
Abstract
Microscopy has made a significant impact on science and is today an integral part of a researcher toolbox and techniques to test scientific hypothesis. However, only recently has the possibility to observe important biological interactions and mechanisms deep inside a tissue, with the ability to focus on a single cell, emerged. New technological discoveries have resulted in broadening the focus of this standard method and promoted novel modalities to be used in several biological contexts and to validate scientific hypothesis. Nowadays, noninvasive methods have shown interesting and groundbreaking applications in tissue engineering, tumor biology, and immunology. Multiphoton microscopy (MPM), second harmonic generation (SHG), fluorescence lifetime imaging microscopy (FLIM), Förster resonance energy transfer (FRET), and intravital microscopy (IVM) have harnessed both endogenous and exogenous fluorophores to uncover new molecular and cellular biology pathways while enabling single cell tracking. By means of these techniques, it is now possible to unveil extracellular matrix density and alignment, perform metabolic analysis, observe complex macromolecular interactions, and validate cell division, death, communication, and fate. Within this chapter, we highlight and showcase the versatility and applicability of different microscopic approaches and their qualitative and quantitative insight into cellular and molecular biology, tissue engineering, immunology, metabolism, and tumor biology.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

1 Introduction
Since its development in the sixteenth century by Hans and Zacharias Janssen, microscopy rapidly found applications in imaging, and during the seventeenth century, works by Robert Hooke and Anton van Leeuwenhoek reported the first high-magnification visualization of cells (Leeuwenhoek 1682; Hooke 1665). Nowadays, microscopic imaging remains a steadfast tool in the life sciences with microscopy techniques routinely applied in the biosciences and tissue engineering research fields. Transmission light, confocal, and fluorescence microscopy are the most commonly used microscopic techniques for cell identification, analysis, and evaluation of cell response to chemical and mechanical stimuli. However, transmission light microscopy gives generic detail of cell responses and is predominantly employed to evaluate cell division and proliferation (Zaritsky et al. 2011). Confocal and fluorescence microscopy often require cell or tissue staining using external fluorophores, either alone or bound to antibodies, rendering it an invasive technique as stained cells are often not compatible for further assays (Gurcan et al. 2009). The use of invasive techniques can require pretreatments or fixation regimes which affect cell and tissue physiology and protein conformation, rendering it a destructive endpoint technique (Mariani et al. 2009). Such necessities have generated an increasing demand for noninvasive methods of fluorescence microscopy in order to diagnose diseases and optimize tissue engineering approaches (Quinn et al. 2012). The use of noninvasive fluorescence-based microscopic techniques can allow real-time analysis of living cells without the requirement of cell fixation, reducing sample manipulation and interference while decreasing costs. In this chapter, we discuss emerging noninvasive fluorescent-based imaging technologies in the evaluation of cell and tissue behavior. These applications enable high-resolution in vitro, in situ, and in vivo imaging of ECM structures and cells in intact and engineered tissues. Uncovering the full potential of noninvasive imaging can provide more accurate results and reduce sample manipulation while avoiding the pitfalls of complex and invasive staining protocols. In particular, we focus on the advanced modality techniques of multiphoton microscopy (MPM) and its applications in second harmonic generation (SHG), fluorescence lifetime imaging microscopy (FLIM), Förster resonance energy transfer (FRET), and phosphorescence lifetime imaging microscopy (PLIM).
2 Principle of Fluorescence
The interaction of light with matter is underpinned by quantum theory which is the basis of most microscopy techniques. Due to the higher mass of protons and their strong electromagnetic interaction in the nucleus of an atom, light interacts preferably with electrons, characterized by their lower mass, existing at the periphery of atoms. The electron of an atom or molecule can absorb the energy of an incident photon of light, promoting a transition to a higher energy state – this energy transfer process is called absorption. The energy associated with a photon is described by the Einstein-Planck equation (1), with h being Planck’s constant, c the speed of light, and λ the photon wavelength. The energy gap that the electron must overcome in order to jump to the increasing energy state depends on the position of the electron, the atom, and the molecular structure of a compound. This is the basis of all fluorescence microscopy, whereby excitation light of a defined wavelength generates fluorescence emission from an excited fluorophore at an emission wavelength.
Before the 1930s, a principle was held that this quantic jump could only occur if the energy of the photon is equal or above the energy gap of the electron transition (Lakowicz 1999). In 1931 this dogma was revised. Maria Goeppert-Mayer described an alternative process in her doctoral dissertation which hypothesized that in specific conditions, such an excitation process could be achieved in two steps (Göppert-Mayer 1931). Specifically, two photons whose combined energy would be equal or higher to the energy gap could be enough to achieve an electronic transition. According to quantum mechanics, a single photon excites the molecule to a virtual intermediate state that by recombination with a second photon brings the molecule to the final excited state and higher energy level. Only in 1961 was this process first confirmed by Wolfgang Kaiser using a CaF2:Eu2+ crystal laser (Kaiser and Garrett 1961). This quantum process is the basis of MPM (Fig. 1).

Energy transitions due to light absorption. This transition occurs from ground state (S0) to a higher energy state (S1) and can be promoted by (a) one-photon, (b) two-photon, or (c) three-photon excitation
3 Multiphoton Microscopy (MPM)
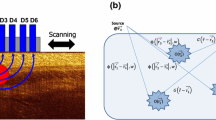
MPM can be modelled further in a time-dependent Schrödinger equation in which the Hamiltonian contains an electric dipole moment:\( {\overrightarrow{E}}_{\gamma}\bullet \overrightarrow{r} \), where \( {\overrightarrow{E}}_{\gamma } \) is the electric field vector of the photons and \( \overrightarrow{r} \)is the position operator. εγ is the photonic energy associated with the electric field vector, and εm is the energy difference between the state m and the ground state. The first-order solution corresponds to the one-photon excitation, and multiphoton transitions are represented by higher-order solutions. Here, the transition probability between the molecular initial state|i⟩, the virtual intermediate state |m⟩, and the final state |f⟩ is given by:
After excitation of a molecule, by a single or multiphoton approach to S1 energy level, the molecule undergoes internal conversion and reaches the lowest vibrational level of the S1 state. From here the energy can be dispersed in a radiative or non-radiative way followed by a return to its ground state. A radiative decay is associated with light emission phenomena, while non-radiative decay is related to the losing of energy by particle vibration and by heat.
Radiative decay can be decomposed further into fluorescence or phosphorescence processes. In detail, fluorescence is characterized by short decay time durations (picoseconds to nanoseconds) and occurs from S1 to S0 energy level (Fig. 2). Phosphorescence has a longer decay time (micro- to milliseconds) due to intersystem crossing that the molecule must undergo to reach the energy state T1 and then convert back to S0.

Simplified Jablonski diagram. Higher vibrational levels are represented by thin lines. hνA/F/P is the energy associated to each photon described by Einstein-Planck’s equation (1). h is related to the Planck’s constant and νA/F/P with the wave frequency of each transition process
The radiative decays of an electron are dependent on the molecular structure and its local environment such as solvent, pH, and temperature. The rate of these decays is measured by quantum yields, whereby a high fluorescence quantum yield results in a higher percentage of energy being released by light, subsequently rendering a brighter emission and higher number of photons being radiated. The absorption and decay pathways that an electron or molecule can undergo are graphically represented in a Jablonski diagram (Fig. 2).
In MPM the absorption step in the Jablonski diagram (Fig. 2) is achieved using light amplification by stimulated emission of radiation (LASER). Wavelengths on the ultraviolet (UV), visible, and near infrared (NIR) range are commonly used as excitation sources in microscopy. When going deeper inside of a sample, light scattering dominates the attenuation of light propagation. However, with the use of nonlinear optical processes such as two-photon excitation, the effects of emission scattering are mitigated. In addition, longer wavelengths can be used which increase light penetration and reduce photobleaching due to the confinement of the excitation volume in a tissue due to the quantic nature of the multiphoton recombination. To accomplish this, intense ultrashort pulse lasers in the femtosecond range (~80–250 fs) are used (Zipfel et al. 2003b; Wilt et al. 2009).
Therefore, wavelengths applied in MPM have lower energies, and less scattering of photons occurs when imaging deep inside biological tissues, while less damage to the samples is produced in comparison with conventional confocal/fluorescence microscopy with higher photostability and higher laser penetration potential .
4 Second Harmonic Generation (SHG)
4.1 Principle of SHG
Falling under the domain of MPM, SHG is a nonlinear optical spectroscopic (NLO) method that has been applied to biological microscopy. It enables direct imaging of anisotropic structures containing large hyperpolarization features (Zipfel et al. 2003a). SHG is a quantic phenomenon in which two photons are converted into a single photon at twice the frequency of the individual exciting photons (Stoller et al. 2003). Therefore, SHG can also be defined as a frequency-doubling procedure (Campagnola 2011). SHG possesses common features with multiphoton microscopy, but it undergoes no energy loss in a sample (in the form of heat and radiation), meaning a 100% conversion of energy resulting with no bleaching or heating effects, therefore allowing a long-term and high-powered observation of samples (Dempsey et al. 2012).
The response to the excitation wavelength depends on the polarization of a material interaction with light. For this, the total polarization, P, for a material interacting with light can be expressed as:
The first term, where χ is the first-order nonlinear susceptibility tensor and E is the electric field vector, is correlated with linear interactions of light such as absorption, scattering, and reflection (Shen 1984). The second term describes SHG, sum, and difference of frequency generation; the third term describes two- or three-photon absorption, third harmonic generation (THG), and stimulated Raman processes and coherent anti-Stokes Raman scattering (CARS) (Chen et al. 2012).
The second-order symmetry nature of SHG enforces specific criteria on the available molecules that can be imaged using this property. Only molecules or regions lacking a center of symmetry (non-centrosymmetric) can be imaged. These molecules are known as harmonophores (Boddupalli and Bratlie 2015). Harmonophores and their environment must be non-centrosymmetric on the scale of the excitation wavelength being used. In addition, they need to possess a phase-matching condition, with the incident and resulting electromagnetic waves travelling at the same velocity as SHG is only efficient in coherent conditions (Vielreicher et al. 2013). Molecules being imaged must have a permanent dipole moment, and for further efficient SHG, they must be aligned within the focal volume of the microscope so that the second term of Eq. 3 is non-zero (Chen et al. 2012).
The principles of SHG facilitate the noninvasive imaging of structures such as type I and II collagen and myosin due to their distinctive molecular structure (Plotnikov et al. 2006; Campagnola 2011). Other isoforms of collagen do not meet these requirements because they lack a fibrillary organization in vivo; therefore, they do not produce sufficient SHG signal to be imaged (Pena et al. 2005) .
4.2 Application of SHG in Biological Imaging
In biomedical applications, multiphoton SHG imaging has been demonstrated as an alternative to invasive microscopy involving tissue removal, fixation, and staining samples. Due to the structural and mechanical role of collagen and elastin in native tissue and tissue-engineered samples, simultaneous imaging of collagen and elastic fibers in tissue-engineered heart valves without resorting to standard histochemical protocols has been of particular interest. With this approach, noninvasive assessment of native tissue structures and quality control of engineered tissue has been achieved (Konig et al. 2005). Conventional ECM imaging requires fixation of samples and embedding in paraffin or cryopreservation and sectioning using microtomes prior to applying a specific staining, for example, picrosirius red (Lattouf et al. 2014). Although cryopreservation can allow storage of a tissue without fixation, rapid freezing can incur damage to the morphology of the tissue (Schenke-Layland et al. 2006). In contrast, paraffin-embedded tissue can maintain a reproducible representation of the tissue. By removing the paraffin and rehydrating the tissue, adequate multiphoton imaging can still be performed without staining protocols. Although if the sample was previously stained, even after following a destaining protocol, some interferences in the MPM/SHG signal will still exist (Monaghan et al. 2016).
Three major requirements to perform both multiphoton and SHG microscopy are a femtosecond mode-locked pulsed laser, a laser-scanning mode microscope, and bandpass filters that can separate SHG signal from multiphoton signal. One possible clinical approach is the acquisition of SHG using standard endoscopes or laparoscopes. Indeed Brown et al. have reported an endoscope capable of MPM/SHG imaging in vivo in unstained organs (Brown et al. 2012)3. Imaging of liver, kidney, and colon tissue revealed no significant differences in image quality between the use of an endoscope and a regular MPM/SHG microscope, and features commonly seen in biopsied histopathology slides from these tissues were observed (Brown et al. 2012). Although significant progress has been made in developing SHG technology for clinical applications, some areas are still undeveloped requiring further improvement and integration of all parts into a viable and easy-to-use instrument. Ultimately, multiphoton microscopy associated with SHG provides important details at sub-micrometer level of ECM and cells in vitro or in vivo while avoiding extensive sample preparation. Furthermore, hallmarks of disease progression, fiber alignment, and biomechanical properties can be assessed. By applying this noninvasive technique, it is possible to decrease surgical trauma, perform analysis in real time, and decrease sample manipulation. In addition, SHG can also be used to diagnose or follow treatment response in vivo by the use of an endoscope.
Fibrous collagen assembles into organized structures beginning in the nanometer range and then ordered into fibers (~50–500 nm) and then fascicles (~50–300 μm) (Puxkandl et al. 2002). A wave phase mismatch between the excitation source and the SHG emission can occur due to the quasicrystalline organization of collagen in biological samples (Prockop and Fertala 1998). This results in a separation of SHG emission into forward and backward components. The difference between both components is dependent on the regularity and the size of fiber assembly. Fibers with an axial size in the same magnitude as the excitation wavelength will exhibit a predominantly forward-direction SHG signal, while fibrils with a smaller cross-section size produce equal levels of backward and forward SHG signals. This implies that for a comprehensive analysis of fiber alignment and collagen quantification, there is a need to capture both SHG signal scattering directions which would entail reflection and transmission paths with detection efficiencies calibrated to perform quantitative assessments .
4.3 Analysis of Pathologies Based on Fibrosis and ECM Remodeling Using SHG
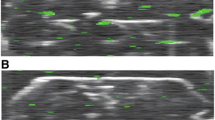
SHG microscopy can facilitate the analysis of deposited collagen, thus allowing diagnosis of fibrotic development of internal organs or tissue (Strupler et al. 2007; Stanciu et al. 2014; Ranjit et al. 2016; Ochoa et al. 2018) (Fig. 3). The use of this technique is more advantageous over standard histology techniques: SHG allows the noninvasive acquisition of 3D image sets in in vivo or in vitro/ex vivo over thick samples (>10 μm) (Brown et al. 2003; Pantazis et al. 2010); and histology protocols can result in an erroneous staining of thin-sliced (1–10 μm) samples due to its effects on tissue morphology, given the rise to misguided interpretations (Strupler et al. 2007). Understanding collagen alignment is an important tool to evaluate disease progression, identification, and response to treatment. Nowadays, SHG is primarily employed as a research tool operated by highly trained researchers. However, using appropriate scoring systems will enable rapid tissue interpretation which will ease the application of this technique outside of the research field while reducing the training needed for new users.

SHG microscopy and applications. (i) Optical sections of collagen obtained in (a) self-assembled collagen; (b) mouse dermis; (c) mouse bone; (d) human ovary. Scale bar: 30 μm. (Reproduced from Chen et al. (2012) with permission from Springer Nature). (ii) Liver section immunolabeled and imaged by two-photon excited fluorescence (TPEF) and SHG in (A) healthy and (B) diseased liver. Unlabeled sections were used as control, and merging between both channels is displayed in white pseudocolor. (Reproduced from Gailhouste et al. (2010) with permission from Elsevier). (iii) Representation of the three TACS measures (A–C) in Wnt-I mouse tumors (a–c) showing tumor environment and correspondent boundary (yellow line). For (A) the intensity of the fluorescent signal relative to x-y location is present. For (B) and (C) histogram with the frequency distribution of fibers. Scale bars, 25 μm. (Reproduced from Provenzano et al. (2006) with permission from Springer Nature)
Fibrosis is a common condition occurring during the development of chronic liver, kidney, heart, or lung diseases. It is characterized by an excess accumulation of freshly synthesized extracellular matrix proteins, such as collagen type I, impairing the normal function of the tissue resulting in more advanced and harder-to-treat pathologies (Bataller and Brenner 2005). The employment of SHG for fibrotic tissue assessment has been reported by Gailhouste et al. in the examination of human liver biopsies (both healthy and cirrhotic). These samples were classified using a METAVIR fibrosis score ranging from F0 (no fibrosis) to F4 (cirrhotic tissue). With SHG, it was possible to formulate a fibrosis-SHG index which allowed for qualification and monitoring of different stages of disease progression while highlighting an easier, standardized methodology with lower sampling bias and able to correlate with the standard applied METAVIR fibrosis scale (Bedossa and Poynard 1996; Gailhouste et al. 2010) (Fig. 3ii). The use of SHG for analysis of fibro-proliferative pathologies is of higher interest for the medical field. Most of fibrotic evaluations need tissue obtained from biopsies. By developing a SHG scoring system (similar to fibrosis-SHG index), it is also possible to reduce the training needed to fruitfully apply this technique in a clinical environment.
Due to rapid cell division and high metabolic activity associated with tumor tissue, an excessive synthesis of ECM can lead to discernable differences in both composition and assembly of collagen in tumor tissue when compared with healthy tissue. This densely packed stroma has a negative impact on the diffuse transport of water and solutes due to the resistance conveyed by the interaction of collagen and proteoglycan components of the ECM resulting in a hindrance of drug treatment responses (Netti et al. 2000). Brown et al. reported the use of SHG in vivo which allowed dynamic imaging and quantification of collagen while rapidly inferring on different types of tumor susceptibility to drug delivery (Brown et al. 2003) .
4.4 Biomechanics
The ultimate goal of tissue engineering is to repair and replace tissues and organs possessing distinct biomechanical functions which should be recapitulated. In addition to the tissue-specific biological requirements, there are criteria that functional engineered tissues should meet: size and mechanical integrity to allow survival under physiological conditions, minimal biomechanical functionality that should progress until the normal tissue function has been restored, maturation, and integration with surrounding host tissues (Robert Lanza 2014).
The alignment and organization of fibrillary collagen have an impact on the mechanical properties of the tissue. In this regard, SHG microscopy has been applied to assess and profile the biomechanics of cartilage (Lilledahl et al. 2011), arthritis disease models (Caetano-Lopes et al. 2009), arteries (Venkatasubramanian et al. 2010), the heart (Sommer et al. 2015), and the cornea (Winkler et al. 2011). The importance of collagen content and orientation for the native tissue function as well as the impact of poor collagen structure on organ function has been delineated using such techniques. The combination of multiphoton microscopy with mechanical stress testing can be also combined allowing a real-time analysis of collagen fiber orientation and alignment response to mechanical stress (Sereysky et al. 2012).
In addition to native tissue, SHG can also be applied to collagen-based (or SHG-responsive) scaffolds proposed for tissue engineering and biomaterial applications (Drury and Mooney 2003). Boddupalli et al. have applied SHG to evaluate collagen production from fibroblasts encapsulated in methacrylate alginate hydrogels. In softer gels, lower collagen production was observed, although their fibers were more aligned in contrast to stiffer gels which promoted higher amounts of collagen with more anisotropic fibers. Both softer and stiffer gels may provide biomedical applications depending on the tissue of native collagen structure and alignment (Boddupalli and Bratlie 2019).
SHG is therefore an advanced microscopic technique that allows clinical, biomedical, and tissue engineering applications. With its increasing usage in research and weighing its features against more standardized procedures, it might be only a factor of developing a more user-friendly hardware, software, and easy-to-interpret results for SHG to be translated into the clinical field .
5 FLIM/PLIM Microscopy
Another advanced noninvasive imaging technology facilitated by multiphoton microscopy is fluorescence lifetime imaging microscopy (FLIM): a microscopy technique that generates an image based on the fluorescence intensity and on the spatial distribution of excited energy states lifetimes of an endogenous or exogenous molecule of interest in live or fixed cells and tissue. FLIM microscopes are designed to measure lifetimes from the nanosecond to microsecond (μs-FLIM or PLIM) range. Within cells there are molecules present with chemical configurations which enable them to undergo fluorescence emission after excitation with a specific UV/Vis wavelength. These components have within their molecular structure an aromatic group and a high number of π-bonds which increases fluorescence quantum yield and the probability of emitting light of higher intensity (Lakowicz 1999). The presence of fluorescent residues (tyrosine and tryptophan) in proteins is standardly measured in both enzymatic and biochemical assays for protein quantification (Stoscheck 1990).
Since the dawn of multiphoton technology, it has been possible to excite a cell’s endogenous fluorophores in order to obtain fluorescence light emission by means of low-energy wavelengths that avoid phototoxic properties characteristic of higher-energy wavelengths (Diffey 1991). This emission is normally designated as cell autofluorescence. The molecules responsible for the autofluorescence emission are aromatic amino acids (tryptophan, tyrosine), serotonin, lipo-pigments (keratin, melanin), NAD(P)H, and FAD (see Table 1 for a more extensive list). In tissues, ECM molecules also contribute for the overall fluorescence of the sample mostly due to the presence of elastin and collagen and their high quantum yield which can be excited to perform SHG.
As described previously, the excitation of a fluorophore can be followed by emission of a photon, and depending on the radiative quantum yield of the fluorophore, this process can occur by fluorescence, phosphorescence, or delayed fluorescence. The predominant emission phenomenon measured in FLIM is fluorescence. The fluorescence lifetime of a molecule is the time – normally pico- to nanoseconds – that a fluorophore remains in an excited state before decaying and emitting a photon. Therefore, fluorophores can be characterized not only by their excitation and emission spectra but also by their unique lifetime. Fluorescence lifetime can be described by Eq. 4:
where If(t) is the fluorescence intensity at a given time t and ǖFC;n is the fraction of the fluorophore that is responsible for a specific τn fluorescence lifetime. The fluorescence lifetime decay of a fluorophore can be decomposed in several fractions depending on the molecule and its environment – mono-exponential or multi-exponential decay. If the fluorophore has a mono-exponential decay, ǖFC;n value is 1, whereas in a multi-exponential decay, the sum of the various fractions ǖFC;n is 1.
Fluorescence microscopy has been one of the most used tools in biological science, and it is mostly based on the fluorescent intensity of the specific fluorophores being analyzed. This technique is dependent on the light detection efficiency of the system and concentration of fluorophores and can result in variable levels of photobleaching and phototoxicity. Contrary to fluorescence microscopy, fluorescence lifetime is minimally affected by the aforementioned variables, although it is sensitive to fluorophore microenvironment such as changes in pH, temperature, or presence of FRET donors/acceptors. Therefore, fluorescence lifetime measurements provide a more focused and profound analysis than measuring the fluorescence intensity (van Munster and Gadella 2005; Provenzano et al. 2009; Ishikawa-Ankerhold et al. 2012) .
5.1 Frequency-Domain and Time-Domain FLIM
Acquisition of FLIM data can be performed in the frequency or time domains. Briefly, in frequency-domain FLIM, a high-frequency modulated laser is used to excite the specimen, and the fluorescence lifetime is determined by the demodulation and phase shift of the fluorescence signal (Schneider and Clegg 1997). Consequently, it is possible to determine the lifetime τ, from the measured phase shift and from the decrease in modulation from the emitted fluorescence compared with the excitation pulse. This will originate two separated lifetimes: τω, lifetime calculated from the phase shift, and τA, the lifetime obtained from the modulation difference. If the fluorophore has a mono-exponential decay, τω = τA. For the analysis of more complex fluorescence lifetime decay patterns, measurements can be repeated using multiple modulation frequencies (Squire and Bastiaens 1999). To appropriately apply frequency-domain FLIM, it is required to have a modulated light source and to be able to extract both the phase and modulation signal from the excitation light and emitted light.
In time-domain FLIM, a short pulse of light is used to excite the sample after which the emitted fluorescence is measured in time resulting in curves described by Eq. 4 (Valentini et al. 2000). These measurements are acquired using a time-correlated single-photon counting (TCSPC) detector where after each excitation pulse, the arrival time of the first photon is monitored at a very high resolution (Phillips et al. 1985). A representative curve of the fluorescence lifetime decay is then obtained by recording the arrival time of the large number of photons being emitted by the sample. This time-domain technique is dependent on the detection sensitivity of the TCSPC unit to avoid bias toward photons with shorter lifetimes. Time-gated FLIM is another time-domain FLIM technique in which photons are collected at a fixed number in discrete intervals of time using a time-gated camera. The sample is excited with a short pulse of light, and the emission light is measured at two (or more) intervals of time during the fluorescence decay opposite to the previous technique where all the photons during a decay are measured. This way, the acquired lifetime is independent of laser intensity fluctuations. Increasing the number of time gates can provide more accurate results for multi-exponential decays (Sytsma et al. 2008).
FLIM measurements can be done in a nonspatially resolved fluorescence spectroscopy by exciting a sample or solution in a cuvette (Boens et al. 2007). For biomedical and tissue engineering fields, it is far more interesting to be able to use FLIM coupled with a microscope allowing morphological analysis and decomposition of complex samples by focusing the excitation volume or having the possibility to 3D reconstruct a sample. Therefore, FLIM has been connected to wide-field (Gadella et al. 1994; Emiliani et al. 2003), confocal (Buurman et al. 1992; Buranachai et al. 2008), multiphoton microscopes (Gratton et al. 2003; Walsh et al. 2013), and light sheet microscopy (Mitchell et al. 2017; Funane et al. 2018)
Typically, the choice for the most adequate FLIM microscopy instrument is dependent on the specific application and spatial-time resolution besides financial consideration, equipment, and expertise available. Most of FLIM systems used in research have been custom-built by their users. Still, there are some considerations to be made between the different FLIM systems: wide-field and confocal microscopy are limited to one-photon excitation, while only confocal and multiphoton microscopy are capable of performing 3D FLIM z-stacks .
5.2 Fluorescence Lifetime Imaging Microscopy (FLIM) to Monitor Cellular Metabolism
All cells require a source of energy to maintain homeostasis and feed energy-consuming processes such as cytoskeletal dynamics, transcription, translation, and DNA repair. Cell metabolism also provides the building blocks for production of nucleotides, phospholipids, and amino acids (Lunt and Vander Heiden 2011). Metabolism in cells has evolved to sense locally available nutrient required to supply the cell to fulfil their biomass needs. When nutrients are scarce, cells halt biomolecular precursor production (amino acids, nucleotides, and acetyl-CoA) and adapt to extract maximum free energy from available resources to survive. Differences in metabolic pathways are dictated by distinct regulatory mechanisms that can control and adjust the cellular metabolism in differentiated and proliferative cells (Vander Heiden et al. 2009). In most mammalian cells, glucose and glutamine are predominantly catabolized and used as sources of carbon, nitrogen, and energy. The breakdown of glucose follows two major pathways: glycolysis and oxidative phosphorylation (OxPhos) to ensure efficient production of functional biomolecules and adenosine 5′-triphosphate (ATP) which is the main component of cellular energy and fuel for important homeostatic reactions happening in the cell (Fig. 4). Glutamine undergoes glutaminolysis which generates biomolecules and helps fuel the Krebs cycle (McKeehan 1982; Dang 2010).

Overview of glycolysis and oxidative phosphorylation. OxPhos was divided into two major pathways: tricarboxylic acid cycle (TCA) and the electron transport chain (ETC). Major by-products of glycolysis and TCA are highlighted as well as a schematic for the ETC process. (Figure was created with BioRender.com)
Glycolysis is the starting point for the breakdown of glucose. It occurs in cell cytoplasm and culminates in the formation of pyruvate. From here, pyruvate is shuttled inside the mitochondria, aided by mitochondrial pyruvate carriers, or can be used as a substrate to produce lactate. In the mitochondria, the starting point of the OxPhos pathway, pyruvate is converted to acetyl-CoA which is used to feed the Krebs cycle by producing two electron carrier species (NADH and FADH2) and important biomolecules for lipid, amino acid, and nucleotide synthesis, such as citrate, malate, oxaloacetate, and α-ketoglutarate. Afterward, both NADH and FADH2 react with enzymes on the electron transport chain (ETC), specifically NADH/ubiquinone oxidoreductase (complex I) and succinate/ubiquinone reductase (complex II), respectively. Here, the passage of electrons between the different protein complexes releases energy which is stored as a proton gradient across the mitochondrial inner membrane ending in the formation of adenosine 5′-triphosphate (ATP) from adenosine 5′-diphosphate (ADP) at complex V or F1F0-ATPase. Overall, the OxPhos pathway yields 36 mol of ATP per mol of glucose consumed, and, in addition, it produces CO2 and reactive oxygen species (ROS) while consuming O2 (Fig. 4). In the cytoplasm, pyruvate conversion to lactate occurs without the consumption of O2 and with only 4 mol of ATP produced per mol of glucose consumed (Folbergrová et al. 1974; Saraste 1999; Vander Heiden et al. 2009; Mookerjee et al. 2017).
Glutamine is imported to the cell and used for the production of glucosamine (in conjugation with glucose), nucleotides, and proteins. Furthermore, it undergoes glutaminolysis and is lysed by glutaminase into glutamate and ammonia. Glutamate is then imported into the mitochondria where glutamine dehydrogenase converts glutamate into α-ketoglutarate generating nicotinamide adenine dinucleotide phosphate (NADPH) while replenishing the Krebs cycle (Fig. 4). The last step of glutamine metabolism makes it an anaplerotic pathway which can be used to feed oxidative phosphorylation if glucose concentration is low (McKeehan 1982; Dang 2010).
Aside from the availability of nutrients in the microenvironment, cellular metabolism can also be affected by other intrinsic or extrinsic factors such as genetic mutations (DeBerardinis et al. 2008), viral/bacterial infection (Thai et al. 2014), hormone or cytokine response (Bauer et al. 2004), cell density (inhibition by contact) (Aldridge and Pye 1976), or chemical metabolic challengers (Daemen et al. 2015). Interestingly, cell metabolism can be more than just a response to a stimuli or a microenvironment condition. An emerging field, known as immunometabolism, has established a definitive relationship between immune cell function and metabolism. It is widely acknowledged that M1-polarized macrophages with pro-inflammatory responses are glycolytic, while anti-inflammatory M2-activated macrophages are characterized by an oxidative phosphorylation-dependent metabolism (O’Neill et al. 2016). Not only is this metabolic preference observed in macrophages, but it is also exhibited in activated effector T cells and activated natural killer (NK) cells compared with their resting state (Zipfel et al. 2003b). In addition to immune cells, stem cells and differentiated cells possess distinct metabolic phenotypes. Stem cells are majorly glycolytic, whereas their differentiated counterpart is more OxPhos. Remarkably, when pluripotency is induced, their metabolic features revert back to a glycolytic state present in native stem cells (Folmes et al. 2012).
The coupling of FLIM measurements with multiphoton microscopy has opened a new modality to probe cellular metabolism in a noninvasive manner. Lakowicz et al. were the first to measure the fluorescence lifetime of free and protein-bound nicotinamide adenine dinucleotide (NADH) uncovering the potential of gauging this endogenous fluorophore and its impact on metabolism (Lakowicz et al. 1992). Since NADH has a single-photon excitation wavelength of 360 nm, it requires powerful and cell-damaging excitation sources. With the use of multiphoton microscopes, this problem is resolved by exciting NADH with 740–760 nm and allowing in vitro and in vivo metabolic profiling (Alfonso-Garcia et al. 2016; Niesner et al. 2004; Skala et al. 2007; Ghukasyan and Kao 2009; Yaseen et al. 2013). Following this, Huang et al. reported fluorescence spectroscopy and multiphoton microscopy of both NADH and FAD which are spectrally distinct at 750 and 900 nm excitation wavelengths. In addition, it is possible to separate both light-emitting species by their emission wavelength provided an adequate 410–490 nm bandpass filter for NADH and 510–560 nm bandpass filter for FAD (Huang et al. 2002).
With FLIM it is possible to uncover the role of these factors on the major cellular metabolic pathways (Fig. 4). Therefore, FLIM microscopy opens a pathway for noninvasive metabolic imaging and analysis that allows deeper understanding of the connections between metabolism and cell function or phenotype while enabling metabolic probing in real time. Furthermore, it can unveil distinctive metabolic features present in diseases (e.g., cancer), consequences of external stimulus (e.g., viral infection), as well as microenvironment nutrient concentration. By combining metabolic modulators (e.g., biomaterials or pathway inhibitors) and FLIM, it is possible to tailor and observe cell metabolism and infer on their impact and improve treatments, immune cell function, or stem cell differentiation while uncovering cell dependence on a specific metabolic profile. Using a multiphoton microscope (MPM) coupled with a fluorescence lifetime imaging microscopy (FLIM) or phosphorescence lifetime imaging microscopy (PLIM) detector, it is possible to evaluate NADH and FAD (Masters et al. 1997; Skala et al. 2007; Digman et al. 2008) and O2 concentrations using exogenous probes (Kondrashina et al. 2012) (Fig. 5). An emerging application of such noninvasive microscopy is cell-specific metabolic analysis which has found applications in tumor biology, immunology, and organoid research fields (Skala et al. 2007; Wallrabe et al. 2018).

Overview of FLIM, PLIM, and FRET imaging. (i) In vivo multiphoton FLIM images representing the continuous effect of doxorubicin on the optical redox ratio of human prostate cancer cell line. (Reproduced from Wallrabe et al. (2018) with permission from Springer Nature). (ii) Functionalization strategies for scaffolds/matrixes (a, c) and dynamics of O2 concentration measurements by PLIM (b). (iii) Intensity-based FRET with estimation of energy transfer efficiency (E%) and distance between probes ® used to identify C/EBPa (CCAAT/enhancer-binding protein a) dimerization. (Reproduced from Wallrabe and Periasamy (2005) with permission from Elsevier)
NADH fluorescence lifetime typically exhibits a double-exponential decay which is decomposed in a short lifetime related to free NADH in the cytoplasm and a long lifetime reflective of enzyme-bound NADH (Lakowicz et al. 1992). Following Eq. 5 is possible to calculate the fraction respective to each of them. In a single-cell type, the enzymes in which NADH reacts are well-conserved regions in a cell, meaning that the majority of the shifts in fluorescence lifetime fractions are connected with the free NADH fraction. Generally, when this fraction increases, the cell metabolic profile is tending toward glycolysis. Conversely, when this fraction decreases, the cell is less dependent on glycolysis (Schaefer et al. 2019). Regarding NADH fluorescence intensity, it has been shown that increasing glucose concentration in the cell culture medium or adding glycolysis inhibitors increases NADH autofluorescence, revealing a higher usage of the OxPhos pathway (Ghukasyan and Kao 2009). However, NADH fluorescence intensity and lifetime have limited capacity for fully grasping the cell metabolic profile; nonetheless, they are well suited for separating distinct cellular populations or following disease progression (Pugh et al. 2013).
The coenzyme nicotinamide adenine dinucleotide phosphate (NADPH) possesses an overlapping excitation and emission spectra with NADH and is hard to be separated (Blacker and Duchen 2016). Although the NADPH contribution is ten times lower than NADH, NADPH still contributes significantly to cell autofluorescence (Blacker et al. 2014). Fluctuations of NADPH levels are intertwined with the long-lived component of the fluorescence lifetime decay – protein-bound NADH. NADPH processes two major biological functions: acting as an electron source for synthesis of fatty acids, steroids, and DNA and a key component in cellular antioxidant systems intertwined with the production of reactive oxygen species (Pollak et al. 2007; Ying 2008). It is still important to understand how the metabolic profile is being affected in the experimental conditions or to perform a biochemical assay to fully understand the impact of NADPH on the cell metabolism.
Assessing other metabolic cofactors besides NADH can add additional information to metabolic profiling. The endogenous molecule flavin adenine dinucleotide (FAD) is commonly used due its autofluorescence. After measuring the fluorescence intensity of both cofactors, it is possible to calculate the redox ratio by following Eq. 5:
This redox ratio has been associated qualitatively with metabolic profiling after challenging of the glycolytic or oxidative phosphorylation pathway (Walsh et al. 2013). During both these metabolic pathways, there is a regeneration of fluorescent NADH, and during OxPhos, the formation of non-fluorescent FADH2. Following Fig. 4, NADH is converted to non-fluorescent NAD+ by complex I and FADH2 to fluorescent FAD+ by complex II of the electron transport chain. When OxPhos is more active, there is more production of FADH2, and therefore more FAD+ production leads to a rise of FAD+ fluorescent intensity culminating in an increase of the redox ratio. On the contrary, when glycolysis is majorly used to obtain energy, NADH is produced increasing its fluorescence emission, and FAD+ regeneration has been hindered resulting in a decrease of the redox ratio (Ostrander et al. 2010). Furthermore, it is possible to combine both fluorescence intensity measurements and lifetimes to originate the optical redox ratio (ORR) which confirms and better describes the shifts in cellular metabolism (Fig. 5i) (Walsh et al. 2012).
While measuring endogenous NADH/FAD (redox or optical metabolic imaging) is highly informative, frequently there is a need in assessing additional parameters of cell metabolism, such as analysis of mitochondrial polarization, oxygen consumption, extracellular acidification, lipid droplet production, and others. The majority of these additional parameters are measured via exogenously introduced or genetically encoded fluoro- and phosphores (Dmitriev 2017).
For example, a number of genetically encoded fluorescent protein biosensors (which require transfection to be introduced into eukaryotic cells) can serve as indicators of redox, peroxide production, or NADH/NAD+ ratio (Mongeon et al. 2016; O’Donnell and Dmitriev 2017). Mitochondrial membrane potential can be assessed by intensity- or FLIM-based measurement, with the help of tetramethylrhodamine methyl ester (TMRM) dye (Brand and Nicholls 2011; Okkelman et al. 2019b). TMRM is a membrane-permeable molecule which accumulates mainly in both the inner mitochondrial membrane and matrix space of live cells due to its charge and solubility. The change in TMRM uptake is linked to shifts in the mitochondria membrane potential which results in variations of the fluorescence intensity. By measuring the fluorescence intensity at 573 and 546 nm, it is possible to estimate the mitochondria membrane potential (Scaduto and Grotyohann 1999). This molecule also has the possibility to be used as an exogenous multiphoton FLIM probe (Okkelman et al. 2019b).
In addition to endogenous or exogenous fluorophores, there is also the possibility to employ genetically encoded fluorescent biosensors. One strategy to create these protein biosensors involves using circularly permuted fluorescent proteins derived from GFPs (Baird et al. 1999). The circular permutation joins the original N and C terminal by a peptide linker, and a specific reporter or binding protein is fused to the new coupled terminal complex originating a conformational coupling between fluorescence emission and binding to a molecule of interest (Hung et al. 2011). This approach has been applied to probe further the glycolysis flux (Shestov et al. 2014), intracellular pH (Tantama et al. 2011), NADH/NAD+ (Masia et al. 2018), and ATP/ADP ratio (Berg et al. 2009).
To further evaluate cell metabolism, an important measurement to be done is oxygen consumption rate (OCR) and/or analysis of relative tissue oxygenation, balanced by oxygen consumption and diffusion. This can be performed by using a number of analytical tools, ranging from labelling with pimonidazole and use of fluorescent proteins to phosphorescence or delayed fluorescence-based approaches (Papkovsky and Dmitriev 2018). Quenched phosphorescence-based detection by using phosphorescence lifetime imaging microscopy (PLIM) is among the popular approaches in tissue engineering, and intravital microscopy and will be discussed later in this chapter (Sect. 5.4).
When performing live-cell imaging, it is necessary to consider possible contributions of the fluorophores described in Table 1, especially where some tissues have fluorophores in abundance such as the brain (serotonin) and epidermal (keratin, melanin) tissue. Nonetheless, by careful selection of excitation wavelengths and bandpass filters, it is possible to ascertain the contribution of each molecule (Huang et al. 2002).
5.3 FLIM-FRET
In the cellular environment, signaling molecules, enzymes, proteins, and other cellular components undergo many dynamic process and interactions that occur in a microsecond or a nanosecond range. Due to the interest in understanding complex cellular pathways with high spatial and temporal specificity, sophisticated imaging technologies such as one- or two-photon Förster resonance energy transfer (FRET) imaging have been developed (Wallrabe and Periasamy 2005).
This imaging technique operates on the transfer of energy (radiative or non-radiative) between a pair of suitable fluorophores (“donors” and “acceptors”) in close proximity (2–10 nm) (Ranjit et al. 2016). For this to occur, both molecules must have sufficient spectral overlap, a favorable dipole-dipole orientation, and a large quantum yield. During the energy transfer process, the donor fluorescence decreases (quenched), and the fluorescence of the acceptor increases, culminating in a decrease in the donor excitation lifetime.
The energy transfer efficiency between a donor and acceptor can be assessed using a confocal or a multiphoton microscopy in fluorescence intensity FRET approaches. Two major approaches of FRET can be used in biomedical research: the first one – qualitative FRET – is used for establishment of molecular colocalization, cellular organization, or conformational changes, where the presence or lack of FRET is a sufficient indicator for the experimental plan. Panyi et al. utilized FRET to observe the presence and colocalization of potassium channels and CD3 molecules in human T lymphocytes by evaluation of fluorescence intensity and selective light emission (Panyi et al. 2003). The second approach to FRET is known as quantitative FRET. Here, values such as the energy transfer efficiency (E%) and distance between fluorophores are estimated. This method was used by Rinnenthal et al. to quantify intracellular Ca2+ in in vivo and ex vivo studies and its relationship with neuronal dysfunction during neuroinflammation (Fig. 5iii) (Rinnenthal et al. 2013).
The spectral overlap needed for an efficient FRET process is also the main cause of experimental errors (Sekar and Periasamy 2003). As it possesses risk of signal contamination, some corrections may have to be performed. The main reason for signal contamination is the spectral overlap between both molecules necessary for FRET to occur, and this is a major pitfall of conventional FRET imaging. To avoid these corrections, FLIM can be applied to measure FRET as it takes in account the change in a donor lifetime in the presence and in the absence of an acceptor (Elangovan et al. 2003; Wallrabe et al. 2003), in contrast to intensity-only-based measurements.
Using FLIM-FRET, Lamond et al. presented a quantitative platform to measure chromatin compaction in live cells (Lleres et al. 2009). In this study, cells genetically engineered to coexpress histone H2B tagged to an enhanced green fluorescence protein or mCherry were used. The FRET process occurred between separate nucleosomes and increased as chromatin became more compact. In the same cell population, varying FRET efficiency levels were observed reflecting distinct compaction levels, spatially distributed.
To summarize, FLIM-FRET microscopy allows real-time imaging with a high resolution (spatial and temporal) of molecular interactions. It has been applied to investigate several biological processes such as signaling pathways, conformational changes, or binding molecules. The use of this technique is increasing with technological advances such as detectors, microscopes, or fluorophores. One interesting advance is the use of quantum dots which could improve both quantitative and qualitative FRET; however, cell toxicity and cell delivery issues need to be solved.
5.4 Measurement of Molecular Oxygen (O2) in Tissue-Engineered Constructs by PLIM and Related Approaches
Since the oxygen-dependent quenching of fluorescence was first described by Kautsky and Hirsch in 1935 (Kautsky and Hirsch 1935), this methodology has evolved to be a popular approach to measure vascular and brain oxygenation; analyze hypoxia in cells, tissues, and aquatic organisms; and study enzyme activity, microbial growth and is exploited in many other areas (Sakadžić et al. 2010; Papkovsky and Dmitriev 2013; Roussakis et al. 2015; Wolfbeis 2015; Yoshihara et al. 2017; Dmitriev and Papkovsky 2018). This methodology is based on the specific collisional quenching of dye phosphorescence (frequently encountered with metalloporphyrins and related metal-coordinated organic complexes) by molecular oxygen, resulting in quantitative decreases of emission intensity and lifetime (typically from 1–2 to hundreds of microseconds). In tissue engineering, the dye can be modified and used in two ways (Fig. 5ii):
-
1.
Become a molecular or supramolecular probe or impregnated in nanoparticles. In this case, the probe or nanosensor has to be delivered into the cell or tissue-engineered construct (multicellular spheroids, islets, or organoids).
-
2.
Impregnate phosphorescent O2 dyes within matrices or scaffold materials surrounding, or in contact with, live tissue – “hybrid scaffold biosensor” approach (see below).
Due to long emission lifetimes and large Stokes shift, phosphorescent probes are highly compatible for multiplexing in spectral and time domains, e.g., with other FLIM probes, antibodies, conventional fluorescent dyes, and protein biosensors. In addition, on a technical level, PLIM is easier to grasp, and due to long emission lifetimes, even microsecond time-gated cameras can be used to perform measurements (Dmitriev et al. 2013). Presently, PLIM is also available on a TCSPC-FLIM and, in frequency modulation-based platforms, with wide-field, confocal, and two-photon excitation modes.
The “hybrid biosensor scaffold” approach enables detection of O2 in proximity of the live tissue, without the need to stain tissue or cells. With high respiratory activity primarily characteristic of live cells and 3D tissue models, the scaffold-based sensing of O2 is very informative and can be used to predict the hypoxia-dependent drug action within engineered tissue, cell differentiation, and wound healing or simply ascertaining the availability of oxygen to the tissue. This approach normally uses electrospinning coating-aided impregnation of dyes or their impregnation in polymer structures due to swelling in organic solvents (Xue et al. 2014, 2015; Jenkins et al. 2015; Yazgan et al. 2017; Roussakis et al. 2019; Schilling et al. 2019). For instance, porous polystyrene-based O2-sensing scaffolds doped with highly photostable PtTFPP O2-sensing dye have demonstrated high compatibility (lack of direct and phototoxic effects and reliable performance in quantitative measurements) with cancer cells and live brain slices and multiplexing with mapping of cell death and mitochondria labelling (Jenkins et al. 2015). Recent work has demonstrated the dual use of impregnated phosphor in bone regeneration process, both for two-photon O2 sensing inside the scaffold and within the area surrounding it, achieved by predictable diffusion/leaching of dendrimeric phosphor from the scaffold (Schilling et al. 2019). It is worth noting that since the amount of the impregnated/scaffold-associated phosphorescent dye can be controlled and remains unchanged during the experiments, the simple fluorescence intensity-based calibration and sensing can be used as alternative to PLIM measurements.
In many applications, researchers strive to measure O2 directly inside cells, and a great variety of intracellular O2 probes, nanosensors, or even dye structures are available for this purpose (Dmitriev and Papkovsky 2015). However, the majority of engineered tissue constructs are three-dimensional which poses a challenge for diffusion-limited delivery of any probe or nanosensor and for imaging, due to limits of light penetration/absorption and scattering. A priori, the best approach is to employ two-photon microscopy although laser scanning confocal or even wide-field imaging of relatively small engineered tissue samples (50~300 μm) can result in successful imaging. Multicellular spheroids are among the most popular models for engineering tissue environment in 3D and were the first model to demonstrate challenges in probe staining for O2 and fluorescence imaging (Dmitriev et al. 2013). These studies reported the multiparameter PLIM of stem cell-derived neurospheres combined with analysis of distribution of hypoxia-specific dyes, cell proliferation, and differentiation markers by immunofluorescence (Dmitriev et al. 2013, 2014). Further on, multiplexed FLIM-PLIM was reported with tumor spheroid and small intestinal organoid models for measurements of oxygenation together with analysis of cell proliferation, temperature imaging, and biosensor scaffold-based analysis of extracellular acidification (Jenkins et al. 2016; Okkelman et al. 2016, 2017; O’Donnell et al. 2018). In principle, O2-PLIM is also compatible with NAD(P)H-FLIM, which was demonstrated by the Rueck group (Kalinina et al. 2016), but only recently has combining NAD(P)H-FLIM with O2-PLIM been applied to analyse tissue-engineered constructs (Okkelman et al. 2019a).
Collectively, PLIM brings advantage of measuring tissue oxygenation in 3D setting, by means of scaffold- and cell staining probe-aided sensing modalities. While the method is still in its infancy in respect to tissue engineering and its advantages are yet to be fully realized, it has already been successfully used in proof-of-concept studies with tumor and stem cell-derived spheroids, organoids, and implanted tissue models. Its emerging applications in tissue engineering are in the multiplexed analysis of cell bioenergetics, control of tissue viability, and studies of therapeutic outcomes under controlled biomaterial oxygenation .
6 Intravital Microscopy (IVM)
Intravital microscopy (IVM) is a powerful optical imaging technique that facilitates continuous monitoring of molecular and cellular processes in vivo. The main principle of IVM is to image and collect information of biomolecular processes occurring in tissue and organ systems while maintaining their native and physiological state. However, accessing the in vivo environment and performing deep-tissue imaging are incompatible with standard microscopic techniques due to light absorbance and scattering. In order to improve tissue penetration, IVM is mostly performed using red or near-infrared excitation wavelengths. In addition, new optics and more flexible instruments are being developed to improve internal organ access (Bullen 2008). To perform quantitative IVM, it is necessary to consider four major components: site preparation for visualization, use of an endogenous or exogenous probe that can be detected by microscopy, a microscope mounted with appropriate detectors, and computer algorithms and mathematical models that can be used to obtain important parameters. Regarding tissue preparation, IVM has three major categories: fitting chronic-transparent windows at the site of interest in the animal model, exteriorized tissue preparations, and in situ preparations (Jain et al. 2002). Most common animal models preferentially subjected to IVM are murine due to its systemic dynamics and zebrafish due to its small size, ex utero development, transparent embryos, and availability to have transparent adult mutants. Transgenic versions of these animal models can also be prepared in which encoding fluorescence proteins in cell lineages or proteins of interest are considered the ideal tools to perform IVM (Progatzky et al. 2013).
One of the major developments in IVM microscopy has been the application of multiphoton microscopy. MPT allows higher resolution and increased tissue penetration, without compromising fluorophore selection, less photodamage and phototoxicity, and better signal-to-noise ratios (Helmchen and Denk 2005; Molitoris and Sandoval 2005; Misgeld and Kerschensteiner 2006; Bullen 2008). However, the cost and the complexity of setting up, adapting to IVM, and maintaining a multiphoton microscopy system has been a barrier to its wider implementation (Zipfel et al. 2003b). Nonetheless, the major advantage of IVM is the possibility to image and analyze pathologies, perform cell tracking, and measure gene or protein expression in real time on a full systemic environment in long-term studies without needing to excise tissue. In addition, IVM can be easily incorporated into other microscopy methods.
6.1 IVM Coupled to Confocal Microscopy
Nomoto et al. used real-time intravital confocal microscopy to evaluate quantitatively polyplexes and polyplex micelles present in the blood circulation with the ultimate goal of uncovering the interaction between non-viral gene vectors and biological components in situ. To achieve this, Cy5 fluorophore was used to label plasmid DNA within polymeric complexes and DyLight 488 conjugated to an anti-GPlbβ to label platelets (Nomoto et al. 2011). Interestingly, another study used confocal microscopy to evaluate platelet dynamics in response to inflammation. Here they evaluated in real time LPS-induced inflammation in phycoerythrin-conjugated CD49b antibody-labelled platelets on the liver, brain, and muscle tissue (Jenne et al. 2011). These examples showcase the ability of using confocal microscopy allied with IVM. Although there are some known limitations of confocal microscopy in terms of tissue penetration, the use of red and far-red fluorophores coupled with antibodies to label specific cells makes it possible to perform cell tracking in regard to distribution, size, and activation in response to internal or external stimulus in different tissues.
6.2 IVM Coupled to Multiphoton Microscopy
Multiphoton microscopy has made it possible to perform deep tissue imaging in animal models for extended durations. It allows the use of endogenous or exogenous fluorophores for specific cell tracking. One field that benefited immensely from this coupling is immunology. Indeed, many important immune cell behaviors occur in densely populated tissues, and it was previously only possible to delineate these activities in vitro. Several studies document the reliability to infer immune cell function in vivo using multiphoton IVM (Mempel et al. 2004; Cavanagh and Weninger 2008; Egen et al. 2008; Hickman et al. 2008; Abtin et al. 2014; Marques et al. 2015). For example, Hickman et al. imaged CD8+ T-cell function in virus-infected cells in the peripheral interfollicular region of lymph nodes. Here, fluorescein isothiocyanate (FITC) and fluorescein-conjugated dextran (500 kilodaltons) were injected to image the lymph node subcapsular sinus and blood vessels, respectively. In addition, complexes of fluorescein-conjugated dextran (70 kilodaltons) and FITC-conjugated antibodies were used to label macrophages and areas of interest (Hickman et al. 2008). This facilitated the identification of virally infected cells, naïve T-cell interaction and activation by dendritic cells in real time which underlined T-cell effector function with dendritic cells, and antiviral response in lymph node periphery. Abtin et al. also uncovered interactions between different immune cell populations in response to a bacterial infection (Abtin et al. 2014). In virally infected tissues, perivascular macrophages release neutrophil chemoattractant signaling molecules promoting the recruitment of neutrophils to the damaged area which was followed in transendothelial and intravascular regions (Abtin et al. 2014). Genetically engineered mice expressing enhanced green fluorescence protein (EGFP), yellow fluorescence protein (YFP), and green fluorescence protein (GFP) were used to track immune cell subpopulations. Evans blue stain and S. aureus expressing red fluorescent protein (RFP were used to identify blood vessels and bacteria inside of the tissues. In addition, SHG was also used to improve epithelial tissue and blood vessel identification by visualization of both collagen and elastin (Abtin et al. 2014). These two studies present an interesting application of multiphoton IVM in immunology field which allowed cell-specific tracking and function inside of tissues and blood vessels on an in vivo and systemic approach.
6.3 IVM Coupled to FLIM
The use of FLIM with IVM synergizes the benefits of multiphoton microscopy while analyzing only endogenous fluorophores such as NADH or FAD which reduces sample manipulation and preparation time. This allows to infer also the in vivo metabolism of specific cell types and tissues in real time (Rinnenthal et al. 2013; Thorling et al. 2013; Hato et al. 2017). Hato et al. performed intravital two-photon FLIM on mouse kidney revealing specific metabolic signatures of different cells. Using FLIM revealed significantly different metabolic profiles of S1 and S2 tubules. However, these differences may have been due to microenvironmental factors such as pH, calcium, and oxygen levels. Interestingly, similar metabolic profiles were uncovered between S1 proximal tubules and distal tubes; therefore, these two segments may share similar metabolites and microenvironments. In addition, by combining two-photon FLIM- IVM on genetically engineered mice expressing DSred in podocytes and endothelial-specific CreERT2 displaying red fluorescence in the endothelium, elusive peritubular capillary endothelium and podocytes were imaged (Hato et al. 2017). This study serves as an example of IVM-FLIM application to image entire or specific healthy and diseased regions in a tissue with subcellular resolution allowing to metabolically profile areas and specific cell types.
6.4 Förster Resonance Energy Transfer (FRET) in IVM
Biomolecular dynamics can also be probed with IVM and FRET. Most FRET (and FLIM-FRET) methods are still applied in vitro. However, in order to achieve an increasing understanding of molecular processes and cellular function in vivo, it could be worthwhile to apply this technique to a highly complex and functional organism (Radbruch et al. 2015). There has been a surge in strategies to incorporate FRET probes within living organisms. One approach is the development of transgenic mice which encode and express fluorescent FRET biosensors, allowing for long-term monitoring of cell and tissue function in healthy and disease models (Thestrup et al. 2014). An alternative and quicker method is lenti- or retroviral transfection of cells in vitro and then transplantation into a living organism (Breart et al. 2008). The main advantage of transplanting genetically modified cells compared to the use of transgenic animal models is the ability to characterize the cells and the FRET biosensors in vitro prior to transplantation (Timpson et al. 2011). Janssen et al. applied IVM-FRET to evaluate tumor cell viability and mitosis during chemotherapy treatment. In this study, the mechanism of action of microtubule-targeting chemotherapeutics belonging to a class of pharmacological molecules called taxanes was compared in both in vitro and in vivo applications. In in vitro studies, the major effect of the treatment observed was induction of apoptosis by mitotic cell death which was previously hypothesized to occur in vivo. However, in vivo the majority of tumor cells died independently of mitotic defects, highlighting a different mechanism occurring in a systemic environment. To observe this difference, caspase-3, a crucial protease for apoptosis induction, was modified with cyan fluorescence protein (CFP) and yellow fluorescent protein (YFP). When caspase-3 is inactive, both fluorophore domains are in close proximity, and, as a result, CFP fluorescence emission decreases as it is transferred to YFP which in turn increases its emission leading to a low CFP to YFP emission ratio. When caspase-3 is activated, a specific motif is cleaved, separating CFP and YFP, rendering a high CFP to YFP ratio (Janssen et al. 2013). This approach illustrates the power of coupling FRET to IVM in order to evaluate biomolecular processes in vivo while comparing to in vitro results .
7 IVM Cell Tracking Studies
IVM can facilitate in vivo study of cellular and molecular dynamics that are limited by in vitro conditions. To fully understand the impact of diseases and drug development in cell biology, there is a need to ensure clinical translatable results and its effect on a complex organism. Tumor biology and immunology are two major fields in which the application of IVM can deepen the understanding of in vivo cellular and molecular processes. Here, labelling cells with exogenous fluorophores or by using endogenous fluorescent molecules provides a window to follow in real time specific cell types, tumor and metastasis development, responses to treatment, and reactions to bacterial/viral infection and cell migration (Fig. 6).

IVM cell tracking in vivo. (i) Mosaic image of myeloma cell division revealing resistance to chemotherapy drugs triggered by highlighted (white circles and arrow) and red cell activation. Scale bar: 500 μm (left) and 100 μm (middle and right). (Reproduced from Lawson et al. (2015) with permission from Nature Publishing Group) (ii) Time-lapse image of neutrophil attraction to site of laser damage in vivo. Scale bar: 25 μm. (Reproduced from Nobis et al. (2017) with permission from Elsevier). (iii) Imaging of cellular contacts between CD4þ T cells (red) and polyclonal CD8þ T cells (green) mediated by pulsed dendritic cell (blue/blue arrows) and medium-pulsed dendritic cells (yellow/yellow arrows). The top row of the right panel shows enlarger views, while the bottom row shows the movement (green) made by CD8þ T cells. Scale bar: left, 30 mm; right, 15 mm. (Reproduced from Castellino et al. (2006) with permission from Springer Nature) (iv) Brainbow expression in genetically engineered mice promote expression of (A) M-CFP in peripheral motor axons and multiple fluorescence proteins in (B) oculomotor nerve and (C) hippocampus (dentate gyrus). (Reproduced from Livet et al. (2007) with permission from Springer Nature)
7.1 Cell Division and Cell Death
Tumor cells are characterized by their ability to undergo and sustain proliferative behavior. Labelling multiple intracellular components can enable the monitoring of mitosis and apoptosis. One approach considers the use of fluorescence tags for histone H2b and α-tubulin, allowing the nuclear morphology to be observed. Orth et al. by using this method demonstrated that cell division occurs similarly in both in vivo and in vitro conditions; however, in vivo it appears to be a far less frequent event (Orth et al. 2011). Several approaches to evaluate cell death in IVM are available. One tactic is a readout based on a red cytoplasmic (CMTMR) and a blue nucleic (Hoechst33342) dye. In this approach, as a cell undergoes apoptosis, there is a measurable variation in the cytoplasm-to-nuclei fluorescent ratio due to the loss of soluble proteins from the cytoplasm and an increase of emission as the nuclei becomes apoptotic (Mempel et al. 2006). Other methods employ FRET fluorophores to follow the triggering of cell death mechanism such as caspase activity or p53 DNA damage. This is an interesting application of IVM to understand autophagy, apoptotic pathways, and response to drug treatments or to follow a certain pathology cycle (Breart et al. 2008; Spencer et al. 2009; Giesen et al. 2011; Lawson et al. 2015) (Fig. 6i). For specific tracking of dividing cells or cell death, IVM on its own, or coupled with FRET, provides multiple approaches that can be used by researchers to understand cell responses to an internal or external stimuli impacting cell fate .
7.2 Cell Migration
Cell mobility is an important feature of cancer invasiveness and immunological responses. This movement is dependent on adhesion, mechanosensing, and cytoskeletal remodeling. With IVM it is possible to image at defined time intervals and follow cell migratory paths, recruitment, velocity, and interaction with other tissue components (Sumen et al. 2004; Nobis et al. 2017). Applying this approach has added a deeper understanding of cell movement in vivo compared to limited in vitro studies. Interestingly, leukocyte movement in vivo relies primarily on the expansion of the actin network and without specific interaction within the extracellular environment; however the in vitro migration is dependent on additional factors such as the anchoring of the cellular membrane by integrins (Lammermann et al. 2008); depending on the cell type, movement is also triggered by chemokine receptor-ligand communications (Okada and Cyster 2007); cells can migrate as one cohesive group primarily due to their cell-cell junctions in a process known as “collective cell migration” (Alexander et al. 2008) or “cell jamming” (Haeger et al. 2014). Furthermore, this technique has been applied to follow both adaptive and innate mobilization of immune cells to inflammation areas (Fig. 6ii) (Auffray et al. 2007; Egen et al. 2011). Applying IVM to survey cell migration in vivo takes in account factors difficult to replicate in in vitro conditions. 3D environments, interaction with soluble factors, and cell and systemic signaling are continuously present in dynamic in vivo environments.
7.3 Cell Communication
Communication between different cells types is established either by chemical signaling, such as the release of cytokines or by physical interactions. Such signaling has an effect on cell proliferation, survival, mobility, and activation of effector functions (Mantovani and Dejana 1989; Goldring and Goldring 1990). The application of IVM to cell-cell interactions is still in its infancy. This is mostly due to the restriction of tracking whole cells, organelles, and macromolecular structures instead of their molecular activities related with production, secretion, and response to effector molecules. Therefore, there is a need to complement current IVM cell-communication experiments with ex vivo studies. Nonetheless, IVM has been used to showcase the complexity of cellular communication and their molecular microenvironment in vivo.
IVM has demonstrated interactions that boost or suppress adaptive immune responses. Here, symbiotic interactions between dendritic cells and CD4 helper T cells result in the release of chemokine signals that promote migration of CCR5+ CD8 T cells (Fig. 6iii) (Castellino et al. 2006). Another IVM cell tracking study has demonstrated that communication between dendritic cells and CD4 regulatory T cells actually suppresses CD4 helper T-cell actions (Tang et al. 2006). In tumor biology, cell communication has been found to have an important role in aggressive tumor microenvironments. It has been showed that perivascular macrophages interact with cancer cells and are hijacked to facilitate cancer cell migration and entry into lymphatic or blood vessels (Wyckoff et al. 2004). SHG coupled with IVM has found that migration of macrophages and tumor cells occurs primarily along collagen fibers which exemplifies the importance of extracellular matrix alignment and density to invasive tumor microenvironment (Roussos et al. 2011). In addition, premetastatic circulating tumor cells have been showed to halt at vascular branch points where they infiltrate the surrounding tissue and promote perivascular growth by angiogenesis or by vascular co-option (Kienast et al. 2010).
In addition to immunological responses, there is a need to continue developing technologies that deepen the ability to probe further into cell-cell communication in a way that surveys interactions at the molecular level .
7.4 Cell Fate
To identify the original cell population in vivo, reference points which allow identification of these cells overtime are needed (Bins et al. 2007). Due to the fast and ever-changing environment of a tissue formation/remodeling site, the use of reference points (e.g., fluorescent tags) that endure long enough to obtain an in-depth analysis of cell division and differentiation is restricted. Therefore, one major limitation of IVM is the ability to track the same cells over long periods of time (weeks) in different regions that the area originally analyzed. Some approaches have been emerging which may overcome this restraint. Two main methodologies have been development and applied which obtain remarkable results: photoswitchable proteins and color-engineered genetic constructs.
Photoactivatable fluorescent proteins (PAFPs) are molecules capable of changing their spectral properties in response to light excitation with a specific wavelength and intensity. Some PAFPs can undergo photoactivation where they convert from a low (dark) to a bright fluorescent state, whereas photoswitchable PAFPs change their emission wavelength depending on the value of the excitation wavelength (Lukyanov et al. 2005). To track cell fate and the development of an initial cell population, one approach is to use genetically engineered mice that express PAFPs. In a study performed by Kendrin et al., cells were modified to encode a photoswitchable protein termed Dendra2 (Kedrin et al. 2008). This protein possesses an emission spectrum similar to GFP before undergoing a photoswitching process. After exposure to one-photon excitation wavelength (~405 nm), it undergoes an irreversible red shift (>150 nm) in both emission and excitation wavelength (Gurskaya et al. 2006). After photoswitching, the red fluorescence emission of Dendra2 increases, and after 5 days of the procedure, the red fluorescence intensity is still observable. Using this approach, heterogeneity of tumor microenvironments was observed. These differences were promoted by the presence of blood vessels within the same tumor which results in varying supplies of nutrients affecting the rate of cell invasion and tumor aggressiveness (Kedrin et al. 2008). Although this has been shown previously, the quantitative analysis of this variability was not compatible with previous techniques (Wyckoff et al. 2007).
The use of color-engineered genetic construct was first introduced by Livet et al. (Fig. 6iv) (Livet et al. 2007). In this study, genetic constructs were introduced into cells that would label clonal populations with distinct colors. This technology denoted Brainbow was based on a recombination system that determines the expression of multiple copies of constructs which code for different fluorescent protein. The process creates a mosaic with up to 90 colors. In this work, Brainbow was used to label neurons and follow their interactions and connectivity over time in vivo. Another variation of these genetic constructs has also been developed (Weber et al. 2011). To follow cell fate in vivo is still elusive; nonetheless, IVM results with PAFPs or genetic cassettes may be able to complement another genetic-based study that use specific molecular identifiers to track cell fate.
8 Conclusions
When considering how recent innovations in the microscopic fields have enabled invasive and noninvasive cell tracking, several common themes become apparent. In the case of multiphoton microscopy, it can increase the depth of imaging in a tissue and allow one to excite endogenous fluorophores without invasive manipulation. SHG can quantify collagen alignment and density, giving an added biomechanical inference to biological dynamics. FLIM captures photons emitted by a sample after laser excitation, resulting in both intensity measurements and fluorescence lifetimes, which can map the microenvironment of light-emitting molecules and cellular metabolism. Last but not least, IVM opens the possibility of in vivo cell and tissue tracking. Here, systemic responses and cell-cell interactions can be evaluated and when coupled with the techniques previously described can benchmark in vivo with in vitro studies revealing compelling and significant outcomes. This renaissance provides the identification of single-cell features, cell-cell communication, and cellular responses to dynamic microenvironments and internal/external stimuli both in vitro and in vivo. This is crucial for a deeper understanding of molecular and cellular biology that is translatable to immunology, cancer research, metabolism, and tissue engineering fields. With this, new technologies and therapeutic developments may arise targeting specific pathways by stimulation or inhibition in a cellular or molecular context. Microscopy is a steadfast technique which has improved and broadened due to several technological advances in the optics and physics fields. “Seeing is believing” is still one of the main arguments that can exert confirmation of a scientific hypothesis. Therefore, interdisciplinary development of imaging technologies will undoubtedly promote a wider comprehension of in vitro and in vivo cell biology with the possibility to modulate cell behavior using tissue engineering and regenerative medicine approaches.
References
Abraham T, Hogg J (2010) Extracellular matrix remodeling of lung alveolar walls in three dimensional space identified using second harmonic generation and multiphoton excitation fluorescence. J Struct Biol 171(2):189–196
Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, Cheng Q, Ng LG, Cavanagh LL, von Andrian UH, Hickey MJ, Firth N, Weninger W (2014) Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol 15(1):45–53
Alam SR, Wallrabe H, Svindrych Z, Chaudhary AK, Christopher KG, Chandra D, Periasamy A (2017) Investigation of mitochondrial metabolic response to doxorubicin in prostate cancer cells: an NADH, FAD and tryptophan FLIM assay. Sci Rep 7(1):10451
Aldridge J, Pye EK (1976) Cell density dependence of oscillatory metabolism. Nature 259(5545):670–671
Alexander S, Koehl GE, Hirschberg M, Geissler EK, Friedl P (2008) Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. Histochem Cell Biol 130(6):1147–1154
Alfonso-Garcia A, Smith TD, Datta R, Luu TU, Gratton E, Potma EO, Liu WF (2016) Label-free identification of macrophage phenotype by fluorescence lifetime imaging microscopy. J Biomed Opt 21(4):46005
Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F (2007) Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317(5838):666–670
Baird GS, Zacharias DA, Tsien RY (1999) Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci U S A 96(20):11241–11246
Bardell D (2004) The biologists’ forum: the invention of the microscope. Bios 75(2):78–84
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115(2):209–218
Bauer DE, Harris MH, Plas DR, Lum JJ, Hammerman PS, Rathmell JC, Riley JL, Thompson CB (2004) Cytokine stimulation of aerobic glycolysis in hematopoietic cells exceeds proliferative demand. FASEB J 18(11):1303–1305
Bedossa P, Poynard T (1996) An algorithm for the grading of activity in chronic hepatitis C. The METAVIR cooperative study group. Hepatology 24(2):289–293
Berg J, Hung YP, Yellen G (2009) A genetically encoded fluorescent reporter of ATP: ADP ratio. Nat Methods 6(2):161–166
Bins AD, van Rheenen J, Jalink K, Halstead JR, Divecha N, Spencer DM, Haanen JB, Schumacher TN (2007) Intravital imaging of fluorescent markers and FRET probes by DNA tattooing. BMC Biotechnol 7:2
Blacker TS, Duchen MR (2016) Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med 100:53–65
Blacker TS, Mann ZF, Gale JE, Ziegler M, Bain AJ, Szabadkai G, Duchen MR (2014) Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun 5:3936
Boddupalli A, Bratlie KM (2015) Multimodal imaging of harmonophores and application of high content imaging for early cancer detection. Mater Discov 1:10–20
Boddupalli A, Bratlie KM (2019) Second harmonic generation microscopy of collagen organization in tunable, environmentally responsive alginate hydrogels. Biomater Sci 7(3):1188–1199
Boens N, Qin W, Basaric N, Hofkens J, Ameloot M, Pouget J, Lefevre JP, Valeur B, Gratton E, vande Ven M, Silva ND Jr, Engelborghs Y, Willaert K, Sillen A, Rumbles G, Phillips D, Visser AJ, van Hoek A, Lakowicz JR, Malak H, Gryczynski I, Szabo AG, Krajcarski DT, Tamai N, Miura A (2007) Fluorescence lifetime standards for time and frequency domain fluorescence spectroscopy. Anal Chem 79(5):2137–2149
Botchway SW, Parker AW, Bisby RH, Crisostomo AG (2008) Real-time cellular uptake of serotonin using fluorescence lifetime imaging with two-photon excitation. Microsc Res Tech 71(4):267–273
Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 435(2):297–312
Breart B, Lemaitre F, Celli S, Bousso P (2008) Two-photon imaging of intratumoral CD8+ T cell cytotoxic activity during adoptive T cell therapy in mice. J Clin Invest 118(4):1390–1397
Brown E, McKee T, diTomaso E, Pluen A, Seed B, Boucher Y, Jain RK (2003) Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat Med 9(6):796–800
Brown CM, Rivera DR, Pavlova I, Ouzounov DG, Williams WO, Mohanan S, Webb WW, Xu C (2012) In vivo imaging of unstained tissues using a compact and flexible multiphoton microendoscope. J Biomed Opt 17(4):040505
Bullen A (2008) Microscopic imaging techniques for drug discovery. Nat Rev Drug Discov 7(1):54–67
Buranachai C, Kamiyama D, Chiba A, Williams BD, Clegg RM (2008) Rapid frequency-domain FLIM spinning disk confocal microscope: lifetime resolution, image improvement and wavelet analysis. J Fluoresc 18(5):929–942
Buurman EP, Sanders R, Draaijer A, Gerritsen HC, van Veen JJF, Houpt PM, Levine YK (1992) Fluorescence lifetime imaging using a confocal laser scanning microscope. Scanning 14(3):155–159
Caetano-Lopes J, Nery AM, Henriques R, Canhão H, Duarte J, Amaral PM, Vale M, Moura RA, Pereira PA, Weinmann P, Abdulghani S, Souto-Carneiro M, Rego P, Monteiro J, Sakagushi S, Queiroz MV, Konttinen YT, Graça L, Vaz MF, Fonseca JE (2009) Chronic arthritis directly induces quantitative and qualitative bone disturbances leading to compromised biomechanical properties. Clin Exp Rheumatol 27(3):475–482
Campagnola P (2011) Second harmonic generation imaging microscopy: applications to diseases diagnostics. Anal Chem 83(9):3224–3231
Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN (2006) Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 440(7086):890–895
Cavanagh LL, Weninger W (2008) Dendritic cell behaviour in vivo: lessons learned from intravital two-photon microscopy. Immunol Cell Biol 86(5):428–438
Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ (2012) Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc 7(4):654–669
Chunqiang L, Pitsillides C, Runnels JM, Côté D, Lin CP (2010) Multiphoton microscopy of live tissues with ultraviolet autofluorescence. IEEE J Sel Topics Quantum Electron 16(3):516–523
Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, Boudreau A, Mroue R, Corson L, O’Brien T, Qing J, Sampath D, Merchant M, Yauch R, Manning G, Settleman J, Hatzivassiliou G, Evangelista M (2015) Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A 112(32):E4410–E4417
Dang CV (2010) Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle 9(19):3884–3886
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20
Dempsey WP, Fraser SE, Pantazis P (2012) SHG nanoprobes: advancing harmonic imaging in biology. BioEssays 34(5):351–360
Diffey BL (1991) Solar ultraviolet radiation effects on biological systems. Phys Med Biol 36(3):299–328
Digman MA, Caiolfa VR, Zamai M, Gratton E (2008) The phasor approach to fluorescence lifetime imaging analysis. Biophys J 94(2):L14–L16
Dimitrow E, Riemann I, Ehlers A, Koehler MJ, Norgauer J, Elsner P, Konig K, Kaatz M (2009) Spectral fluorescence lifetime detection and selective melanin imaging by multiphoton laser tomography for melanoma diagnosis. Exp Dermatol 18(6):509–515
Dmitriev RI (2017) Multi-parametric live cell microscopy of 3D tissue models. Springer, Cham
Dmitriev RI, Papkovsky DB (2015) Intracellular probes for imaging oxygen concentration: how good are they? Methods Appl Fluoresc 3(3):034001
Dmitriev RI, Papkovsky DB (2018) Quenched-phosphorescence detection of molecular oxygen: applications in life sciences. Royal Society of Chemistry, Cambridge
Dmitriev RI, Zhdanov AV, Nolan YM, Papkovsky DB (2013) Imaging of neurosphere oxygenation with phosphorescent probes. Biomaterials 34(37):9307–9317
Dmitriev RI, Kondrashina AV, Koren K, Klimant I, Zhdanov AV, Pakan JM, McDermott KW, Papkovsky DB (2014) Small molecule phosphorescent probes for O 2 imaging in 3D tissue models. Biomater Sci 2(6):853–866
Dowling K, Dayel MJ, Lever MJ, French PM, Hares JD, Dymoke-Bradshaw AK (1998) Fluorescence lifetime imaging with picosecond resolution for biomedical applications. Opt Lett 23(10):810–812
Drury JL, Mooney DJ (2003) Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials 24(24):4337–4351
Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN (2008) Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity 28(2):271–284
Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN (2011) Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity 34(5):807–819
Ehlers A, Riemann I, Stark M, Konig K (2007) Multiphoton fluorescence lifetime imaging of human hair. Microsc Res Tech 70(2):154–161
Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M, Periasamy A (2003) Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods 29(1):58–73
Emiliani V, Sanvitto D, Tramier M, Piolot T, Petrasek Z, Kemnitz K, Durieux C, Coppey-Moisan M (2003) Low-intensity two-dimensional imaging of fluorescence lifetimes in living cells. Appl Phys Lett 83(12):2471–2473
Folbergrová J, Ljunggren B, Norberg K, Siesjö BK (1974) Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res 80(2):265–279
Folmes CD, Dzeja PP, Nelson TJ, Terzic A (2012) Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11(5):596–606
Funane T, Hou SS, Zoltowska KM, van Veluw SJ, Berezovska O, Kumar ATN, Bacskai BJ (2018) Selective plane illumination microscopy (SPIM) with time-domain fluorescence lifetime imaging microscopy (FLIM) for volumetric measurement of cleared mouse brain samples. Rev Sci Instrum 89(5):053705
Gadella TW Jr, Arndt-Jovin DJ, Jovin TM (1994) Visualization of lipid-receptor interactions on single cells by time-resolved imaging fluorescence microscopy. J Fluoresc 4(4):295–298
Gailhouste L, Le Grand Y, Odin C, Guyader D, Turlin B, Ezan F, Desille Y, Guilbert T, Bessard A, Fremin C, Theret N, Baffet G (2010) Fibrillar collagen scoring by second harmonic microscopy: a new tool in the assessment of liver fibrosis. J Hepatol 52(3):398–406
Ghukasyan VV, Kao F-J (2009) Monitoring cellular metabolism with fluorescence lifetime of reduced nicotinamide adenine dinucleotide. J Phys Chem C 113(27):11532–11540
Giesen U, Langner F, Mielke C, Mosconi M, Dirks WG (2011) Online imaging of initial DNA damages at the PTB microbeam. Radiat Prot Dosim 143(2–4):349–352
Goldring MB, Goldring SR (1990) Skeletal tissue response to cytokines. Clin Orthop Relat Res 258:245–276
Göppert-Mayer M (1931) Über Elementarakte mit zwei Quantensprüngen. Ann Phys 401(3):273–294
Gratton E, Breusegem S, Sutin J, Ruan Q, Barry N (2003) Fluorescence lifetime imaging for the two-photon microscope: time-domain and frequency-domain methods. J Biomed Opt 8(3):381–390
Gurcan MN, Boucheron LE, Can A, Madabhushi A, Rajpoot NM, Yener B (2009) Histopathological image analysis: a review. IEEE Rev Biomed Eng 2:147–171
Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA (2006) Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol 24(4):461–465
Haeger A, Krause M, Wolf K, Friedl P (2014) Cell jamming: collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochim Biophys Acta 1840(8):2386–2395
Hato T, Winfree S, Day R, Sandoval RM, Molitoris BA, Yoder MC, Wiggins RC, Zheng Y, Dunn KW, Dagher PC (2017) Two-photon intravital fluorescence lifetime imaging of the kidney reveals cell-type specific metabolic signatures. J Am Soc Nephrol 28(8):2420–2430
Helmchen F, Denk W (2005) Deep tissue two-photon microscopy. Nat Methods 2(12):932–940
Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, Barber GN, Bennink JR, Yewdell JW (2008) Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol 9(2):155–165
Hooke R (1665) Micrographia: or some physiological descriptions of minute bodies made by magnifying glasses. With Observations and Inquiries Thereupon. Great Britain, The Royal Society.
Huang S, Heikal AA, Webb WW (2002) Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J 82(5):2811–2825
Hung YP, Albeck JG, Tantama M, Yellen G (2011) Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab 14(4):545–554
Ishikawa-Ankerhold HC, Ankerhold R, Drummen GP (2012) Advanced fluorescence microscopy techniques–FRAP, FLIP, FLAP, FRET and FLIM. Molecules 17(4):4047–4132
Jain RK, Munn LL, Fukumura D (2002) Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer 2(4):266–276
Janssen A, Beerling E, Medema R, van Rheenen J (2013) Intravital FRET imaging of tumor cell viability and mitosis during chemotherapy. PLoS One 8(5):e64029
Jenkins J, Dmitriev RI, Morten K, McDermott KW, Papkovsky DB (2015) Oxygen-sensing scaffolds for 3-dimensional cell and tissue culture. Acta Biomater 16:126–135
Jenkins J, Borisov SM, Papkovsky DB, Dmitriev RI (2016) Sulforhodamine nanothermometer for multiparametric fluorescence lifetime imaging microscopy. Anal Chem 88(21):10566–10572
Jenne CN, Wong CH, Petri B, Kubes P (2011) The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PLoS One 6(9):e25109
Jyothikumar V, Sun Y, Periasamy A (2013) Investigation of tryptophan-NADH interactions in live human cells using three-photon fluorescence lifetime imaging and Forster resonance energy transfer microscopy. J Biomed Opt 18(6):060501
Kaiser W, Garrett CGB (1961) Two-photon excitation in CaF2:Eu2+. Phys Rev Lett 7(6):229–231
Kalinina S, Breymayer J, Schäfer P, Calzia E, Shcheslavskiy V, Becker W, Rück A (2016) Correlative NAD (P) H-FLIM and oxygen sensing-PLIM for metabolic mapping. J Biophotonics 9(8):800–811
Kautsky H, Hirsch A (1935) Nachweis geringster. Sauerstoffmengen durch Phosphoreszenztilgung 222(2):126–134
Kedrin D, Gligorijevic B, Wyckoff J, Verkhusha VV, Condeelis J, Segall JE, van Rheenen J (2008) Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods 5(12):1019–1021
Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, Winkler F (2010) Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16(1):116–122
Kierdaszuk B, Gryczynski I, Modrak-Wojcik A, Bzowska A, Shugar D, Lakowicz JR (1995) Fluorescence of tyrosine and tryptophan in proteins using one- and two-photon excitation. Photochem Photobiol 61(4):319–324
Kondrashina AV, Dmitriev RI, Borisov SM, Klimant I, O'Brien I, Nolan YM, Zhdanov AV, Papkovsky DB (2012) A phosphorescent nanoparticle-based probe for sensing and imaging of (intra)cellular oxygen in multiple detection modalities. Adv Funct Mater 22(23):4931–4939
Konig K, Schenke-Layland K, Riemann I, Stock UA (2005) Multiphoton autofluorescence imaging of intratissue elastic fibers. Biomaterials 26(5):495–500
Lakowicz JR (1999) Principles of fluorescence spectroscopy. Kluwer academic, New York
Lakowicz JR, Maliwal BP (1983) Oxygen quenching and fluorescence depolarization of tyrosine residues in proteins. J Biol Chem 258(8):4794–4801
Lakowicz JR, Szmacinski H, Nowaczyk K, Johnson ML (1992) Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci U S A 89(4):1271–1275
Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, Keller M, Forster R, Critchley DR, Fassler R, Sixt M (2008) Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453(7191):51–55
Lattouf R, Younes R, Lutomski D, Naaman N, Godeau G, Senni K, Changotade S (2014) Picrosirius red staining: a useful tool to appraise collagen networks in normal and pathological tissues. J Histochem Cytochem 62(10):751–758
Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, Kaplan W, Paton-Hough J, Fellows C, Pettitt JA, Neil Dear T, Van Valckenborgh E, Baldock PA, Rogers MJ, Eaton CL, Vanderkerken K, Pettit AR, Quinn JM, Zannettino AC, Phan TG, Croucher PI (2015) Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun 6:8983
Leeuwenhoek Av (1682) Observationes microscopicae Antonii Lewenhoeck, circa particulas liquorum globosa et animalia. Leipzig, Acta Eruditorum.
Lilledahl MB, Pierce DM, Ricken T, Holzapfel GA, Davies Cde L (2011) Structural analysis of articular cartilage using multiphoton microscopy: input for biomechanical modeling. IEEE Trans Med Imaging 30(9):1635–1648
Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, Lichtman JW (2007) Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450(7166):56–62
Lleres D, James J, Swift S, Norman DG, Lamond AI (2009) Quantitative analysis of chromatin compaction in living cells using FLIM-FRET. J Cell Biol 187(4):481–496
Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV (2005) Innovation: photoactivatable fluorescent proteins. Nat Rev Mol Cell Biol 6(11):885–891
Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27:441–464
Lutz V, Sattler M, Gallinat S, Wenck H, Poertner R, Fischer F (2012) Impact of collagen crosslinking on the second harmonic generation signal and the fluorescence lifetime of collagen autofluorescence. Skin Res Technol 18(2):168–179
Mantovani A, Dejana E (1989) Cytokines as communication signals between leukocytes and endothelial cells. Immunol Today 10(11):370–375
Mariani MM, Lampen P, Popp J, Wood BR, Deckert V (2009) Impact of fixation on in vitro cell culture lines monitored with Raman spectroscopy. Analyst 134(6):1154–1161
Marques PE, Oliveira AG, Chang L, Paula-Neto HA, Menezes GB (2015) Understanding liver immunology using intravital microscopy. J Hepatol 63(3):733–742
Masia R, McCarty WJ, Lahmann C, Luther J, Chung RT, Yarmush ML, Yellen G (2018) Live cell imaging of cytosolic NADH/NAD(+) ratio in hepatocytes and liver slices. Am J Physiol Gastrointest Liver Physiol 314(1):G97–G108
Masters BR, So PT, Gratton E (1997) Multiphoton excitation fluorescence microscopy and spectroscopy of in vivo human skin. Biophys J 72(6):2405–2412
McKeehan W (1982) Glycolysis, glutaminolysis and cell proliferation. Cell Biol Int Rep 6(7):635–650
Mempel TR, Scimone ML, Mora JR, von Andrian UH (2004) In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr Opin Immunol 16(4):406–417
Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, von Andrian UH (2006) Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity 25(1):129–141
Misgeld T, Kerschensteiner M (2006) In vivo imaging of the diseased nervous system. Nat Rev Neurosci 7(6):449–463
Mitchell CA, Poland SP, Seyforth J, Nedbal J, Gelot T, Huq T, Holst G, Knight RD, Ameer-Beg SM (2017) Functional in vivo imaging using fluorescence lifetime light-sheet microscopy. Opt Lett 42(7):1269–1272
Molitoris BA, Sandoval RM (2005) Intravital multiphoton microscopy of dynamic renal processes. Am J Physiol Renal Physiol 288(6):F1084–F1089
Monaghan MG, Kroll S, Brucker SY, Schenke-Layland K (2016) Enabling multiphoton and second harmonic generation imaging in paraffin-embedded and histologically stained sections. Tissue Eng Part C Methods 22(6):517–523
Mongeon R, Venkatachalam V, Yellen G (2016) Cytosolic NADH-NAD+ redox visualized in brain slices by two-photon fluorescence lifetime biosensor imaging. Antioxid Redox Signal 25(10):553–563
Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 292(17):7189–7207
Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK (2000) Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res 60(9):2497–2503
Niesner R, Peker B, Schlusche P, Gericke KH (2004) Noniterative biexponential fluorescence lifetime imaging in the investigation of cellular metabolism by means of NAD(P)H autofluorescence. ChemPhysChem 5(8):1141–1149
Nobis M, Herrmann D, Warren SC, Kadir S, Leung W, Killen M, Magenau A, Stevenson D, Lucas MC, Reischmann N, Vennin C, Conway JRW, Boulghourjian A, Zaratzian A, Law AM, Gallego-Ortega D, Ormandy CJ, Walters SN, Grey ST, Bailey J, Chtanova T, Quinn JMW, Baldock PA, Croucher PI, Schwarz JP, Mrowinska A, Zhang L, Herzog H, Masedunskas A, Hardeman EC, Gunning PW, Del Monte-Nieto G, Harvey RP, Samuel MS, Pajic M, McGhee EJ, Johnsson AE, Sansom OJ, Welch HCE, Morton JP, Strathdee D, Anderson KI, Timpson P (2017) A RhoA-FRET biosensor mouse for intravital imaging in normal tissue homeostasis and disease contexts. Cell Rep 21(1):274–288
Nomoto T, Matsumoto Y, Miyata K, Oba M, Fukushima S, Nishiyama N, Yamasoba T, Kataoka K (2011) In situ quantitative monitoring of polyplexes and polyplex micelles in the blood circulation using intravital real-time confocal laser scanning microscopy. J Control Release 151(2):104–109
O’Donnell N, Dmitriev RI (2017) Three-dimensional tissue models and available probes for multi-parametric live cell microscopy: a brief overview. In: Multi-parametric live cell microscopy of 3D tissue models. Springer, Cham, pp 49–67
O’Donnell N, Okkelman IA, Timashev P, Gromovykh TI, Papkovsky DB, Dmitriev RI (2018) Cellulose-based scaffolds for fluorescence lifetime imaging-assisted tissue engineering. Acta Biomater 80:85–96
O’Neill LA, Kishton RJ, Rathmell J (2016) A guide to immunometabolism for immunologists. Nat Rev Immunol 16(9):553–565
Ochoa LF, Kholodnykh A, Villarreal P, Tian B, Pal R, Freiberg AN, Brasier AR, Motamedi M, Vargas G (2018) Imaging of murine whole lung fibrosis by large scale 3D microscopy aided by tissue optical clearing. Sci Rep 8(1):13348
Okada T, Cyster JG (2007) CC chemokine receptor 7 contributes to Gi-dependent T cell motility in the lymph node. J Immunol 178(5):2973–2978
Okkelman IA, Dmitriev RI, Foley T, Papkovsky DB (2016) Use of fluorescence lifetime imaging microscopy (FLIM) as a timer of cell cycle S phase. PLoS One 11(12):e0167385
Okkelman IA, Foley T, Papkovsky DB, Dmitriev RI (2017) Live cell imaging of mouse intestinal organoids reveals heterogeneity in their oxygenation. Biomaterials 146:86–96
Okkelman IA, Neto N, Papkovsky DB, Monaghan MG, Dmitriev RI (2019a) A deeper understanding of intestinal organoid metabolism revealed by combining fluorescence lifetime imaging microscopy (FLIM) and extracellular flux analyses. Redox Biol 30:101420
Okkelman IA, Papkovsky DB, Dmitriev RI (2019b) Estimation of the mitochondrial membrane potential using fluorescence lifetime imaging microscopy. Cytometry A. https://doi.org/10.1002/cyto.a.23886
Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ (2011) Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res 71(13):4608–4616
Ostrander JH, McMahon CM, Lem S, Millon SR, Brown JQ, Seewaldt VL, Ramanujam N (2010) Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status. Cancer Res 70(11):4759–4766
Pantazis P, Maloney J, Wu D, Fraser SE (2010) Second harmonic generating (SHG) nanoprobes for in vivo imaging. Proc Natl Acad Sci U S A 107(33):14535–14540
Panyi G, Bagdany M, Bodnar A, Vamosi G, Szentesi G, Jenei A, Matyus L, Varga S, Waldmann TA, Gaspar R, Damjanovich S (2003) Colocalization and nonrandom distribution of Kv1.3 potassium channels and CD3 molecules in the plasma membrane of human T lymphocytes. Proc Natl Acad Sci U S A 100(5):2592–2597
Papkovsky DB, Dmitriev RI (2013) Biological detection by optical oxygen sensing. Chem Soc Rev 42(22):8700–8732
Papkovsky DB, Dmitriev RI (2018) Imaging of oxygen and hypoxia in cell and tissue samples. Cell Mol Life Sci 75(16):2963–2980
Pena AM, Boulesteix T, Dartigalongue T, Schanne-Klein MC (2005) Chiroptical effects in the second harmonic signal of collagens I and IV. J Am Chem Soc 127(29):10314–10322
Phillips D, Drake RC, Oconnor DV, Christensen RL (1985) Time correlated single-photon counting (Tcspc) using laser excitation. Anal Instrum 14(3–4):267–292
Plotnikov SV, Millard AC, Campagnola PJ, Mohler WA (2006) Characterization of the myosin-based source for second-harmonic generation from muscle sarcomeres. Biophys J 90(2):693–703
Pollak N, Dölle C, Ziegler M (2007) The power to reduce: pyridine nucleotides – small molecules with a multitude of functions. Biochem J 402(2):205–218
Prockop DJ, Fertala A (1998) The collagen fibril: the almost crystalline structure. J Struct Biol 122(1–2):111–118
Progatzky F, Dallman MJ, Lo Celso C (2013) From seeing to believing: labelling strategies for in vivo cell-tracking experiments. Interface Focus 3(3):20130001
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ (2006) Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med 4(1):38–54
Provenzano PP, Eliceiri KW, Keely PJ (2009) Multiphoton microscopy and fluorescence lifetime imaging microscopy (FLIM) to monitor metastasis and the tumor microenvironment. Clin Exp Metastasis 26(4):357–370
Pugh TD, Conklin MW, Evans TD, Polewski MA, Barbian HJ, Pass R, Anderson BD, Colman RJ, Eliceiri KW, Keely PJ, Weindruch R, Beasley TM, Anderson RM (2013) A shift in energy metabolism anticipates the onset of sarcopenia in rhesus monkeys. Aging Cell 12(4):672–681
Puxkandl R, Zizak I, Paris O, Keckes J, Tesch W, Bernstorff S, Purslow P, Fratzl P (2002) Viscoelastic properties of collagen: synchrotron radiation investigations and structural model. Philos Trans R Soc Lond Ser B Biol Sci 357(1418):191–197
Quinn KP, Bellas E, Fourligas N, Lee K, Kaplan DL, Georgakoudi I (2012) Characterization of metabolic changes associated with the functional development of 3D engineered tissues by non-invasive, dynamic measurement of individual cell redox ratios. Biomaterials 33(21):5341–5348
Radbruch H, Bremer D, Mothes R, Gunther R, Rinnenthal JL, Pohlan J, Ulbricht C, Hauser AE, Niesner R (2015) Intravital FRET: probing cellular and tissue function in vivo. Int J Mol Sci 16(5):11713–11727
Ranjit S, Dobrinskikh E, Montford J, Dvornikov A, Lehman A, Orlicky DJ, Nemenoff R, Gratton E, Levi M, Furgeson S (2016) Label-free fluorescence lifetime and second harmonic generation imaging microscopy improves quantification of experimental renal fibrosis. Kidney Int 90(5):1123–1128
Rinnenthal JL, Bornchen C, Radbruch H, Andresen V, Mossakowski A, Siffrin V, Seelemann T, Spiecker H, Moll I, Herz J, Hauser AE, Zipp F, Behne MJ, Niesner R (2013) Parallelized TCSPC for dynamic intravital fluorescence lifetime imaging: quantifying neuronal dysfunction in neuroinflammation. PLoS One 8(4):e60100
Robert Lanza RLAJV (2014) Principles of tissue engineering. Elsevier, London
Roussakis E, Li Z, Nichols AJ, Evans CLJACIE (2015) Oxygen-sensing methods in biomedicine from the macroscale to the microscale. Angew Chem Int Ed 54(29):8340–8362
Roussakis E, Ortines RV, Pinsker BL, Mooers CT, Evans CL, Miller LS, Calderón-Colón XJB (2019) Theranostic biocomposite scaffold membrane. Biomaterials 212:17–27
Roussos ET, Condeelis JS, Patsialou A (2011) Chemotaxis in cancer. Nat Rev Cancer 11(8):573–587
Sakadžić S, Roussakis E, Yaseen MA, Mandeville ET, Srinivasan VJ, Arai K, Ruvinskaya S, Devor A, Lo EH, Vinogradov SA (2010) Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nat Methods 7(9):755
Saraste M (1999) Oxidative phosphorylation at the fin de siecle. Science 283(5407):1488–1493
Scaduto RC, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76(1):469–477
Schaefer PM, Kalinina S, Rueck A, von Arnim CAF, von Einem B (2019) NADH autofluorescence-a marker on its way to boost bioenergetic research. Cytometry A 95(1):34–46
Schenke-Layland K, Madershahian N, Riemann I, Starcher B, Halbhuber KJ, Konig K, Stock UA (2006) Impact of cryopreservation on extracellular matrix structures of heart valve leaflets. Ann Thorac Surg 81(3):918–926
Schilling K, El Khatib M, Plunkett S, Xue J, Xia Y, Vinogradov SA, Brown E, Zhang X (2019) Electrospun fiber mesh for high-resolution measurements of oxygen tension in cranial bone defect repair. ACS Appl Mater Interfaces 11:33548–33558
Schneider PC, Clegg RM (1997) Rapid acquisition, analysis, and display of fluorescence lifetime-resolved images for real-time applications. Rev Sci Instrum 68(11):4107–4119
Sekar RB, Periasamy A (2003) Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol 160(5):629–633
Sereysky JB, Andarawis-Puri N, Jepsen KJ, Flatow EL (2012) Structural and mechanical effects of in vivo fatigue damage induction on murine tendon. J Orthop Res 30(6):965–972
Shear JB, Xu C, Webb WW (1997) Multiphoton-excited visible emission by serotonin solutions. Photochem Photobiol 65(6):931–936
Shen YR (1984) The principles of nonlinear optics. Wiley, New York
Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, Kim D, Le A, Yellen G, Albeck JG, Locasale JW (2014) Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. eLife 3:e03342. https://doi.org/10.7554/eLife.03342
Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, Ramanujam N (2007) In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci U S A 104(49):19494–19499
Sommer G, Schriefl AJ, Andra M, Sacherer M, Viertler C, Wolinski H, Holzapfel GA (2015) Biomechanical properties and microstructure of human ventricular myocardium. Acta Biomater 24:172–192
Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK (2009) Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459(7245):428–432
Squire A, Bastiaens PI (1999) Three dimensional image restoration in fluorescence lifetime imaging microscopy. J Microsc 193(Pt 1):36–49
Stanciu SG, Xu S, Peng Q, Yan J, Stanciu GA, Welsch RE, So PT, Csucs G, Yu H (2014) Experimenting liver fibrosis diagnostic by two photon excitation microscopy and bag-of-features image classification. Sci Rep 4:4636
Stoller P, Celliers PM, Reiser KM, Rubenchik AM (2003) Quantitative second-harmonic generation microscopy in collagen. Appl Opt 42(25):5209–5219
Stoscheck CM (1990) Quantitation of protein. Methods Enzymol 182:50–68
Strupler M, Pena AM, Hernest M, Tharaux PL, Martin JL, Beaurepaire E, Schanne-Klein MC (2007) Second harmonic imaging and scoring of collagen in fibrotic tissues. Opt Express 15(7):4054–4065
Sumen C, Mempel TR, Mazo IB, von Andrian UH (2004) Intravital microscopy: visualizing immunity in context. Immunity 21(3):315–329
Sytsma J, Vroom JM, de Grauw CJ, Gerritsen HC (2008) Time-gated fluorescence lifetime imaging and microvolume spectroscopy using two-photon excitation. J Microsc 191(1):39–51
Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA (2006) Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol 7(1):83–92
Tantama M, Hung YP, Yellen G (2011) Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J Am Chem Soc 133(26):10034–10037
Thai M, Graham NA, Braas D, Nehil M, Komisopoulou E, Kurdistani SK, McCormick F, Graeber TG, Christofk HR (2014) Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab 19(4):694–701
Thestrup T, Litzlbauer J, Bartholomaus I, Mues M, Russo L, Dana H, Kovalchuk Y, Liang Y, Kalamakis G, Laukat Y, Becker S, Witte G, Geiger A, Allen T, Rome LC, Chen TW, Kim DS, Garaschuk O, Griesinger C, Griesbeck O (2014) Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat Methods 11(2):175–182
Thorling CA, Liu X, Burczynski FJ, Fletcher LM, Roberts MS, Sanchez WY (2013) Intravital multiphoton microscopy can model uptake and excretion of fluorescein in hepatic ischemia-reperfusion injury. J Biomed Opt 18(10):101306
Timpson P, McGhee EJ, Anderson KI (2011) Imaging molecular dynamics in vivo-from cell biology to animal models. J Cell Sci 124(Pt 17):2877–2890
Valentini G, D’Andrea C, Comelli D, Pifferi A, Taroni P, Torricelli A, Cubeddu R, Battaglia C, Consolandi C, Salani G, Rossi-Bernardi L, De Bellis G (2000) Time-resolved DNA-microarray reading by an intensified CCD for ultimate sensitivity. Opt Lett 25(22):1648–1650
van Munster E.B., Gadella T.W.J. (2005) Fluorescence Lifetime Imaging Microscopy (FLIM). In: Rietdorf J. (eds) Microscopy Techniques. Advances in Biochemical Engineering, vol 95. Springer, Berlin, Heidelberg. https://doi.org/10.1007/b102213
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033
Venkatasubramanian RT, Wolkers WF, Shenoi MM, Barocas VH, Lafontaine D, Soule CL, Iaizzo PA, Bischof JC (2010) Freeze-thaw induced biomechanical changes in arteries: role of collagen matrix and smooth muscle cells. Ann Biomed Eng 38(3):694–706
Vielreicher M, Schurmann S, Detsch R, Schmidt MA, Buttgereit A, Boccaccini A, Friedrich O (2013) Taking a deep look: modern microscopy technologies to optimize the design and functionality of biocompatible scaffolds for tissue engineering in regenerative medicine. J R Soc Interface 10(86):20130263
Wallrabe H, Periasamy A (2005) Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol 16(1):19–27
Wallrabe H, Elangovan M, Burchard A, Periasamy A, Barroso M (2003) Confocal FRET microscopy to measure clustering of ligand-receptor complexes in endocytic membranes. Biophys J 85(1):559–571
Wallrabe H, Svindrych Z, Alam SR, Siller KH, Wang T, Kashatus D, Hu S, Periasamy A (2018) Segmented cell analyses to measure redox states of autofluorescent NAD(P)H, FAD & Trp in cancer cells by FLIM. Sci Rep 8(1):79
Walsh A, Cook RS, Rexer B, Arteaga CL, Skala MC (2012) Optical imaging of metabolism in HER2 overexpressing breast cancer cells. Biomed Opt Express 3(1):75–85
Walsh AJ, Cook RS, Manning HC, Hicks DJ, Lafontant A, Arteaga CL, Skala MC (2013) Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res 73(20):6164–6174
Weber K, Thomaschewski M, Warlich M, Volz T, Cornils K, Niebuhr B, Tager M, Lutgehetmann M, Pollok JM, Stocking C, Dandri M, Benten D, Fehse B (2011) RGB marking facilitates multicolor clonal cell tracking. Nat Med 17(4):504–509
Wilt BA, Burns LD, Wei Ho ET, Ghosh KK, Mukamel EA, Schnitzer MJ (2009) Advances in light microscopy for neuroscience. Annu Rev Neurosci 32:435–506
Winkler M, Chai D, Kriling S, Nien CJ, Brown DJ, Jester B, Juhasz T, Jester JV (2011) Nonlinear optical macroscopic assessment of 3-D corneal collagen organization and axial biomechanics. Invest Ophthalmol Vis Sci 52(12):8818–8827
Wolfbeis OSJB (2015) Luminescent sensing and imaging of oxygen: fierce competition to the Clark electrode. BioEssays 37(8):921–928
Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64(19):7022–7029
Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J (2007) Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 67(6):2649–2656
Xue R, Behera P, Xu J, Viapiano MS, Lannutti JJ (2014) Polydimethylsiloxane core–polycaprolactone shell nanofibers as biocompatible, real-time oxygen sensors. Sensors Actuators B Chem 192:697–707
Xue R, Ge C, Richardson K, Palmer A, Viapiano M, Lannutti JJ (2015) Microscale sensing of oxygen via encapsulated porphyrin nanofibers: effect of indicator and polymer “core” permeability. ACS Appl Mater Interfaces 7(16):8606–8614
Yaseen MA, Sakadzic S, Wu W, Becker W, Kasischke KA, Boas DA (2013) In vivo imaging of cerebral energy metabolism with two-photon fluorescence lifetime microscopy of NADH. Biomed Opt Express 4(2):307–321
Yazgan G, Dmitriev RI, Tyagi V, Jenkins J, Rotaru G-M, Rottmar M, Rossi RM, Toncelli C, Papkovsky DB, Maniura-Weber K (2017) Steering surface topographies of electrospun fibers: understanding the mechanisms. Sci Rep 7:158. https://doi.org/10.1038/s41598-017-00181-0
Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10(2):179–206
Yoshihara T, Hirakawa Y, Hosaka M, Nangaku M, Tobita S (2017) Oxygen imaging of living cells and tissues using luminescent molecular probes. J Photochem Photobiol C: Photochem Rev 30:71–95
Zaritsky A, Natan S, Horev J, Hecht I, Wolf L, Ben-Jacob E, Tsarfaty I (2011) Cell motility dynamics: a novel segmentation algorithm to quantify multi-cellular bright field microscopy images. PLoS One 6(11):e27593
Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW (2003a) Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci U S A 100(12):7075–7080
Zipfel WR, Williams RM, Webb WW (2003b) Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 21(11):1369–1377
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this entry

Cite this entry
Neto, N., Dmitriev, R.I., Monaghan, M.G. (2020). Seeing Is Believing: Noninvasive Microscopic Imaging Modalities for Tissue Engineering and Regenerative Medicine. In: Gimble, J., Marolt Presen, D., Oreffo, R., Wolbank, S., Redl, H. (eds) Cell Engineering and Regeneration. Reference Series in Biomedical Engineering(). Springer, Cham. https://doi.org/10.1007/978-3-319-08831-0_40
Download citation
DOI: https://doi.org/10.1007/978-3-319-08831-0_40
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-08830-3
Online ISBN: 978-3-319-08831-0
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences