P450 CYP17A1-mediated androgen formation: evidence for a reactive peroxoanion intermediate. J Am Chem Soc 135:16245–16247
Akhtar M, Wright JN, Lee-Robichaud P (2011) A review of mechanistic studies on aromatase (CYP19) and 17alpha-hydroxylase-17,20-lyase (CYP17). J Steroid Biochem Mol Biol 125:2–12
Lipscomb JD, Sligar SG, Namtvedt MJ, Gunsalus IC (1976) Autooxidation and hydroxylation reactions of oxygenated cytochrome P-450cam. J Biol Chem 251:1116–24
Denisov IG, Hung S-C, Weiss KE, McLean MA, Shiro Y, Park S-Y, Champion PM, Sligar SG (2001) Characterization of the oxygenated intermediate of the thermophilic cytochrome P450 CYP119. J Inorg Biochem 87:215–226
Lambeir AM, Appleby CA, Dunford HB (1985) The formation and decay of the oxyferrous forms of the cytochromes P-450 isolated from Rhizobium japonicum. Rapid spectral scan and stopped flow studies. Biochim Biophys Acta 828:144–150
Zhang H, Myshkin E, Waskell L (2005) Role of cytochrome b5 in catalysis by cytochrome P450 2B4. Biochem Biophys Res Commun 338:499–506
Grinkova YV, Denisov IG, Waterman MR, Arase M, Kagawa N, Sligar SG (2008) The ferrous-oxy complex of human aromatase. Biochem Biophys Res Commun 372:379–382
Lambeir AM, Dunford HB (1985) Oxygen binding to dithionite-reduced chloroperoxidase. Eur J Biochem 147:93–96
Tejero J, Biswas A, Wang ZQ, Page RC, Haque MM, Hemann C, Zweier JL, Misra S, Stuehr DJ (2008) Stabilization and characterization of a heme-oxy reaction intermediate in inducible nitric-oxide synthase. J Biol Chem 283:33498–33507
Abu-Soud HM, Gachhui R, Raushel FM, Stuehr DJ (1997) The ferrous-dioxy complex of neuronal nitric oxide synthase. Divergent effects of L-arginine and tetrahydrobiopterin on its stability. J Biol Chem 272:17349–17353
Davydov R, Kofman V, Nocek JM, Noble RW, Hui H, Hoffman BM (2004) Conformational substates of the oxyheme centers in alpha and beta subunits of hemoglobin as disclosed by EPR and ENDOR studies of cryoreduced protein. Biochemistry 43:6330–6338
Gasyna Z (1979) Intermediate spin-states in one-electron reduction of oxygen-hemoprotein complexes at low temperature. FEBS Lett 106:213–218
Davydov RM, Chauhan N, Thackray SJ, Anderson JL, Papadopoulou ND, Mowat CG, Chapman SK, Raven EL, Hoffman BM (2010) Probing the ternary complexes of indoleamine and tryptophan 2,3-dioxygenases by cryoreduction EPR and ENDOR spectroscopy. J Am Chem Soc 132:5494–5500
Davydov R, Osborne RL, Shanmugam M, Du J, Dawson JH, Hoffman BM (2010) Probing the oxyferrous and catalytically active ferryl states of Amphitrite ornata dehaloperoxidase by cryoreduction and EPR/ENDOR spectroscopy. Detection of compound I. J Am Chem Soc 132:14995–15004
Davydov RM, Yoshida T, Ikeda-Saito M, Hoffman BM (1999) Hydroperoxy-heme oxygenase generated by cryoreduction catalyzes the formation of.alpha.-meso-hydroxyheme as detected by EPR and ENDOR. J Am Chem Soc 121:10656–10657
Davydov R, Sudhamsu J, Lees NS, Crane BR, Hoffman BM (2009) EPR and ENDOR characterization of the reactive intermediates in the generation of NO by cryoreduced oxy-nitric oxide synthase from Geobacillus stearothermophilus. J Am Chem Soc 131:14493–14507
{kind=link}
Acknowledgments
Our work on the mechanisms of cytochrome P450 has spanned four decades and has been made possible by a large number of talented students, post-doctoral associates, research associates, and technical staff. We particularly wish to acknowledge two long-term National Institutes of Health grants that have supported this work, GM31756 (formerly GM24976) and GM33775.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Denisov, I., Sligar, S. (2015). Activation of Molecular Oxygen in Cytochromes P450. In: Ortiz de Montellano, P. (eds) Cytochrome P450. Springer, Cham. https://doi.org/10.1007/978-3-319-12108-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-12108-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-12107-9
Online ISBN: 978-3-319-12108-6
eBook Packages: MedicineMedicine (R0)