
Abstract
In this chapter, we review HDL’s role in reverse cholesterol transport and point out to evidence supporting that HDL acts as a shuttle to transport cholesterol to other lipoproteins, with important roles that are not restricted to the cardiovascular system. We then discuss the atheroprotective roles of HDL in light of the several recent randomized clinical trials that were aimed at raising HDL cholesterol but failed to improve cardiovascular outcomes, and conclude that low HDL cholesterol is a marker, and not a mediator of cardiovascular disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cholesterol
- Familial hypercholesterolemia
- LDL
- HDL
- VLDL
- FH
- LDLR
- Cholesterol disease
- Increased cholesterol levels
- Lipoprotein
- Cardiovascular disease
- Gene
- Clinical signs
- Lipid
- APOAI:
-
Apolipoprotein A-I
- ApoB:
-
Apolipoprotein B
- CVD:
-
Cardiovascular disease
- FH:
-
Familial hyperlipidemia
- HDL:
-
High-density lipoprotein
- IDL:
-
Intermediate-density lipoprotein
- LCAT:
-
Lecithin-cholesterol acetyltransferase
- LDL:
-
Low-density lipoprotein
- LDL-C:
-
Low density lipoprotein- cholesterol
- LDLR:
-
Low density lipoprotein receptor
- VLDL:
-
Very-low-density lipoprotein
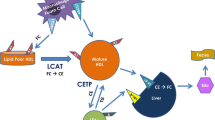
It is important to understand that HDL particles carry HDL cholesterol, and that changes in HDL cholesterol may not indicate changes to HDL particle numbers. Indeed, some pharmacotherapies that increased HDL cholesterol (for example CETP inhibitors) increase the cholesterol content of HDL (by increasing the size of the particle) without increasing particle numbers. This distinction is important as we discuss below the many functions of HDL.
I Reverse Cholesterol Transport
Reverse cholesterol transport was first defined by John Glomset [1] in the early 1960s as the capacity of HDL particles to transport cholesterol from the periphery to the liver for excretion. This concept of reverse cholesterol transport was mostly based on in vitro studies examining the ability of Apolipoprotein A-I to esterify cholesterol through LCAT and has been used to explain the protective properties of HDL. The idea gained wide spread acceptance with the publication of the initial Framingham study revealing that greater HDL cholesterol levels were associated with decreased CVD risk [2]. Over the last 50 years, several studies emerged supporting a different role for Apo A-I and HDL in cardiovascular disease. HDL forms after the secretion of disc-shaped apo A-I containing particle by hepatocytes and enterocytes, known prebeta-1 HDL. Apo A-I on this nascent HDL particle activates the adenosine triphosphate (ATP)–binding cassette (ABC) protein, ABCA1, on the surface of peripheral cells, including macrophages. Once activated, the ABCA 1 protein transports unesterified cholesterol and phospholipids from the cell onto the nascent HDL particle. On the surface of HDL particle, the cholesterol is esterified by lecithin-cholesterol acyl transferase (LCAT) and its cofactor, apo A-I. As it circulates, nascent HDL particles are transformed into a mature, spherical HDL particle that contains cholesteryl ester in its core. The current dogma suggests that HDL returns the majority of circulating cholesterol to the liver and thus acts as the good cholesterol favoring reverse cholesterol transport. Indeed, over the last 10 years, several large randomized control trials have been designed on this promise that raising HDL cholesterol lowers CVD risk. The results of the majority of these studies do not support the concept that raising HDL cholesterol reduces CVD risk. These recent findings have suggested the need to reexamine our understanding of HDL biology. In this chapter, I present evidence from both basic and clinical studies that suggest a cholesterol shuttle function for HDL particles.
II. HDL functions
-
1.
HDL particles shuttle cholesterol between lipoproteins and are a source for LDL cholesterol: Emerging evidence suggests that HDL particles acquire and exchange a substantial amount of cholesterol from and to other lipoproteins and not just peripheral tissues [3, 4]. It has been shown that cellular unesterified cholesterol is initially taken up by lipid-poor (pre-beta-1 HDL) [5, 6]. However, the majority of this cholesterol is subsequently transferred to plasma LDL and only a small proportion (5 %) of it is esterified on HDL before it reaches LDL [7]. This explains the higher amount of cholesterol that LDL particles carry. Moreover, patients with Tangier disease who have a defect in ABCA-1 and do not form HDL cholesterol have reduced levels ( 50 % decrease) in their LDL cholesterol content [8].
-
2.
Prebeta-1 HDL particles are not exclusive acceptors of cellular cholesterol, and VLDL particles are critical for cholesterol return to the liver: The HDL centric reverse cholesterol hypothesis is based on prebeta-1 HDL initially acquiring free cholesterol from peripheral tissues to be esterified and later circulated to the liver for excretion. Recent studies support a major role for LDL and VLDL as cholesterol acceptors in the fasting and post prandial state respectively. It has long been demonstrated that red blood cells are major storage sites of the circulating cholesterol pool [9, 10]. Four to five hours after fatty meal, the efflux of un-esterified cholesterol from red blood cells (RBCs) increases; the most potent acceptor of RBC cholesterol are chylomicrons and VLDL, while low levels of HDL do not limit cholesterol efflux from the cells [11]. More recently, infusion of reconstituted HDL in humans was shown to “shuttle” cholesterol into VLDL particles, where VLDL metabolism became the primary mechanism for the ability of plasma to move cholesterol from cells to the liver [12]. In agreement, a kinetic study of lipoprotein metabolism demonstrated that the majority ( 70 %) of cholesterol ester transfers back to the liver on VLDL [13].
-
3.
LDL particles are a major storage site for cholesterol esters in plasma: In fasting plasma, LDL un-esterified cholesterol was the only source of cholesterol for esterification. Based on these findings it appears that regardless whether the initial acceptors are prebeta1 HDL, chylomicrons or VLDL, cellular un-esterified cholesterol passes through LDL [3].
-
4.
HDL cholesterol sources and fates: The HDL cholesterol pool is primarily a function of the ABCA-1, ABCG1, SR-BI transporters and CETP activity. ABCA-1 activity in the liver, intestine and adipose tissue [14] substantially contribute to the formation of lipidated Apo A-I and thus formation of HDL cholesterol. On the other hand, steroidogenic organs such as the ovaries and the adrenals expressing high levels of SR-BI transporter can exchange HDL cholesterol. In metabolic diseases such as obesity, CETP is a major factor in shuttling cholesterol between LDL and HDL particles. Importantly, macrophage cholesterol constitutes less than 5 % of HDL’s cholesterol [15]. Thus, changes in HDL cholesterol levels may not imply that cholesterol is moving out of macrophages, and changes in macrophage cholesterol efflux may not result in increases in HDL cholesterol levels. Although HDL is involved in returning cholesterol to the liver, this likely constitutes a minor HDL function, and perhaps a minor mechanism for reverse cholesterol transport in humans. The increased ratio of phospholipid to cholesterol on the HDL surface suggests that HDL’s role in cholesterol transport is through facilitate cholesterol delivery to the adrenals and steriodogenic organs to maintain steroid and sex hormone synthesis, perhaps through SR-BI receptors. One example is through a study by Vergeer et al [16] showing that carriers of a genetic defect in the SR-BI transporter with increased concentrations of HDL cholesterol have impaired adrenal and platelet functions.
-
5.
Apo A-I, the major HDL protein, protects against vascular inflammation and oxidation of LDL particles: Infusing rabbits with increasing doses of Apo A-I reduces markers of vascular inflammation (ICAM and VCAM) [17], whereas infusion of recombinant HDL reduced both measures of atherosclerosis and these markers of inflammation [18]. There are human studies suggestive that low HDL-C is associated with increases in vascular measures of inflammation, and that HDL isolated from research participants following the ingestion of a polyunsaturated diet can reduce these markers of inflammation [19]. In addition to its capacity to reduce vascular inflammation, Apo A-1 or HDL appear to reduce the susceptibility to LDL oxidation. Infusion of human apoA-I into human recipients results in LDLs becoming resistant to oxidation and being less effective in inducing monocyte chemotactic activity in a human artery wall co-culture [20]. However, it is important to note that studies infusing Apo-I or HDL and demonstrating beneficial clinical outcomes in humans are to date lacking.
-
6.
Complexity of the HDL proteome suggests pleiotropic HDL functions: Recent proteomic studies suggest that HDL particles are highly complex [21–23]. For example, HDL enriched with complement proteins implicate functions related to the immune system. One particular example is illustrated by ApoL1. Patients lacking ApoL1 in their HDL’s are at an increased risk of developing a Trypansomal infection [24]. Enrichment of HDL with SAA after inflammation assists in clearing of cellular cholesterol after macrophage induced cytotoxicity of microbial cells [25]. In addition, SAA enriched HDL has a strong affinity to SR-BI (highly expressed in the adrenals) [26] suggesting a mechanism for supplying cholesterol to assist with increased cortisol production during periods of stress.
Important Advances
-
A.
Lessons learned from CETP inhibition:
CETP evolved with higher species (rabbits to primates) perhaps as a mechanism to accommodate an increase in cholesterol turnover by facilitating cholesterol exchange between HDL and VLDL particles. The rationale for inhibiting CETP was based on that CETP inhibition raised HDL cholesterol, lowered LDL cholesterol and that in some families loss of function mutations in CETP activity was associated with longevity. To date, interventional studies using three CETP inhibitors did not demonstrate improvements in cardiovascular risk. Three CETP inhibitor trials to date did not reveal CVD benefit, and in fact one of these trials revealed harm (Illuminate trial [27]). CETP inhibition represents the fallacy of good and bad cholesterol concept. As discussed before, a key kinetic study revealed that around 70 % of cholesterol is returned to the liver via VLDL by CETP mediated transfer. Thus, inhibiting CETP will force reverse cholesterol transport through HDL, a mechanism that may not be very efficient with concomitant use of statins that are known to upregulate the liver LDL receptor (in anticipation for cholesterol getting returned to the liver on VLDL and LDL). Moreover, increasing HDL-C by CETP inhibition will favor SR-BI uptake not only in the liver, but also in the adrenals, or any tissues expressing high levels of SR-BI. Thus, it is not surprising that in the Illuminate trial [27] blood pressure was elevated in the Trocetrapib arm. This could represent an increase in HDL mediated activation of the steroidogenic aldosterone pathway in the adrenals that preferentially express SR-BI receptors.
-
B.
The LDL receptor and the ABCA-1 transporters in atherosclerosis:
Insights from Tangier’s Disease and Familial Hyperlipidemia (FH) underpin important differences of HDL and LDL functions on atherosclerosis. FH is a common genetic disorder characterized by a defect in the LDL-R. Homozygous mutations lead to atherosclerosis in early childhood, whereas heterozygous mutations are more common and lead to atherosclerosis later childhood and into adulthood if untreated. The loss of the liver LDL receptor function is a bottle neck for reverse cholesterol transport as the majority of cholesterol esters are returned to the liver through the LDL-receptor. There is a futile increase in CETP activity in FH that compounds the defect in apoB clearance. In this situation, CETP activity favors increased HDL catabolism and explains the lower HDL-C levels seen in FH [28]. Thus, the liver LDL receptor is critical in LDL cholesterol re uptake for catabolism. Macrophages can take up LDL cholesterol without relying on the LDL receptor to (through scavenger receptor A and pinocytosis) [29]. The excess in circulating lipoproteins as a result of cholesterol clearance favors foam cell formation and atherosclerosis development. These events clearly demonstrate that excess cholesterol “exposure” as a function of defective liver clearance is atherogenic, and mechanisms to reduce atherosclerosis may depend on improving cholesterol clearance. In contrast to the importance of the liver LDL-receptor in atherosclerosis, the link between ABCA-1 activity and atherosclerosis is less clear. A recent study by the Parks Lab [30] suggested that ABCA-1 deletion in the liver did not accelerate atherosclerosis despite major reductions in HDL cholesterol content. This has implications on any strategy that raises HDL cholesterol through inhibiting its liver metabolism such as the newer anti-mir33b therapies. Mir-33 is a microRNA that inhibits ABCA-1 expression and regulates liver cholesterol metabolism [31]. Inhibiting mir-33 in the liver leads to increases in HDL cholesterol and decreases in triglycerides [32] and currently in the pipeline for human studies. However, it is not clear that increasing HDL cholesterol by making more liver HDL is atheroprotective, given the fact that HDL cholesterol exchanges its cholesterol with LDL without an overall effect that alters cholesterol excretion. Patients with Tangier’s Disease have a complete loss-of-mutation in the ABCA-1 transporter, a critical membrane protein for lipidating Apo A-I and thus forming HDL. However, patients with Tangier have decreased LDL cholesterol reflecting the important concept that HDL-C substantially contributes to LDL-C content. Thus, the net effect is “less” exposure to cholesterol compared to the environment of FH. Clinically, patients with Tangier’s Disease have a modest increase in the risk of premature CVD compared to FH, appearing later in life [8]. They have neurologic deficits and characteristic orange colored tonsils that likely reflect cholesterol accumulation in macrophages [8]. Moreover, the “less severe” loss-of-function mutations in ABCA-1 transporter that are associated with decreases in HDL-C are not associated with increased CVD risk [33] in one large prospective Danish population study. These findings suggest that changes in HDL cholesterol per se may not be critical to the development of atherosclerosis, and perhaps a marker of other diseases (such as metabolic diseases in case of increased CETP function).
-
C.
Is raising HDL cholesterol a good target for decreasing heart disease risk?
To date, two randomized clinical trials involving CETP inhibitors [27, 34], and two niacin trials [35, 36] did not translate into decreased CVD outcomes. These findings raise an important question on the futility of raising HDL cholesterol. Indeed, deleting the major transporters known to contribute to macrophage cholesterol efflux and reverse cholesterol transport does not change HDL cholesterol levels. The macrophage cholesterol content is a very minor contributor to the HDL cholesterol pool [15, 37]. Another example of the discrepancy between HDL cholesterol levels and atherosclerosis is through studies involving the SR-BI receptor. SR-BI overexpression increases RCT through increased HDL particle uptake into the liver. In animal studies, the result is decreased atherosclerosis coupled with a decrease in HDL cholesterol [38]. Thus, mechanisms that increase HDL cholesterol by blocking its liver uptake may retard reverse cholesterol transport. The implications of the above mentioned studies are particularly relevant to niacin. Niacin increases HDL cholesterol, possibly by inhibiting HDL uptake into the liver, and to a lesser extent by increasing Apo A-I levels. This mechanism may not imply that cholesterol is being transported out of macrophages. However, niacin can lower LDL-C and triglyceride levels and these are potentially be atheroprotective changes. Recent clinical studies (AIM HIGH [35] and HPS-2 thrive [36]) with niacin added on top of statins highlight our knowledge gap in the role of raising HDL-C as a target for therapy to lessen the CVD burden. Recent studies have highlighted a potential role for cholesterol efflux as a metric of HDL functions representing a better index of reverse cholesterol transport than HDL cholesterol [39, 40]. However, the determinants of cholesterol efflux are not yet fully understood.
-
D.
Apo A-I raising therapies:
An alternative strategy for raising HDL cholesterol is through increasing Apo A-I levels. As discussed above, animal and early clinical human studies clearly demonstrate that Apo A-I protects against atherosclerosis through its effect on modulation vascular inflammation and reducing LDL oxidation [17–20]. Over the last 10 years, Apo A-I and HDL infusion therapies [41–43] were shown to reduce the severity of atherosclerosis using intravascular ultrasound studies of coronary arteries (IVUS). In addition to Apo A-I infusion therapies, promoters of apoA-I synthesis (RX208) [44] and Apo A-I mimetic peptides were developed [45]. We are still awaiting randomized clinical trials of these Apo A-I therapies using clinical endpoints. The main barriers to the progression of these studies have been their toxicity profiles [46] or their methods of administration.
Summary
Raising HDL cholesterol as a sole target of therapy appears to be a futile strategy. Most of the cholesterol on HDL comes from the liver and intestine. Raising it adds more cholesterol into the system, and shuttling this cholesterol into LDL particles can be atherogenic. Macrophages do not store much cholesterol. Therapies that can increase cholesterol efflux out of macrophages are unlikely to change HDL cholesterol unless they simultaneously move cholesterol out of the major cholesterol stores. One major gap in the field is a reliable marker of macrophage reverse cholesterol transport, and cholesterol efflux might represent such a metric. Development of such an index can help inform us on interventions favor an atheroprotective cholesterol transport. Thus, low HDL cholesterol is a marker and not a mediator of cardiovascular disease. Apo A-I has atheroprotective activities, but the developments of such therapies have been hampered with toxicity profiles or the methods of administration. It is likely that raising HDL cholesterol by increasing Apo A-I is atheroprotective, but more definitive answers await randomized clinical trials.
References
Glomset JA. Physiological role of lecithin-cholesterol acyltransferase. Am J Clin Nutr. 1970;23(8):1129–36.
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease: the Framingham Study. Am J Med. 1977;62(5):707–14.
Dobiášová M, Frohlich JJ. Advances in understanding of the role of lecithin cholesterol acyltransferase (LCAT) in cholesterol transport. Clinica chimica acta. 1999;286(1):257–71.
Huang Y, von Eckardstein A, Assmann G. Cell-derived unesterified cholesterol cycles between different HDLs and LDL for its effective esterification in plasma. Arterioscler Thromb Vasc Biol. 1993;13(3):445–58.
Castro GR, Fielding CJ. Early incorporation of cell-derived cholesterol into pre-. beta.-migrating high-density lipoprotein. BioChemistry. 1988;27(1):25–9.
Francone OL, Royer L, Haghpassand M. Increased prebeta-HDL levels, cholesterol efflux, and LCAT-mediated esterification in mice expressing the human cholesteryl ester transfer protein (CETP) and human apolipoprotein AI (apoA-I) transgenes. J Lipid Res. 1996;37(6):1268–77.
Miida T, Fielding C, Fielding P. Mechanism of transfer of LDL-derived free cholesterol to HDL subfractions in human plasma. Biochemistry. 1990;29(46):10469–74.
Serfaty-Lacrosniere C, Civeira F, Lanzberg A, Isaia P, Berg J, Janus ED, et al. Homozygous Tangier disease and cardiovascular disease. Atherosclerosis. 1994;107(1):85–98. PubMed PMID: 7945562.
Hung KT, Berisha SZ, Ritchey BM, Santore J, Smith JD. Red blood cells play a role in reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32(6):1460–5.
Ho Y, Brown M, Goldstein J. Hydrolysis and excretion of cytoplasmic cholesteryl esters by macrophages: stimulation by high density lipoprotein and other agents. J Lipid Res. 1980;21(4):391–8.
Chung BH, Franklin F, Cho BS, Segrest J, Hart K, Darnell BE. Potencies of lipoproteins in fasting and postprandial plasma to accept additional cholesterol molecules released from cell membranes. Arterioscler Thromb Vasc Biol. 1998;18(8):1217–30.
Chan DC, Hoang A, Barrett PHR, Wong AT, Nestel PJ, Sviridov D, et al. Apolipoprotein B-100 and ApoA-II kinetics as determinants of cellular cholesterol efflux. J Clin Endocrinol Metab. 2012;97(9):E1658–E66.
Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45(9):1594–607.
Zhang Y, McGillicuddy FC, Hinkle CC, O’Neill S, Glick JM, Rothblat GH, et al. Adipocyte modulation of high-density lipoprotein cholesterol. Circulation. 2010;121(11):1347–55. PubMed PMID: 20212278. Pubmed Central PMCID: 2925122.
Haghpassand M, Bourassa PA, Francone OL, Aiello RJ. Monocyte/macrophage expression of ABCA1 has minimal contribution to plasma HDL levels. J Clin Invest. 2001;108(9):1315–20. PubMed PMID: 11696576. Pubmed Central PMCID: 209438.
Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, et al. Genetic variant of the scavenger receptor BI in humans. N Engl J Med. 2011;364(2):136–45.
Puranik R, Bao S, Nobecourt E, Nicholls SJ, Dusting GJ, Barter PJ, et al. Low dose apolipoprotein AI rescues carotid arteries from inflammation in vivo. Atherosclerosis. 2008;196(1):240–7.
Nicholls SJ, Dusting GJ, Cutri B, Bao S, Drummond GR, Rye K-A, et al. Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation. 2005;111(12):1543–50.
Calabresi L, Gomaraschi M, Villa B, Omoboni L, Dmitrieff C, Franceschini G. Elevated soluble cellular adhesion molecules in subjects with low HDL-cholesterol. Arterioscler Thromb Vasc Biol. 2002;22(4):656–61.
Navab M, Imes S, Hama S, Hough G, Ross L, Bork R, et al. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest. 1991;88(6):2039.
Yassine H, Borges CR, Schaab MR, Billheimer D, Stump C, Reaven P, et al. Mass spectrometric immunoassay and MRM as targeted MS-based quantitative approaches in biomarker development: potential applications to cardiovascular disease and diabetes. Proteomics Clin Appl. 2013;7(7–8):528–40. PubMed PMID: 23696124.
Yassine HN, Jackson AM, Borges CR, Billheimer D, Koh H, Smith D, et al. The application of multiple reaction monitoring and multi-analyte profiling to HDL proteins. Lipids Health Dis. 2014;13(1):8.
Vaisar T, Pennathur S, Green PS, Gharib SA, Hoofnagle AN, Cheung MC, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117(3):746–56. PubMed PMID: 17332893. Epub 2007/03/03. eng.
Vanhollebeke B, Truc P, Poelvoorde P, Pays A, Joshi PP, Katti R, et al. Human Trypanosoma evansi infection linked to a lack of apolipoprotein LI. N Engl J Med. 2006;355(26):2752–6.
Malle E, Steinmetz A, Raynes JG. Serum amyloid A (SAA): an acute phase protein and apolipoprotein. Atherosclerosis. 1993;102(2):131–46. PubMed PMID: 7504491.
Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem. 2005;280(4):2954–61.
Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356(13):1304–16. PubMed PMID: 17387129. Epub 2007/03/28. eng.
Bellanger N, Orsoni A, Julia Z, Fournier N, Frisdal E, Duchene E, et al. Atheroprotective reverse cholesterol transport pathway is defective in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2011;31(7):1675–81.
Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52(1):223–61.
Bi X, Zhu X, Duong M, Boudyguina EY, Wilson MD, Gebre AK, et al. Liver ABCA1 deletion in LDLrKO mice does not impair macrophage reverse cholesterol transport or exacerbate atherogenesis. Arterioscler Thromb Vasc Biol. 2013;33(10):2288–96.
Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328(5985):1570–3.
Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121(7):2921.
Frikke-Schmidt R, Nordestgaard BG, Stene MC, Sethi AA, Remaley AT, Schnohr P, et al. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008;299(21):2524–32.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99. PubMed PMID: 23126252. Epub 2012/11/07. eng.
Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–67. PubMed PMID: 22085343. Epub 2011/11/17. eng.
Group H-TC. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203.
Aiello RJ, Brees D, Bourassa PA, Royer L, Lindsey S, Coskran T, et al. Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arterioscler Thromb Vasc Biol. 2002;22(4):630–7. PubMed PMID: 11950702.
Zhang Y, Da Silva J, Reilly M, Billheimer J, Rothblat G, Rader D. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–4.
Khera AV, Cuchel M, de la Llera-MoyaM, Rodrigues A, Burke MF, Jafri K, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364(2):127–35.
Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, et al. HDL cholesterol efflux capacity and incident cardiovascular events. New England J Med. 0(0):null. PubMed PMID: 25404125.
Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290(17):2292–300.
Tardif J-C, Grégoire J, L’Allier PL, Ibrahim R, Lespérance J, Heinonen TM, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297(15):1675–82.
Waksman R, Torguson R, Kent KM, Pichard AD, Suddath WO, Satler LF, et al. A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. 2010;55(24):2727–35.
Bailey D, Jahagirdar R, Gordon A, Hafiane A, Campbell S, Chatur S, et al. RVX-208A small molecule that increases apolipoprotein AI and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol. 2010;55(23):2580–9.
Watson CE, Weissbach N, Kjems L, Ayalasomayajula S, Zhang Y, Chang I, et al. Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J Lipid Res. 2011;52(2):361–73.
Tardif J, Gregoire J, L’Allier P, Ibrahim R, Lesperance J, Heinonen T, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–82.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Yassine, H. (2015). Is Low HDL Cholesterol a Marker or a Mediator of Cardiovascular Disease?. In: Yassine, H. (eds) Lipid Management. Springer, Cham. https://doi.org/10.1007/978-3-319-11161-2_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-11161-2_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11160-5
Online ISBN: 978-3-319-11161-2
eBook Packages: MedicineMedicine (R0)