
Abstract
Metabolic network analysis based on stoichiometric and constraint-based methods has become one of the most popular and successful modeling approaches in network and systems biology. Although these methods rely solely on the structure (stoichiometry) of metabolic networks and do not require extensive knowledge on mechanistic details of the involved reactions, they enable the extraction of important functional properties of biochemical reaction networks and deliver various testable predictions. This chapter gives an introduction on basic concepts and methods of stoichiometric and constraint-based modeling techniques. The mathematical foundations of the most important approaches—including graph-theoretical analysis, conservation relations, metabolic flux analysis, flux balance analysis, elementary modes, and minimal cut sets—will be presented, and applications in biology and biotechnology will be discussed. It will be shown that network problems arising in the context of metabolic network modeling are related to different fields of applied mathematics such as graph and hypergraph theory, linear algebra, linear programming, and combinatorial optimization. The methods presented herein are discussed in light of biological applications; however, most of them are generally applicable and useful to analyze any chemical or stoichiometric reaction network.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Metabolic networks
- Reaction networks
- Stoichiometric models
- Constraint-based modeling
- Metabolic engineering
- Systems biology
5.1 Introduction
Systems biology is a relatively young and interdisciplinary research area that emerged as a logical consequence of the accumulating factual biological knowledge and the huge amounts of experimental biological data generated through novel measurement technologies. There are many different definitions of systems biology, but two important key features of almost all definitions are (i) a shift from reductionism to a systemic (holistic) perspective on biological systems and (ii) the synergistic combination and iterative use of experimental work (“wet lab”) and mathematical modeling (“dry lab”) to achieve this goal.
Biological systems—here we will focus on the cellular scale—show an inherent complexity both in the number and structure of its components (DNA, RNA, proteins, metabolites, etc.) and in the way these compounds interact with each other. Moreover, even in simple organisms like bacteria many interwoven processes take place concurrently in the cell including metabolism, signal transduction, gene regulation, DNA replication, and growth. It is therefore not surprising that a great variety of mathematical approaches has been employed to model the diversity of biological (sub)systems and phenomena. Many of those methods are well known from and frequently used in other fields, for example, differential equations for mechanistic and dynamic modeling of networks of interacting compounds. However, particular features of biological systems and of experimental biological data often require and enforce the development of novel, more tailored modeling approaches. For example, biological systems modeling is typically hampered by a great level of uncertainty: the data are notoriously noisy, and mechanistic details and kinetic parameters of biochemical reactions are often not known. In fact, what is often available to the modeler is qualitative biological knowledge (e.g., the network topology of interactions) and qualitative or semiquantitative trends from experimental data (e.g., increased concentration of a metabolite after deletion of a gene). Accordingly, qualitative or semiquantitative methods that provide meaningful biological insights and allow reasoning and predictions under such a knowledge base have attracted increased attention [11].
Systems analysis naturally implies the analysis of networks; sometimes, the terms networks and systems are even used as synonyms. However, there is a tendency to employ the term network when emphasizing the invariant structure of relationships between the components of a system. Based on their function, cellular networks can be divided into three major classes: (i) metabolic networks; (ii) signal transduction networks; and (iii) gene regulatory networks. Metabolic networks are responsible for uptake and degradation of substrates and nutrients and for synthesis of building blocks and energy needed for assembling all constituents of the cell. Signaling networks sense environmental signals and the internal state of the cell and induce appropriate responses, for example, by up- and down-regulating the expression of certain genes. Finally, gene regulatory networks can be seen as an abstraction of signaling networks; they capture causal links between genes. For example, the protein P A encoded in a gene G A may serve as a regulator for gene G B , that is, the expression of gene G B (and thus the abundance of protein P B ) depends on the activity of gene G A .
Obviously, the three networks do not operate in isolation, and there are many links between them. For example, the concentrations of certain metabolites serve as trigger for signaling pathways. However, signaling and gene regulatory networks mainly consist of proteins or/and genes that mutually activate or deactivate each other thereby generating signal or information flows. In contrast, metabolic networks are composed of metabolites and the metabolic reactions between them. A metabolic reaction is normally catalyzed by an enzyme and converts a set of reactants into a set of products. Accordingly, metabolic networks generate mass (or material) flows. Clearly, at the lowest level, almost all interactions in metabolic and signaling or regulatory networks take place by the action of biochemical reactions. The dynamic behavior of (bio)chemical reaction networks and their mass flows can be described by a class of ordinary differential equations (ODEs) having a particular structure (see Eq. (1) in Sect. 5.2). In this representation, one would not formally distinguish between the three types of networks. However, signaling or regulatory processes are often represented in a different way (not as reactions), especially if one analyzes the static network structure [69]. Therefore, signal and mass flows often imply different network representations and thus different techniques for their analysis.
This chapter is devoted to methods for stoichiometric modeling of metabolic reaction networks. Such methods rely solely on the structure (stoichiometry) of metabolic networks and do not require extensive knowledge on mechanistic and kinetic details of the involved reactions. As we will see, although purely based on network topology, stoichiometric modeling allows one to study important functional properties of metabolic networks and to derive various testable predictions. For this reason, stoichiometric modeling, in particular the large subclass of constraint-based modeling approaches [29, 34, 82, 92], has become one of the most popular and successful modeling frameworks in systems biology.
The main goal of this chapter (which largely extends an earlier contribution [72]) is to give an introduction on basic concepts and methods of stoichiometric modeling techniques for the computer-aided analysis of metabolic networks. We will discuss the mathematical foundations of the most important approaches and outline their applications in biology and biotechnology. From the mathematical point of view, stoichiometric network analysis uses methods from different fields of applied mathematics such as linear algebra, linear programming, combinatorial optimization, or graph and hypergraph theory. Whereas we will illustrate how biological questions in metabolic networks can be formalized mathematically (e.g., as a linear programming problem), we will not thoroughly describe how they are solved computationally (e.g., by the simplex algorithm) and assume that appropriate tools are available. Some algorithms and mathematical theory relevant to problems discussed herein are described in more detail in chapter Combinatorial Optimization: The Interplay of Graph Theory, Linear and Integer Programming Illustrated on Network Flow in this book. It should also be noted that although the methods presented herein are discussed in light of biological applications, most of them are generally applicable and useful to analyze any chemical or stoichiometric reaction network: Metabolites can be exchanged by arbitrary chemical substances and biochemical reactions by any chemical conversion. Hence, whenever we speak in the following about metabolic (reaction) networks, we may substitute “chemical” for “metabolic.”
Due to the large number of methods that have been developed and deployed for metabolic network analysis over the last 10–15 years, it is impossible to provide a complete review on all relevant methods. The given references, in particular reviews such as [29, 82, 92], should provide suitable links for further reading. A branch of theory that cannot be touched herein since it would easily fill another chapter is chemical reaction network theory (CRNT) and related approaches [17, 24, 32]. These methods aim at predicting qualitative dynamic properties (e.g., the existence of multiple steady states) from reaction network structure alone, and applications thereof can also be found in biology as described elsewhere [18, 24, 124].
5.2 Stoichiometric Models of Metabolic Networks
Metabolic reaction networks consist of metabolites and metabolic reactions connecting them by interconversions. Biochemical reactions are characterized by the following properties:
-
Stoichiometry: The stoichiometry of a reaction is captured in the reaction equation and specifies the participating species (reactants and products) and the molar ratios (stoichiometric coefficients) in which they are consumed or produced.
-
Reversibility: In principle, all chemical reactions are thermodynamically reversible. However, some metabolic reactions can be considered to be practically irreversible because they (nearly) exclusively proceed in one direction under biological conditions. Irreversible reactions reduce the potential behaviors a network can exhibit.
-
Gene–enzyme-reaction associations: Almost all biochemical reactions are catalyzed by enzymes. The connections between reactions and enzymes do not have to be unique because several enzymes (isoenzymes) may catalyze the same reaction, whereas multifunctional enzymes have the ability to catalyze several distinct reactions. Furthermore, each enzyme has one or several associated genes by which it is encoded (enzyme complexes are composed by several subunits, which may be encoded in separate genes). The resulting gene-enzyme-reaction associations [34, 135] thus allow one to relate properties of the reaction network to genomic information. Conversely, knowing the genes of an organism can be of great help and is often the main information source to build organism-specific metabolic network models (see below).
-
Reaction kinetics: Reaction kinetics describes the dynamics of the reaction based on the reaction mechanism and enzyme properties (including allosteric effectors). In many cases, these characteristics of a reaction are, at least in parts, unknown.
Stoichiometric analysis of metabolic networks is mainly based on the first three (static) properties, whereas reaction kinetics is usually not considered. One exception are certain thermodynamic data that are readily available and can be taken into account for some analyses (e.g., change of Gibbs free energy under standard conditions or upper/lower boundaries of selected reaction rates).
5.2.1 Tools and Databases for Reconstructing Metabolic Networks
Several resources, in addition to primary literature and review papers, have been made available during the last two decades to support the process of building stoichiometric models of metabolic networks. First, databases have been established to collect information about metabolic parts and capabilities of different organisms. Shortly after, computational tools have been developed to automate and standardize the procedure of reconstruction metabolic networks from this information. These tools typically use a whole genome sequence as input and search for genes that encode enzymes. Based on the findings and with the help of pathway reference maps, whole metabolic pathways are then compiled.
There are two prominent databases each of which covers metabolic networks of many different species: the BioCyc collection [15] and KEGG (Kyoto Encyclopedia of Genes and Genomes [63]). These databases have been developed to compile and store genome-wide networks of metabolic reactions and, to different extents, also regulatory and signaling processes. KEGG is an integrated database resource comprising genome, chemical, and network information. One of its most useful features is the collection of manually constructed reference pathway maps. KEGG derives orthologous groups of reactions through sequence comparison in the genomes of currently over 1300 organisms and thus makes it possible to easily compare their metabolic capabilities.
The BioCyc collection comprises more than 1900 organism-specific pathways and genomes. It started in 1996 with EcoCyc, which is now the BioCyc instance of Escherichia coli (E. coli). In 2000, the MetaCyc database [65] was established, which serves as a pathway reference database, and by now contains more than 1790 experimentally elucidated metabolic pathways from different organisms. In conjunction with the Pathway Tools [66], MetaCyc can be used to derive a new BioCyc instance from the annotated genome of an organism. A recent feature of the Pathway Tools is the generation of flux-balance analysis models ([79]; cf. Sect. 5.5) from a BioCyc database. This allows for a convenient conversion of the database content into a mathematical form that can then be used to support the reconstruction process (e.g., through the identification of blocked reactions; Sect. 5.5).
Two additional important resources for network reconstruction(s) are BiGG (Biochemically, Genetically, and Genomically structured genome-scale metabolic network reconstructions [111]) and Model SEED [51]. The BiGG database contains stoichiometric models derived from metabolic network reconstructions that have been extensively validated and curated. All models in BiGG are available in SBML format (see Sect. 5.6) for academic use. In contrast to BiGG, which relies on manual curation, Model SEED uses a largely automated pipeline for generating draft metabolic models of an organism starting from an assembled genome sequence. Several hundred network reconstructions have been generated through this pipeline.
Reconstructed genome-scale networks typically comprise between several hundred up to several thousand reactions and metabolites [34, 92]. For eukaryotic organisms, compartments within the cell (mitochondria, chloroplasts, etc.) need often to be considered, which increases network size. A list of available reconstructed metabolic models that can directly be used for stoichiometric network analysis can be found at http://gcrg.ucsd.edu/InSilicoOrganisms/OtherOrganisms. A more detailed survey and comparison of metabolic databases can be found in [64]. Additional information about (automatic) metabolic network reconstruction and constraint-based modeling is presented in [47].
Many of the resources described above focus on genome-scale reconstructions of metabolic networks. Nevertheless, depending on the question at hand, it can be sufficient to study medium-scale core models, which typically concentrate on the central metabolism. Pathways, whose evidence or function is unclear or which are less important for certain aspects, are then excluded. For example, models of the often studied central metabolism typically contain 80–150 reactions.
5.2.2 Formal Description of Metabolic Networks
Having compiled all components of a metabolic reaction network, the network structure can be formally described as follows:
-
m: number of species (metabolites).
-
q: number of reactions (if available, gene-enzyme-reaction-associations can be stored as Boolean relationships for each reaction [135]).
-
N: m×q stoichiometric matrix: each row corresponds to one species, and each column to one of the reactions. The matrix element n ij stores the stoichiometric coefficient of species i in reaction j; it is negative if the metabolite i is consumed, positive if it is produced, and zero if it is neither consumed nor produced in the reaction. If a reaction is reversible (see below), then it is necessary to specify forward and backward directions and to assign the stoichiometric coefficients. with respect to the forward direction.
-
Rev: the set of reversible reactions
-
Irrev: the set of irreversible reactions (Rev∩Irrev=∅)
It is convenient to directly include processes such as transport (e.g., substrate uptake or exchange of metabolites between different compartments) and biomass synthesis in this formalism by treating them as pseudo reactions. The biomass synthesis reaction is often contained in the stoichiometric matrix and describes the (cumulative) molar requirements of energy (ATP) and building blocks such as amino acids, fatty acids, nucleotides, etc. needed to build the major constituents (macromolecules such as proteins, DNA, RNA, lipids, etc.) of one gram biomass dry weight.
Important characteristics of network models are the boundaries and the connections to the environment. Related to this issue is the notion of internal and external metabolites (or species). Internal species are explicitly balanced in the network model, and, hence, they are included in N. In contrast, external species are thought to be sinks or sources, which in most cases lie physically outside the system (for example, substrates or products) but could also be located inside the cell (a typical example would be water). For completeness, external species can be included in the stoichiometric matrix; however, for most analyses (in particular, for those that rely on steady state; see Sect. 5.5), their corresponding rows in N will be removed.
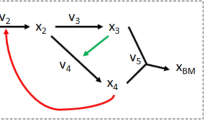
Figure 1 depicts a simple example network, which we call N1 throughout this chapter, and its corresponding variables. This network comprises six internal metabolites, four external species (external “substrate” A(ext) and external products P(ext), D(ext) and E(ext)), and ten reactions (of which R7 is considered to be reversible). As described above, only the internal species were included in N. Notably, when excluding external metabolites it may happen that a reaction (column) in N contains no positive (e.g., R2, R3, R10) or no negative (R1) stoichiometric coefficients.

Example network N1: graphical representation and stoichiometric matrix
5.2.3 Reaction Networks Are Hypergraphs
Most reactions in metabolic networks are bi- or even trimolecular, that is, in general, a reaction connects a set of reactants with a set of products. For this reason, metabolic networks are a special class of directed hypergraphs [77] and can therefore not per se be treated as graphs (see also Sect. 5.3). A directed hypergraph ℋ is a tuple ℋ=(V,E) with a set V of vertices and a set E of directed hyperedges. Directed hyperedges are also called hyperarcs, and each hyperarc h consists in turn of a set of start nodes (the tail X) and a set of end nodes (the head Y): h=(X,Y) with X,Y⊂V. Directed graphs are special cases of directed hypergraphs where X and Y contain exactly one node for each arc limiting the scope to 1:1-relationships, whereas directed hypergraphs can represent arbitrary n:m-relationships. For example, for a stoichiometric reaction 2A+B→C+3D+E, we have X={A,B} and Y={C,D,E}. This formalism describes correctly the sets of reactants and products; however, it would not account for the stoichiometric coefficients. One can extend this representation by adding to each hyperarc two functions assigning the stoichiometric coefficients for the nodes in X and Y, respectively [77]. However, in practice it is more convenient to use the stoichiometric matrix as introduced above, which in fact represents the incidence matrix of the spanned hypergraph.
5.2.4 Linking Network Structure and Dynamics
The stoichiometric matrix N is fundamental not only for stoichiometric but also for dynamic modeling of metabolic networks. Generally, the changes of the species’ concentrations over time can be described by the following system of differential equations:
The m×1 vector c(t) contains the metabolite concentrations, typically in mmol per gram cell dry weight, mmol/gDW. The q×1 vector r(t) comprises the (net) reaction rates at time t, normally in units of mmol/(gDW⋅h). The vector r(t) is also called a flux vector or flux distribution and is usually a function of the metabolite concentrations and a parameter vector p:
As mentioned above, the uncertainties in describing a metabolic system dynamically are concentrated within the kinetic description f of the reaction rates, whereas N, the structural invariant of system (1), is usually known. As long as the available data and knowledge base allow kinetic modeling of a metabolic system, the modeling approach having potentially the highest predictive and explanatory power will be the preferred. However, due to limited knowledge, predictive kinetic models of metabolic networks comprise rarely more than 20 state variables. In larger systems, one therefore has to restrict the analysis on static network properties. However, structural relationships captured in N are clearly of fundamental importance and impose constraints for the dynamic behavior. A typical example is conservation relations limiting the feasible space of the trajectories; see Sect. 5.4. Furthermore, chemical reaction network theory and related approaches [17, 18, 24, 32] demonstrate that important dynamic properties of reaction networks (such as the ability to exhibit bistable behavior) can sometimes be excluded by network structure alone.
5.3 Graph-Theoretical Analysis of Metabolic Networks
Statistical network theory approaches seek to identify emergent topological properties and dynamical regularities of large-scale networks and have frequently been applied to networks from diverse fields such as the Internet, social networks, or traffic networks [1, 91, 131]. For example, one key result found was that many real-world networks exhibit a small-world or/and scale-free topology [1, 3, 131, 147] These studies on global network architectures are usually based on graph-theoretical measures of the network topology. Three general key measures are the following:
-
(i)
Connectivity and degree distribution: The connectivity (or degree) k of a node is the number of links it is attached to, and P(k) is the degree distribution of the graph. For example, in statistically homogeneous networks (Erdős–Réyni random graphs), the connectivity follows a Poisson distribution, implying that nodes with many more edges than the average degree are extremely rare [131]. In contrast, scale-free networks have a higher probability to contain (few) dominating hubs with very high degrees resulting in a power-law distribution of connectivities with parameter γ: P(k)∼k −γ [1].
-
(ii)
(Shortest) Path length: A path is a sequence of nonrepeating edges connecting a start node with an end node, and its length is the number of involved edges. Two particular network measures are the maximum and the average shortest path length between all pairs of nodes. Both measures are relatively small in small-world and scale-free networks when compared to standard random networks of the same size.
-
(iii)
Clustering: In clustered networks there is a high probability that two neighbors of a given node are also connected by an edge.
Biological networks have also been analyzed based on these graph-theoretical measures, and it has been shown that many of them show a scale-free structure, including metabolic networks [4, 59]. However, in the case of metabolic networks, the question arises how their structure can be treated as graphs at all. As already discussed in the previous section, metabolic networks are hypergraphs where the reactions are hyperedges connecting sets of start (reactant) nodes with sets of end (product) nodes. In a graph this is not allowed, an edge connects exactly one start with one end node. For example, reaction R9 in Fig. 1 is not compatible with a graph. Thus, before applying graph-theoretical tools, a transformation of metabolic reaction networks from their hypergraph into a graph representation is necessary. Different transformations are possible; the most frequently used ones are the following two:
-
(1)
Substrate (compound) graph: Each metabolite becomes a node. A directed edge is introduced between two metabolites A and B if A is a reactant in a reaction where B is a product (sometimes, alternatively, an edge between A and B is introduced if both metabolites participate in the same reaction).
-
(2)
Bipartite graph: Both metabolites and reactions are nodes and each directed edge connects either a metabolite with a reaction (if the metabolite is a reactant of this reaction) or a reaction with a metabolite (if the latter is a product in this reaction).
Figure 2(a) shows a simple reaction network with its associated representations as substrate graph and bipartite graph. A disadvantage of the substrate graph is that different reaction hypergraphs can have the same substrate graph representation, whereas bipartite graphs can be uniquely reconverted to the original hypergraph.

(a) Example network in hypergraph, substrate graph, and bipartite graph representations and (b) after removal of reaction R2
Analysis of graph representations of genome-scale metabolic networks revealed that these topologies have scale-free character and possess the small-world property [4, 59]. This is rather intuitive since most metabolites are only weakly connected, whereas a few dominating hubs such as the metabolic cofactors ADP, ATP, NAD(P), NAD(P)H or very central carbon metabolites (like pyruvate) exist. The overall topology and the major hubs of these networks are well conserved among species [4, 59]. In addition, it was found that the average (shortest) path length is quite low (and almost identical in all considered organisms) proving the small-world property. As one consequence of all these findings, the network topology of metabolic networks has been shown to be robust against random removal of nodes; only when central hubs are deleted, network fragmentation occurs. Scale-free networks, whose topology emerges by the preferential attachment of edges to nodes with higher connectivity, give also an intuitive explanation how metabolic networks could have been evolved to large-scale networks.
A somewhat different perspective on the global architecture of metabolic networks was presented in [25]. This study highlights the bow-tie structure of the metabolism: a core (central metabolism) of relatively few intermediate common currencies (ATP as energy and NAD(P)H as reduction equivalents; 12 precursor metabolites serving as building blocks) allows the cell to take up a wide range of nutrients and to produce a large variety of products and complex macromolecules. The authors also argue that metabolic networks are rather scale-rich than scale-free.
The results obtained from a graph-theoretical perspective are helpful for understanding the global organization of metabolic networks. However, simplifying the hypergraphical structure of metabolic networks to graphs may strongly limit the interpretability of the results, in particular when studying functional properties [8, 77]. Look again at Fig. 2(a). In the hypergraph, we can easily see that four reactions are required to produce E from A. However, in the two graph representations, we find a connection (path) via three edges or three reactions, respectively. Furthermore, in Fig. 2(b), we deleted reaction R2 mimicking a knock-out of the gene encoding the catalyzing enzyme of R2. Clearly, from the hypergraph representation we can conclude that synthesis of E from A is then not possible anymore. However, if we searched for paths in the graph representations, then both in the substrate and in the bipartite graph, we would find a (shortest) path connecting A with E wrongly indicating that E could still be produced from A. Here, the AND relationship for reaction R2 needs to be accounted for (species B AND C are needed). Generally, short path lengths in the graph prove neither that synthesis pathways between substrates and products exist nor that they are short. Instead, shortest paths in the graph representation rather indicate shortest “influence paths” between nodes along which a perturbation of a metabolite’s concentration could spread over the network and affect the concentration of another metabolite [77]. In fact, a concentration change of one of the two reactants in bimolecular reaction will affect the reaction rate even if the other reactant remains constant.
Graph analysis of metabolic networks can thus be useful to get a quick overview on the global network topology; however, the hypergraphical structure must explicitly be taken into account when studying network function. All techniques described in the following sections fulfill this requirement.
5.4 Stoichiometric Conservation Relations
Conservation relations (CRs) are weighted sums of metabolite concentrations that remain constant in (an ODE model of) a reaction network, irrespective of the chosen reaction kinetics in Eq. (2). A typical example for metabolic network models is [NADH]+[NAD+]=CONST (brackets indicate species concentrations). NADH is known to serve as an electron carrier in the cell. In many redox-coupled reactions, two electrons from a donor are taken up by the oxidized form NAD+ yielding NADH: NAD++H++2e−→NADH. In other reactions, NADH in turn serves as donor of electrons thereby getting back to the NAD+ state (the reverse equation above). Thus, whenever NAD+ is consumed, NADH is produced, and vice versa. Accordingly, the sum of both concentrations remains constant whatever the dynamic concentration changes are. If one of the two metabolites participates in a reaction, then the other does so as well but on the opposite side of the reaction equation. Therefore, the corresponding row of NAD+ in the stoichiometric matrix N is exactly the same as for NADH, except that it is multiplied by −1. This implies that these two rows are linearly dependent. In fact, linear dependencies between rows (species) in N uniquely characterize CRs [49]. To show this, we identify a CR by an m×1 vector y and observe that, by definition of CRs, y fulfills, at all time points t,
with fixed constant S. Differentiation of both sides of the equation with respect to t and substituting the right-hand side of Eq. (1) for \(\dot{\mathbf{c}}(t)\) yields
Since we demand that the last equation must hold for any t and for any chosen kinetic rate law, it follows that
or, equivalently, after transposing the system,
with 0 being the q×1 zero vector. Hence, each CR y corresponds to a set of linearly dependent rows (species) in the stoichiometric matrix, and the coefficients of y are determined in such a way that the resulting linear combination of the species rows yields 0. In other words, a CR y lies in the left null space of N or, equivalently, in the right null space (or kernel) of the transpose of N. According to basic rules of linear algebra [130], the dimension of the left null space of N is \(m-\operatorname{rank}(\mathbf{N})\), that is, conservation relations only exist if \(\operatorname{rank}(\mathbf{N})< m\). Then, \(m-\operatorname{rank}(\mathbf{N})\) linearly independent CRs—forming a basis of the left null space—can be found completely characterizing the space of CRs.
Network N1 (Fig. 1) does not contain any CR since \(\operatorname{rank}(\mathbf{N})=m=6\). This is a consequence of not explicitly considering external metabolites; would we include the four external metabolites in N (yielding then a system with 10 reactions and 10 metabolites), matrix N would have rank 9, resulting in one CR simply stating that the sum of all species concentrations remains constant. Such an “overall” CR is typical for systems with proper mass balances.
For further illustration, we consider now an even simpler example network with four metabolites A,B,C,D and just one reaction: A+B→C+2D. In this case, we have
and, hence, three linearly independent CRs exist because \(m-\operatorname{rank}(\mathbf{N})=4-1=3\). They can be found by searching for linearly independent solutions y that solve
yielding a basis for the space of CRs, which we arrange as columns in a matrix Y. One possible instance could be
The three columns express the following CRs: (1) [A]−[B]=S1=CONST; (2) [A]+[C]=S2=CONST; (3) 2[B]+[D]=S3=CONST. Furthermore, each linear combination of these CRs forms another CR, for example, (1)+(2)=2[A]−[B]+[C]=S1+S2=S4=CONST. The space of CRs is completely described by \(\operatorname{span}(\mathbf{Y})\), that is, all linear combinations of columns in Y yield valid CRs, and each CR corresponds to a unique combination of the basis vectors in Y. This property is independent of the chosen basis Y. However, sometimes one is interested in support-minimal CRs, that is, in CRs with a minimal number of involved species [49]. For the example above, [A]−[B]=CONST is such a support-minimal CR, whereas 2[A]−[B]+[C]=CONST is not since a subset of the three involved species A, B, C already spans a CR. Furthermore, nonnegative CRs (where all nonzero coefficients are positive) are also of special importance since they indicate so-called conserved moieties. The case of NADH and NAD+ is such an example where the NAD+ molecule is the conserved moiety (NADH consists of the scaffold of NAD+ plus one proton and two electrons). Enumerating support-minimal or/and signed CRs is mathematically the same problem as computing elementary modes lying in the right null space of N (see Sect. 5.5), and the algorithm outlined there can be applied here as well.
Identifying the CR subspace is a simple task but brings important benefits also beyond detecting conserved moieties [20, 49, 105]. CRs provide a nice example how stoichiometric relations affect systems dynamics: CRs confine the possible dynamic behavior of the species in a given reaction network (Eq. (1)) to a subspace with \(m-\operatorname{rank}(\mathbf{N})\) dimensions. The value of any CR cannot change, irrespective of the chosen kinetics. In our small example above, if we had [A]−[B]=6 at the beginning, then the system could never reach a state where the difference of [A] and [B] is unequal to 6. For this reason, CRs express systems redundancies that can be exploited for model reduction. Generally, one can remove \(m-\operatorname{rank}(\mathbf{N})\) state variables from the ODE system (1) without losing any relevant information: the removed species can, at any time point, be calculated from the remaining state variables by using the algebraic relationships captured by the CRs (see also chapter Introduction to the Geometric Theory of ODEs with Applications to Chemical Processes of this book). In our example above, we could thus remove three species, for example, B, C, D and model only A explicitly as a state variable. With the initial concentrations of all species (by which we can compute the constants for all CRs) we can replace the differential equations for B, C, and D and derive their concentrations from A at any time point by using the algebraic relationships of the CR [105]. This type of model reduction is often routinely done for ODE models of reaction networks; for some analyses, it is even necessary to avoid a singular Jacobian of system (1).
5.5 Steady-State and Constraint-Based Modeling
5.5.1 Steady-State Flux Distributions and the Null Space of N
The cellular metabolism usually involves fast reactions and a high turnover (i.e., small turnover times) of metabolites when compared to regulatory events. Therefore, analysis of metabolic networks is often based on the approximation that, on longer time-scales and under constant external conditions, metabolite concentrations and reaction rates do not change. Applying this steady-state assumption to Eq. (1) leads to the central steady-state or (metabolite) balancing equation
This homogeneous system of linear equations expresses the algebraic consequence of steady-state, namely that, for each metabolite, the sum of reaction rates weighted with the metabolite’s stoichiometric coefficients must sum up to zero. In other words, production and consumption of each metabolite are equal. This equation is very similar to Kirchhoff’s first law for electric circuits, see chapter Mathematical Modeling and Analysis of Nonlinear Time-Invariant RLC Circuits of this book. The latter is based on the incidence matrix of the underlying graph spanned by the circuit. Here, N fulfills the same role as the incidence matrix of the reaction network.
Apart from setting the derivatives of the concentrations to zero in Eq. (1), there is an important change how we treat the reaction rates: the latter depend normally on metabolite concentrations and kinetic parameters (Eq. (2)) but are now considered as an independent variable r. In this way, we “get rid” of the unknown kinetic relationships and consider all flux vectors that solve Eq. (10) as potential solutions. Clearly, in the real (dynamic) system, only a small subset of those rate vectors might be attainable, but it is nevertheless convenient and useful to consider the complete space of potential solutions of Eq. (10). It worth noting that Eq. (10) is fulfilled also in oscillating systems (see, e.g., [150]) for the averaged reaction rates.
The trivial solution r=0 always fulfills Eq. (10). However, since this would represent thermodynamic equilibrium, we are obviously interested in other solutions. Here it becomes clear why we distinguish between external and internal metabolites (Sect. 5.2): would we include external substrates and products in N, then flux distributions where the cell converts substrates into products would not be part of the null space. It is therefore reasonable to demand the steady-state condition only for the internal metabolites.
Since the number of reactions q in real networks is usually much larger than the number m of internal metabolites, an infinite number of flux distributions r usually solves the system of equations (10). From linear algebra it is known that all solutions are contained in a linear subspace called the (right) null space (or kernel) of N (in contrast to the left null space studied in the context of conservation relations; Sect. 5.4). The dimension of the null space, the nullity, is \(q-\operatorname{rank}(\mathbf{N})\), which equals the number of linearly independent solutions that can be found for Eq. (10) [130]. Similar as for conservation relations, we can thus easily compute \(q-\operatorname{rank}(\mathbf{N})\) basis vectors of the null space and arrange them in a kernel matrix K. Each column in K represents a steady-state flux distribution, and all other steady-state rate vectors r can then be constructed by a unique linear combination a of the columns in K:
Notably, whereas infinite many kernel matrices K exist if the null space dimension is larger than zero, the solution a in Eq. (11) is unique for given K and r.
For illustration, Fig. 3 shows a simple reaction network (called N2) together with its formal representation. The null space has dimension \(q-\operatorname{rank}(\mathbf{N})=4-2=2\). Accordingly, the kernel matrix must have two columns, and one possible instance reads:
One particular steady-state flux vector in this network would be r=(2,1,1,2)T, which can be constructed from K by using a=(2,−1)T.

Example network 2 (N2) with its stoichiometric matrix
Although the kernel matrix is not unique, some important general network properties can be derived from a null space basis as discussed next.
5.5.2 Uncovering Basic Network Properties from the Kernel Matrix
It may happen that a reaction must have a zero flux if a network is in steady state; we call such reactions blocked reactions. A simple example is a reaction in which a “dead-end” metabolite participates, that is, if the stoichiometric coefficient of this metabolite is zero in all other reactions. It follows immediately that the flux through this reaction must be zero since otherwise the metabolite cannot reach a steady state (see Fig. 4(a)).

Examples of blocked and coupled reactions. (a) Reactions R2 and R3 are blocked because of dead-end metabolite D. (b) Reaction R1 is blocked. (c) Reactions R1, R2, and R3 are coupled
Other reactions may become blocked because they are in a pathway leading to a dead-end metabolite (as R2 in Fig. 4(a)). There can also be more complicated cases as shown in Fig. 4(b): reaction R1 would produce a metabolite B from A. However, there is only one pathway consuming B in which it will in turn be recycled. For this reason, reaction R1 is blocked in steady state since otherwise B would accumulate.
With Eq. (11) we can derive a criterion for identifying blocked reactions since the latter must have a corresponding zero row in the kernel matrix. Then, any linear combination of the columns in K will yield a zero for the rate of this reaction. Blocked reactions often indicate reconstruction errors (which are sometimes not easy to find in networks with thousands of reactions), for example, due to missing elements. One can then search for appropriate corrections or remove blocked reactions when further analyzing the network with steady-state methods.
Another network feature that can be uncovered by the kernel matrix is coupled reactions (also called enzyme subsets or correlated reaction sets). For any steady-state flux vector, coupled reactions operate with a fixed ratio in their rates [13, 97], that is, there is a strong dependency between the fluxes. Typical examples are reactions in a linear pathway as R1, R2, and R3 in Fig. 4(c), which must have identical rates in steady state. The same holds for R3 and R9 in network N1 (Fig. 1), which are also coupled with a rate ratio of 1. In N2 (Fig. 3), reactions R1 and R4 are coupled, again with a ratio of 1, demonstrating that coupled reactions are not necessarily a sequence of conversion steps. Coupled reactions can again be found by the kernel matrix: their corresponding rows in K differ only by a (scalar) factor (indicating the constant ratio of their rates). Often, one can find sets of coupled reactions (if a reaction R1 is coupled with reaction R2 and reaction R2 with another reaction R3, then also R1 with R3). In fact, each reaction belongs to one equivalence class of coupled reactions (many reactions are the only member of their own class). Finding coupled reactions has some benefit: it is expected and has been observed that those reactions are commonly regulated [94, 119]. Moreover, a reaction will become blocked if one of its coupled partner reactions is removed from the network.
Other important conclusions can be drawn if K is block-diagonalizable. Then, certain subnetworks can be identified in the system that are either completely disconnected or whose steady-state fluxes are completely independent from the fluxes in the rest of the network [49].
The kernel matrix thus enables one to quickly analyze some basic properties of the network. However, apart from the nonuniqueness of K, a major disadvantage of the kernel matrix is that the reversibilities of the reactions (i.e., sign restrictions on some reaction rates) are not taken into account. For example, since reaction R2 in N2 is irreversible, the second column of K in (12) is not a valid flux distribution in this network because of the negative sign for R2. Furthermore, unblocked reactions may become blocked and uncoupled reactions coupled (or hierarchically coupled, see [13, 26]) under the reversibility constraints. It can even happen that the null space has a large dimension although nothing than the trivial (zero) flux distribution is feasible in the network. Hence, the “real” degrees of freedom can only roughly be estimated from the dimension of K. As we will see later in this section, these shortcomings will be overcome by constraint-based approaches and methods of pathway analysis.
5.5.3 Metabolic Flux Analysis
The aim of metabolic flux analysis (MFA) is to determine a specific steady-state flux distribution of a metabolic network, for example, from an experiment. Since Eq. (10) is underdetermined (the dimension of the null space quantifies the degrees of freedom), one needs measurements of at least some reaction rates to calculate some or even all unknown rates. Whereas internal fluxes can usually not directly be measured experimentally, it is often possible to quantify several uptake rates (of substrates and oxygen) and excretion rates (of products such as carbon dioxid, fermentative products, etc.). For this purpose, microorganisms or cell cultures are cultivated under controlled steady-state or pseudo-steady-state conditions, for example, in a bioreactor. Moreover, the growth rate μ (normally given in [h −1]) of the biomass can often be determined experimentally. One can therefore divide the steady-state equation (10) into the known/measured (index k) and unknown (index u) part, possibly after rearranging the columns in N and elements in r:
This leads to the central equation for MFA:
With l measured rates (in r k ), the number of unknown rates in r u is s=q−l. Since N k and r k are known, their product becomes a vector, and, hence, Eq. (14) forms a standard inhomogeneous system of linear equations. The general solution for r u is given by [74]
\(\mathbf{N}_{u}^{\#}\) is the Penrose pseudo inverse of N u . It has dimension l×m and exists for any matrix and gives a (particular) least-squares-solution for Eq. (14). K u denotes the kernel matrix of N u . K u solves the homogeneous variant of Eq. (14), and linear combinations of the columns of K u (expressed by a) therefore characterize the degrees of freedom for r u . In the simplest case, N u is an m×m square matrix with full rank, where \(\mathbf{N}_{u}^{\#}\) coincides with the standard inverse \(\mathbf{N}_{u}^{-1}\), and where K u is the zero vector. One can then compute a unique and exact solution for all unknown rates. In general, however, based on the rank of N u , the scenario equation (14) has to be classified with respect to two characteristics [74, 143]: (i) determinacy: a scenario is either determined (\(\operatorname{rank}(\mathbf{N}_{u})=s\)) or underdetermined (\(\operatorname{rank}(\mathbf{N}_{u})< s\)); (ii) redundancy: a scenario is either redundant (\(\operatorname{rank}(\mathbf{N}_{u})< m\)) or nonredundant (\(\operatorname{rank}(\mathbf{N}_{u})=m\)). Since these two properties are independent, four possible cases can be distinguished. The case where pseudo-inverse and standard inverse coincide (\(m=s=\operatorname{rank}(\mathbf{N}_{u})\) is a determined and nonredundant system. If a scenario is underdetermined, not all unknown rates can be determined uniquely, but some could be calculable, namely those rates that have a corresponding zero row in K u [74]. If a system is redundant (which is possible for the determined and undetermined case), then it usually contains inconsistencies with respect to the measured rates, which can be balanced by statistical approaches before computing the uniquely calculable rates [129, 143]. In this context, large inconsistencies will point to gross measurement or modeling errors.
We discuss an example of an MFA scenario for network N1 (Fig. 5). Suppose that the rates R1=5, R2=2, and R3=1 were measured. This results in a nonredundant and underdetermined system where the rates R6=3, R8=1, R9=1, and R10=2 would be uniquely calculable. In contrast, R4, R5, and R7 cannot be determined since they make up two parallel pathways whose fluxes cannot be resolved from the measurements. If we measured in addition R10=0, we would have an underdetermined redundant scenario, and the given rates would indicate some degree of inconsistency.

Example for metabolic flux analysis: stationary rates of R1, R2, and R3 were measured (blue bold arrows). Using this information, one can determine the fluxes of R6, R8, R9, and R10 (green dashed arrows). The other rates remain unknown (thin arrows)
MFA has become a standard method in microbiology and bioprocess engineering [129]. It is routinely used to characterize and quantify flux distributions in the central metabolism of microbes and also higher eukaryotic cells grown under controlled conditions. A general problem of MFA is that, even if all exchange rates are measured, not all internal rates can be determined uniquely. This problem is induced by parallel pathways or internal cycles in metabolic networks leading to dependencies in N u that cannot be resolved by measuring exchange fluxes only [74]. Then, further assumptions must be made, or isotopic (13C) tracer experiments could be employed to deliver further constraints, whose experimental and mathematical treatment is, however, much more complicated [149]. In genome-scale networks, neither MFA nor 13C-MFA can be used due to the large number of degrees of freedom (often several hundreds).
Again, we note that MFA as described above does not account for the sign restrictions of irreversible reactions. An alternative approach for MFA that includes these constraint is flux variability analysis introduced in a later subsection.
5.5.4 Constraint-Based Modeling and Flux Balance Analysis
5.5.4.1 Principles of Constraint-Based Modeling
As we have seen in a previous section, the assumption of steady state reduces the space of relevant flux distributions in a reaction network from “everything is possible” to the null space of N. The basic idea of the constraint-based modeling approach is to incorporate additional well-defined physicochemical and biological constraints that further limit the space of feasible stationary flux vectors [82, 102, 103]. The most important standard constraints considered can be expressed by linear equations or/and inequalities:
Definition 1
(Standard constraint-based problem for metabolic reaction networks)
Standard constraint-based problems for metabolic reaction networks are imposed by the following linear constraints (or subsets thereof):
- (C1):
-
Steady state: Nr=0
- (C2):
-
Capacity/Reversibility: α i ≤r i ≤β i
Generally, upper or lower boundaries for fluxes are often known for exchange (uptake/excretion) reactions; for internal reactions, the v max value might be available from biochemical studies, which can be helpful to specify flux boundaries. For irreversible reactions, one usually sets α i =0. If the boundaries are unknown, then one may set them to large absolute values or even infinity (±∞). Notably, for some reactions, one may have positive lower boundaries, for instance, for the nongrowth associated ATP demand for maintenance processes in the cell (often included as a pseudo reaction in metabolic network models). C2 can be simplified to the following pure reversibility constraint when capacity values are not known or not of interest:
- (C2′):
-
Reversibility: r i ≥0 (∀i∈Irrev)
- (C3):
-
Measurements: r i =m i (for measured/known rates i)
- (C4):
-
Optimality: maximize r w T r=w 1 r 1+w 2 r 2+⋯+w q r q
The linear objective function is defined by a q-dimensional vector w specifying the linear combination of reaction rates to be maximized.
By this definition, null space and metabolic flux analysis can be seen as special constraint-based methods operating on the constraints C1 and C1+C3, respectively. We also note that the constraint-based problem as stated above can be seen as a generalization of the LP formulation of the maximum network flow problem presented in chapter Combinatorial Optimization: The Interplay of Graph Theory, Linear and Integer Programming Illustrated on Network Flow of this book. Basically, in the latter, the graph incidence matrix replaces the stoichiometric matrix of the hypergraphical metabolic network, and the source (s) and target (t) nodes are treated as external “metabolites.”
Constraints C1 and C2′ are solely defined by network structure and are the basic constraints taken into account by virtually all constraint-based methods. The set  of all flux vectors r obeying the two constraints
of all flux vectors r obeying the two constraints

form a convex polyhedral cone [9, 109], which is, in stoichiometric studies, often referred to as a flux cone. As it arises from C1 and C2′, this cone is an intersection of the null space with the positive half-spaces of the irreversible reactions. An example of a two-dimensional polyhedral cone in a three-dimensional space (network with three reactions) is shown in Fig. 6. As suggested by this picture, the edges of such a cone are of eminent importance; they are subject to pathway analysis (Sect. 5.5.5). The constraints C2, C3, and C4 further restrict the flux cone to a smaller subset of flux vectors yielding, in general, a polyhedron, which can be bounded (then also called a polytope) or unbounded. Note that the optimality condition C4 is not always considered as a constraint. However, one may treat it as such since the optimality criterion reduces the space of relevant flux vectors similar to the other constraints. The optimality condition C4 is central to the approach of flux balance analysis, which is introduced next.

Example of a convex polyhedral cone for a minimalistic network with one metabolite and three reactions. The cone is spanned by convex combinations of E1 and E3 (the extreme rays) and is unbounded in this (open) direction. E1, E2, and E3 correspond to the elementary modes of the system (see Sect. 5.5.5)
5.5.4.2 Flux Balance Analysis
Flux balance analysis (FBA) seeks to identify particular flux distributions that keep the network in steady state (constraint C1), are feasible with respect to reversibility and capacity (C2), and maximize a linear objective function (C4), optionally in the context of some known or measured rates (constraint C3). The characteristic and necessary assumption of FBA is optimality (constraint C4). Together with the other constraints, it gives rise to a standard linear optimization (or linear programming) problem (see [9] and chapter Combinatorial Optimization: The Interplay of Graph Theory, Linear and Integer Programming Illustrated on Network Flow of this book). The most frequently used objective function is maximization of biomass synthesis (growth), which seems to be a physiologically realistic cellular objective, at least for some micro- and unicellular organisms growing under certain (e.g., substrate-limiting) conditions [31, 34, 55]. Importantly, since the substrate uptake rate or its upper boundary must be set to a finite value (as the problem would otherwise be unbounded), the optimal flux distribution with respect to growth rate delivers the largest amount of biomass that can be produced by it, that is, what is then effectively optimized is the biomass yield. Another meaningful objective function mimicking “natural objectives” is maximization of ATP (cf. [115], where different objective functions where tested and validated). For biotechnological applications, one is typically interested in the maximal yield of a certain product that can be produced from a given substrate [145]. The vector w in the linear objective function used in C4 encodes the optimization criterion and weights the reaction rates. For maximizing the biomass yield, for example, only the coefficient corresponding to the growth rate is set to one, and all others to zero.
As an example for an FBA problem, suppose that we want to maximize the amount of P synthesized from substrate A in network N1 (Fig. 1), that is, we maximize the rate of reaction R2. Assuming that the network can maximally “metabolize” two units of A per unit of time, the variables and constraints for the resulting FBA problem (cf. Definition 1) read:
We see that all α i =0 except α 7=−∞ because R7 is the only reversible reaction. β 1 was set to the maximal uptake rate of A, and only w 2 is nonzero since we want to maximize R2. Using standard computer routines like the simplex algorithm or more sophisticated computational methods ([9], chapter Combinatorial Optimization: The Interplay of Graph Theory, Linear and Integer Programming Illustrated on Network Flow of this book), one can easily solve such a linear optimization problem. In our example, one could get a solution as displayed in Fig. 7, showing that the maximal rate of R2 (synthesis of P) is two, that is, the maximum yield P(ext)/A(ext)=r R2/r R1 is unity.

Optimal flux distribution for producing maximal amount of P from A in N1 (see FBA problem (17))
5.5.4.3 Applications of Flux Balance Analysis
FBA has become the most popular method of the constraint-based approach, and sometimes both terms are used as synonyms. In the following, we outline major application areas of the standard FBA formulation, whereas advanced variants of FBA for more specific questions are discussed later on.
Predicting Optimal Behavior and Reaction Essentialities
As already mentioned above, some microorganisms such as E. coli have been shown to behave stoichiometrically optimal with respect to biomass yield, at least under substrate-limiting conditions [31, 33, 34, 55]. In the case of genetically modified organisms or after changing the environmental conditions, the optimal state is often reached after adaptive evolution, where many consecutive generations are cultivated under selective pressure [37, 55]. In both cases, it is straightforward to use FBA to calculate the optimal (maximal) biomass yield and thus the expected optimal behavior.
The effect of genetic modifications on the optimal behavior can also be assessed with FBA. For example, the deletion of certain reaction(s) in the network by corresponding gene knock-out(s) can be incorporated as constraint C3 (the respective reaction rate is set to zero). After reoptimization one can check whether the maximal growth rate is reduced; it can never increase since the FBA problem of the mutant has more constraints than the wild type. Moreover, FBA can also identify reaction deletions that completely block growth, that is, where the maximal growth rate becomes zero. In this way, one can predict which reactions/genes are essential and which are (potentially) dispensable for growth or for any other network function. In many studies, it was shown that FBA predictions of mutant viability correlate well with the observed phenotypes of microorganisms (see, e.g., [30, 36]). A false negative prediction (a cell can perform a certain function (such as growth) in an experiment although FBA predicted the opposite) implies a falsification of the network structure since some alternative pathway(s) must be missing. Conversely, for a false positive prediction (a network predicted to be functional by FBA is nonfunctional in an experiment), one cannot exclude that this mismatch was caused by unknown capacity or regulatory constraints. Thus, FBA predicts the potential capability of the reaction network to tolerate a knock-out.
Flux Coupling Analysis and Blocked Reactions
FBA can be used to detect coupled and blocked reactions [13, 26]. For example, to identify all blocked reactions, one maximizes and minimizes each reaction rate separately with the constraints C1 and C2. A blocked reaction fulfills that both its minimum and maximum rate is zero. In contrast to the analysis of the kernel matrix described in an earlier section, reversibility constraints are explicitly considered, which may result in more blocked/coupled reactions as found by the kernel matrix alone. Moreover, hierarchical couplings can be detected where one reaction is used when another reaction is active, but not necessarily vice versa [13, 26]. Such a relationship holds, for instance, in N1: a nonzero flux through reaction R9 needs a nonzero flux through R6, but not vice versa. Such investigations help to identify implicit structural constraints, which may also impose constraints for the regulation of coupled reactions or pathways.
Determining Optimal Product Yields and Searching for Intervention Strategies
FBA enables one to predict potential production capabilities of a metabolic network. In principle, given a substrate, FBA can compute the maximally achievable yield for any metabolite in the network. Such predictions are useful for biotechnological applications and metabolic engineering [129, 145]. Moreover, as explained in detail in Sect. 5.5.6, certain FBA approaches can serve as a tool to search for suitable intervention strategies for targeted (re)design of metabolic networks.
The usefulness of FBA has been proven in many applications, in particular for microbial model organisms [82, 103]. However, the standard form of FBA has also some limitations with respect to its predictive power. First, FBA critically depends on the optimality criterion applied. This is rather unproblematic as long as we explore the potential capabilities of a metabolic network. But it can become critical if we want to predict the actual cellular behavior with the often assumed objective of optimal growth: not all cells, and bacterial cells not under all circumstances, behave stoichiometrically optimal [120]. A second issue is related to uniqueness. Whereas the optimal value of the objective function is unique and an optimal solution will normally be found quickly also in larger networks, the calculated optimal flux distribution (maximizing the objective function) may not be unique resulting in a set of optimal solutions [67]. For illustration, look again at our FBA example in Fig. 7, where we found an optimal solution that produces the maximally possible amount of P from A (with a maximal yield of one unit P per unit A). However, we can easily find another optimal flux distribution that also realizes this optimal yield, for example, the one depicted in Fig. 8(a). Furthermore, any convex linear combination (a linear combination λ 1 v 1+λ 2 v 2+⋯+λ n v n is convex if λ i ≥0 and ∑λ i =1) of this solution with the one in Fig. 7—here with a factor of 0.5 for both—results in another optimal flux distribution shown in Fig. 8(b). Hence, infinitely many optimal flux distributions exist in this small network. This is true for any FBA problem as soon as at least two optimal solutions have been found. Therefore, in most cases, albeit the FBA constraints C2 and especially C4 reduce the solution space considerably, infinite many alternate solutions can remain, and FBA in its standard form delivers always one particular optimal solution. Thus, even if optimality is assumed, it may happen that only little can be said about the internal behavior, that is, how the fluxes are distributed inside the cell [85].

Two further optimal flux distributions for producing P from A in N1. (Note: the arrow of the reversible reaction R7 switched its direction due to the negative rate of R7)
One may try to enumerate all qualitatively distinct optimal solutions (as the two in Figs. 7 and 8(a)) for a given FBA problem. This can be done by mixed-integer linear programming [107], vertex enumeration methods [67], or, in smaller networks (as described in Sect. 5.5.5), by metabolic pathway analysis.
A simpler approach is to identify at least those reaction rates that are fixed for all optimal solutions. For the optimization problem (17) we defined for N1, just by inspection of Figs. 7 and 8(a) we can conclude that R3, R6, R9, and R10 must be zero for optimal behavior since they are involved in side-production of E and D. Furthermore, R1, R2, and R8 must carry a fixed flux of two. Thus, only R4, R5, and R7 remain variable. Fixed and variable rates in an FBA problem can be identified by flux variability analysis as described in the following section.
5.5.4.4 Flux Variability Analysis
Given an FBA problem, the goal of flux variability analysis (FVA, [85]) is to quantify the variability (the feasible range) of each reaction rate. This characterization of variability is less precise than enumerating all qualitatively distinct solutions but is often sufficient in many applications, and it can easily be computed in very large networks.
We consider an FBA scenario as in Definition 1, initially without objective function (constraint C4), that is, only with steady-state (C1) and capacity or reversibility constraints (C2), possibly in combination with measurements (C3). The solution space of C1–C3 gives rise to a polyhedron, and as long as this polyhedron is not a single point, multiple solutions r exist, implying that at least some fluxes must be variable. To identify the range for a rate r i , we now use “constraint” C4 of the FBA problem to first minimize and then to maximize rate r i . If we repeat this procedure for all other (free) reaction rates, we get the feasible ranges of all unknown reaction rates. Importantly, if the minimum and maximum rates of a reaction coincide, then a unique rate value can be concluded for this reaction.
FVA is a simple yet very useful technique for constraint-based analysis. In principle, FVA can be seen as a variant of metabolic flux analysis “featured” by FBA methods. In contrast and as an advantage to “classcial” MFA, reversibility (and capacity) constraints can directly be included (in addition to measurements), which may drastically reduce the solution space and possibly lead to uniquely resolvable reaction rate values not detectable by MFA. Therefore, FVA has been widely employed as a network and flux analysis tool for underdetermined systems, and examples can be found in [14, 46, 107]. FVA may only get problems (and requires methods from classical MFA) if the defined scenario is redundant (see Sect. 5.5.3), which is, however, unlikely in larger networks.
We now come back to the problem of multiple optimal solutions in FBA problems. FVA facilitates the identification of fixed and variable reaction rates in optimal flux distributions by a two-step procedure [85]: We first determine the optimal value v opt of the objective function w T r. In a second step, we incorporate w T r=v opt as an additional constraint of type C3. In the case of growth (yield) optimization, this means to fix the growth rate to its optimal value. In a second step, we now apply FVA, that is, we determine the feasible range of all reaction rates for the optimal behavior. Applying this procedure to the example scenario in (17) and Figs. 7 and 8, we would identify the uniquely resolvable rates R3=R6=R9=R10=0 and R1=R2=R8=2, whereas R4 and R5 are variable in a range of [0,2], and R7 in [−2,2].
5.5.4.5 Extensions and Variants of FBA
As pointed out several times, FBA proved to be a very suitable and flexible modeling approach since it allows one to study various important functional properties of medium- and genome-scale metabolic networks from network structure. It is therefore not surprising that basic principles of FBA have been utilized also in specialized or generalized variants of FBA, resulting in a variety of methods (for a comprehensive review, see [82]). The main drivers for extending classical FBA were (i) the integration of data, in particular of gene expression and metabolite concentration data [10, 106], (ii) the integration of regulatory events, (iii) an improved prediction of the effects of gene perturbations, (iv) the description of dynamic (transient) changes of metabolic fluxes, and (v) the use of FBA for metabolic engineering. Some of these methods also require an extension of the formalism since they transform a linear programming (LP) into a mixed integer linear programming (MILP) problem. We give here a brief overview on selected methods for (i)–(iv), FBA for metabolic engineering will be discussed in detail in Sect. 5.5.6.
FBA with Regulation
An approach to combine (transcriptional) regulatory networks with FBA models was presented in [21]. The idea is to put a Boolean network of gene regulatory events on top of the metabolic FBA model. This so-called rFBA model is used to predict the on/off effect of environmental signals (e.g., Gene/Reaction A is active IF substrate S is available AND oxygen NOT) on the expression of certain metabolic genes and thus on the availability of certain pathways. Although rFBA considers only Boolean logic and can get problems if the latter contains causal cycles (feedback loops), it can improve the predictive power of FBA models [22, 126]. A more sophisticated and data-driven approach was proposed by the PROM (probabilistic regulation of metabolism) framework, where data and prior knowledge on (candidate) regulators are used to generate a probabilistic representation of transcriptional regulatory networks, which is eventually combined with the FBA model [16].
FBA with Metabolite Concentration Data and Advanced Thermodynamic Constraints
The assumption of steady state is central to FBA. The advantage is that the metabolite concentrations and their dynamic behavior need not to be taken into account. However, this advantage turns into a disadvantage when experimental metabolite concentration data are available since their inclusion in FBA is not straightforward. One suitable approach to include metabolite concentration data is based on the thermodynamic constraint that the Gibb’s free energy change must be negative for any reaction to proceed in forward direction (or positive for the backward direction). The Gibb’s free energy change ΔG of the ith reaction is described by
where c M denotes the concentration of metabolite M, and n M its stoichiometric coefficient in reaction i; S i and P i are the sets of substrates (reactants) and products of the ith reaction, R is the universal gas constant, and T the absolute temperature; \(\varDelta G^{0}_{i}\) is the Gibb’s energy change of reaction i under standard conditions (where each reactant has a concentration of 1 M), which can be determined from estimated Gibb’s energy of formation of participating reactants and is listed for a large number of metabolic reactions [50, 57]. Measured (or estimated) metabolite concentrations (or concentration ranges) then allow one to predict the sign of ΔG i of reaction i and thus the direction (reversibility) of the reaction flux r i since it must hold by thermodynamic laws that \(\operatorname{sgn}(\varDelta G_{i}) = - \operatorname{sgn}(r_{i})\). Even if the direction of a reaction cannot uniquely be resolved, certain sign patterns in the flux vectors can often be excluded since no realistic metabolite concentration vector would exist supporting this pattern. Hence, integrating thermodynamic constraint in the FBA formulation reduces the solution space [52]. However, the solution space is not convex anymore, and searching for valid flux vectors becomes technically more complicated.
Related to these considerations are efforts to incorporate constraints that exclude thermodynamically infeasible cycles (without explicit consideration of Gibb’s free energy changes). Infeasible cycles would produce a steady-state net flux in a closed network without consumption of external sources or energy. Since the thermodynamic driving forces around such a metabolic loop must add up to zero, no feasible flux distribution should produce a net flux in such a cycle. This is equivalent to Kirchhoff’s second law for electric circuits. For example, a thermodynamically feasible flux vector in network N2 (Fig. 3) will exclude a net flux in the cycle spanned by R3 (backward) and R2, that is, a negative flux for R3 and positive flux for R2 cannot take place at the same time in steady state. Again, MILP formulations are appropriate to include such constraints in FBA problems [112].
FBA with Gene Expression Data
Gene expression data are nowadays also frequently available due to the advances of transcriptomic measurement technologies. Although it has been shown that there is no simple relationship between a reaction flux and the expression level of a gene encoding the catalyzing enzyme, in a simplified approach, one can assume that low expression implies that there is a close-to-zero flux, whereas high expression values suggest high fluxes. In this way, gene expression data can be used to shrink the solution space eventually enabling one to predict tissue- or context-specific fluxes on the basis of gene expression values as in the iMAT approach [127]. A more advanced variant of this approach was presented in [142], and reviews on related methods for integrating expression data in FBA studies can be found in [10, 106].
FBA for Predicting Effects of Genetic Modifications
Predicting the flux changes as a consequence of gene/reaction deletions is one important application of FBA. Even if the wild type grows optimally, mutants may not necessarily behave optimally with respect to their retained resources. Instead, one could postulate that they adjust their metabolism with minimal effort [123]. This assumption suggests that the cell searches for the “nearest” solution in the new (reduced) feasible space of steady-state flux distributions, which is part of the wild-type solution space. The approach of minimization of metabolic adjustment (MoMA, [123]) formalizes this assumption resulting in the following optimization problem, where r opt represents the optimal flux vector of the wild type, and d the index of the deleted reaction whose rate is set to zero:
The first three lines correspond to C1–C3 in the usual FBA, whereas the fourth term leads to a quadratic programming problem whose handling, however, is mathematically straightforward. For mutants of the bacterium Escherichia coli, this approach led to better predictions than FBA [123] (see also [125] presenting another variant of this approach). However, MoMA and related methods need at first the flux distribution from the wild type, which is also assumed to be optimal and, hence, determined by FBA. Therefore, MoMA faces the problem of nonunique optimal flux distributions in the wild type, which can result in nonunique solutions for the mutant [85]. Hence, for MoMA, it is essential to identify the real flux distribution in the wild type under a given environment.
Analyzing a large set of metabolic flux data by multiobjective optimization theory, a recent paper [116] suggests that the metabolism operates under different objectives. Moreover, the authors argue that bacteria might evolve under a trade-off of two principles, namely (i) FBA-like optimality for the current condition and (ii) a MoMA-like principle by which the cells can quickly adjust their metabolism under changing conditions.
FBA and Dynamic Fluxes
Several efforts have been undertaken to simulate also dynamic profiles of (selected) metabolite concentrations and metabolic fluxes in FBA models. Regarding the concentration of biomass and external metabolites (substrates, byproducts), this is straightforward and has been used by several related approaches [21, 86, 144]: FBA with steady-state assumption for the internal metabolites is used to predict exchange fluxes (sometimes, selected exchange fluxes are also explicitly modeled by kinetic rate equations) by which the time course of biomass and external species can be computed through integration over discrete time intervals (similar to Euler method). Such an approach was suitable, for example, to describe the sequential utilization of substrates during diauxic growth of E. coli on different substrates [21, 86].
An advanced approach (integrated FBA, iFBA) was presented in [23], where FBA with Boolean regulatory constraints (rFBA) was coupled with differential equations for selected internal and external metabolite concentrations. This work demonstrated a strategy how existing modules of ODE/Boolean representations of metabolic/regulatory processes can be integrated with FBA models.
5.5.5 Metabolic Pathway Analysis
Metabolic pathway analysis deals with the discovery and analysis of reaction sequences (pathways) in metabolic networks that have some meaningful functional interpretation. There were some early efforts to define chemical and metabolic reaction pathways in a mathematically rigorous way (e.g., [81, 88]; see also [139]). This preliminary work resulted in the development of two related concepts for metabolic pathways—elementary flux modes [117, 118] and extreme pathways [114]—which have become the most accepted and most successful approaches. Since the two concepts are very similar (in many cases even identical; for comparison, see [71, 95]), we will focus here on elementary flux modes (or, shortly, elementary modes). As we will see, elementary modes fit well into the constraint-based modeling framework and provide a suitable concept to study a number of functional and combinatorial properties of (metabolic) reaction networks. Since elementary modes are strongly related to extreme rays of convex cones, they build a bridge from metabolic network analysis to discrete and combinatorial geometry.
5.5.5.1 Definition and Properties of Elementary Modes
Elementary modes (EMs) have been defined as feasible steady-state flux vectors that use a support-minimal (irreducible) set of reactions [117, 118]. The notion of support is a key for the concept of EMs. The support \(\operatorname{supp}(\mathbf{v})\) of a vector v is the set of indices of nonzero entries: \(\operatorname{supp}(\mathbf{v})=\{i| v_{i} \ne 0\}\).
Definition 2
(Elementary modes)
An elementary mode (EM) is a flux vector e fulfilling the following three conditions [117, 118]:
-
(i)
Steady state: Ne=0
-
(ii)
Reaction reversibility: e i ≥0 (∀i∈Irrev)
-
(iii)
Support-minimality (Elementarity, Nondecomposability): there is no vector \(\tilde{\mathbf{e}}\) that fulfills (i) and (ii) and \(\operatorname{supp}(\tilde{\mathbf{e}}) \subsetneqq \operatorname{supp}(\mathbf{e})\).
An EM e is called reversible if \(\operatorname{supp}(\mathbf{e}) \cap \mathit{Irrev} = \emptyset\) and irreversible otherwise.
Note that conditions (i) and (ii) are identical to constraints C1 and C2′ in the general constraint-based problem formulation (Definition 1). Recall also that these two constraints form the flux cone (16) containing all feasible steady-state flux distributions. The third condition (iii), which is sometimes also called genetic independence, ensures that an EM uses a minimal number of reactions, that is, no proper subset of EM’s reactions can constitute a nontrivial feasible flux distribution. It is this property by which EMs form pathway- or cycle-like structures (see below).
Conditions (i)–(iii) completely define the set of EMs of a network up to a scaling factor for each EM. If e is an EM, then, obviously, e′=λ e (λ>0) also defines an EM (in case of reversible EMs, one can also choose a negative scaling factor λ). We consider e and e′ as equivalent representations of one and the same EM since they have the same support. Would we normalize each EM with respect to an appropriate norm, only one representative per EM equivalence class would remain.
Figure 9 displays the five EMs of network N1. The involved reactions (the support) of each EM are indicated by thick blue arrows together with their relative fluxes (uninvolved reactions have zero flux). The EMs were normalized so that the smallest flux through a reaction is unity. One can easily verify that the three EM properties in definition (2) are fulfilled for each EM. One can also recognize the pathway-like structure of EMs; they represent minimal connected subnetworks that convert a set of external substrates (here A(ext)) into external products (here D(ext), P(ext), and E(ext)) while keeping the internal metabolites in a balanced state—a key difference to paths computed in graph representations of reaction networks (cf. Sect. 5.3). As already mentioned above, EMs may also constitute internal cycles. For example, R2 and R3 (in backward direction) make up such a cyclic EM in network N2 (Fig. 3).

The elementary modes of network N1. The participating reactions of each mode are indicated by thick blue edges. The numbers show the relative flux through the involved reactions
Elementary modes possess a number of important theoretical properties, which turned out to be very useful for metabolic network analysis.
Property 1: EMs as Vectors or Sets
An EM can be represented as a vector e as in Definition 2. However, an EM is uniquely defined already by its support; hence, an EM can be represented by the set E of its involved reactions \(E=\operatorname{supp}(\mathbf{e})\). In the following, when listing the support of an EM, we will write Ri (instead of i) for indicating that the ith reaction is part of an EM. We thus have, for example, EM3={R1,R6,R10} in Fig. 9. The representation as a set is preferred when dealing with combinatorial properties of EMs. Note that the relative fluxes e i of an EM represented as a vector e (important for certain applications) can be easily computed from the set representation and the stoichiometric matrix [42].
Property 2: “Surviving” EMs After Reaction Deletions
When a reaction in a network is deleted, the new set of EMs in the remaining network is immediately given by all those (surviving) original EMs that do not involve the deleted reaction. Thus, would we delete reaction R8 in N1 (Fig. 9), then EM3, EM4, and EM5 would constitute the complete set of EMs in the reduced network.
Property 3: EMs Generate the Flux Cone
Linear combinations of elementary modes (with nonnegative coefficients for irreversible EMs and arbitrary coefficients for reversible EMs) generate the flux cone  , providing thus an alternative description to Eq. (16):
, providing thus an alternative description to Eq. (16):

Property 3 emphasizes the relationship between EMs and the extreme rays of the flux cone  . An extreme ray of a cone is a one-dimensional face of the cone; its direction is represented by a vector v [9, 40, 67]; see also chapter Combinatorial Optimization: The Interplay of Graph Theory, Linear and Integer Programming Illustrated on Network Flow of this book. An extreme ray cannot be constructed by a conic (nonnegative) linear combination of other vectors of the cone, and each ray corresponds to an edge of the cone (see Fig. 6). From (21) it follows that the extreme rays must be contained in the set of EMs (otherwise they could not be generated by the EMs). The set of EMs may, however, also contain vectors that can be constructed by other EMs. This may happen due to negative rates of reversible reactions. Hence, for generating all vectors of
. An extreme ray of a cone is a one-dimensional face of the cone; its direction is represented by a vector v [9, 40, 67]; see also chapter Combinatorial Optimization: The Interplay of Graph Theory, Linear and Integer Programming Illustrated on Network Flow of this book. An extreme ray cannot be constructed by a conic (nonnegative) linear combination of other vectors of the cone, and each ray corresponds to an edge of the cone (see Fig. 6). From (21) it follows that the extreme rays must be contained in the set of EMs (otherwise they could not be generated by the EMs). The set of EMs may, however, also contain vectors that can be constructed by other EMs. This may happen due to negative rates of reversible reactions. Hence, for generating all vectors of  , a subset of EMs might be sufficient, which is called a generating set. The simple example network in Fig. 6 has three irreversible EMs: E1=(0,−1,1)T, E2=(1,0,1)T, and E3=(1,1,0)T, which are also shown in the cone representation. Whereas E1 and E3 are extreme rays, E2 is not since it can be constructed by a nonnegative (conic) linear combination of E1 and E3. However, for most applications of EMs, it is important to consider all nondecomposable flux vectors and, hence, all EMs [71]. For example, Property 2 would not hold if we would consider only the generating vectors E1 and E3. Furthermore, by splitting all reversible reactions into two irreversible ones (one in forward and one in backward direction) one can transform the original network into a fully irreversible network. The linear constraints are then in standard form (all free variables are nonnegative), which is often used in combinatorial and computational geometry:
, a subset of EMs might be sufficient, which is called a generating set. The simple example network in Fig. 6 has three irreversible EMs: E1=(0,−1,1)T, E2=(1,0,1)T, and E3=(1,1,0)T, which are also shown in the cone representation. Whereas E1 and E3 are extreme rays, E2 is not since it can be constructed by a nonnegative (conic) linear combination of E1 and E3. However, for most applications of EMs, it is important to consider all nondecomposable flux vectors and, hence, all EMs [71]. For example, Property 2 would not hold if we would consider only the generating vectors E1 and E3. Furthermore, by splitting all reversible reactions into two irreversible ones (one in forward and one in backward direction) one can transform the original network into a fully irreversible network. The linear constraints are then in standard form (all free variables are nonnegative), which is often used in combinatorial and computational geometry:

Importantly, up to the trivial cycles composed by a forward and backward reaction of a formerly reversible reaction, the extreme rays of this cone can uniquely be mapped to the EMs of the original cone (21); see [42]. In fact, this relationship can be used to compute EMs by the double description method, which is well known from computational geometry [40]. This procedure uses a tableau and applies iteratively Gaussian combinations to generate new candidate vectors, which, as the hard step, need to be checked for elementarity (property (iii) in Definition 2). The enumeration of EMs (rays) is a combinatorial problem and can become challenging as millions of EMs can easily arise in networks of larger size (>100 reactions). There are some particular algorithmic improvements that have been achieved in the context of metabolic pathway analysis by which now up to several hundreds of millions of EMs may become computable [42, 75, 134, 141]. However, it is often still not possible to enumerate EMs in genome-scale networks. In those cases, shortest EMs might be computed [27], or projection methods applied [61, 87].
As a refined definition a cone is pointed if it does not contain a line. A line arises if a vector v and its negative −v are both contained in the cone. The set of all lines gives rise to the lineality space. If the lineality space is empty, the cone is pointed since the zero point is then an extreme point of the cone (similarly as for extreme rays, an extreme point cannot be generated by conic combinations of points of the cone). Hence, a flux cone is pointed if it does not contain any reversible EM, that is, if all α j in Eq. (21) are nonnegative. This is fulfilled in most realistic biochemical networks. We note that in case of a pointed cone, the set of generating vectors is unique. We will not further consider generating vectors, but some alternative descriptions of flux cones are based on them [114].
It is also worth noting that all feasible steady state flux vectors r can be generated by the set of EMs (Eq. (21)); however, for a given vector r, the decomposition in EMs is, in general, not unique. This is a major difference to the basis of the null space. For some applications of EMs, it would be useful to have a unique decomposition, and some heuristics have been proposed for this purpose [56, 148].
We finally want to mention the relationship of EMs to another field of combinatorics. Property (iii) of EMs in Definition 2 implies that EMs form a set of minimally linearly dependent columns (reactions) in N. Dependent and independent sets are objects studied in the theory of matroids [93]. In fact, EMs correspond to the so-called circuits representing minimally dependent sets of a matroid. This relationship has not been intensively studied yet in the context of metabolic network analysis and could provide an interesting topic for future research.
5.5.5.2 Applications of Elementary Modes and Metabolic Pathway Analysis
EMs represent minimal functional units of a reaction network and proved to be a very useful and practical concept to analyze numerous functional and structural properties of metabolic networks. Whereas FBA usually concentrates on particular (optimal) steady-state flux vectors, EM analysis seeks to exhaustively explore the solution space (flux cone) based on a finite set of distinct vectors effectively describing the stoichiometric capabilities of a (bio)chemical reaction network. Many aspects that can be studied by FBA, as discussed above, can therefore often be tackled more exhaustively and more systematically by EMs. On the other hand, applications of EMs are limited to networks of moderate size since their computation is normally intractable in genome-scale networks. Furthermore, FBA is often better suited if several inhomogeneous constraints (C2 and C3 in Definition 1) have to be taken into account.
We give here an overview on applications of EMs; a more detailed review on this topic can be found in [139].
Identification of Functional Pathways and Cycles
Since EMs correspond to pathways or cycles, they can be used to identify—in an unbiased way—functional metabolic reaction routes. In this way, hitherto unknown mechanisms might be identified in metabolic models [62, 122]. As long as all relevant cellular metabolites are included as (internal) species in the metabolic model, almost all EMs convert external substrates to external products. As discussed in Sect. 5.5.4.3, cyclic EMs without consumption of external sources represent thermodynamically infeasible loops [101] and could thus be used to correct, for example, reaction reversibilities.
Overall Stoichiometry and Yields
Each EM has its specific stoichiometry with respect to external metabolites, though different EMs may have identical overall conversions. For this reason, EMs can be grouped into (equivalence) classes with respect to their net stoichiometry. In N1, for example, the overall stoichiometry of EM1 and EM2 is 1A(ext)→1P(ext), for EM3, we get 1A(ext)→1D(ext), and for EM4 and EM5, 2A(ext)→1P(ext)+1E(ext) (see Fig. 9). In this way, EMs allow the determination of the complete conversion capabilities of a metabolic network. This is helpful to understand how the cell may synthesize its own components. It becomes also very useful for biotechnological applications since we can immediately derive what are the optimal (or close-to-optimal) yields of certain products of interest and what are the pathways that generate these yields. Recall the example of finding optimal flux vectors for maximal synthesis of product P(ext) from substrate A(ext) (Eq. (17)). We have seen that the optimal yield is one and that infinitely many optimal flux distributions with this yield exist. This can immediately be seen by the overall stoichiometry of EMs: EM1 and EM2 are the two qualitatively distinct solutions with the optimal yield of one, and the complete space (subcone) of optimal solution is therefore spanned by nonnegative linear combinations of these two EMs (compare EM1 and EM2 in Fig. 9 with Figs. 7 and 8(a)).
Reaction Importance, Phenotype Predictions and Coupled Reactions
Since pathway analysis yields all possible routes from which all other flux vectors can be generated, the importance of single reactions for certain network behaviors can be analyzed. For instance, we can conclude that reactions R1, R3, R6, and R9 in N1 are essential (indispensable) for production of E since they are required in all EMs synthesizing E (EM4 and EM5). Hence, removing any of these reactions would delete these EMs, and only EM1–EM3 could survive at all (whether they remain operational or do not depend on the deleted reaction). Furthermore, not surprisingly, we see that reaction R1 (substrate uptake) is essential for all flux vectors since it is utilized in all EMs. We could thus “predict” a nonfunctional network in the absence of A(ext) or after deletion of R1. Such predictions can conveniently be made by EM analysis, e.g., for the viability of mutants [128] (they will be identical to the predictions made by FBA). The (relative) number of EMs, in which a reaction is involved, can thus generally be seen as an importance measure of this reaction for performing a certain function. In [128], it was shown that reaction participations correlate well with relative expression values of genes encoding the respective metabolic enzymes.
Reaction couplings can also be identified conveniently by EMs by simply searching for strict or hierarchical cooccurrences of certain reactions in the EMs. Finally, blocked reactions are identifiable as those that do not occur in any EM.
Network Flexibility
Generally, the number of EMs available for a given function quantifies the flexibility (and, to a certain degree, robustness) of the network with respect to this function. The more EMs are available the more combinations of reactions form functional pathways. This in turn means that a failure or removal of one or several reactions can be easier compensated if a large set of EMs is available [6, 128].
Another application of EMs is the computation and subsequent analysis of minimal cut sets as detailed in the following section.
5.5.5.3 Minimal Cut Sets
We have seen that the effects of reaction removals can be easily and immediately predicted when having the EMs at hand. With this property, one can even systematically search for combinations of interventions (reaction deletions) that block certain network functions. This leads to the notion of minimal cut sets, which, as we will see, does not only provide a suitable approach for assessing network robustness and targeted network redesign but also establishes a fundamental dual relationship between function and dysfunction in metabolic networks. In the following, we denote by  the complete set of EMs of a network. We assume that each EM
the complete set of EMs of a network. We assume that each EM  is represented as a set and, hence, by the support \(E=\operatorname{supp}(\mathbf{e})\) of its vector representation e.
is represented as a set and, hence, by the support \(E=\operatorname{supp}(\mathbf{e})\) of its vector representation e.
The basic idea of minimal cut sets is to block (undesired) capabilities of a network by removing an appropriate set of reactions (which can then be mapped to a set of gene knock-outs). We first need a suitable formalism to specify our intervention goal, that is, the function(s) or flux vector(s) to be disabled. In the original work [70], an undesired function was identified by one (or several) objective reaction(s), and a cut set had to block all steady-state flux vectors in the set
However, a more general and convenient approach to specify an intervention goal (or a set of target flux vectors) is to define a set 𝒯 of target modes,  , subsuming all the undesired behaviors to be repressed. In network N1, for example, our intention could be to block the synthesis of D and E, and we therefore select all those EMs in 𝒯 that export these metabolites; hence, 𝒯={EM3,EM4,EM5} (Fig. 9). In principle, we can imagine that 𝒯 spans an undesired region (a subcone) of the flux cone
, subsuming all the undesired behaviors to be repressed. In network N1, for example, our intention could be to block the synthesis of D and E, and we therefore select all those EMs in 𝒯 that export these metabolites; hence, 𝒯={EM3,EM4,EM5} (Fig. 9). In principle, we can imagine that 𝒯 spans an undesired region (a subcone) of the flux cone  whose flux vectors we want to disable. We can now define the property of a cut set.
whose flux vectors we want to disable. We can now define the property of a cut set.
Definition 3
(Cut set)
A cut set C is a set of reactions “hitting” all target modes, that is,

With this definition, removing or blocking the reactions contained in a cut set from the network will disable the operation of all target modes since, by the definition of an EM, no subset of the reactions of an EM can realize a nonzero steady-state flux distribution. Similarly as for EMs, we demand a cut set to be minimal.
Definition 4
(Minimal cut set)
A minimal cut set (MCS) is a cut set C where no proper subset of C is a cut set, that is, no subset of C hits all target modes.
From this definition it follows that MCSs are the so-called minimal hitting sets of the target modes (the attribute “hitting” reflecting property (24); [69]). Minimal hitting sets are well-known objects from the theory of undirected hypergraphs [7]. Undirected hypergraphs can be seen as a family of subsets from a ground set (each subset forms a hyperedge). The set of target modes gives rise to an undirected hypergraph: its ground set corresponds to the set of reactions, and the EMs in 𝒯 represent the hyperedges. Several algorithms have been proposed to enumerate minimal hitting sets (here, MCSs) for a given hypergraph (here, a given set of target modes) and it turned out that, for computing MCSs in metabolic networks, the Berge algorithm [7] performed best [48]. An alternative algorithm was recently presented in [60].
Coming back to our example, the seven MCSs blocking synthesis of E and D (i.e., the minimal hitting sets of 𝒯={EM3,EM4,EM5}) are depicted in Fig. 10. One can easily verify the required property of an MCS: each of the seven MCSs hits all target modes, whereas no subset of any MCS would do so.

Minimal cut sets blocking all flux vectors synthesizing D or E in N1. They are the minimal hitting sets of EM3, EM4, and EM5 in Fig. 9
As a natural application, MCSs offer a systematic framework for computing intervention strategies, for example, to combat the metabolism of pathological organisms or to genetically design production strains for biotechnological applications as detailed in Sect. 5.5.6 (see also constrained MCSs introduced in Definition 5).
Interpreting MCSs as minimal failure modes of a network (function), they are useful to assess the robustness or fragility of a network (function) [6, 69, 70]: fragility would be indicated by MCSs with low cardinality. Interestingly, computing MCSs blocking growth of the bacterium E. coli, one finds very different spectra of MCSs, depending on the chosen substrate. Hence, network robustness or fragility strongly depends on environmental conditions. Similarly to EMs—but here from another perspective—we can evaluate the importance of single reactions for certain network functions. For example, essential reactions are MCSs of size 1.
MCSs are potentially also useful as a diagnosis tool. Suppose that an organism’s metabolism is in a pathological state (e.g., due to gene mutations) and that it can therefore not produce a certain metabolite. The set of MCSs gives us a complete set of minimal failure modes that may have caused this observed behavior.
Finally, MCSs can also be used to identify all sets of measurements (of reaction rates) through which other reaction rates become uniquely calculable in metabolic flux analysis [69].
5.5.5.4 Duality Between EMs and MCSs
We have seen that the MCSs blocking a certain functionality can be computed as the minimal hitting sets of those (target) modes that realize this function. This already indicates a strong relationship between the minimal functional units (EMs) enabling a certain function and the minimal set of interventions (MCSs) blocking it. In fact, as summarized in Fig. 11, there are even more relationships between MCSs and EMs, which originate from an inherent duality between both concepts. We can first observe that the minimal hitting set property of MCSs with respect to the EMs holds also in the reverse direction: the EMs are the minimal hitting sets of the MCSs (as a lesson, one may verify this property for the MCSs in Fig. 10). Thus, if we could calculate the MCSs independently of the EMs, then we could, in a second step, compute the EMs as minimal hitting sets of the MCSs. In fact, a brute-force approach to directly determine the MCSs without the bypass via the (target) EMs is to use FBA to test consecutively all single, double, triple, etc. reaction knock-out combinations whether they block a given target functionality or not. Alternatively, mixed integer linear programming techniques can be used for a targeted search of minimal knock-out sets [132]. In principle, all MCSs could be identified in this way, however, even in medium-sized networks, enumerating MCSs by such approaches becomes computationally quickly prohibitive. However, this relationship demonstrates that MCSs can be characterized and computed without knowing the corresponding target modes. EMs and MCSs are equivalent descriptions of a network’s function but from two different perspectives.

Duality principles for elementary modes and minimal cut sets
As shown in [2], there is another type of dualities between EMs and MCSs (Fig. 11): for a network with a given functionality, one can construct a dual network in which EMs (MCSs) correspond to the MCSs (EMs) of the original (primal) network. To derive a representation of the dual network, we need another way to describe the target functionality of the primal network:
The nonzero elements of t characterize the target function. For example, if “growth” is the target function of interest, then t will be a vector containing zeros except a nonzero value for the growth rate (this representation requires that all reactions with a nonzero value in t are irreversible, possibly by splitting reversible ones into two irreversible ones). To characterize more complex target functions, one may replace t T r≥1 by the more general expression
with T being a matrix [2]. For simplicity, we focus here on the simpler case. Using the famous Farkas lemma ([9]; see also chapter Combinatorial Optimization: The Interplay of Graph Theory, Linear and Integer Programming Illustrated on Network Flow of this book and the theory of irreducible inconsistent subsystems [43], one can show that the MCSs of system (25) can be identified as a particular set of extreme rays of the (dual) cone spanned by
The (primal) MCSs correspond to those extreme rays (EMs) of this cone where w>0 and which have minimal support in v. The matrix I∈R q×q is the identity matrix, and \(\bar{\mathbf{I}}_{\mathrm{Irrev}} \in \mathbf{R}^{q\times |\mathit{Irrev}|}\) is the identity matrix for irreversible reactions filled with zero rows for the reversible reactions. N dual is the stoichiometric matrix of the dual network that contains basically the transposed constraints of the primal description (25) (inequalities multiplied by −1) plus the (q×q) identity matrix I from which the MCSs will be identified via the v part. Since the stoichiometric matrix of the primal system is transposed in the dual, reactions of the primal system become metabolites in the dual, and, likewise, metabolites become reactions. A simple example for finding MCSs in the primal as EMs in the dual network is shown in Fig. 12. The two MCSs (blocking synthesis of P) in the primal network can be identified by the two EMs in the dual network where w participates and which have minimal support in the v part (the ith element of elements v corresponds to the ith primal reaction). Conversely, the MCSs in the dual network (blocking the w part by cutting exclusively the v reactions) correspond to the primal EMs.

Example for computing MCSs in the primal and in the dual network
The duality principles of (bio)chemical networks have important implications for functional analysis of reaction networks. They tell us that the sets of EMs and MCSs are two dual but equivalent representations of stoichiometric capabilities of the network and their roles are interchanged in a dual network. Furthermore, the duality framework offers novel algorithmic approaches to compute EMs or MCSs, or even both (a concurrent calculation procedure for EMs and MCSs was presented in [48], which is based on the joint-generation algorithm of Fredman and Khachiyan [39]). It will depend on the application which path of calculation turns out to be most effective. As shown in [2], the dual network representation (27) simplifies the integration of inhomogeneous constraints when specifying (target) flux vectors. In this way, MCSs become directly computable also for (possibly, bounded) flux polyhedra.
5.5.5.5 Constrained Minimal Cut Sets
For some applications, especially those related to intervention strategies, one is interested in MCSs that not only disable certain functionalities but also keep other network functions operable. It is then useful to generalize the approach of MCS to constrained MCSs [45]. To motivate this extension, suppose that we want to synthesize product P in network N1 with optimal yield (via EM1 or/and EM2, Fig. 9). It would hence be reasonable to block all other pathways (EM3, EM4, EM5) thus leading to the MCSs as given in Fig. 10. One of the identified MCSs for this problem is MCS1 (removing substrate uptake reaction). Clearly, this MCS cannot be a suitable knockout candidate for the enhanced production of P since is destroys not only the target modes but also, as a side effect, all EMs synthesizing P.
We therefore demand not only that the MCSs hit all target modes in 𝒯 but that they additionally preserve a minimum number n of EMs with desired functions. Desired EMs can be specified by a set  . In realistic applications, there is usually no MCS that hits all target modes and not any of the desired modes; hence, we allow that only a subset of the desired modes “survives” an MCS: n≤|𝒟| (often one uses n=1). For a given MCS C, we collect in 𝒟C all desired EMs that are not hit by C:
. In realistic applications, there is usually no MCS that hits all target modes and not any of the desired modes; hence, we allow that only a subset of the desired modes “survives” an MCS: n≤|𝒟| (often one uses n=1). For a given MCS C, we collect in 𝒟C all desired EMs that are not hit by C:

With this notation, we can now give a definition of constrained MCSs.
Definition 5
(Constrained minimal cut set)
An MCS C is a constrained MCS (cMCS) if it satisfies the constraint

The set of cMCSs is uniquely defined by 𝒟, 𝒯, and n, and arbitrary combinations are possible. Clearly, well-posed intervention problems fulfill 𝒟∩𝒯=∅. It is allowed that some EMs are neither in 𝒟 nor in 𝒯, that is, the union of 𝒟 and 𝒯 does not necessarily cover  . We do not care about those “neutral” EMs; they may survive or not when removing the reactions of a cMCS.
. We do not care about those “neutral” EMs; they may survive or not when removing the reactions of a cMCS.
Since they form a subset of the complete set of MCSs, constrained MCSs can be identified after calculation of the (unconstrained) MCSs in a postprocessing step by discarding all MCSs violating constraint (29). In many cases, however, it is advantageous to drop candidate MCSs violating the side constraints already during the computation of cMCSs. An adapted Berge algorithm implementing this strategy has been proposed in [45]. An alternative strategy for computing cMCSs was presented in [60].
With this generalized definition of MCSs, we now come back to the example with network N1, where the goal was to identify intervention strategies that would disrupt all EMs except those that produce P with optimal yield from A(ext) (Fig. 1). Accordingly, the set of target modes reads 𝒯={EM3,EM4,EM5}, and the set of desired modes reads 𝒟={EM1,EM2}. If we demand to keep at least one desired EM (n=1), then only four of the MCSs in Fig. 10 would be retained as constrained MCSs, namely MCS2, MCS4, MCS5, and MCS7. If we demand that all desired EMs must survive (n=2), then the set of cMCSs would further reduce to MCS2, MCS4, and MCS5.
The use of cMCSs in designing realistic and complex intervention strategies for targeted optimization of production strains is described in the following section.
5.5.6 Metabolic Engineering and Computation of Rational Design Strategies
The production of industrially relevant compounds from renewable resources using biological systems becomes more and more attractive, not only for economical but also for sustainability reasons. Metabolic engineering as an enabling technology for this process aims at developing new experimental and theoretical methodologies for the targeted improvement of metabolic pathways in suitable production hosts. A large variety of theoretical approaches for strain and process optimization has been developed [84]. Many of them rely on constraint-based modeling approaches [139, 152] on which we will focus in the following. Constraint-based models cannot only be used to compute the (potential) maximum yield of a product but also to search for suitable interventions that redirect the fluxes toward the product to eventually achieve a yield that is close to the optimum. As shown below, clever design strategies aim to couple biological (growth) and economical (product) objectives. Several successfully engineered production strains demonstrate the potential of model-driven metabolic design [31, 38, 55, 58, 136, 137, 140, 151].
5.5.6.1 Principles of Model-Based Strain Design
The targeted optimization of metabolic networks intends to redirect steady-state fluxes of production hosts (which are typically microbial organisms but sometimes also, for instance, mammalian cell lines) in a way that synthesis of a desired compound is increased. Desired qualities of the constructed producer strains are: (i) high product yield, (ii) high productivity, and (iii) strain stability. Most stoichiometric optimization techniques address (i), a few also (ii). The majority of stoichiometric methods delivers reaction (or gene) targets to be knocked out to increase product yield. Some methods suggest also enhancement of certain reaction fluxes, which can then be implemented by targeted overexpression of the respective metabolic enzymes. Some optimization approaches propose also indirect interventions to redistribute flux changes by knocking out/overexpressing certain regulators. Such approaches usually require stoichiometric models that are coupled with Boolean rules describing regulatory events (see rFBA introduced above and [68]).
The general objectives of metabolic engineering strategies are visualized in Fig. 13. The left-hand side shows a phenotypic phase plane, which is basically a projection of the flux cone (or flux polyhedron when inhomogeneous flux boundaries are considered) onto two characteristic key quantities: the biomass yield (x-axis) and the product yield (y-axis). The yellow region shows all attainable combinations of these yields in steady-state flux vectors of the network. There are two extreme points: one with optimal biomass yield (which often corresponds to the behavior of the wild type; this is the basic assumption of FBA) and one with maximal product yield, where the substrate would be completely converted to the product, and no biomass would be produced. The desired phenotype is indicated by the blue area: flux vectors that exhibit a relatively high product yield while still allowing a reasonable biomass yield. Accordingly, intervention strategies seek either to redistribute fluxes into the desired space or, typically realized by knockouts, to cut away undesired regions (right-hand side of Fig. 13). The red dot indicates a flux vector, where the optimal biomass yield in the remaining space of feasible flux vectors is coupled to high product yield.

Objectives of metabolic engineering. Left: the phenotypic space of a metabolic network showing all achievable product/biomass yield combinations in steady-state flux vectors. From an engineering point of view, the blue area shows the desired phenotype, whereas the cell will often be close to the growth-yield optimal state. One possible engineering strategy is to search for knockouts that cut away undesired regions while retaining a space similar to the one shown on the right-hand side
To obtain a desired phenotypic space as shown in Fig. 13, one may follow two basic strategies as illustrated in Fig. 14 (in reality, one often uses combinations of both). A simple and straightforward approach could be to delete reactions that are on pathways with low yields or leading to undesired products, possibly in combination with the overexpression of reactions that are on pathways connecting substrate(s) with product(s). Obviously, reactions/genes that are required for building biomass components cannot be removed. In the extreme case, only pathways to the desired product and biomass precursors would be retained. However, with such a strategy, it is not ensured that the cell will really use the pathway leading to the product since it may not be its primary objective. For example, in the network in Fig. 14(a), we might be interested in overproducing metabolite P. With the strategy described above we could delete reactions R2, R4, and R11 to avoid suboptimal product yield and synthesis of an undesired metabolite (Fig. 14(b)). The remaining pathways in the network synthesize biomass and P via optimal but separated routes. It may thus happen (and is likely) that the mutant will adjust its metabolism so that only the pathway for biomass synthesis is activated and no product is excreted at all.

Coupling of product and biomass synthesis. (a) The wild-type network is given on top. (b) A knockout strategy deleting of reactions R2, R4, and R11 leads to yield-optimal but decoupled pathways for synthesis of metabolite P and biomass. (c) Deletion of R3, R7, and R11 leads to obligatory excretion of metabolite P when biomass is synthesized
A more complex concept that overcomes these problems is the coupling of product synthesis and cell growth as illustrated in Fig. 14(c). In this scenario, one deletes reactions R3, R7, and R11. With the remaining set of reactions, it is obligatory that, whenever the organism synthesizes biomass, it has to produce and excrete metabolite P since the precursor E required for biomass can only be synthesized via this route. In that way, the product excretion is coupled to biomass formation.
A typical (indirect) example of coupling product and biomass synthesis is the fermentative production of organic acids under anaerobic heterotrophic growth conditions: the excretion of the latter becomes essential for growth since only in this way reducing equivalents (NAD(P)H) can be balanced (as oxygen is not available as terminal electron acceptor under anaerobic conditions).
Coupling of product synthesis and growth has two main advantages. The first is that a certain minimal yield can be guaranteed whenever the cells grow. The second advantage is that, in microbial organisms, the productivity can be increased by adaptive evolution [100]: when mutations have been implemented, the first generations of the cells may perform suboptimal with respect to growth as their regulatory system is disturbed. Over time they will adaptively evolve toward higher growth rates again. As they are thereby forced to excrete the desired product, they will increase also the product yield.
Constraint-based metabolic design algorithms have been developed based on FBA, elementary modes, and minimal cut sets, and, as we will see in the following, many of them seek to derive network redesign strategies that lead to coupled product and biomass synthesis.
5.5.6.2 FBA-Based Approaches for Metabolic Engineering
FBA is naturally well suited for metabolic network optimization since it relies on an objective function to be optimized. FBA is frequently used to explore potential production capabilities of metabolic networks. OptKnock [12] was the first FBA-based optimization method proposed for a directed search of targets in metabolic networks. The basic idea is to consider the two competing objectives of chemical overproduction and biomass maximization with the help of a bilevel optimization problem (Table 1). The inner problem is similar to classical FBA formulation and describes the biological objective (typically, biomass-yield maximization) together with other constraints (steady-state, maximal substrate uptake rate, ATP maintenance demand, etc.). In contrast, the outer optimization searches for suitable reaction knockouts that, under the given inner biological objective, maximize product synthesis. This approach thus directly aims at a coupling approach. Solving this bi-level optimization problem is more complicated than standard FBA since it requires mixed integer linear optimization (MILP) techniques [12].
The OptKnock approach was successfully applied to realistic problems [38, 151] and initiated the development of extended or modified versions (for an overview, see [152]) that allow, for example, the inclusion of regulatory constraints or the consideration of heterologous reactions (genes). These methods include OptStrain [99], OptReg [98], OptOrf [68], OptForce [104], and RobustKnock [133]. In the latter, the outer objective was adapted to maximize the minimal production of the chemical of interest. Only by this reformulation, product excretion becomes really obligatorily coupled to cell growth, which is not the case for the original OptKnock formulation. The key characteristics of OptKnock and RobustKnock are depicted in Fig. 15. A knockout strategy computed with the RobustKnock approach could lead to the phenotypic phase plane that is surrounded by the dashed red line (mutant B). Here, at biomass-yield optimal conditions (the red point), the minimal product yield is relatively high, though smaller as the maximally achievable product yield in a mutant delivered by OptKnock. However, in the OptKnock strategy (blue dashed line; mutant A), the minimally possible product yield of the mutant at growth-optimal state can be much lower than the (guaranteed) product yield resulting from the RobustKnock approach. A drawback of both strategies is that the coupling can be quite sensitive to the assumption of biomass-yield optimality. If the organism behaves suboptimally with respect to growth yield, the minimal guaranteed product yield can quickly drop to small values or even down to zero. As the assumption of growth-optimal behavior is not always fulfilled [120], it would therefore be desirable to achieve coupling of product and biomass synthesis also when this biological objective is not maximized. Furthermore, all the algorithms mentioned above deliver in each run exactly one solution; multiple solutions have to be computed iteratively by including the found solutions as constraints such that they will not be detected again.

Example of the remaining phenotypic solution space after applying the OptKnock (blue dashed line) and RobustKnock (red dashed line) knock-out strategies
Solving MILPs imposed by OptKnock and similar methods may become challenging for multiple (more than three) knockouts in genome-scale networks. To speed up the calculation, OptGene [96] and GDLS [83] apply evolutionary optimization or a heuristic local search algorithm, respectively. Although they cannot guarantee that the global optimum will be identified, their application may become favorable compared to global search methods.
5.5.6.3 Computing Intervention Strategies Based on EMs
Elementary-modes analysis is a suitable tool for metabolic engineering strategies [139], especially because the effects of deletions can be easily predicted (see Property 2 of EMs, Sect. 5.5.5). A particular EM-based metabolic engineering method that has been successfully applied in a number of case studies is the approach of minimal metabolic functionality (MMF) [137]. It starts with selecting (few) optimal EMs reflecting a desired behavior. Then, in each loop of the heuristic algorithm, a reaction is selected as a suitable knockout candidate whose deletion will eliminate as many EMs as possible while retaining all (or at least some) EMs of the desired behavior (high product yield). By sequential application of this procedure all EMs except those with desired functionalities are deleted. In principle, obligatory coupling of product and biomass synthesis can be enforced within this concept by keeping appropriate remaining functionalities. MMF shares some properties with cMCSs (see below), but the algorithm delivers only one solution (i.e., one cMCS), which is not necessarily the one with the lowest number of interventions.
The first application of MMF was to identify and implement six knockout targets that led to an E. coli mutant exhibiting an increased biomass yield [137]. The approach was also used to design mutants that overproduce different products of the central metabolism (e.g., ethanol [136, 138]) or, as a representative of a secondary metabolite, carotenoids [140].
A second example of an EM-based engineering approach is a simple correlation analysis [89]. This approach analyzes correlations in normalized EMs between reaction fluxes and product synthesis. Positively correlated reactions are suggested as overexpression candidates and negatively correlated reactions as knockout candidates.
CASOP (Computational Approach for Strain Optimization aiming at high Productivity, [44]) provides an alternative heuristic approach, which also identifies both knockout and overexpression targets. A difference to most other methods is that it directly aims at increasing the productivity of a producer strain. CASOP evaluates the spectrum of conversion routes (EMs) to assess the importance of each reaction (for product yield and network capacity) when the fluxes are redirected to the product (while keeping lowered biomass synthesis feasible). As a result, CASOP delivers a reaction ranking suggesting gene knockout and overexpression candidates.
5.5.6.4 Design Strategies Based on Constrained Minimal Cut Sets
As introduced in a subsection above, constrained MCSs provide a particular EM-based approach to enumerate intervention strategies that block undesired and keep desired functionalities. By a concrete example we will briefly illustrate the use of cMCSs for metabolic design problems and demonstrate how this concept enables the enumeration of all knockout solutions that robustly couple biomass and product synthesis (cf. [45]). The case study is on anaerobic ethanol production by E. coli for growth on glucose as carbon source. The first step is to compute the EMs, which, in this case, was done in the network presented by Trinh et al. [138]. It is very useful to plot the (5010) EMs in the phenotypic phase plane showing for each EM (= one dot) its specific growth and ethanol yield (Fig. 16). One can see that there is already a coupling of product and biomass synthesis established for growth-optimal behavior—a peculiarity of anaerobic conditions where ethanol is naturally produced by E. coli as a fermentative product. However, assume that we are interested in higher yields than the one already reached by the wild type. As described in Sect. 5.5.5.5, the next step is therefore to specify the set of target modes 𝒯 and desired modes 𝒟. Figure 16 (left) shows that we marked all EMs with an ethanol yield below 1.4 (mol ethanol per mol glucose) as target modes (red dots). Desired modes (blue dots) must lie above this threshold, and, in addition, we demand that they have a minimal biomass yield of 0.02 to enable reasonable growth in the strain to be constructed. There are some EMs (black dots in the upper left corner) that are neither target nor desired modes; there would be no problem if they were deleted; “survival” of some of them can be accepted because (i) they have a high product yield and (ii) a larger biomass yield is guaranteed to be feasible since we demand that at least one desired mode must remain intact (n=1). Computing now the cMCSs results in 1988 different knockout solutions that solve this engineering task. The minimum number of knockouts required is five. The remaining phenotype of an exemplary quintuple mutant is shown in Fig. 16 (right), which reflects all desired properties: whenever the cell metabolizes glucose, it must produce ethanol with high yield. This holds, in particular, when the cell grows with optimal biomass yield, but also if it does not. Hence, the minimal product yield is independent of the assumption of growth-yield optimality (Fig. 16(b)). However, one can also construct cMCSs where one assumes that the cell evolves toward the optimal growth state (coupled with product synthesis). This will reduce the number of knockouts to be invested but becomes problematic if the cell behaves not as optimal as initially assumed.

Example of using constrained minimal cut sets for designing knockout strategies for ethanol overproduction by E. coli grown under anaerobic condition on glucose. Left: phenotypic phase plane with all EMs of the wild type (blue dots: desired EMs; red dots: target EMs; black dots: EMs neither desired nor target EM). Right: Remaining phenotypic space of a designed mutant corresponding to an exemplary cMCS with five knockouts. Growth yield is given in [g biomass per mmol substrate] and ethanol yield in [mmol ethanol per mmol substrate]
Two major advantages of the cMCSs approach are (i) the extremely convenient and flexible approach to define and solve an intervention problem via target and desired EMs and (ii) the full enumeration of all equivalent knockout strategies enables the selection of the best (most practical) strategy from the complete set of solutions. With the full enumeration of cMCSs, one may also identify important properties, such as knockouts being essential to achieve a given intervention goal. The high flexibility of the approach is also proved by the fact that several MILP-based procedures (including OptKnock and RobustKnock) and the MMF approach mentioned above can be reformulated as special cMCSs problem delivering then all solutions. Certainly, the flexibility and completeness have their price as the computation and, therefore, application of cMCSs (and EMs) are currently still restricted to medium-scale networks.
5.6 Software Tools
Here we give a brief overview of software packages that provide tools and computational methods facilitating metabolic network analysis as described in this chapter. Note that tools and databases for metabolic network reconstruction were already described in Sect. 5.2. In the following, we will focus on software for network visualization and, in particular, for metabolic network analysis.
Generally, metabolic network models can be represented in the Systems Biology Markup Language (SBML; [54]), a common model format used to store and exchange models of biological systems. Most software tools dealing with metabolic networks provide an SBML importer/exporter, although certain features relevant for some methods (e.g., objective function for FBA) cannot be conveyed yet in this format. Furthermore, as a standard for describing biochemical network diagrams, the Systems Biology Graphical Notation (SBGN; [80]) was established.
Different software packages for visualization of metabolic (and other biological) networks are available, including, JDesigner [110], CellDesigner [41], Cell Illustrator [90], GLAMM [5], or Vanted [78]. Some of them do not only allow direct model construction and graph drawing but also facilitate visualization of experimental data in the context of a given network. One particular tool tailored for metabolic network visualization is Omix [28], which has its strengths in visualizing metabolic fluxes and data from isotopic tracer experiments. Further, it can visualize metabolome data and display networks at different abstraction levels.
Apart from visualization tools, there is a large collection of software packages facilitating computational analysis of stoichiometric and metabolic networks (see also Copeland et al. [19]). The majority of them focuses on constraint-based techniques, in particular, flux analysis, FBA, flux variability analysis, and pathway analysis based on elementary modes. Most software packages provide a graphical user interface (GUI) and/or command line functions, some are web-based. The tools may also differ in their functionality regarding available LP/MILP solvers, dependencies on other programs/environments (especially, MATLAB), and licensing issues. An overview of some main characteristics of selected popular tools for metabolic network analysis (excluding visualization tools) is given in Table 2. The command-line oriented COBRA toolbox for MATLAB [113] provides arguably the largest collection of functions for FBA-related studies; it also comprises implementations of FBA-based metabolic engineering algorithms. The application of metabolic engineering algorithms is the particular focus of OptFlux [108], a GUI-based stand-alone software package. YANA [121] focuses on computing and analyzing elementary modes. Metatool [146] and EFMtool [134] are almost exclusively devoted to calculate elementary modes with EFMtool being currently the fastest implementation available for this purpose. CellNetAnalyzer [73, 76] is a MATLAB package for biological (metabolic and signaling) network analysis that can either be used within a GUI or from command line. It provides several functions for FBA-related studies and offers a comprehensive set of tools and algorithms for EM-based network analysis, including also calculation and exploration of (constrained) minimal cut sets. FASIMU [53] provides another toolbox with functions for FBA studies, also allowing the consideration of thermodynamic constraints. Finally, MicrobesFlux [35] is a web-based platform that allows reconstruction of metabolic networks directly from the KEGG database and subsequent FBA-related analysis.
References
Albert, R., Barabasi, A.-L.: Statistical mechanics of complex networks. Rev. Mod. Phys. 74, 47–97 (2002)
Ballerstein, K., von Kamp, A., Klamt, S., Haus, U.-U.: Minimal cut sets in a metabolic network are elementary modes in a dual network. Bioinformatics 28, 381–387 (2012)
Barabasi, A.-L., Bonabeau, E.: Scale-free networks. Sci. Am. 288, 60–99 (2003)
Barabasi, A.-L., Oltvai, Z.: Network biology. Nat. Rev. Genet. 5, 101–113 (2004)
Bates, J.T., Chivian, D., Arkin, A.P.: GLAMM: genome-linked application for metabolic maps. Nucleic Acids Res. 39, W400–W405 (2011)
Behre, J., Wilhelm, T., von Kamp, A., Ruppin, E., Schuster, S.: Structural robustness of metabolic networks with respect to multiple knockouts. J. Theor. Biol. 252, 433–441 (2008)
Berge, C.: Hypergraphs. Combinatorics of Finite Sets. North-Holland, Amsterdam (1989)
Bernal, A., Daza, E.: Metabolic networks: beyond the graph. Curr. Comput.-Aided Drug Des. 7, 122–132 (2011)
Bertsimas, D., Tsitsiklis, J.N.: Introduction to Linear Optimization. Athena Scientific, Belmont (1997)
Blazier, A.S., Papin, J.A.: Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 3, 299 (2012)
Bornholt, S.: Less is more in modeling large genetic networks. Science 310, 449–451 (2005)
Burgard, A.P., Pharkya, P., Maranas, C.D.: Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 84, 647–657 (2003)
Burgard, A.P., Nikolaev, E.V., Schilling, C.H., Maranas, C.D.: Flux coupling analysis of genome-scale metabolic network reconstructions. Genome Res. 14, 301–312 (2004)
Bushell, M., Sequeira, S., Khannapho, C., Zhao, H., Chater, K., Butler, M., Kierzek, A., Avignone-Rossa, C.: The use of genome scale metabolic flux variability analysis for process feed formulation based on an investigation of the effects of the ZWF mutation on antibiotic production in Streptomyces coelicolor. Enzyme Microb. Technol. 39, 1347–1353 (2006)
Caspi, R., Altman, T., Dreher, K., Fulcher, C.A., Subhraveti, P., Keseler, I.M., Kothari, A., Krummenacker, M., Latendresse, M., Mueller, L.A., Ong, Q., Paley, S., Pujar, A., Shearer, A.G., Travers, M., Weerasinghe, D., Zhang, P., Karp, P.D.: The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 40, 742–753 (2012)
Chandrasekaran, S., Price, N.D.: Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. 107, 17845–17850 (2010)
Clark, B.L.: Stoichiometric network analysis. Cell Biophys. 12, 237–253 (1988)
Conradi, C., Flockerzi, D.: Multistationarity in mass action networks with applications to ERK activation. J. Math. Biol. 65, 107–156 (2012)
Copeland, W.B., Bartley, B.A., Chandran, D., Galdzicki, M., Kim, K.H., Sleight, S.C., Maranas, C.D., Sauro, H.M.: Computational tools for metabolic engineering. Metab. Eng. 14, 270–280 (2012)
Cornish-Bowden, A., Hofmeyr, J.H.: The role of stoichiometric analysis in studies of metabolism: an example. J. Theor. Biol. 216, 179–191 (2002)
Covert, M.W., Schilling, C.H., Palsson, B.O.: Regulation of gene expression in flux balance models of metabolism. J. Theor. Biol. 213, 73–88 (2001)
Covert, M.W., Knight, E.M., Reed, J.L., Herrgard, M.J., Palsson, B.O.: Integrating high-throughput and computational data elucidates bacterial networks. Nature 429, 92–96 (2004)
Covert, M.W., Xiao, N., Chen, T.J., Karr, J.R.: Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 24, 2044–2050 (2008)
Craciun, G., Tang, Y., Feinberg, M.: Understanding bistability in complex enzyme-driven reaction networks. Proc. Natl. Acad. Sci. 103, 8697–8702 (2006)
Csete, M., Doyle, J.: Bow ties, metabolism and disease. Trends Biotechnol. 22, 446–450 (2004)
David, L., Marashi, S.A., Larhlimi, A., Mieth, B., Bockmayr, A.: FFCA: a feasibility-based method for flux coupling analysis of metabolic networks. BMC Bioinform. 12, 236 (2011)
de Figueiredo, L.F., Podhorski, A., Rubio, A., Kaleta, C., Beasley, J.E., Schuster, S., Planes, F.J.: Computing the shortest elementary flux modes in genome-scale metabolic networks. Bioinformatics 25, 3158–3165 (2009)
Droste, P., Miebach, S., Niedenführ, S., Wiechert, W., Nöh, K.: Visualizing multi-omics data in metabolic networks with the software Omix: a case study. Biosystems 105, 154–161 (2011)
Durot, M., Bourguignon, P.Y., Schachter, V.: Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol. Rev. 33, 164–190 (2009)
Edwards, J.S., Palsson, B.O.: The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. 97, 5528–5533 (2000)
Edwards, J.S., Ibarra, R.U., Palsson, B.O.: In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19, 125–130 (2001)
Feinberg, M.: Chemical reaction network structure and the stability of complex isothermal reactors—I. The deficiency zero and deficiency one theorems. Chem. Eng. Sci. 42, 2229–2268 (1987)
Feist, A.M., Palsson, B.O.: The biomass objective function. Curr. Opin. Microbiol. 13, 344–349 (2010)
Feist, A.M., Herrgard, M.J., Thiele, I., Reed, J.L., Palsson, B.O.: Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 7, 129–143 (2009)
Feng, X., Xu, Y., Chen, Y., Tang, Y.J.: MicrobesFlux: a web platform for drafting metabolic models from the KEGG database. BMC Syst. Biol. 6, 94 (2012)
Foerster, J., Famili, I., Palsson, B.O., Nielsen, J.: Large-scale evaluation of in silico gene deletions in Saccharomyces cerevisiae. Omics. J. Integr. Biol. 7, 193–202 (2003)
Fong, S.S., Palsson, B.O.: Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat. Genet. 36, 1056–1058 (2004)
Fong, S.S., Burgard, A.P., Herring, C.D., Knight, E.M., Blattner, F.R., Maranas, C.D., Palsson, B.O.: In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol. Bioeng. 91, 643–648 (2005)
Fredman, M.L., Khachiyan, L.: On the complexity of dualization of monotone disjunctive normal forms. J. Algorithms 21, 618–628 (1996)
Fukuda, K., Prodon, A.: Double description method revisited. In: Deza, M., Euler, R., Manoussakis, I. (eds.) Combinatorics and Computer Science, vol. 1120, pp. 91–111. Springer, Berlin (1996)
Funahashi, A., Matsuoka, Y., Jouraku, A., Morohashi, M., Kikuchi, N., Kitano, H.: CellDesigner 3.5: a versatile modeling tool for biochemical networks. Proc. IEEE 96, 1254–1265 (2008)
Gagneur, J., Klamt, S.: Computation of elementary modes: a unifying framework and the new binary approach. BMC Bioinform. 5, 175 (2004)
Gleeson, J., Ryan, J.: Identifying minimally infeasible subsystems of inequalities. ORSA J. Comput. 2, 61–63 (1990)
Hädicke, O., Klamt, S.: CASOP: a computational approach for strain optimization aiming at high productivity. J. Biotechnol. 147, 88–101 (2010)
Hädicke, O., Klamt, S.: Computing complex metabolic intervention strategies using constrained minimal cut sets. Metab. Eng. 13, 204–213 (2011)
Hädicke, O., Grammel, H., Klamt, S.: Metabolic network modeling of redox balancing and biohydrogen production in purple nonsulfur bacteria. BMC Syst. Biol. 5, 150 (2011)
Haggart, C.R., Bartell, J.A., Saucerman, J.J., Papin, J.A.: Whole-genome metabolic network reconstruction and constraint-based modeling. Methods Enzymol. 500, 411–433 (2011)
Haus, U.-U., Klamt, S., Stephen, T.: Computing knock-out strategies in metabolic networks. J. Comput. Biol. 15, 259–268 (2008)
Heinrich, R., Schuster, S.: The Regulation of Cellular Systems. Chapman & Hall, New York (1996)
Henry, C.S., Broadbelt, L.J., Hatzimanikatis, V.: Thermodynamics-based metabolic flux analysis. Biophys. J. 92, 1792–1805 (2007)
Henry, C.S., DeJongh, M., Best, A.B., Frybarger, P.M., Linsay, B., Stevens, R.L.: High-throughput generation and optimization of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982 (2010)
Hoppe, A., Hoffmann, S., Holzhuetter, H.G.: Including metabolite concentrations into flux-balance analysis: thermodynamic realizability as a constraint on flux distributions in metabolic networks. BMC Syst. Biol. 1, 23 (2007)
Hoppe, A., Hoffmann, S., Gerasch, A., Gille, C., Holzhütter, H.G.: FASIMU: flexible software for flux-balance computation series in large metabolic networks. BMC Bioinform. 12 (2011)
Hucka, M., Finney, A., Sauro, H.M., Bolouri, H., Doyle, J.C., Kitano, H., et al.: The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531 (2003)
Ibarra, R.U., Edwards, J.S., Palsson, B.O.: Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420, 186–189 (2002)
Ip, K., Colijn, C., Lun, D.S.: Analysis of complex metabolic behavior through pathway decomposition. BMC Syst. Biol. 5, 91 (2011)
Jankowski, M.D., Henry, C.S., Broadbelt, L.J., Hatzimanikatis, V.: Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 95, 1487–1499 (2008)
Jantama, K., Haupt, M.J., Svoronos, S.A., Zhang, X., Moore, J.C., Shanmugam, K.T., Ingram, L.O.: Combining metabolic engineering and metabolic evolution to develop nonrecombinant strains of Escherichia coli C that produce succinate and malate. Biotechnol. Bioeng. 99, 1140–1153 (2008)
Jeong, H., Tombor, B., Albert, R., Oltvai, Z.N., Barabasi, A.L.: The large-scale organisation of metabolic networks. Nature 407, 651–654 (2000)
Jungreuthmayer, C., Zanghellini, J.: Designing optimal cell factories: integer programming couples elementary mode analysis with regulation. BMC Syst. Biol. 6, 103 (2012)
Kaleta, C., de Figueiredo, L.F., Schuster, S.: Can the whole be less than the sum of its parts? Pathway analysis in genome-scale metabolic networks using elementary flux patterns. Genome Res. 19, 1872–1883 (2009)
Kaleta, C., de Figueiredo, L.F., Werner, S., Guthke, R., Ristow, M., Schuster, S.: In silico evidence for gluconeogenesis from fatty acids in humans. PLoS Comput. Biol. 7, e1002116 (2011)
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., Tanabe, M.: KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, 109–114 (2012)
Karp, P.D., Caspi, R.: A survey of metabolic databases emphasizing the MetaCyc family. Arch. Toxicol. 85, 1015–1033 (2011)
Karp, P.D., Riley, M., Saier, M., Paulsen, I.T., Paley, S.M., Pellegrini-Toole, A.: The EcoCyc and MetaCyc databases. Nucleic Acids Res. 28, 56–59 (2000)
Karp, P.D., Paley, S.M., Krummenacker, M., Latendresse, M., Dale, J.M., Lee, T.J., Kaipa, P., Gilham, F., Spaulding, A., Popescu, L., Altman, T., Paulsen, I., Keseler, I.M., Caspi, R.: Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief. Bioinform. 11, 40–79 (2010)
Kelk, S.M., Olivier, B.G., Stougie, L., Bruggeman, F.J.: Optimal flux spaces of genome-scale stoichiometric models are determined by a few subnetworks. Sci. Rep. 2, 580 (2012)
Kim, J., Reed, J.L.: OptORF: optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst. Biol. 4, 53 (2010)
Klamt, S.: Generalized concept of minimal cut sets in biochemical networks. Biosystems 83, 233–247 (2006)
Klamt, S., Gilles, E.D.: Minimal cut sets in biochemical reaction networks. Bioinformatics 20, 226–234 (2004)
Klamt, S., Stelling, J.: Two approaches for metabolic pathway analysis? Trends Biotechnol. 21, 64–69 (2003)
Klamt, S., Stelling, J.: Stoichiometric and constraint-based modeling. In: Szallasi, Z., Stelling, J., Periwal, V. (eds.) System Modeling in Cellular Biology, pp. 73–96. MIT Press, Cambridge (2006)
Klamt, S., von Kamp, A.: An application programming interface for CellNetAnalyzer. Biosystems 105, 162–168 (2011)
Klamt, S., Schuster, S., Gilles, E.D.: Calculability analysis in underdetermined metabolic networks illustrated by a model of the central metabolism in purple nonsulfur bacteria. Biotechnol. Bioeng. 77, 734–751 (2002)
Klamt, S., Gagneur, J., von Kamp, A.: Algorithmic approaches for computing elementary modes in large biochemical reaction networks. Syst. Biol. 152, 249–255 (2005)
Klamt, S., Saez-Rodriguez, J., Gilles, E.D.: Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol. 1, 2 (2007)
Klamt, S., Haus, U.-U., Theis, F.: Hypergraphs and cellular networks. PLoS Comput. Biol. 5, e1000385 (2009)
Klukas, C., Schreiber, F.: Integration of -omics data and networks for biomedical research with VANTED. J. Integr. Bioinform. 7, 112 (2010)
Latendresse, M., Krummenacker, M., Trupp, M., Karp, P.D.: Construction and completion of flux balance models from pathway databases. Bioinformatics 3, 388–396 (2012)
Le Novère, N., Hucka, M., Mi, H., Moodie, S., Schreiber, F., Sorokin, A., et al.: The systems biology graphical notation. Nat. Biotechnol. 27, 735–741 (2009)
Leiser, J., Blum, J.J.: On the analysis of substrate cycles in large metabolic systems. Cell Biophys. 11, 123–138 (1987)
Lewis, N.E., Nagarajan, H., Palsson, B.O.: Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305 (2012)
Lun, D.S., Rockwell, G., Guido, N.J., Baym, M., Kelner, J.A., Berger, B., Galagan, J.E., Church, G.M.: Large-scale identification of genetic design strategies using local search. Mol. Syst. Biol. 5, 296 (2009)
Maertens, J., Vanrolleghem, P.A.: Modeling with a view to target identification in metabolic engineering: a critical evaluation of the available tools. Biotechnol. Prog. 26, 313–331 (2010)
Mahadevan, R., Schilling, C.H.: The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 5, 264–276 (2003)
Mahadevan, R., Edwards, J.S., Doyle, F.J.: Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 83, 1331–1340 (2002)
Marashi, S.A., David, L., Bockmayr, A.: Analysis of metabolic subnetworks by flux cone projection. Algorithms Mol. Biol. 7, 17 (2012)
Mavrovouniotis, M.L., Stephanopoulos, G., Stephanopoulos, G.: Computer-aided synthesis of biochemical pathways. Biotechnol. Bioeng. 36, 1119–1132 (1990)
Melzer, G., Esfandabadi, M.E., Franco-Lara, E., Wittmann, C.: Flux design: in silico design of cell factories based on correlation of pathway fluxes to desired properties. BMC Syst. Biol. 3, 120 (2009)
Nagasaki, M., Saito, A., Jeong, E., Li, C., Kojima, K., Ikeda, E., Miyano, S.: Cell Illustrator 4.0: a computational platform for systems biology. In Silico Biol. 10, 5–26 (2010)
Newman, M.E.J.: The structure and function of complex networks. SIAM Rev. 45, 167–256 (2003)
Oberhardt, M.A., Palsson, B.O., Papin, J.A.: Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 5, 320 (2009)
Oxley, J.G.: Matroid Theory. Oxford University Press, Oxford (2004)
Papin, J.A., Price, N.D., Palsson, B.O.: Extreme pathway lengths and reaction participation in genome-scale metabolic networks. Genome Res. 12, 1889–1900 (2002)
Papin, J.A., Stelling, J., Price, N.D., Klamt, S., Schuster, S., Palsson, B.O.: Comparison of network-based pathway analysis methods. Trends Biotechnol. 22, 400–405 (2004)
Patil, K.R., Rocha, I., Förster, J., Nielsen, J.: Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinform. 6, 308 (2005)
Pfeiffer, T., Sanchez-Valdenebro, I., Nuno, J.C., Montero, F., Schuster, S.: METATOOL: for studying metabolic networks. Bioinformatics 15, 251–257 (1999)
Pharkya, P., Maranas, C.D.: An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 8, 1–13 (2006)
Pharkya, P., Burgard, A.P., Maranas, C.D.: OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 14, 2367–2376 (2004)
Portnoy, V.A., Bezdan, D., Zengler, K.: Adaptive laboratory evolution—harnessing the power of biology for metabolic engineering. Curr. Opin. Biotechnol. 22, 590–594 (2011)
Price, N.D., Famili, I., Beard, D.A., Palsson, B.O.: Extreme pathways and Kirchhoff’s second law. Biophys. J. 83, 2879–2882 (2002)
Price, N.D., Papin, J.A., Schilling, C.H., Palsson, B.O.: Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 21, 162–169 (2003)
Price, N.D., Reed, J.L., Palsson, B.O.: Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2, 886–897 (2004)
Ranganathan, S., Suthers, P.F., Maranas, C.D.: OptForce: an optimization procedure for identifying all genetic manipulations leading to targeted overproductions. PLoS Comput. Biol. 6(4), e1000744 (2010)
Reder, C.: Metabolic control theory: a structural approach. J. Theor. Biol. 135, 175–201 (1986)
Reed, J.L.: Shrinking the metabolic solution space using experimental datasets. PLoS Comput. Biol. 8, e1002662 (2012)
Reed, J.L., Palsson, B.O.: Genome-scale in silico models of E. coli have multiple equivalent phenotypic states: assessment of correlated reaction subsets that comprise network states. Genome Res. 14, 1797–1805 (2004)
Rocha, I., Maia, P., Evangelista, P., Vilaca, P., Soares, S., Pinto, J.P., Nielsen, J., Patil, K.R., Ferreira, E.C., Rocha, M.: OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 4, 45 (2010)
Rockafellar, R.T.: Convex Analysis. University Press (1970)
Sauro, H.M., Hucka, M., Finney, A., Wellock, C., Bolouri, H., Doyle, J., Kitano, H.: Next generation simulation tools: the systems biology workbench and BioSPICE integration. Omics. J. Integr. Biol. 7, 355–372 (2004)
Schellenberger, J., Park, J.O., Conrad, T.M., Palsson, B.O.: BiGG: a biochemical genetic and genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinform. 11, 213 (2010)
Schellenberger, J., Lewis, N.E., Palsson, B.O.: Elimination of thermodynamically infeasible loops in steady-state metabolic models. Biophys. J. 100, 544–553 (2011)
Schellenberger, J., Que, R., Fleming, R.M., Thiele, I., Orth, J.D., Feist, A.M., Zielinski, D.C., Bordbar, A., Lewis, N.E., Rahmanian, S., Kang, J., Hyduke, D.R., Palsson, B.O.: Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 6, 1290–1307 (2011)
Schilling, C.H., Letscher, D., Palsson, B.O.: Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J. Theor. Biol. 203, 229–248 (2000)
Schuetz, R., Kuepfer, L., Sauer, U.: Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 3, 119 (2007)
Schuetz, R., Zamboni, N., Zampieri, M., Heinemann, M., Sauer, U.: Multidimensional optimality of microbial metabolism. Science 336, 601–604 (2012)
Schuster, S., Hilgetag, C.: On elementary flux modes in biochemical reaction systems at steady state. J. Biol. Syst. 2, 165–182 (1994)
Schuster, S., Fell, D.A., Dandekar, T.: A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 18, 326–332 (2000)
Schuster, S., Klamt, S., Weckwerth, W., Moldenhauer, F., Pfeiffer, T.: Use of network analysis of metabolic systems in bioengineering. Bioprocess Biosyst. Eng. 24, 363–372 (2002)
Schuster, S., Pfeiffer, T., Fell, D.A.: Is maximization of molar yield in metabolic networks favoured by evolution? J. Theor. Biol. 252, 497–504 (2008)
Schwarz, R., Musch, P., von Kamp, A., Engels, B., Schirmer, H., Schuster, S., Dandekar, T.: YANA—a software tool for analyzing flux modes, gene-expression and enzyme activities. BMC Bioinform. 6, 135 (2005)
Schwender, J., Goffman, F., Ohlrogge, J.B., Shachar-Hill, Y.: Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature 432, 779–782 (2004)
Segre, D., Vitkup, D., Church, G.M.: Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. 99, 15112–15117 (2002)
Shinar, G., Feinberg, M.: Structural sources of robustness in biochemical reaction networks. Science 327, 1389–1391 (2010)
Shlomi, T., Berkman, O., Ruppin, E.: Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc. Natl. Acad. Sci. 24, 7695–7700 (2005)
Shlomi, T., Eisenberg, Y., Sharan, R., Ruppin, E.: A genome-scale computational study of the interplay between transcriptional regulation and metabolism. Mol. Syst. Biol. 3, 101 (2007)
Shlomi, T., Cabili, M., Herrgard, M., Palsson, B.O., Ruppin, E.: Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 26, 1003–1010 (2008)
Stelling, J., Klamt, S., Bettenbrock, K., Schuster, S., Gilles, E.D.: Metabolic network structure determines key aspects of functionality and regulation. Nature 420, 190–193 (2002)
Stephanopoulos, G.N., Aristidou, A.A., Nielsen, J.: Metabolic Engineering. Academic Press, San Diego (1998)
Strang, G.: Linear Algebra and Its Applications. Academic Press, New York (1980)
Strogatz, S.H.: Exploring complex networks. Nature 410, 268–276 (2001)
Suthers, P.F., Zomorrodi, A., Maranas, C.D.: Genome-scale gene/reaction essentiality and synthetic lethality analysis. Mol. Syst. Biol. 5, 301 (2009)
Tepper, N., Shlomi, T.: Predicting metabolic engineering knockout strategies for chemical production: accounting for competing pathways. Bioinformatics 26, 536–543 (2010)
Terzer, M., Stelling, J.: Large-scale computation of elementary flux modes with bit pattern trees. Bioinformatics 24, 2229–2235 (2008)
Thiele, I., Palsson, B.O.: A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121 (2010)
Trinh, C.T., Srienc, F.: Metabolic engineering of Escherichia coli for efficient conversion of glycerol to ethanol. Appl. Environ. Microbiol. 75, 6696–6705 (2009)
Trinh, C.T., Carlson, R., Wlaschin, A., Srienc, F.: Design, construction and performance of the most efficient biomass producing E. coli bacterium. Metab. Eng. 8, 628–638 (2006)
Trinh, C.T., Unrean, P., Srienc, F.: Minimal Escherichia coli cell for the most efficient production of ethanol from hexoses and pentoses. Appl. Environ. Microbiol. 74, 3634–3643 (2008)
Trinh, C.T., Wlaschin, A., Srienc, F.: Elementary mode analysis: a useful metabolic pathway analysis tool for characterizing cellular metabolism. Appl. Microbiol. Biotechnol. 81, 813–826 (2009)
Unrean, P., Trinh, C.T., Srienc, F.: Rational design and construction of an efficient E. coli for production of diapolycopendioic acid. Metab. Eng. 12, 112–122 (2010)
Urbanczik, R., Wagner, C.: An improved algorithm for stoichiometric network analysis: theory and applications. Bioinformatics 21, 1203–1210 (2005)
Van Berlo, R.J., de Ridder, D., Daran, J.M., Daran-Lapujade, P.A., Teusink, B., Reinders, M.J.: Predicting metabolic fluxes using gene expression differences as constraints. IEEE/ACM Trans. Comput. Biol. Bioinform. 8, 206–216 (2011)
Van der Heijden, R.T.J.M., Heijnen, J.J., Hellinga, C., Romein, B., Luyben, K.Ch.A.M.: Linear constraint relations in biochemical reaction systems: I. Classification of the calculability and the balanceability of conversion rates. Biotechnol. Bioeng. 43, 3–10 (1994)
Varma, A., Palsson, B.O.: Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl. Environ. Microbiol. 60, 3724–3731 (1994)
Varma, A., Boesch, B.W., Palsson, B.O.: Biochemical production capabilities of Escherichia coli. Biotechnol. Bioeng. 42, 59–73 (1993)
von Kamp, A., Schuster, S.: Metatool 5.0: fast and flexible elementary modes analysis. Bioinformatics 22, 1930–1931 (2006)
Watts, D.J., Strogatz, S.H.: Collective dynamics of ‘small-world’ networks. Nature 393, 409–410 (1998)
Wiback, S.J., Mahadevan, R., Palsson, B.O.: Reconstructing metabolic flux vectors from extreme pathways: defining the alpha-spectrum. J. Theor. Biol. 224, 313–324 (2003)
Wiechert, W.: 13C metabolic flux analysis. Metab. Eng. 3, 195–206 (2001)
Wolf, J., Passarge, J., Somsen, O.J.G., Snoep, J.L., Heinrich, R., Westerhoff, H.V.: Transduction of intracellular and intercellular dynamics in yeast glycolytic oscillations. Biophys. J. 78, 1145–1153 (2000)
Yim, H., et al.: Metabolic engineering of Escherichia coli for direct production of 1, 4-butanediol. Nat. Chem. Biol. 7, 445–452 (2011)
Zomorrodi, A.R., Suthers, P.F., Ranganathan, S., Maranas, C.D.: Mathematical optimization applications in metabolic networks. Metab. Eng. (2012). doi:10.1016/j.ymben.2012.09.005
Acknowledgement
This work was partially supported by the Federal State of Saxony-Anhalt (Research Center “Dynamic Systems: Biosystems Engineering”) and by the the German Federal Ministry of Education and Research (e:Bio project CYANOSYS II (FKZ 0316183D); Biotechnologie 2020+ project CASCO2 (FKZ: 031A180B)).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Klamt, S., Hädicke, O., von Kamp, A. (2014). Stoichiometric and Constraint-Based Analysis of Biochemical Reaction Networks. In: Benner, P., Findeisen, R., Flockerzi, D., Reichl, U., Sundmacher, K. (eds) Large-Scale Networks in Engineering and Life Sciences. Modeling and Simulation in Science, Engineering and Technology. Birkhäuser, Cham. https://doi.org/10.1007/978-3-319-08437-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-08437-4_5
Publisher Name: Birkhäuser, Cham
Print ISBN: 978-3-319-08436-7
Online ISBN: 978-3-319-08437-4
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)