
Abstract
In order to understand the importance of the structural properties of compound semiconductors for the operation of a thin-film solar cell, this section aims to explain the operation principle using the example of a Cu(In,Ga)Se2 (CIGSe) thin-film solar cell. For detailed information the reader is kindly referred to the literature for a more extensive overview of the recent developments [1], device operation [2] and material preparation [3].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
CIGSe thin-film solar cells are made of a stack of metal and semiconductor thin films in the following sequence: a molybdenum back contact (metal), a polycrystalline CIGSe absorber layer (p-type semiconductor), CdS buffer layer (n-type semiconductor), and ZnO front contact (n-type semiconductor). Together, the CdS and the ZnO are often referred to as the ‘window’ of the device. The core of the device is the p-n-heterojunction between the p-type absorber layer and the n-type window layers. The resulting energy band line-up and a cross sectional scanning electron microscope view of a complete device are shown in Fig. 5.1. Due to the high doping of the ZnO front contact layer the field, which develops upon contact of the n- and p-type materials in the interface region, is located almost entirely inside the absorber layer. In comparison to homojunctions, the heterojunction has the advantage that the n-type component can be chosen such that its band gap is large and the photoactive band-gap Eg of the solar-cell device, determined by the band gap of the absorber-layer material, is optimized to reach high conversion-efficiencies.

Energy band diagram of a standard Cu(In,Ga)Se2 thin film solar cell (left) in correlation to the cross sectional view of a complete solar cell device as seen in the scanning electron microscope (right)
Figure 5.2 illustrates the basic working principle of a CIGSe thin-film solar cell. Electron-hole pairs are generated by light absorption within the absorber thin-film. Absorption of photons with an energy higher than Eg results in the loss of excess energy via thermalization. If an electron-hole pair is excited within the depletion region near the font interface of the device, it is immediately spatially separated by the electric field that is present. If generation happens outside the depletion region, the minority charge carrier has to diffuse into the depletion region in order to contribute to the photo current. Recombination of the electron-hole pairs can occur by a number of processes: radiative and non-radiative, band-to-band or via one or several defect levels located in the band gap of the absorber material, within the bulk of the thin-film material, within the depletion region, and at the front interface of the device or possibly at the grain boundaries of the polycrystalline CIGSe thin-film. The nature of grain boundaries in CIGSe thin-films however, has proven to be considerably more benign than in other semiconductor materials such as for example silicon, the exact reasons for which are still under discussion [4].

Working principle of a CIGSe thin film solar cell: electron-hole pairs are excited by the absorption of incident light; within the depletion region of the p-n-heterojunction they are immediately spatially separated; charge carriers, which are generated in the bulk of the thin film, can diffuse into the depletion region. When hv > Eg excess energy is lost by thermalization. A number of recombination channels are present in the bulk, in the depletion region and at the interfaces
CIGSe absorbers for photovoltaic application are mostly fabricated slightly Cu-poor and with an overall Ga content of [Ga]/([Ga] + [In]) ~ 0.3. The resulting CIGSe material is a p-type semiconductor material (Eg ~ 1.15 eV), which is highly compensated. This means that there are acceptor- and donor-type defects present within the material and p-type conductivity is established due to the exceptionally-low formation energy of Cu vacancies. In addition, the formation of defect complexes, such as 2V −Cu + In 2+Cu , seems to play an important role in terms of the electronic properties of the material, and also regarding phase formation and crystal structure [5] (Figs. 5.2 and 5.3).

Elemental flux and substrate-temperature profiles for a typical 3-stage co-evaporation process
For most applications rigid soda-lime glass is used as a substrate material, but flexible metal or polyimide foils have been used successfully. Working on soda-lime glass, it has been established that Na plays an important role in increasing the carrier concentration within the CIGSe absorber [6]. It diffuses at elevated process temperatures from the glass substrate through the Mo back contact into the growing CIGSe thin-film [7], and also has an effect on the morphology and material interdiffusion in the growing layer [8, 9]. On samples, which do not intrinsically contain sodium, it has to be externally supplied.
While ZnO and Mo are usually sputtered, CdS is most widely deposited via chemical-bath deposition. As the use of Cd-containing components is viewed critically, Cd-free alternatives are in development and, in some cases, already incorporated in the commercially-available product [10]. The methods for CIGSe thin-film fabrication can be grouped into two main categories: co-evaporation and sequential processing [10]. The method of co-evaporation relies on the more or less simultaneous evaporation of the elements Cu, In, Ga, and Se to form a CIGSe thin-film on a heated, Mo-coated substrate in vacuum. For sequential processing on the other hand, a so-called precursor layer in either metallic, binary, or nanoparticle form is treated in a reactive atmosphere in order to make the CIGSe thin-film absorber. The current record conversion-efficiency of 20.3 % [11] is reached by a CIGSe thin-film device that was deposited via 3-stage co-evaporation [12].
The 3-stage co-evaporation process for CIGSe deposition leads to a thin film, which has a characteristic compositional in-depth profile, shown in Fig. 5.4. While Ga accumulates near the Mo/CIGSe interface during the inter-diffusion of Cu-Se in stage 2, the last stage of the process also provides for a slightly increased Ga content near the surface. Of course the latter depends on the Ga flux during stage 3. The Ga profile is of relevance to the resulting solar-cell device, as the Ga content determines the energy band-gap of the CIGSe material. A graded compositional profile is therefore equivalent with an in-depth band-gap grading within the device. Compositional gradients, as shown in Fig. 5.4, can be observed even within single grains. Not only does the Ga content have an impact on the resulting energy band-gap of CIGSe, but also on the Cu-deficiency, in particular near the absorber surface. This is possibly caused by the nature of the last stage of the 3-stage process, which can affect its band gap. Cu-deficient CIGSe phases, such as Cu(In,Ga)3Se5, show slightly-larger band gaps than the stoichiometric compound. The presence of such Cu-poor phases at the front interface has been argued to be of relevance for efficient CIGSe thin-film devices [13]. Other evidence points towards entirely Cu-free film surfaces, which ensure favourable interface formation between the CIGSe and the buffer layer [14]. It is most likely that the exact process-parameters determine which of the different scenarios holds in each case. CIGSe thin-films from other manufacturing routines, such as the sequential processing, will have grown under very different thermodynamic and kinetic growth conditions and, so far, only little is known of the extent to which the resulting thin films can be considered identical.

In order to understand the correlation between growth conditions, material properties and final device quality, much has to be understood regarding the basic material properties of the absorber material in question. Topics such as the occupation density of the different sites within the crystal lattice, i.e. defect formation under certain growth conditions, at certain material compositions, or in the presence of foreign elements are areas where neutron diffraction can provide valuable input for the design of growth models, analytical material science, and also for computational methods.
2 Material Properties of Chalcopyrite-Type Compound Semiconductors
\( {\text{CuB}}^{\text{III}} {\text{C}}_{2}^{\text{VI}} \left( {{\text{C}}^{\text{VI}} = {\text{Se}},{\text{ S}}} \right) \) compound semiconductors are part of the chalcopyrite (ch) family and are located in the middle of the ternary system Cu–BIII–CVI, on the pseudo-binary section \( {\text{Cu}}_{ 2} {\text{C}}^{\text{VI}}{-}{\text{B}}_{3}^{\text{III}} {\text{C}}_{2}^{\text{IV}} \) (see Fig. 5.5). The band gap (Eg) ranges from 1.0 to 1.5 eV for a single junction thin-film solar cell. The \( {\text{CuB}}^{\text{III}} {\text{C}}_{2}^{\text{VI}} \) compounds crystallize in the chalcopyrite-type crystal structure, named after the mineral CuFeS2. This tetragonal crystal structure (space group \( I\bar{4}2d \)) consists of two specific cation-sites. The monovalent cations are sited on the 4a (0 0 0) and the trivalent cations (In3+, Ga3+) on 4b (0 0 ½) position. All cations are tetrahedrally coordinated by the anions (8d (x ¼ \( {\raise0.7ex\hbox{$\text{1}$} \!\mathord{\left/ {\vphantom {\text{1} \text{8}}}\right.\kern-0pt} \!\lower0.7ex\hbox{$\text{8}$}} \))) and vice versa.

The ternary system Cu-BIII-CIV and the pseudo-binary tie line Cu2CVI-BIII 2CVI 3
A closer examination of the pseudo-binary tie-line reveals a stability of the ch-phase over a defined compositional-range. That means, the ch-phase accepts a deviation from ideal stoichiometry (\( {\text{CuB}}^{\text{III}} {\text{C}}_{2}^{\text{VI}} \)) by maintaining the crystal structure, and without the formation of any secondary phase. The compound Cu1−yInySe0.5+y is single phase in the region of 0.513 ≤ y ≤ 0.543 and contains within this area only the chalcopyrite-type phase [15]. The common highly-efficient Cu(In,Ga)Se2 thin-film solar devices all exhibit an overall off-stoichiometric composition, due to the multi-stage process applied to grow these absorbers. Such deviations from stoichiometry always cause structural inhomogeneities and charge mismatches, which influence the material properties. One effect is the generation of point defects, which influences the electronic and optical properties of the compound semiconductor. In general 12 intrinsic point-defects can exist within the ch-type crystal structure.
-
3 vacancies: on the two cation and one anion sites (VCu, V IIIB , V VIC )
-
6 anti-site defects: \( {\text{B}}_{\text{Cu}}^{\text{III}} ,{\text{ Cu}}_{\text{B}}^{\text{III}} ,{\text{ C}}_{\text{Cu}}^{\text{VI}} ,{\text{ Cu}}_{\text{C}}^{\text{VI}} ,{\text{ B}}_{\text{C}}^{{{\text{III}}\,\,\,{\text{VI}}}} ,{\text{ C}}_{\text{B}}^{{{\text{VI}}\,\,\,{\text{III}}}} \)
-
3 interstitial defects: \( {\text{Cu}}_{\text{i}} ,{\text{ B}}_{\text{i}}^{\text{III}} ,{\text{ C}}_{\text{i}}^{\text{VI}} \)
These intrinsic point-defects cause different defect levels in the energy gap of the semiconductor (see Table 5.1) and therefore influence the electronic and optical properties, sensitively. Consequently, it is of great importance to know where the atoms are.
In addition to the generation of point defects, the anion position (x(CVI)) of chalcopyrite crystallites is also affected by off stoichiometry. A change of the anion position is proposed to be directly correlated with a change in Eg. The x-parameter controls the position of the valence-band maximum and conduction-band minimum, and therefore Eg. Current studies have shown that Eg decreases for the Cu-poor composition in CuInSe2 caused by a change in x(Se), which is weakly dependent on the concentration of copper vacancies (VCu) [17]. The interplay between the crystal structure and the optical and electronic properties is a fundamental problem, which has to be understood when tailoring high efficiency thin-film devices with a compound semiconductor as absorber layer. For instance, an uncontrolled change in Eg within an absorber layer is undesirable because it is less optimal for absorption of the incoming sunlight.
It is difficult to identify and quantify very small changes in the crystal structure, such as point defects or changes in atomic positions by imaging techniques. Therefore, it is preferred to study such effects by diffraction methods. The method of Rietveld refinement is applied to refine the crystal structure using an X-ray or neutron powder diffraction pattern of the off-stoichiometric compound in detail. This method provides information about the cation distribution and the position of the atoms within the structure with high accuracy. In the following section we discuss the reasons for the preferred use of neutrons in the description of structural changes in detail, and how point defects in compound semiconductors can be identified.
3 Structural Analysis of Off-Stoichiometric Chalcopyrites
Among various experimental possibilities, structural analysis of compound semiconductors by diffraction techniques using X-rays or neutrons has become a technique of choice. The reasons are various, and here we place the focus on the use of neutrons as a radiation source.
The highest efficiency thin-film devices consist of a Cu-poor Cu(In,Ga)Se2 absorber layer. The interplay of structural with electronic and optical properties is therefore interesting to study for this quaternary compound and the corresponding ternaries (CuGaSe2 and CuInSe2). In the case of CuGaSe2 or Cu(In,Ga)Se2 the cations Cu+ and Ga3+ have an identical number of electrons (28). This is a problem for the differentiation of these two cations by conventional X-ray diffraction, where the diffraction is at the electron shell of the atoms. Since atomic scattering form factors f for X-rays are proportional to the atomic number Z, the positions of the unit cell atoms of similar atomic number and the fractional occupation of the Wyckoff sites are difficult to distinguish. Hence, a differentiation of these cations in the atomic structure by conventional X-ray diffraction is impossible. In the case of neutron diffraction, the scattering is at the nucleus and the neutron-scattering lengths of copper and gallium are different (bCu = 7.718(4) fm, bGa = 7.288(2) fm [18]). Moreover, in the case of X-rays, destructive interference effects lead to a decrease of the scattering amplitude with angle. In contrast to diffraction at the electron shell, the atomic nuclei cross-sections are very small and the interference effects are also very small. Therefore, neutron-scattering amplitudes do not decrease rapidly with angle, resulting in the advantage of high intensities of Bragg-reflections observed even at high Q-values. This is especially important for the determination of atomic positions and atomic site occupations. For the analysis of point defects in compound semiconductors and position parameters, for instance of the anion, neutron powder diffraction is advantageous.
3.1 Rietveld Refinement
To get detailed information about the cation and anion distribution within the materials, the neutron powder diffraction data has to be analysed by the Rietveld refinement method [19, 20]. To obtain reliable results it is important to perform the Rietveld procedure in a physically-reasonable sequence. In the case of the chalcopyrite-type compound semiconductors the chalcopyrite-type crystal structure was used as basis model. The free structural parameters of the fit were the lattice constants, the anion position parameter x, the cation site occupancy factors (SOF) of the 4a and 4b site (SOF4a , SOF4b ) and atomic displacement parameters (ADP). The following example sequence of free parameters can be applied for the analysis of Cu(In,Ga)(Se,S)2 compounds:
-
1.
Refined parameters in the first step were the global parameters, zero shift and the scaling factor. Profile parameters like the u, v, w, x and y parameters, defining the full width at half maximum (FWHM), were fixed at the values of a previously-measured standard sample such as Y2O3. Also the background values were fixed in the beginning of the refinement. Other fixed parameters were the structure parameters: lattice constants, ADPs, SOFs, and the atomic position parameters. The SOFs were fixed at values according to the chemical composition as known from chemical analysis. The isotropic ADPs \( (b_{iso}^{A} ; \, A = 4a, 4b; 8d) \) were kept at 1.0
-
2.
Subsequently, lattice constants were refined, the u, v, w, x and y parameters remained fixed, as well as ADPs, SOFs, and position parameters.
-
3.
The \( b_{iso}^{A} \) of 4a, 4b, and 8d positions are refined, keeping the SOFs of the respective positions fixed.
-
4.
The cation site occupancy factors (SOF4a , SOF4b ) are then refined whilst b iso were fixed to their previously-refined values.
-
5.
Step 3 and 4 are repeated until no change of parameters occurs. This procedure has been expanded with the use of anisotropic ADPs, \( b_{ii}^{A} \), (A = 4a, 4b; i = 1–3), since the tetragonal system leads to the need for anisotropic atomic displacement.
-
6.
The \( b_{ii}^{A} \) and the SOFs are refined simultaneously until convergence.
-
7.
If necessary, background and profile parameters: u, v, w, x and y as well as asymmetry values are refined.
This refinement strategy results in reliable values for SOFs of the respective species. Using the following method of average neutron-scattering length [22], it is possible to determine the point defect concentration in complex off-stoichiometric compounds over a large sample volume.
3.2 Point-Defect Analysis by the Method of Average Neutron-Scattering Length
The real crystal structure of a compound is defined by a deviation of the average structure caused by different types of defects. Extended defects, such as grain boundaries and dislocations influence the structure on a grand scale, without a variation on the level of individual unit cells. Small defects like single, isolated point defects, or correlated defects forming small defect-clusters, generally cause systematic variations of unit cells, which can be detected by diffraction methods. The method of average neutron-scattering length is an approach that gives a possible solution of point defects. By this method it is possible to determine the cation distribution within the material for a wide range of structure types, and will be explained here using CuInSe2 and CuGaSe2 as examples.
The approach is based on the fact, that vacancies (VCu and VIn), as well as anti-site defects (InCu and CuIn), will change the neutron-scattering length of the cation sites 4a (copper site) and 4b (indium site) in the chalcopyrite-type structure significantly [15]. In a neutron diffraction experiment, the neutron interacts with the atomic nucleus and the neutron-scattering lengths of copper and indium are different (bCu = 7.718 (4) fm, bIn = 4.065 (2) fm, bV = 0 [18]). Thus, the distribution of copper and indium on both cation sites of the structure can be revealed from the SOFs determined by Rietveld analysis of the neutron diffraction data.
If different species like Cu, In, and vacancies occupy the same structural site j, the average neutron-scattering length of this site is defined by:
Here, N is the fraction of the species on the corresponding site and b are the neutron-scattering lengths. As an additional requirement the full occupation of the site j has to be taken into account, which is achieved when \( \mathop \sum \nolimits N_{i} = 1 (i = {\text{Cu}},{\text{In}},{\text{V}}) \). In an example case the chalcopyrite-type crystal structure is used as basis model for the Rietveld refinement. Thereby, Cu occupies the 4a and In the 4b site. An experimental average neutron-scattering length for the two sites using the SOFs can be calculated by:
where SOF4a and SOF4b are the cation site occupancy factors of the chalcopyrite-type crystal structure from the Rietveld analysis. The evaluation of the experimental average neutron-scattering length of a series of different Cu/In ratios, and therefore different degrees of off-stoichiometry, reveals a decrease of \( \bar{b}_{4a}^{exp} \) (see Fig. 5.6) with decreasing Cu/In. Taking into account b V < b In < b Cu, a decrease of \( \bar{b}_{4a}^{exp} \) can only be caused by the presence of vacancies (VCu) or anti-site defects of type InCu. Since the decrease of \( \bar{b}_{4a}^{exp} \) becomes stronger with decreasing Cu/In, an increase of the particular defect concentration can be assumed. Clearly, the value for \( \bar{b}_{4b}^{exp} \) increases with decreasing Cu/In ratio. This can only be due to the presence of anti-site defects of type InCu (b Cu > b In).

Experimental average neutron-scattering length as a function of Cu/In ratio
The experimental average neutron-scattering lengths have to be compared with theoretical values. In a first step, these values are derived from a cation distribution model built on the basis of the known chemical composition of the material. The general formula for the calculation of the cation distribution is given by Eq. (5.1). A simultaneous comparison of \( \bar{b}_{j} \) with \( \bar{b}_{j}^{exp} \) during variation of the cation-distribution model leads to the corresponding amounts of isolated point-defects.
Using this approach it was possible to determine the cationic point-defect concentration for various chalcopyrite-type compounds. It was clearly shown that CuInSe2 tends, when being Cu-poor, to form VCu, InCu, and CuIn defects, resulting in a partially disordered chalcopyrite-type crystal structure. In contrast to CuInSe2, CuGaSe2 exhibits the same crystal structure, but due to the small ionic radii of Ga3+ [23] the material tends to form interstitial defects of type Gai [24]. These differences sensitively influence the properties of a final solar device. The kind of point defects present in CuInSe2 allow a neutralization of isolated point-defects by clustering together forming a neutral defect complex of type (2VCu + InCu), which has a considerable binding-energy [16]. Such a neutralization of point defects cannot be assumed for CuGaSe2. This crucial difference is one reason why it is possible to design a thin-film solar cell with a very off-stoichiometric absorber and high defect-concentration, like CuInSe2, but maintain considerable efficiency. Tailoring high efficiency devices with a CuGaSe2 absorber layer is still a challenging task due to different problems. One aspect is the difference in the presence of intrinsic point-defects in Cu-poor CuGaSe2 discovered by neutron powder diffraction.
4 Low-Temperature Thermal Expansion in Chalcopyrite-Type Compound Semiconductors
Solids whose crystal structures are based on tetrahedrally-coordinated ions may show the intriguing property of negative thermal-expansion.
The ternary \( {\text{A}}^{\text{I}} {\text{B}}^{\text{III}} {\text{C}}_{2}^{\text{VI}} \) semiconductors (A = Ag, Cu; B = Al, Ga, In; C = S, Se, Te), exhibit such a tetrahedral coordination (see Fig. 5.7). The coordination tetrahedron around an anion (sulfur or selenium) consists of two monovalent and two trivalent cations. The chemical bonds within such a tetrahedron are of mixed covalent and ionic character, whereby the ionicity of the bonds is different for the AI–CVI and BIII–CVI bonds. These different interactions result in different bond lengths (RAC ≠ RBC) as well as bond angles and lead to a displacement of the anions from the ideal tetrahedral site by a quantity u = ∣x − ¼∣ (where x is the anion x coordinate).

The tetragonal chalcopyrite-type crystal structure (left). This structure consists of a periodic sequence of tetrahedra, each one built of a central anion surrounded by 4 cations (right)
The linear thermal-expansion coefficients are closely related to the Grüneisen parameters γ of lattice vibrations [25]. The occurrence of a negative thermal-expansion can be understood using the notation of a balance between acoustic shear and compression modes of the observed crystal structure. The Grüneisen parameters of the shear modes show a tendency to negative values, while those of the compression modes are positive [25–27]. Hence, the temperature dependence of the thermal expansion is determined by the degree of excitations of the various modes and can change its sign when the relative thermal-population of the modes varies.
In \( {\text{A}}^{\text{I}} {\text{B}}^{\text{III}} {\text{C}}_{2}^{\text{VI}} \) chalcopyrite-type semiconductors the thermal-expansion behaviour is described by the independent linear thermal-expansion coefficients \( \alpha_{a} \) and \( \alpha_{c} \) with
The uniaxial chalcopyrite-type structure comes with two independent Grüneisen parameters \( \gamma_{a} \) and \( \gamma_{c} \), which are related to \( \alpha_{a} \) and \( \alpha_{c} \) according to [28]:
Here V m is the molar volume, C p the molar specific-heat at constant pressure and c ij are the adiabatic elastic-stiffnesses. With increasing ionicity the Grüneisen parameter should become more negative [29]. Thus, the covalent character of the chemical bond is expected to strongly affect the Grüneisen parameter.
The determination of linear thermal-expansion coefficients by dilatometry or X-ray diffraction [30–33] has shown that \( \alpha_{a} \) and \( \alpha_{c} \) vary independently with temperature. This is caused by the axial symmetry of the chalcopyrite-type crystal structure and the difference in strength of the Cu-CVI and BIII-CVI cation-anion bonds.
The investigation of the negative thermal-expansion is conveniently achieved by neutron powder diffraction. One aspect for the use of neutrons is again the high intensity in the diffraction pattern at high Q-values, important for an exact determination of the chalcogen position. It is important to monitor the change of this position at low temperatures to describe the bond stretching during cooling. The negative thermal-expansion has been studied for several chalcopyrite-type compounds, whereby the focus now lies on Cu(InxGa1−x)Se2 once with high (x = 0.918) and once with low indium content (x = 0.096), to show the effect of different bond ionicities on the negative thermal-expansion. Neutron powder diffraction patterns were collected for temperatures between 1.5 K > T > 300 K and structures refined by the Rietveld method according to the previously-described sequence. The ionicity can be calculated following Phillip’s definition [34]:
with X A and X B the electronegativity of the elements A and B (X Cu = 1.9; X Ga = 1.81; X In = 1.78). According to Phillip’s definition the bond ionicities increase from Cu–Se (f i = 0.1002) to In–Se (f i = 0.115) and Ga–Se (f i = 0.128). Thus the ionicity of the BIII–Se cation-anion bond is increasing with increasing substitution of indium by gallium. From this it follows that with a high amount of gallium the difference in bond ionicity between the Cu–Se and BIII–Se cation-anion bond increases, which lead to an increased anisotropy.
The higher anisotropy affects the change of lattice parameters with decreasing temperature, which is stronger for the gallium-rich Cu(In,Ga)Se2 and pure CuGaSe2 than for indium-rich Cu(In,Ga)Se2 (see Fig. 5.8). Applying a third-order polynomial fit to the lattice parameters the thermal-expansion coefficients \( \alpha_{a} \) and \( \alpha_{c} \), can be derived. The temperature at which the linear thermal-expansion becomes negative (T0), is seen to vary with the chemical composition (see Table 5.2).

Lattice constants a and c as a function of temperature for a the In-rich sample with In/(In + Ga) = 0.918 b the Ga-rich sample with In/(In + Ga) = 0.096 and c pure CuGaSe2
The variation of T0 with chemical composition should be discussed in context with the bond ionicity of the BIII-Se bonds, which increase with increasing substitution of indium by gallium. Thus, with increasing gallium content the ionicity increases and the temperature, for which the linear thermal-expansion coefficient changes its sign, increases. This is observed for Cu(InxGa1−x)Se2 with different x-values as summarized in Table 5.2.
The In/(In + Ga) ratio strongly influences the character of the covalent-ionic BIII–Se cation-anion bond, and therefore the behaviour of the linear thermal-expansion coefficients of the two lattice constants \( \alpha_{a} \) and \( \alpha_{c} \).
Also, the x-parameter of the selenium anion as a function of temperature is strongly affected by the different bond ionicities. In the Ga-rich sample the tetragonal deformation u = 0.25–x(Se) strongly tends to zero with decreasing temperature, whereas it stays almost constant for the In-rich sample (Fig. 5.9). This effect is explained by the higher bond-ionicity for the Ga–Se bond compared to the In–Se bond.

The tetragonal deformation u as a function of temperature for the indium-rich and gallium-rich Cu(InxGa1−x)Se2 compounds
The change of the tetragonal distortion and the anion position parameter x(Se) is reflected by the change in the average cation-anion bond distances and angles, which change markedly for the Ga-rich sample compared to In-rich sample (see Fig. 5.10).

The cation-anion-cation bond angles as a function of temperature for a Ga-rich and b In-rich Cu(InxGa1−x)Se2
5 Novel Materials for In-Reduced Thin Film Solar-Cell Absorbers
Chalcopyrite-based thin-film solar cells with the semiconductor Cu(In,Ga)(S,Se2) (CIGSe) as absorber material show the highest efficiencies among thin-film photovoltaics in the laboratory as well as in module production (see Part 1). This indium based thin-film technology has a huge potential for low-cost photovoltaic production, but the scarcity of indium (indium’s abundance in the continental crust is estimated to be approximately 0.05 part per million). Indium can be jointly refined from trace concentrations in leading ores as zinc, copper, and lead or the material comes from recycled scrap. Increasing prices of indium could easily limit the production growth. There is also an increased demand for indium for use in other technologies, for example items such as: flat panel displays, solders, thermal-interface materials, batteries, compound semiconductors and light-emitting diodes. On the other hand, the worldwide reserve of viable indium is approximately 16 kton.Footnote 1 Nevertheless, the price of indium has varied dramatically in recent years from US$94/kg in 2002 to over US$1,000/kg in 2006. The price in 2012/2013 was about US$580/kg [35]. This reflects the scarcity of supply and the dependence on the small number of production facilities worldwide.
In order to secure the long term development of compound semiconductor based thin-film solar cells, the search for the replacement of indium is advisable. Semiconductors suitable as absorber materials in thin-film solar cells should fulfil a number of requirements, the most important amongst them being a high absorption-coefficient (in the range of 105 cm−1) and a band-gap energy in the optimal range of about 1.4 eV. Here multinary compounds, like solid solutions between non-isotype binary II-VI and ternary I-III-VI2 compounds, are potential candidates. Moreover, there are a variety of chalcogenide minerals available as a source of novel, indium-free absorber materials. Replacing indium in CuIn(S,Se)2 by the abundant elements zinc and tin yields the quaternary compound Cu2ZnSn(S,Se)4, which is also a direct band-gap p-type semiconductor with an absorption coefficient higher than 104 cm−1 [36].
5.1 Cation Distribution in 2(ZnX)-CuBIIICVI 2 Mixed Crystals
The formation of solid solutions between the non-isotype compounds ZnC and CuBIIIC2 (B-Al, Ga, In; C-S,Se) enables the band-gap energy to be changed from the large value of the binary wide-gap semiconductor ZnC to the band gap of the chalcopyrite-type end member CuInC2 [37]. The possible application of 2(ZnS)-CuInS2 mixed crystals as absorbers in thin-film solar cells was introduced by Bente et al. [38]. However, these solid-solution series are characterized by a relatively-large miscibility gap, where a tetragonal and a cubic phase coexists [37, 39–41], thus the applicable range is limited.
For tetragonal Zn2x(CuB)1−xC2 mixed crystals, the question of the distribution of the three cations Zn2+, Cu+ and B3+ on the two cation positions of the chalcopyrite-type structure arises. Like in CIGSe, the cation distribution influences the opto-electronic properties of the solid-solution compounds. The Cu–Zn differentiation problem existing in X-ray diffraction due to a nearly-equal atomic form-factor f for Cu+ and Zn2+, can be solved by neutron diffraction because of the different neutron scattering cross-sections for Cu and Zn, which are based on the different neutron scattering lengths (bZn = 5.68 fm, bCu = 7.718 fm [18]).
The most detailed analysis was performed for chalcopyrite-type Zn2x(CuIn)1−xS2 mixed crystals [42]. It should be noted that the study was performed using powder samples with stoichiometric composition (the chemical composition was determined by wavelength-dispersive X-ray spectroscopy using an electron microprobe system). For the interpretation of the average neutron-scattering lengths of the cation sites 4a and 4b (\( \bar{b}_{4a}^{exp} \) and \( \bar{b}_{4b}^{exp} \)), derived from the cation SOFs determined by Rietveld analysis of neutron powder diffraction data, the principle of the conservation of tetrahedral bonds (CTB) for ternary ABC2 chalcopyrites [43] was applied. In the chalcopyrite-type structure a displacement of the anion from its ideal position (¼, i.e. the middle of the cation tetrahedron) by u − ¼ (u is the anion x-coordinate) can be observed. Hence different bond lengths RAC ≠ RBC result, which in turn cause different-sized anion tetrahedra AC4 and BC4, resulting in a tetragonal deformation η = c/2a parallel to the crystallographic c-axis. The bond lengths are [43]
The parameters η and u are considered as the degrees of freedom of the chalcopyrite-type structure [Jaffe, Zunger 84]. The Abrahams-Bernstein relation [44]
correlates the tetragonal distortion u with the lattice parameters a and c. However, there is a limitation: only one of the anion tetrahedra is assumed to be deformed, whereas the other is taken as regular.
According to the CTB, the degrees of freedom (η and u) would attain values that simultaneously minimize the difference between the bond lengths RAC and RBC and the sums of the elemental radii as
By applying Eqs. (5.6) and (5.7) the solutions for Eq. (5.8) can be written as
Here \( \upalpha \) is the bond-mismatch parameter and β the mean-square-bond
The CTB model can be extended to quaternary chalcopyrite-type compounds, assuming the covalent radii in the equations above as the average radius of the cations on the two cation positions, according to
Here ZnA, CuA, and InA are the mole fractions of the cations on the Wyckoff position 4a (A) and 4b (B) according to the cation-distribution model. These fractions correspond to the total amount of Zn, Cu, and In in (2ZnS)x(CuInS2)1–x (i.e. ZnA + ZnB = 2x). Thus the average cation-radii are influenced by the cation distribution.
For the calculation of the average neutron-scattering lengths of the cation sites 4a and 4b (\( \bar{b}_{4a}^{calc} \) and \( \bar{b}_{4b}^{calc} \)), a certain cation distribution has to be assumed. A first comparison with the experimentally-determined average neutron-scattering lengths show, that Zn is not statistically distributed on the sites 4a and 4b. Thus three different aspects have to be taken into account for modelling the cation distribution:
-
(i)
Zn is non-statistically distributed
-
(ii)
If Zn prefers the 4a position a CuIn anti-site is enforced and if Zn prefers the 4b position an InCu anti site is enforced (enforced anti-sites)
-
(iii)
Independent of the Zn distribution, CuIn and InCu anti-sites may exist (spontaneous anti-sites)
The evaluation criteria for the cation distribution in tetragonal (2ZnS)x(CuInS2)1−x mixed crystal were formulated as:
-
(1)
u(exp) = u(calc) (u(exp) is determined by Rietveld analysis of the powder diffraction data, u(calc) applying the CTB rule)
-
(2)
\( \bar{b}_{4a}^{exp} = \bar{b}_{4a}^{calc} \) and \( \bar{b}_{4b}^{exp} = \bar{b}_{4b}^{calc} \)
Applying both criteria, and taking into account the aspects (i)–(iii), the cation distribution was evaluated in two steps. First, the possible cation distributions fulfilling criterion (1) were derived. As can be seen from Fig. 5.11, a variety of different cation distributions are possible.

Possible cation distributions fulfilling the criterion u(exp) = u(calc) for the sample Zn0.18Cu0.92In0.90S2. Closed symbols refer to the 4a position, open symbols to the 4b position
In the second step criterion (2) is also taken into account. The graphical solution is shown in Fig. 5.12. It becomes clear, that Zn occupies the 4b site preferentially, enforcing ~1.8–4.5 % InCu. Moreover, there is a small fraction of spontaneous Cu-In anti-sites (i.e. CuIn and InCu).

Average neutron-scattering lengths calculated for possible cation distributions taking into account a non-statistical Zn distribution as well as enforced and/or spontaneous Cu–In anti-sites. Both limits (all Zn would be ZnCu or ZnIn) narrow the possible cation distributions. The dotted lines give the calculated average neutron-scattering length for the sites 4a and 4b for different cation distributions (with the experimental error), taking into account both enforced and spontaneous Cu-In anti-sites. The blue lines mark the distributions which fulfil criterion (1). Both criteria only meet within the region marked by the green vertical lines
Taking into account the experimental error of the average neutron-scattering lengths \( \bar{b}_{4a}^{exp} \) and \( \bar{b}_{4b}^{exp} \) it can be deduced, that 27.5 % of the Zn occupies the 4a site, whereas the rest of the Zn occupies the 4b site. This leads to 3.6 % enforced InCu anti-site. These results are in a good agreement with the cation distribution evaluated from X-ray powder diffraction data ((Zn + Cu)Cu = 0.91(3); InCu = 0.09(3); (Zn + Cu)In = 0.14 and InIn = 0.86(3)), but here Cu and Zn could not be distinguished. The cation distribution in Zn2x(CuIn)1−xS2 mixed crystal is summarized in Table 5.3.
The CTB rule was only applied for the sulfide mixed-crystals due to the relatively-well known cation radii in sulfides [23]. For the selenide and telluride Zn2x(CuIn)1−xC2 mixed crystals the differences between \( \bar{b}_{4a}^{exp} \) and \( \bar{b}_{4a}^{calc} \) as well as \( \bar{b}_{4b}^{exp} \) and \( \bar{b}_{4b}^{calc} \), assuming a statistical Zn distribution in the calculation of the average neutron-scattering lengths, were considered [42, 45]. Here, a non-statistical distribution of Zn on the both cation sites was also found. With increasing ZnX-content in Cu0.5In0.5X there is a propensity for a more statistical distribution of the cations, indicating a tendency for disorder in the cation substructure.
It can be assumed that the non-statistical distribution of Zn and the associated Cu-In anti-sites are related to the limited solubility of ZnX in Cu0.5B0.5X. This fact can be discussed within the framework of formation energies of intrinsic point-defects in copper chalcopyrites. The Cu-In anti-site occupancy, resulting in CuIn and InCu, are the defects with the lowest formation energies (in CuInSe2: 1.3 eV for CuIn and 1.4 eV for InCu [46]). Thus, these defects can be formed relatively easily. Nevertheless, the formation energies of ZnCu or ZnIn are not known. If one of the occupancies were energetically unfavourable, the solubility would be affected.
Using the approach of the average neutron-scattering length and the CTB rule it was possible to determine the cation distribution for various stoichiometric chalcopyrite-type Zn2x(CuB)1−xC2 compounds. It was clearly shown that Zn tends to occupy the 4b site preferentially, resulting in the formation of InCu and CuIn defects, resulting in a partially disordered chalcopyrite-type crystal structure.
5.2 Point Defects in Cu2ZnSn(S,Se)4 Kesterite-Type Semiconductors
Substitution of indium by zinc and tin in CuIn(S,Se)2 leads to the quaternary compound semiconductor Cu2ZnSn(S,Se)4 (CZTSSe). The record efficiency of thin-film solar cells using a CZTSSe absorber layer is above 10 % [47].
Both compounds, CZTS and CZTSe, belong to the family of tetrahedrally-coordinated adamantine semiconductors [48]. Here each anion is tetrahedrally coordinated by four cations (two copper, one zinc and one tin), whereas each cation is coordinated by four anions (sulfur or selenium). Thus, the structure is characterized by a well-defined framework of tetrahedral bond arragnements, which is advantageous for the properties of the material.
For the quaternary \( {\text{A}}_{2}^{\text{I}} {\text{B}}^{\text{II}} {\text{C}}^{\text{IV}} {\text{X}}_{4}^{\text{VI}} \) chalcogenides with A-Cu; B-Zn; C-Si, Ge, Sn and X-S, Se, different crystal structures are discussed in literature: the kesterite- type structure (space group \( I\bar{4} \)), the stannite-type structure (space group \( I\bar{4}2m \)), as well as the wurtz-stannite (space group \( Pmn2_{1} \)) and the wurtz-kesterite type structure (space group \( Pc \)). The same tetrahedral metal-coordination (2Cu, one II- and one IV-element surrounding each S-atom) is possible in all four space groups. A clear decision can only be made by a detailed structure analysis for each compound.
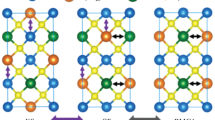
At room temperature CZTS adopts the space group \( I\bar{4} \). The structure can be described as a cubic close-packed array of sulfur atoms, with metal atoms occupying one half of the tetrahedral voids. CZTSe was reported to crystallize in the space group \( I\bar{4}2m \) in a topologically-identical structure, but different cation distribution of AI and BII among the positions (0, 0, 0), (0, ½, ¼), and (0, ¼, ¾) [49]. Both structures are represented in Fig. 5.13.

Kesterite-type (a) and stannite-type (b) crystal structure (blue Cu, orange Zn, red Sn, yellow S,Se)
Neutron powder diffraction and the method of the average neutron-scattering length were used to clarify possible differences of the cation distribution in CZTS and CZTSe, especially with respect to the electronically-similar elements copper and zinc [50, 51].
According to the general formula for the calculation of the average neutron-scattering length (see also Sect. 5.3.2)
where A and B are two different cations, V represent possible vacancies and j stands for the Wyckoff position, the following equations were derived for the calculation of the experimental average neutron-scattering lengths \( \bar{b}_{j}^{exp} \)
- Kesterite type structure (j = 2a, 2c, 2d)::
-
$$ \begin{gathered} \overline{b}_{2a}^{\exp } = occ_{2a} \cdot b_{\text{Cu}} \hfill \\ \bar{b}_{2c}^{\exp } = occ_{2c} \cdot b_{\text{Zn}} \hfill \\ \bar{b}_{2d}^{\exp } = occ_{2d} \cdot b_{\text{Cu}} \hfill \\ \end{gathered} $$(5.14)
- Stannite type structure (j = 2a, 4d)::
-
\( \begin{gathered} \bar{b}_{2a}^{\exp } = occ_{2a} \cdot b_{{\text{Zn}}} \hfill \\ \bar{b}_{4d}^{\exp } = occ_{4d} \cdot b_{\text{Cu}} \hfill \\ \end{gathered} \)
The cation site-occupancy values occ j resulted from the Rietveld analysis of the neutron diffraction data. Because the study was performed using stoichiometric powder samples (the chemical composition of the samples was determined by wavelength-dispersive X-ray spectroscopy), vacancies were not taken into account.
The Rietveld analysis of the neutron diffraction data was performed by applying different cation-distribution models within both structure types as the starting model for the crystal structure (see Table 5.4). The cation site occupancies were taken as free parameters in the refinement. It was found that the refined cation site-occupancy values differ from their nominal values (Table 5.4), with the exception of the tin. Thus, one can conclude that the site is not only occupied by the initially-supposed model cation, but also by a mixture of different elements. For example, a decrease of the average neutron-scattering length of the 2d site in the kesterite-type structure can be attributed to a partial Zn occupation due to b Cu > b Zn.
The refined cation site occupancies were used to calculate the experimental average neutron-scattering lengths \( \bar{b}_{j}^{exp} \) (see Fig. 5.14). The first result obtained was a confirmation of the occupancy of the 2b site by tin in both compounds. Concerning the sites 2a, 2c and 2d for the space group \( I\bar{4} \) as well as 2a and 4d for the space group \( I\bar{4}2m \), a similar picture for both compounds appears. Irrespective of the structure model used in the Rietveld analysis, the average neutron-scattering length of the 2a position indicates that this position is occupied by copper only. The average neutron-scattering lengths of the sites 2c and 2d indicate a mixed occupancy of these both sites by copper and zinc. Approximately 50 % of the zinc site 2c is occupied by copper and vice versa concerning the copper site 2d, forming CuZn and ZnCu anti-sites. Nevertheless, this disorder is limited to the positions 2c and 2d and hence to the lattice planes at z = 1/4 and 3/4. The 2a site is always occupied by copper only, and there are no indications of zinc occupying that site.

Experimental average neutron-scattering length of the cation sites (the 2b site is excluded) evaluated from the corresponding site occupancy values determined by Rietveld analysis for CZTS (a) and CZTSe (b)
These facts bring us to the conclusion that both compounds, CZTS and CZTSe, adopt the kesterite-type structure, but exhibit Cu-Zn disorder in the (00l) lattice planes at z = 1/4 and 1/3. This disorder creates CuZn and ZnCu anti-site defects. These results are supported by ab initio calculations carried out on these systems [52, 53]. The authors report that the CuZn anti-site acceptor can be predicted as the most probable defect and thus can be easily formed. Moreover, the kesterite-type structure was indicated as the ground-state structure for both CZTS and CZTSe, though the stannite-type structure has only a slightly-lower binding energy (CZTS: 2.9 meV/atom [52], 1.3 meV/atom [53], CZTSe: 3.8 meV/atom [52], 3.3 meV/atom [53]).
Using the approach of the average neutron-scattering length, and applying different structural models with different cation distributions in the structural description in the Rietveld refinement procedure, the crystal structure of CZTS and CZTSe was determined and intrinsic point-defects were identified experimentally. In contradiction with earlier X-ray diffraction studies, the kesterite-type structure was also proved for CZTSe.
Notes
- 1.
Mineral Commodities Summary 2008, United States Geological Survey (2008); later versions (2009–2012) do not give a number for In reserves.
References
T. Unold, C.A. Kaufmann, in Chalcopyrite Thin-Film Materials and Solar Cells, vol. 1, 1st edn. Comprehensive Renewable Energy, vol 1. Photovoltaic Solar Energy (Elsevier, Oxford, 2012)
R. Scheer, H.W. Schock, Chalcogenide Photovoltaics: Physics, Technologies, and Thin Film Devices (Wiley-VCH, Weinheim, 2011)
S.R. Kodigala, Chapter 8—Cu(In1−xGax)Se2 and CuIn(Se1−xSx)2 thin film solar cells, in Thin Films and Nanostructures, vol. 35, ed. by K. Subba Ramaiah (Academic Press, New York, 2010), pp. 505–679
S. Siebentritt, M. Igalson, C. Persson, S. Lany, Prog. Photovoltaics 18, 390 (2010)
S.B. Zhang, S.H. Wei, A. Zunger, H. Katayama-Yoshida, Phys. Rev. B 57, 9642 (1998)
S.H. Wei, S.B. Zhang, A. Zunger, J. Appl. Phys. 85, 7214 (1999)
R. Caballero, C.A. Kaufmann, V. Efimova, T. Rissom, V. Hoffmann, H.W. Schock, Prog. Photovoltaics 21, 30 (2012)
M. Bodegard, O. Lundberg, J. Malmstrom, L. Stolt, High voltage Cu(In,Ga)Se-2 devices with Ga-profiling fabricated using co-evaporation, in Conference Record of the Twenty-Eighth IEEE Photovoltaic Specialists Conference—2000 (2000), pp. 450–453
D. Rudmann, D. Bremaud, A.F. da Cunha, G. Bilger, A. Strohm, M. Kaelin, H. Zogg, A. Tiwari, Thin Solid Films 480, 55 (2005)
S. Niki, M. Contreras, I. Repins, M. Powalla, K. Kushiya, S. Ishizuka, K. Matsubara, Prog. Photovoltaics 18, 453 (2010)
P. Jackson, D. Hariskos, E. Lotter, S. Paetel, R. Wuerz, R. Menner, W. Wischmann, M. Powalla, Prog. Photovoltaics 19, 894 (2011)
A.M. Gabor, J.R. Tuttle, D.S. Albin, M.A. Contreras, R. Noufi, A.M. Hermann, Appl. Phys. Lett. 65, 198 (1994)
D. Schmid, M. Ruckh, F. Grunwald, H.W. Schock, J. Appl. Phys. 73, 2902 (1993)
H. Monig, C.H. Fischer, R. Caballero, C.A. Kaufmann, N. Allsop, M. Gorgoi, R. Klenk, H.W. Schock, S. Lehmann, M.C. Lux-Steiner, I. Lauermann, Acta Mater. 57, 3645 (2009)
C. Stephan, S. Schorr, M. Tovar, H.W. Schock, Appl. Phys. Lett. 98, 091901 (2011)
S.B. Zhang, S.H. Wei, A. Zunger, Phys. Rev. Lett. 78, 4059 (1997)
L. Gütay, D. Regesch, J.K. Larsen, Y. Aida, V. Depredurand, A. Redinger, S. Caneva, S. Schorr, C. Stephan, J. Vidal, S. Botti, S. Siebentritt, Phys. Rev. B 86, 045216 (2012)
A.J. Dianoux, G. Lander, Neutron Data Booklet (Institut Laue-Langevin, Grenoble, 2001)
H.M. Rietveld, J. Appl. Crystallogr. 2, 65 (1969)
H.M. Rietveld, Acta Crystallogr. 22, 151 (1967)
L.B. McCusker, R.B. Von Dreele, D.E. Cox, D. Louer, P. Scardi, J. Appl. Crystallogr. 32, 36 (1999)
A. Furrer, J. Mesot, T. Strässle, in Neutron Scattering in Condensed Matter Physics, vol. 4. Series on Neutron Techniques and Applications (World Scientific, London, 2009)
R.D. Shannon, Acta Crystallogr. A 32, 751 (1976)
C. Stephan, T. Scherb, C.A. Kaufmann, S. Schorr, H.W. Schock, Appl. Phys. Lett. 101, 101907 (2012)
T.H.K. Barron, J.G. Collins, G.K. White, Adv. Phys. 29, 609 (1980)
M. Blackman, Philos. Mag. 3, 831 (1958)
A. Bienenstock, G. Burley, J. Phys. Chem. Solids 24, 1271 (1963)
H. Neumann, P. Deus, R.D. Tomlinson, G. Kuhn, B. Hintze, Phys. Status Solidi A 84, 87 (1984)
T.F. Smith, G.K. White, J. Phys. C. Solid State 8, 2031 (1975)
H.G. Bruhl, H. Neumann, G. Kuhn, Solid State Commun. 34, 225 (1980)
P. Deus, H. Neumann, G. Kuhn, B. Hinze, Phys. Status Solidi A 80, 205 (1983)
H. Neumann, Cryst. Res. Technol. 18, 659 (1983)
S. Schorr, D. Sheptyakov, J. Phys. Condens. Matter 20, 104245 (2008)
J.C. Phillips (ed.), Bonds and Bands in Semiconductors (Academic, New York, 1973)
www.geodexminerals.com. Accessed 4 March 2013
K. Ito, T. Nakazawa, Jpn. J. Appl. Phys. 27, 2094 (1988)
S. Schorr, G. Wagner, J. Alloy. Compd. 396, 202 (2005)
K. Bente, T. Döring, Chemie der Erde 51 (1991)
S. Schorr, Thin Solid Films 515, 5985 (2007)
G. Wagner, S. Lehmann, S. Schorr, D. Spemann, T. Doering, J. Solid State Chem. 178, 3631 (2005)
L. Roussak, G. Wagner, S. Schorr, K. Bente, J. Solid State Chem. 178, 3476 (2005)
S. Schorr, M. Tovar, N. Stusser, K. Bente, Phys. B 350, E411 (2004)
J.E. Jaffe, A. Zunger, Phys. Rev. B 29, 1882 (1984)
S.C. Abrahams, J. Bernstei, J. Chem. Phys. 55, 796 (1971)
S. Schorr, M. Tovar, D. Sheptyakov, L. Keller, G. Geandier, J. Phys. Chem. Solids 66, 1961 (2005)
J. Klais, Untersuchung intrinsischer Fehlstellen in nicht-stöchiometrischen CuInSe2 Kristallen durch Positronenlebensdauer- und elektrische Messungen. Technische Universität Bergakademie Freiberg (1999)
D.A.R. Barkhouse, O. Gunawan, T. Gokmen, T.K. Todorov, D.B. Mitzi, Prog. Photovoltaics 20, 6 (2012)
B.R. Pamplin, J. Phys. Chem. Solids 25, 675 (1964)
I.D. Olekseyuk, L.D. Gulay, I.V. Dydchak, L.V. Piskach, O.V. Parasyuk, O.V. Marchuk, J. Alloy. Compd. 340, 141 (2002)
S. Schorr, H.J. Hoebler, M. Tovar, Eur. J. Mineral. 19, 65 (2007)
S. Schorr, Sol. Energ. Mat. Sol. C 95, 1482 (2011)
S.Y. Chen, X.G. Gong, A. Walsh, S.H. Wei, Appl. Phys. Lett. 96, 021902 (2010)
C. Persson, J. Appl. Phys. 107, 053710 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Schorr, S., Stephan, C., Kaufmann, C.A. (2015). Chalcopyrite Thin-Film Solar-Cell Devices. In: Kearley, G., Peterson, V. (eds) Neutron Applications in Materials for Energy. Neutron Scattering Applications and Techniques. Springer, Cham. https://doi.org/10.1007/978-3-319-06656-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-06656-1_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-06655-4
Online ISBN: 978-3-319-06656-1
eBook Packages: EnergyEnergy (R0)