
Abstract
Stressors may be major contributors to cardiovascular disease. The systemic adaptive response to stressors, referred as “stress response”, is primarily mediated by the activation of the hypothalamic-pituitary-adrenal (HPA) axis and the catecholaminergic system. At the cellular level, cardiomyocytes have developed various mechanisms to counterbalance stressors affecting their contractile, metabolic and other functions. These mechanisms include remodeling and activation of the fetal gene program. Here we describe the fundamentals of the stress response, we review the implications of stressors such as oxidative, endoplasmic and metabolic on heart function and we discuss the effects of specific hormonal mediators of the stress response in heart function. We particularly refer to the cardiovascular effects of Corticotropin Releasing Hormone and its related peptides in supporting the adaptive response, a potentially therapeutic strategy to overcome stressors threatening homeostasis and driving disease development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Stress
- Homeostasis
- Adaptive response
- Hypothalamic-pituitary-adrenal (HPA) axis
- Corticotropin-releasing hormone (CRH)
1 Introduction: Definition of Stress
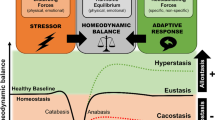
Living organisms survive by maintaining a complex dynamic and harmonious equilibrium, or homeostasis, that is constantly challenged by intrinsic or extrinsic disturbing forces or stressors. In this context, stress may be defined as a ‘state of disharmony, or threatened homeostasis’. Although minimum at normal levels, stress is an important part of normal daily life, as it enables the organism to cope with changes in the environment. However, excess stress, either physical or psychological, is a cause of harm [1].
As already described in Chap. 6, Hans Selye, who was the first to define stress, conceived the General Adaptation Syndrome, as “the non-specific response by the body to any demand”, physical or psychological [2]. The General Adaptation Syndrome represents a “chronological development of the stress response to stressors when their action is prolonged”. It consists of an initial “alarm reaction” or “shock” phase, a second “resistance” phase and a final “exhaustion” phase. According to Selye, during the second phase the body is allowed to compensate and restore homeostasis. In addition, Selye distinguished eustress (healthy stress) from distress (pathogenic stress). Distress resulting from an excessive or inappropriate response to a stressor can lead to stress-induced maladies [3]. The adaptive responses can be either specific to the stressor or nonspecific. Along these lines, Chrousos and Gold described the activity of the stress system as a sigmoidal dose-response curve with stressor potency and stress response related in a nonlinear manner. This concept can be modified to show stressor potency as an independent variable and the stress response as the dependent variable. The activity of the stress response will need a threshold to be activated, beyond which a quasilinear relationship commences. As expected, beyond a given point, the activity of the stress response does not increase despite further increase in stressor potency. This dose-response sigmoid curve is individual-specific [4]. The latter is the core of the theory of “allostasis”, which is defined as the “stability obtained through changes”. This concept which was developed by Sterling and Eyer refers to the active processes via which the body responds to daily events to maintain homeostasis [5]. The concept of allostasis suggests that when an organism is threatened, effector systems are activated to promote adaptation to a new equilibrium, rather than a return to the previous, no-longer-existing condition. Various allostatic mechanisms may be triggered by any given condition. Depending on the actual mechanisms triggered some sort of adaptation will be affected; this is the allostatic load to which the organism is submitted. Under normal conditions, allostasis is the mechanism that protects the body, promotes improved health and guarantees survival. When the allostatic load is excessively strong or lasts for a long period, it becomes allostatic overload, with the continued activation of the effector systems, which is extremely harmful, resulting in maladaptation and the appearance of stress related diseases [4]. Stress increases the susceptibility of the body to infection, autoimmune diseases, chronic fatigue syndrome, psychiatric illnesses, cancer, and chronic diseases. Depending on the individual situation and predisposition, stress can be a major contributor to the development of cardiovascular disorders (CVD), such as ischemic heart disease, arrhythmia and sudden death [6].
2 Hormonal Regulation of the Adaptive Response
The stress system is organized into central, peripheral, and cellular systems within a network of anatomical regions that work together to keep the body both informed and ready to react to encountered changes in either external or internal cellular environments [1]. The hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS) constitute the main effector pathways of the stress system [7, 8].
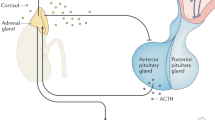
The HPA axis is the principal endocrine axis regulating the stress response [4]. Stressors of different origins activate the hypothalamic corticotropin-releasing hormone (CRH), which stimulates pituitary ACTH secretion and finally release of glucocorticoid from the adrenal gland [9]. In parallel, the catecholaminergic system is activated both at the locus coeruleus and the adrenal medulla, leading in the release of norepinephrine and epinephrine. The HPA axis operates via forward positive feedback system, so that following its activation, glucocorticoid, the end-product of its activation, exerts negative feedback on CRH and ACTH. CRH has anxiogenic properties and coordinates the adaptive, behavioral, and physical changes that occur during stress. Glucocorticoid, the final mediator of the HPA axis, plays a crucial role in mounting the adaptive response to stress and are crucial for the appropriate termination of every stress response. Glucocorticoid, via their receptors, act on positive or negative glucocorticoid responsive elements (GREs) to activate or suppress the expression of various genes, which are either directly or indirectly implicated in vital metabolic pathways [10, 11]. Cortisol, the primary glucocorticoid in humans, is continuously secreted by the adrenal cortex and its release increases significantly during environmental stressors [12].
The SNS, the other principal effector component of the stress system, provides a mechanism which allows rapid regulation of vital functions, such as cardiovascular, respiratory, gastrointestinal, renal, and endocrine functions [13, 14]. Cytokines, in particular IL-6, are potent activators of the central stress response, forming a feedback loop through which the immune/inflammatory system communicates with the brain [15].
When stress, which moderates the proteome of the cytosol, endoplasmic reticulum (ER) and mitochondria, is sensed in one tissue, it can activate responses in different tissues in a non-autonomous way. This way, mechanisms that protect against and mechanisms that promote disease progression rely on coordination and communication between cells and tissues. The control of intercellular communication and cell-autonomous regulation is subject to neuroendocrine regulation through HPA activation. Chronic activation of the stress system, together with its associated catabolic, anti-reproductive, antigrowth and immunosuppressive effects, may prove detrimental for the organism. A cellular stress response may thus develop into a systemic stress response through activation of the hypothalamic-pituitary-adrenal (HPA) axis and release of glucocorticoid. Results of chronic stress include behavioral changes affecting physical activity and dietary habits leading to weight gain and the metabolic syndrome, and thus to abnormalities of glucose and lipid metabolism [16], induction of chronic hypercortisolemia that can in the long run cause visceral fat accumulation, decreased lean body mass, and insulin resistance [17, 18], and increased sympathoadrenal system activity. The latter contributes to impaired glucose tolerance and to increased risk for acute cardiovascular events [19].
3 Stress and Cardiovascular Disease
The concept of stress being involved as risk factor for CVD dates back more than 100 years ago and has been thoroughly investigated using a combination of epidemiological, mechanistic, psychophysiological experiments and clinical studies. Adaptation to stressors is an integral part of cardiovascular physiology, whereas stress can be a major contributor in the development of cardiovascular disorders, such as ischemic heart disease, arrhythmia and sudden death [6]. Chronic stress, either at early life or adulthood, has been associated with a 40–60 % excess risk of cardiovascular disorder [20].
Cardiomyocytes have developed various mechanisms to deal with stressors affecting their contractile performance and/or energy supply. The success of these mechanisms depends to a large extent on the nature and duration of the stress. At the cellular level, stress results in adverse rearrangement of cytoskeletal structures, a process known as remodeling (REM), together with the accumulation of dysfunctional mitochondria and ER. Success or failure of these cellular adaptive mechanisms, which will be discussed in detail in the following paragraphs, may lead to repair of the injury or propagation of the damage often progressing to heart failure (HF).
3.1 Cardiomyocyte Remodeling in Response to Stress
At the cellular level, REM causes stress to the cardiomyocyte as a whole, by altering energy metabolism, the structure of the contractile apparatus and the cytoskeleton. Thus, cardiomyocytes lose the characteristics of mature (differentiated) cardiomyocytes and re-express genes from embryonic or fetal developmental stages REM is intimately connected to cellular dedifferentiation. It has been proposed that by dedifferentiation or sarcomeric disassembly, cardiomyocytes gain resistance to altered metabolic and/or hypoxic stress [21–24]. Dedifferentiation provides cardiomyocytes with a plasticity which allows the myocardium to cope much better with hypoxia-induced or overload-induced cell death, preventing the progression from compensated hypertrophy to HF [25]. Parallel activation of the fetal gene program is characterized by the up-regulation of early cardiac-specific transcription factors, including MEF2c, NFAT, GATA and Nkx2.5 [26], as well as the stem cell markers c-kit, Runx1 and Dab2. Numerous other genes, including destrin, a-SM-actin, the natriuretic peptides ANP and BNP, smooth-muscle actin (ACTN1), moiesin, and oncostatin M (OSM) are also re-expressed. The fetal gene program also shows induced expression of ER chaperones such as BiP/GRP78, GRP94 and calreticulin during early development, and also triggers significant changes in metabolic programs [27]. These changes are also described in Chap. 17.
In diseased states, compensatory REM is accompanied by certain maladaptive processes including inflammatory infiltration, increased interstitial connective tissue (increasing the oxygen diffusion distance and wall stiffness), reduction of cardiomyocyte cell contacts, and apoptotic cell death, eventually reducing the ventricular ejection capacity [28–30]. The degree of cardiomyocyte degeneration and loss of differentiation markers can be correlated with the extent of inflammatory infiltration [23, 31].
Myocyte dedifferentiation, including loss of myoglobin, reduces oxygen-dependent generation of ATP, but maintains the ability of heart cells to generate energy by glycolysis. Cardiac REM and metabolic adaptation are closely interconnected, as reflected by the characteristic switch from fatty acid oxidation to glucose utilization in stressed cardiomyocytes. Interestingly, although changes in mitochondrial metabolism seem initially beneficial to sustain contractility and survival, increasing evidence suggests that metabolic reprogramming might eventually result in mitochondrial dysfunction and cardiac failure [32].
A good method to study and understand compensatory REM in animal models is cardiac ischemia, as it comprises response mechanisms caused by the lack of oxygen and includes secondary inflammatory infiltration. Importantly, REM affects both ischemic and non-ischemic regions of the ventricle leading to changes in chamber size, shape and function. Smaller damages may only lead to hypertrophy of remaining cardiomyocytes in order to normalize wall stress, while large infarcts cause significant changes in heart architecture [33–35].
3.2 Cardiomyocytes and Oxidative Stress
The accumulation of dysfunctional mitochondria is characteristic of a wide spectrum of cardiac diseases. Mitochondria are important cellular energy components. In addition to producing energy through respiration, mitochondria regulate cellular metabolism and produce reactive oxygen species (ROS) through electron transport chains (ETCs). Although the adrenergic system plays a central role in stress signaling and stress is often associated with increased production of ROS, ROS overproduction generates oxidative stress, a key part of various common pathological conditions, including the metabolic syndrome and numerous cardiovascular diseases, such as coronary artery disease, heart failure, left ventricular hypertrophy, diabetic cardiomyopathy and hyperkinetic arrhythmias.
ROS are reactive chemical units involving two main categories: (a) free radicals such as superoxide (O2 −), hydroxyl (OH) and nitric oxide (NO); and (b) non-radical derivatives of O2 such as hydrogen peroxide (H2O2) and peroxynitrate (ONOO−) [36]. Under normal conditions, ROS control several physiological processes, including host defense, hormone biosynthesis, fertilization and cell signaling. Oxidative stress causes protein, lipid and DNA damage leading to cellular dysfunction. Excessive O2 − may decrease nitric oxide (NO) availability, leading to endothelial dysfunction and endothelium-dependent vasodilation decrease [37]; while oxidative protein modification may result in the formation of nitrotyrosine that represents a powerful and autonomous marker of cardiovascular diseases. O2 − is also implicated in the generation of oxidized LDL, a key initiator of atherosclerosis (ATH) [38].
ROS can cause posttranslational protein modifications to regulate signaling pathways. Redox modifications may affect mitochondrial function either directly or indirectly by acting on mediators involved in most mitochondrial activities. For example, STAT3 seems to preserve ETC activity by preventing ROS leakage at complex I. In mice, cardiac-specific deletion of STAT3 results in the development of cardiac inflammatory fibrosis, dilated cardiomyopathy and heart failure with advancing age; while lack of STAT3 eliminates the cardioprotective effects of ischemic preconditioning, contrary to its overexpression in cardiomyocytes, which protects mice against doxorubicin toxicity that involves mitochondrial dysfunction [39]. Expression of a recombinant form of STAT3 that targets mitochondria and lacks the DNA-binding domain protected the heart from ischemic damage by decreasing mitochondrial ROS (mROS) production and attenuating cytochrome c release in a model of ischemia. mROS is of a dual nature. Very high quantities of mROS directly damage proteins, lipids and nucleic acids. Lower levels, however, function as signaling molecules to adapt to stress, and even lower amounts of mROS are required for normal cell function. For example, mROS are involved in cardioprotective preconditioning pathways, and antioxidants render ischemic preconditioning ineffective [40].
Emerging evidence points to the role of NAPDH oxidase (NOX) family of enzymes, a major source of ROS in both metabolic and cardiovascular dysfunction [41]. As very well established, β-adrenergic signaling is markedly attenuated in conditions such as heart failure, with downregulation and desensitization of the receptors and their uncoupling from adenylyl cyclase. Transgenic activation of β2-adrenoreceptor leads to elevation of NADPH oxidase activity, with greater ROS production and p38 MAPK phosphorylation. Inhibition of NADPH oxidase or ROS significantly reduces the p38 MAPK signaling cascade. βAR stimulation antagonizes the protective effect of the Akt pathway through inhibition of hypoxia-inducible factor 1-alpha (Hif1a) and Sirt1 induction, key elements in cell survival [42]. Sirtuins, a family of seven highly conserved class III histone deacetylases that regulate a wide range of cellular processes, such as transcription, inflammation, apoptosis, and aging [43], are involved in modulating the cellular stress response directly by deacetylation of some factors. Lately, the role of Sirt1 (also called NAD+ – dependent protein deacetylase) as the most important factor in the pathway of mitochondrial biogenesis and a key regulator of cellular defense and survival has been recognized [44]. Sirt1 increases cellular stress resistance, by induction of insulin sensitivity, decrease in circulating free fatty acids and insulin-like growth factor 1 (IGF-1), induction of 5’ AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1a) activity, and mitochondrial genesis. Resveratrol, a therapeutic agent and an activator of SIRT1, suppresses ROS production and induces the expression of the mitochondrial protein NDUFA13 [45, 46].
3.3 Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) is an intracellular organelle with major contribution to protein synthesis and metabolism of nutrients and toxic metabolic products. ER stress developing in states of overwhelming load of the ER is associated with a variety of states such as oxidative stress, ROS production, viral infection, environmental toxins, heat, drugs, inflammatory cytokines, lipotoxicity, Ca2+ depletion, metabolic starvation and aging. ER stress has been also identified as a key contributor in the pathogenesis of cardiac and vascular diseases [27, 47–50]. Tight control of metabolic homeostasis that is necessary for normal function of all tissues is particularly important for the cardiac tissue and involves Ca2+, K+, Na+, Cl−, oxygen, glucose, amino acids, and fatty acids. Disruption of metabolic homeostasis can lead to ER stress. For example, disruption of Ca2+ homeostasis is directly responsible for ER stress and activation of cardiac REM, leading to cardiac hypertrophy. Cardiomyocytes monitor Ca2+ homeostasis in regard to excitation-contraction versus ER stress by, at least in part, the regulation of store-operated Ca2+ entry (SOCE) and an ER membrane-associated Ca2+ sensor, stromal interaction molecule 1 (STIM1) [51]. In rats, SOCE is abundant in neonatal cardiomyocytes, while it is absent in adult cardiomyocytes [52], correlating with STIM1 mRNA and protein expression. Stress-triggered STIM1 re-expression and consequent SOCE activation are critical elements in the upstream, Ca2+ -dependent control of pathological cardiac hypertrophy [52]. In states of thoracic aortic constriction (TAC) and in neonatal cardiomyocytes under induced hypetrophy, STIM1 protein and mRNA levels are significantly increased. The above is an example of the molecular pathways mediating mobilization of Ca2+ -signaling molecules and pathways and their interconnection to the preservation of ER function, all contributing to support normal cardiac function in normal and adverse conditions.
There is a constant, interactive relation between active inflammation and ER stress, underlying a spectrum of diseases with insulin resistance the most widely studied [53]. Increase of the unfolded protein response (UPR), ROS production, Ca2+ release from the ER, and the activation of the nuclear factor κ light-chain enhancer of activated B cells (NF-κB) and of Jun N-terminal kinase (JNK) can trigger the inflammatory response associated with ATH and CVD [54–56]. Briefly, upon tissue damage, inflammatory cells such as neutrophils and macrophages are recruited to the site of damage, initiating inflammatory cytokine production and ROS generation, protein folding disruption and ER stress triggering. When ER stress is prolonged, NF-κB initiates apoptosis, thereby shifting the outcome of the compensatory mechanism from an adaptive to a maladaptive one. NF-κB activation is initiated via the action of IκB kinase (IκBK), which phosphorylates and inactivates IκB-α. Its inactivation in turn results in NF-κB nuclear translocation and in the expression of inflammation-related genes, such as those encoding the potent inflammatory cytokines TNFα and IL-6 [54, 56]. Specific to cardiomyocytes, NFκB deletion promotes an adaptive response to ER stress, whereas once NFκB is activated, cardiomyocytes undergo inflammation, fibrosis and apoptosis. The angiogenic factor MCP-1 is linked to inflammation and the activation of autophagy in the heart. During chronic inflammation, increased MCP-1 expression induces oxidative stress by upregulating monocyte chemoattractant protein-induced protein (MCPIP), a novel zinc finger protein, which causes oxidative stress via iNOS activation and ROS production, followed by the induction of ER stress-responsive genes and autophagy [57]. Gene-silencing experiments have demonstrated that MCPIP reduction decreases MCP-1 induced cell death in cardiac myoblasts. MCP-1 recruits monocytes and differentiates monocytic lineage cells into endothelium-like cells, thus promoting vascular growth and angiogenesis [58].
ER stress is highly associated with various CVD. Myocardial infarction (MI) provides a classic example; it results in severe complications associated with hypoxia and fuel starvation in the muscle tissue, generation of a buildup of misfolded protein, and thus disruption of ER homeostasis and generation of ER stress. Furthermore, during ischemia, ER stress results in the degeneration of cardiac myocytes and over time in cell death. However, if preconditioning, such as mild heat stress, is applied, damage may be prevented, most possibly due to the activation of an adaptive coping response, consequently limiting the amount of damage generated during the subsequent insult. Similarly, hypertension or aortic construction or stenosis, also resulting in pressure overload, induce ER stress. The list of CVD implicating ER stress is growing rapidly. Congenital dilated cardiomyopathy (CDCM) is linked to a mutation in the KDEL receptor, which leads to a buildup of misfolded proteins and results in ER stress [59]. Reports show that disruptions in the excitation-contraction coupling, with abnormal Ca2+ homeostasis, result in ER stress and congestive HF. Atherosclerosis results from the accumulation of misfolded proteins due to the oxidation of lipids, the upregulation of homocysteine in vascular cells, and inclusion of large amounts of cholesterol esters in macrophages. ER stress may also play a role in cardiac amyloidosis. It is thus obvious that ER stress and CVD are intimately intertwined and therefore therapeutic interventions to cope with ER stress provide a promising venue in the fight against detrimental chronic diseases [60].
3.4 Effects of the Hormonal Mediators of the Stress Response in the Cardiovascular System
HPA axis is the principal endocrine axis regulating the stress response. Stressors of different origins activate the hypothalamic CRH, which stimulates pituitary ACTH secretion and finally release of glucocorticoid from the adrenal gland. In parallel, the catecholaminergic system is activated both at the locus coeruleus and peripheral sites leading to the release of norepinephrine and epinephrine. The HPA axis operates via forward positive feedback system, while the end-product, glucocorticoid exerts negative feedback regulation of CRH and ACTH. All different components of the HPA axis affect, directly or indirectly, heart function and may precipitate disease development. A summary of HPA axis and CRH regulation is shown in Fig. 14.1. Below, we give a brief summary of the effects of glucocorticoid and the CRH family of peptides in the regulation of cardiovascular function.

HPA axis is the principal endocrine axis regulating the stress response. Stressors of different origins activate the hypothalamic CRH, which stimulates pituitary ACTH secretion and finally release of glucocorticoid from the adrenal gland. The HPA axis operates via forward positive feedback system, while the end-product, glucocorticoid exerts negative feedback regulation of CRH and ACTH. In addition to its expression in the central nervous system, several functions in peripheral sites have been identified. Genetic ablation of CRH (Crh knockout, Crh−/− mouse) in the heart negatively affects cardiac function, through the Erk1/2, Akt and NFκB signaling pathways
3.4.1 Glucocorticoid
Cardiomyocytes participate actively in the adaptive stress response via initiating a cascade of pathways to secure homeostasis. Failure to do so, results in extensive tissue damage, disease development and eventually HF. Glucocorticoid act by synchronizing the metabolic, autonomic, psychological, hemostatic and cardiovascular components of the stress response via its multiple tissue-specific genomic and non-genomic effects. These actions facilitate the vascular and metabolic effects of other stress hormones, such as catecholamines, glucagon and angiotensin-II, through stimulation of α1 adrenergic and angiotensin II receptor expression, and increase in the affinity and binding capacity of β-adrenergic receptors. In parallel, glucocorticoid suppresses associated processes such as inflammation, cellular proliferation and tissue repair processes. Finally, glucocorticoid prepare the organism for prolonged nutritional deprivation by facilitating proteolysis and support development of insulin resistance at the muscle level, while inducing gluconeogenesis and lipolysis. This pleiotropic action of glucocorticoid may be harmful for the cardiovascular system because of increased blood pressure and insulin resistance. Corticosteroid excess is associated with adverse cardiovascular outcomes. Patients with Cushings’s syndrome and primary aldosteronism, two conditions characterized by glucocorticoid excess and inappropriately high aldosterone production respectively, have increased risk for CVD. Prior to treatment of Cushing’s syndrome, it has not been uncommon for patients to experience early death from MI or stroke. In animal models, excess glucocorticoid can induce ATH. In humans, it has been suggested that corticosteroid-treated patients with rheumatoid arthritis and lupus erythematosus develop significantly more ATH than those not treated with steroids and the risk of ATH is related to the cumulative dose of corticosteroid. There have also been cases of adverse outcomes of steroid-dependent rheumatoid arthritis patients treated with thrombolytic agents for acute MI, possibly due to increased risk of myocardial rupture. However, there has been no consensus on the proposed adverse effects of glucocorticoid given acutely in MI [61].
On the other hand, glucocorticoid deficiency may result in hypotension, weight loss, hypoglycemia and death, particularly during stress; while glucocorticoid excess can lead to hypertension, insulin resistance, hyperglycemia and weight gain [62–64]. The vascular effects of glucocorticoid seem to be achieved by enhancing adrenergic stimulation, angiotensin II and possibly endothelin-1. Glucocorticoid upregulate angiotensin II type I receptor expression and α1 adrenergic receptors in rat vascular smooth muscle cells and potentiate the vasoconstrictive actions of angiotensin-II and norepinephrine in animals [65]
3.4.2 Crh and CRH-Related
In addition to its expression in the central nervous system, CRH has been identified in peripheral sites with functions not completely elucidated yet. So far, a dual, both anti- and pro- proinflammatory role for peripheral CRH has been identified. Its significant expression in inflamed human and rodent tissues due to the corresponding neutrophil and polymorphonuclear cell accumulation has been shown [66], and its effects have been confirmed in transgenic animal models [67–70]. Angiogenic effects of CRH, both in vivo and in vitro, have been shown [71, 72].
CRH belongs to an extended family of CRH-related peptides, which includes urocortins (UCN) (I, II and III). These peptides share significant homology with CRH, while they all bind the same family of receptors (CRHR1 and CRHR2), with CRH and UCN I binding both receptors, although with different affinities, and UCN II and III binding CRHR2 exclusively. UCNs and CRHRs are also widely expressed in a variety of tissues. CRH receptors belong to the G-protein coupled receptors (GPCRs) family and upon activation stimulate various intracellular pathways, in a time- and tissue-dependent manner. Expression of both CRHRs and UCN have been identified in the endothelium and the heart with CRHR2 robustly [73] and CRHR1 minimally if at all expressed [74]. Activation of phosphoinositide-3-kinase (PI3K)/Akt (protein kinase B/Akt) and mitogen-activated protein kinases (MAPK) in endothelial cells by CRHR2 links its expression with specific biological effects, while protective effects in cardiomyocytes during experimental ischemia have been shown [75, 76]. Furthermore we have recently found stimulation of VEGF by CRH via CRHR2 in endothelial cells [72, 77].
Cardioprotective effects of UCNs have been well documented in several experimental systems. In general, urocortins exhibit potent vasodilatory effects in arteries of different species through regulation of intracellular Ca2+ levels, increase heart contractility and evoke positive inotropic and lusitropic effects. More specifically, UCNs protect the heart from ischemia and reperfusion injury via improvement of post-ischemic cardiac performance, such as cardiac contractility, prevention from Ca2+ overload and reduction of cardiac cell death. These effects involve the activation of several signaling pathways targeting both cytoplasmic and mitochondrial processes. In particular UCN1, by initially binding to GPCRs, initiates the activation of PI3K, protein kinase A (PKA), Akt, protein kinase C (PKC) and MAPKs. Activation of these kinase pathways alters the activity of various channels including the mitochondrial permeability transition pore (MPTP), which is involved in the induction of cell death [78, 79], as described in Sects. 15.2.1 and 15.2.2. Apart from their acute effects, UCNs exert more prolonged, modulatory actions, such as transcriptional and translational effects on mitochondrial ATP-sensitive potassium channel (KATP), calcium insensitive phospholipase A2 (iPLA2) and protein kinase C epsilon (PKCε), all three involved in cardioprotection [80–84]. UCN1 acts via a p42/p44 MAPK (Erk1/2)-dependent signaling pathway in protecting both in vitro primary cell models and ex vivo and in vivo rodent heart from reperfusion injury. Erk1/2 phosphorylation and activation is mediated by the MAP kinase MEK1/2 following UCN1 treatment; while its inhibition disrupts UCN1-mediated cardioprotection [85, 86]. PI3K and its downstream effector Akt are crucial for UCN-stimulated increase in survival of cardiomyocytes [87]. Both MEK1/2 and PI3K are responsible for preventing pro-caspases -9 and -3 from being cleaved into their active forms [88, 89]. UCN2 and UCN3 also act through the PI3K pathway [75]. In addition to the protective effects, UCN treatment of both neonatal and adult rat cardiomyocytes stimulates the secretion of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), both of which are markers of hypertrophy [90, 91]. Another significant contribution of UCN1-mediated cardioprotection involves de novo protein synthesis. For example, expression of the cardioprotective heat shock protein 90 (HSP90) has been reported to be induced by UCN1, a specific effect blocked by the MK1/2 inhibitor PD98059 [92]. The expression of cardiotrophin-1 (CT-1), an additional cardioprotective peptide, is also increased upon exposure of cells to UCN 1 [93].
Based on the above experimental evidence for the cardioprotective role of urocortins and on the reported role of CRH on various peripheral sites, a possible role for CRH in the cardiac adaptive response to stressors has recently been explored by our group. The absence of CRH in mice leads to low levels of ACTH and corticosterone and a blunted HPA response. Thus, CRH regulates adrenal function as well as responses to stress mediated via the HPA axis. Genetic ablation of CRH (Crh-knockout, Crh−/−, mice) leads to inability of the cardiovascular system to cope with a mild stressor, resulting in death. Similar to those described for UCNs, signaling pathways involved in this process, include Erk1/2, Akt, and AMPK. Crh deletion also results in increased infiltration of the heart tissue with inflammatory cells and apoptosis. Metabolic dysregulation evident by significantly compromised ability for fatty acid oxidation may underlie, at least, the latter, and possibly drive the compromised function of the Crh−/− heart [94].
4 Conclusion
Heart is a tissue where different pathways activated by various stressors convey and alter its functions, often resulting in remodeling, chronic inflammation, metabolic dysfunction and ultimately, heart failure. Given the active participation of the heart in the adaptive response to stressors, and the effects of specific mediators of the adaptive response on these processes, the possibility they represent promising targets for novel therapeutic schemes is raised. New models taking into account the contribution of these factors at both the cell and systemic responses and proposing novel therapeutic strategies need to be validated.
Abbreviations
- AMPK:
-
5’ AMP-activated protein kinase
- ANP:
-
Atrial natriuretic peptide
- ATH:
-
Atherosclerosis
- BNP:
-
Brain natriuretic peptide
- CDCM:
-
Congenital dilated cardiomyopathy
- CRH:
-
Cortioctropin-releasing hormone
- CT-1:
-
Cardiotrophin-1
- CVD:
-
Cardiovascular disorder
- ER:
-
Endoplasmic reticulum
- ETCs:
-
Electron transport chains
- GPCRs:
-
G-protein coupled receptors
- GREs:
-
Glucocorticoid responsive elements
- Hif1α:
-
Hypoxia-inducible factor 1-alpha
- HPA:
-
Hypothalamic-pituitary-adrenal
- HSP90:
-
Heat shock protein 90
- IGF-1:
-
Insulin-like growth factor 1
- iPLA2 :
-
Calcium insensitive phospholipase A2
- IκBK:
-
IκB kinase
- JNK:
-
jun N-terminal kinase
- KATP :
-
ATP-sensitive potassium channel
- MAPK:
-
Mitogen activated protein kinase
- MCPIP:
-
Monocyte chemoattractant protein-induced protein
- MI:
-
Myocardial infarction
- MPTP:
-
Mitochondrial permeability transition pore
- mROS:
-
Mitochondrial ROS
- NFκB:
-
Nuclear factor κ light-chain enhancer of activated B cells
- PGC1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PI3K:
-
Phosphoinositide-3-kinase
- PKCε:
-
Protein kinase C epsilon
- REM:
-
Remodeling
- ROS:
-
Reactive oxygen species
- SNS:
-
Sympathetic nervous system
- SOCE:
-
Store-operated Ca2+ entry
- STIM1:
-
Stromal interaction molecule 1
- UCN:
-
Urocortin
- UPR:
-
Unfolded protein response
References
Cuesta JM, Singer M. The stress response and critical illness: a review. Crit Care Med. 2012;40:3283–9.
Selye H. What is stress? Metabolism. 1956;5:525–30.
Selye H. The stress concept. Can Med Assoc J. 1976;115:718.
Chrousos PG, Gold PW. The concepts of stress and stress system disorders. JAMA. 1992;267:1244–52.
Sterling P, Eyer J. Biological basis of stress-related mortality. Soc Sci Med E. 1981;15:3–42.
Davis AM, Natelson BH. Brain-heart interactions. The neurocardiology of arrhythmia and sudden cardiac death. Tex Heart Inst J. 1993;20:158–69.
Gold PW, Goodwin F, Chrousos GP. Clinical and biochemical manifestations of depression: relationship to the neurobiology of stress, part 1. N Engl J Med. 1988;319:348–53.
Gold PW, Goodwin F, Chrousos GP. Clinical and biochemical manifestations of depression: relationship to the neurobiology of stress, part 2. N Engl J Med. 1988;319:413–20.
Dallman MF, Yates FE. Dynamic asymmetries in the corticosteroid feedback path and distribution-metabolism-binding elements of the adrenocortical system. Ann N Y Acad Sci. 1969;156:696–721.
De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301.
Boumpas DT, Chrousos GP, Wilder RL, Cupps TE, Balow JE. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann Intern Med. 1993;119:1198–208.
Mastorakas G, Chrousos GP, Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in human. J Clin Endocrinol Metab. 1993;77:1690–4.
Kvetnansky R, Sabban EL, Palkovits M. Catecholaminergic system in stress: structural and molecular genetic approaches. Physiol Rev. 2009;89:535–606.
Gilbey MP, Spyer KM. Essential organization of the sympathetic nervous system. Baillieres Clin Endocrinol Metab. 1993;7:259–78.
Turnbull AV, Rivier CL. Sprague-Dawley rats obtained from different vendors exhibit distinct adrenocorticotropin responses to inflammatory stimuli. Neuroendocrinology. 1999;70:186–95.
Adam TC, Epel ES. Stress, eating and the reward system. Physiol Behav. 2007;91:449–58.
Björntorp P. Do stress reactions cause abdominal obesity and comorbidities? Obes Rev. 2001;2:73–86.
Rosmond R, Dallman MF, Björntorp P. Stress-related cortisol secretion in men: relationships with abdominal obesity and endocrine metabolic and hemodynamic abnormalities. J Clin Endocrinol Metab. 1998;83:1853–9.
Grippo AJ, Johnson AK. Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress. 2009;12:1–21.
Steptoe A, Kivimäki M. Stress and cardiovascular disease. Nat Rev Cardiol. 2012;9:360–70.
Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–70.
Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian Cardiomyocytes. Genes Dev. 2005;19:1175–87.
Kubin T, Pöling J, Kostin S, Gajawada P, Hein S, Rees W, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. 2011;9:420–32.
Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. 2010;1188:191–8.
Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–91.
Zhang Y, Li TS, Lee ST, Wawrowsky KA, Cheng K, Galang G, et al. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS One. 2010;5:e12559.
Groenendyk J, Sreenivasaiah PK, Kim DH, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–97.
Mann N, Rosenzweig A. Can exercise teach us how to treat heart disease? Circulation. 2012;126:2625–35.
Hou J, Kang YJ. Regression of pathological cardiac hypertrophy: signaling pathways and therapeutic targets. Pharmacol Ther. 2012;135:337–54.
Kostin S, Hein S, Arnon E, Scholtz D, Schaper J. The cytoskeleton and related proteins in the human failing heart. Heart Fail Rev. 2000;5:271–80.
Pöling J, Gajawada P, Lörchner H, Polyakova V, Szibor M, Böttger T, et al. Tha Janus face of OSM-mediated cardiomyocyte dedifferentiation during cardiac repair and disease. Cell Cycle. 2012;11:439–45.
Verdejo HE, del Campo A, Troncoso R, Gutierrez T, Toro B, Quiroga C, et al. Mitochondria, myocardial remodeling, and cardiovascular disease. Curr Hypertens Rep. 2012;14:532–9.
Seino Y, Ikeda U, Takahashi M, Hojo Y, Irokawa M, Kasahara T, et al. Expression of monocyte chemoattractant protein-1 in vascular tissue. Cytokine. 1995;7:575–9.
Apostolakis S, Lip GY, Shantsila E. Monocytes in heart failure: relationship to a deteriorating immune overreaction or a desperate attempt for tissue repair? Cardiovasc Res. 2010;85:649–60.
van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–29.
Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31 Suppl 2:S170–80.
Huang PL. Unraveling the links between diabetes, obesity, and cardiovascular disease. Circ Res. 2005;96:1129–31.
Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–37.
Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–34.
Wojtovich AP, Nadtochiy SM, Brookes PS, Nehrke K. Ischemic preconditioning: the role of mitochondria and aging. Exp Gerontol. 2012;47:1–7.
Fortuno A, San José G, Moreno MU, Beloqui O, Díez J, Zalba G. Phagocytic NADPH oxidase overactivity underlies oxidative stress in metabolic syndrome. Diabetes. 2006;55:209–15.
Khan M, Mohsin S, Avitabile D, Siddiqi S, Nguyen J, Wallach K, et al. β-Adrenergic regulation of cardiac progenitor cell death versus survival and proliferation. Circ Res. 2013;112:476–86.
Pantazi E, Zaouali MA, Bejaoui M, Folch-Puy E, Ben Abdennebi H, Rosellό-Catafau J. Role of sirtuins in ischemia-reperfusion injury. World J Gastroenterol. 2013;19:7594–602.
Motta MC, Divacha N, Lemieux M, Kamel C, Chen D, Gu W, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–63.
Hosoda R, Kuno A, Hori YS, Ohtani K, Wakamiya N, Oohiro A, et al. Differential cell-protective function of two resveratrol (trans-3, 5, 4’- trihydroxystilbene) glucosides against oxidative stress. J Pharmacol Exp Ther. 2013;344:124–32.
Li YG, Zhu W, Tao JP, Xin P, Liu MY, Li JB, et al. Resveratrol protects cardiomyocytes from oxidative stress through SIRT1 and mitochondrial biogenesis signaling pathways. Biochem Biophys Res Commun. 2013;438:270–6.
Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007;101:975–84.
Kimata Y, Kohno K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr Opin Cell Biol. 2011;23:135–42.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102.
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90.
Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533.
Luo X, Hojayev B, Jiang N, Wang ZV, Tandan S, et al. STIM1-dependent store-operated Ca2+ entry is required for pathological cardiac hypertrophy. J Mol Cell Cardiol. 2012;52:136–47.
Hasnain SZ, Lourie R, Das I, Chen AC, McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. 2012;90:260–70.
Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: model of NF-κB- and MAP kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763–72.
Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19.
Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–62.
Kolattukudy PR, Niu J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ Res. 2012;110:174–89.
Niyama H, Kai H, Yamamoto T, Shimada T, Sasaki K, et al. Roles of endogenous monocyte chemoattractant protein-1 in ischemia-induced neovascularization. JAm Coll Cardiol. 2004;44:661–6.
Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007–17.
Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–82.
Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157:545–59.
Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–61.
Dostert A, Heinzel T. Negative glucocorticoid receptor response elements and their role in glucocorticoid action. Curr Pharm Des. 2004;10:2807–16.
Degawa-Yamauchi M, Moss KA, Bovenkerk JE, Shankar SS, Morrison CL, Lelliott CJ, et al. Regulation of adiponectin expression in human adipocytes: effects of adiposity, glucocorticoids, and tumour necrosis factor alpha. Obes Res. 2005;13:662–9.
Ht L, Long CS, Gray MO, Rokosh DG, Honbo NY, Karliner JS. Cross talk between angiotensin AT1 and alpha 1-adrenergic receptors: angiotensin II downregulates alpha 1a-adrenergic receptor subtype mRNA and density in neonatal rat cardiac myocytes. Circ Res. 1997;81:396–403.
Karalis K, Sano H, Redwine J, Listwak S, Wilder RL, Chrousos GP. Autocrine or paracrine inflammatory actions of corticotropin-releasing hormone in vivo. Science. 1991;254:421–3.
Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–62.
Karalis KP, Kontopoulos E, Muglia LJ, Majzoub JA. Corticotropin-releasing hormone deficiency unmasks the proinflammatory effect of epinephrine. Proc Natl Acad Sci U S A. 1999;96:7093–7.
Venihaki M, Dikkes P, Carrigan A, Karalis KP. Corticotropin-releasing hormone regulates IL-6 expression during inflammation. J Clin Invest. 2001;108:1159–66.
Benou C, Wang Y, Imitola J, VanVlerken L, Chandras C, Karalis KP, et al. Corticotropin-releasing hormone contributes to the peripheral inflammatory response in experimental autoimmune encephalomyelitis. J Immunol. 2005;174:5407–13.
Arbiser JL, Karalis K, Viswanathan A, Koike C, Anand-Apte B, Flynn E, et al. Corticotropin-releasing hormone stimulates angiogenesis and epithelial tumor growth in the skin. J Invest Dermatol. 1999;113:838–42.
Im E, Rhee SH, Park YS, Fiocchi C, Taché Y, Pothoulakis C. Corticotropin-releasing hormone family of peptides regulates intestinal angiogenesis. Gastroenterology. 2010;138:2457–67.
Kimura Y, Takahashi K, Totsune K, Muramatsu Y, Kaneko C, Darnel AD, et al. Expression of urocortin and corticotropin-releasing factor receptor subtypes in the human heart. J Clin Endocrinol Metab. 2002;87:340–6.
Baigent SM, Lowry PJ. mRNA expression profiles for corticotrophin-releasing factor (CRF), urocortin, CRF receptors and CRF-binding protein in peripheral rat tissues. J Mol Endocrinol. 2000;25:43–52.
Brar BK, Jonassen AK, Egorina EM, Chen A, Negro A, Perrin MH, et al. Urocortin-II and urocortin-III are cardioprotective against ischemia reperfusion injury: an essential endogenous cardioprotective role for corticotropin releasing factor receptor type 2 in the murine heart. Endocrinology. 2004;145:24–35.
Huang M, Kempuraj D, Papadopoulou N, Kourelis T, Donelan J, Manola A, et al. Urocortin induces interleukin-6 release from rat Cardiomyocytes through p38 MAP kinase, ERK and NF-kappaB activation. J Mol Endocrinol. 2009;42:397–405.
Chaniotou Z, Giannogonas P, Theocharis S, Teli T, Gay J, Savidge T, et al. Corticotropin-releasing factor regulates TLR4 expression in the colon and protects mice from colitis. Gastroenterology. 2010;139:2083–92.
Townsend PA, Davidson SM, Clarke SJ, Khaliulin I, Carroll SJ, Scarabelli TM, et al. Urocortin prevents mitochondrial permeability transition in response to reperfusion injury indirectly by reducing oxidative stress. Am J Physiol Heart Circ Physiol. 2007;293:H928–38.
Baines CP. The mitochondrial permeability transition pore as a target of cardioprotective signaling. Am J Physiol Heart Circ Physiol. 2007;293:H903–4.
Lawrence KM, Chanalaris A, Scarabelli T, Hubank M, Pasini E, Townsend PA, et al. K(ATP) channel gene expression is induced by urocortin and mediates its cardioprotective effect. Circulation. 2002;106:1556–62.
Lawrence KM, Scarabelli TM, Turtle L, Chanalaris A, Townsend PA, Carroll CJ, et al. Urocortin protects cardiac myocytes from ischemia/reperfusion injury by attenuating calcium-insensitive phospholipase A2 gene expression. FASEB J. 2003;17:2313–5.
Lawrence KM, Townsend PA, Davidson SM, Carroll CJ, Eaton S, Hubank M, et al. The cardioprotective effect of urocortin during ischaemia/reperfusion involves the prevention of mitochondrial damage. Biochem Biophys Res Commun. 2004;321:479–86.
Lawrence KM, Kabir AM, Bellahcene M, Davidson S, Cao XB, McCormick J, et al. Cardioprotection mediated by urocortin is dependent on PKCepsilon activation. FASEB J. 2005;19:831–3.
Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12:181–8.
Schulman D, Latchman DS, Yellon DM. Urocortin protects the heart from reperfusion injury via upregulation of p42/p44 MAPK signaling pathway. Am J Physiol Heart Circ Physiol. 2002;283:H1481–8.
Latchman DS. Urocortin protects against ischemic injury via a MPK-dependent pathway. Trends Cardiovasc Med. 2001;11:167–9.
Brar BK, Stephanou A, Knight R, Latchman DS. Activation of protein kinase B/ Akt by urocortin is essential for its ability to protect cardiac cells against hypoxia/ reoxygenation-induced cell death. J Mol Cell Cardiol. 2002;34:483–92.
Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21.
Terada K, Kaziro Y, Satoh T. Analysis of Ras-dependent signals that prevent caspase-3 activation and apoptosis induced by cytokine deprivation in hematopoietic cells. Biochem Biophys Res Commun. 2000;267:449–55.
Chanalaris A, Lawrence KM, Townsend PA, Davidson S, Jamshidi Y, Stephanou A, et al. Hypertrophic effects of urocortin homologous peptides are mediated via activation of the Akt pathway. Biochem Biophys Res Commun. 2005;328:442–8.
Railson JE, Liao Z, Brar BK, Buddle JC, Pennica D, Stephanou A, et al. Cardiotrophin-1 and urocortin cause protection by the same pathway and hypertrophy via distinct pathways in cardiac myocytes. Cytokine. 2002;17:243–53.
Brar BK, Railson J, Stephanou A, Knight RA, Latchman DS. Urocotin increases the expression of heat shock protein 90 in rat cardiac myocytes in a MEK1/2-dependent manner. J Endocrinol. 2002;172:283–93.
Janjua S, Lawrence KM, Ng LL, Latchman DS. The cardioprotective agent urocortin induces expression of CT-1. Cardiovasc Toxicol. 2003;3:255–62.
Tzanavari T, Varela A, Theocharis S, Pantos C, Cokkinos D, Karalis K. CRH regulates cardiac function in normal and inflammatory states. In: Abstract of XXII Journées Européennes de la Societé Française de Cardiologie, Paris, 12–14 Jan 2012.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tzanavari, T., Karalis, K.P. (2015). Stress Proteins and the Adaptive Response of the Heart. In: Cokkinos, D. (eds) Introduction to Translational Cardiovascular Research. Springer, Cham. https://doi.org/10.1007/978-3-319-08798-6_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-08798-6_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-08797-9
Online ISBN: 978-3-319-08798-6
eBook Packages: MedicineMedicine (R0)