
Abstract
Perfluorooctanoic acid (PFOA) is an emerging persistent organic pollutant receiving increasing attention due to its global occurrence and resistance to most conventional degradation methods. Some special techniques such as ultrasonication, aqueous electron reduction, persulfate photolysis, and phosphotungstic-acid photocatalysis have been developed to decompose PFOA. However, these methods are not satisfactory due to their harsh reaction conditions or/and high energy consumption. Heterogeneous photocatalysis based on TiO2, an effective, mild method for the treatment of most waters contaminated with organic compounds, is ineffective in degrading PFOA. Recently, we found other semiconductor materials possess higher PFOA degradation activities than TiO2, such as β-Ga2O3 and In2O3. This paper provides an overview of some recent advances in the photocatalytic degradation of PFOA: (1) mechanism for photocatalytic degradation of PFOA by In2O3, (2) the photocatalytic performance of different In2O3 nanostructures, (3) photocatalytic degradation of PFOA by β-Ga2O3 nanomaterials, (4) potential applications in wastewater treatment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Photocatalytic Activity
- Photocatalytic Decomposition
- Photogenerated Hole
- Diffuse Reflectance Infrared Fourier Transform Spectroscopy
- Perfluorooctanoic Acid
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
3.1 Introduction
Perfluorocarboxylic acids (PFCAs, C n F2n+1COOH) are widely used as industrial surfactants, additives, firefighting foams, coatings, and lubricants owing to their versatile surface activities, and high chemical and thermal stability. Among them, perfluorooctanoic acid (PFOA, C7F15COOH) is the most commonly used industrial PFCAs. A large amount of PFOA is manufactured through the electrochemical fluorination (ECF) and telomerization process. PFOA may also be the breakdown product of other perfluoroalkyl substances (PFASs) [1–4]. Direct or indirect emissions of PFOA during manufacture, use, and disposal have resulted in its widespread occurrence in the environment. Over the past decade, PFOA has been detected in various environmental matrices such as water, sediments, domestic sludge, dust, and biotic samples such as fish, human blood, and liver tissue [5–9].
As an emerging persistent organic pollutant, PFOA is the subject of increasing regulatory interest because of its bioaccumulation, environmental persistence, and growing evidence of its toxicity to humans. Significant levels of PFOA have been detected in serum samples in USA [10], China [11], and Japan [12]. The PFOA concentrations detected in 2,094 human serum samples from 2003 to 2004 in USA National Health and Nutrition Examination Survey (NHANES) ranged from 0.1 to 77.2 ng/mL with a median of 4.0 ng/mL [13]. In general, the longer the carbon chain length, the longer they can stay in the body. For example, perfluorobutane sulfonate, which has four carbons, has a half-life for elimination in a little over 1 month in humans, while PFOA, so called C8 compounds, has a half-life of 3.8 years [14].
Various in vivo and in vitro models have been used to assess the potential bioeffects of PFOA over the past decades. Distribution of PFOA in human tissues is unclear, but in vivo studies revealed the high likelihood of its accumulation in, primarily, the liver, kidney, and blood [15]. It was also found that exposure to PFOA could arrest cell cycle replication [16], alter peroxisomal and MAPK-related signaling pathways [17], and induce oxidative DNA damage in mammalian cells [18, 19]. The U.S. Environmental Protection Agency (EPA) has considered PFOA to be a “likely carcinogen.”
Perfluorinated compounds (PFCs) have unique physiochemical properties gained by replacing the majority of the hydrogens in an organic molecule with fluorine. Organic poly- and perfluorination yields a more thermally stable (i.e., C2H5–H of 101 kcal/mol vs. C2F5–F of 127 kcal/mol, and CF3–CF3 of 99 kcal/mol vs. CH3–CH3 of 89 kcal/mol), more resistant to oxidation (i.e., F + e− → F−, E 0 = 3.6 V), and weakly polarizable organic compound [20].
Unlike most persistent and bioaccumulative organic pollutants, PFOA is water-soluble and does not strongly adsorb to soil or sediments; thus, it has a high migration potential and poses a serious threat to ground and surface water resources. PFOA discharged or emitted from fluorochemical plants could pose potential threats on surrounding ecosystems due to contamination of food webs [21]. The consumption of contaminated drinking water and groundwater has been determined to be an important human exposure route in PFCs-contaminated areas. In January 2009, the U.S. EPA issued provisional health advisories for PFOA and perfluorooctanesulfonate (PFOS) in drinking water at 400 and 200 ng/L respectively after several revisions [22].
PFCs are particularly recalcitrant and environmentally persistent. The physiochemical properties of PFCs make them difficult to treat by most conventional methods. Due to its inherent resistance to chemical and microbiological treatment, many technologies have been developed to decompose PFOA, such as ultrasonication, aqueous electron reduction, direct photolysis, persulfate photolysis, and phosphotungstic-acid photocatalysis.
PFOA has strong absorption from deep UV region to 200 nm, thus it can be directly decomposed by UV with wavelength <200 nm. Phosphotungstic acid, H3PW12O40, has been reported to be utilized for photocatalytic degradation of PFOA [23]. H3PW12O40 is a heteropolyacid or polyoxometalate that has been used as an electron shuttle. Hori et al. proposed that H3PW12O40 photocatalysis of PFOA involves a photo-Kolbe type mechanism. Hori et al. also reported that sulfate radical anions (SO4 •−) produced by S2O8 2− photolysis can decompose PFOA and other shorter-chain PFCAs via an electron transfer from PFOA to SO4 •− radical [24, 25]. In other oxidative processes, a similar decomposition mechanism was proposed, i.e., a PFOA molecule first loses an electron and then it is decarboxylated to form a perfluoroheptyl radical [23, 26, 27]. On the other hand, PFOA can be reductively decomposed by aqueous electrons (eaq −) via a defluorination step [28].
However, these methods are not satisfactory due to their harsh reaction conditions or/and high energy consumption. Heterogeneous photocatalysis is an effective, mild method for the treatment of various organic compound-contaminated waters. The advantages of the photocatalytic processes (operation under ambient temperature and pressure, high stability and low cost of the catalyst, complete mineralization without selectivity restrictions, and possibility of using solar light as energy source) open a wide range of environmentally friendly applications that are either under investigation or already in the market. Both the technological and economic relevance of photocatalysis has considerably increased over the past decade. TiO2, In2O3 and β-Ga2O3 have been investigated as photocatalysts for PFOA decomposition.
3.2 Photocatalytic Degradation of PFOA by In2o3 and TiO2
The applications of semiconductor photocatalysis are usually based on its properties of oxidation, reduction, sterilization, and super-hydrophilicity or a combination thereof. Figure 3.1 schematically shows oxidizing species which are usually involved in the photocatalytic process. The oxidation of surface OH− or H2O to hydroxyl radicals (OH•) takes place in the photogenerated holes; on the other hand, the photogenerated electrons reduce adsorbed oxygen to superoxide radical anion (O2 •−), which can be further transformed into hydroperoxyl radical (•OOH), hydrogen peroxide (H2O2), and hydroxyl radicals (OH•). In the presence of organic compounds, parent compounds and subsequent intermediates will be oxidized through several steps before final mineralization to carbon dioxide, water, and inorganic acids by the highly oxidizing species or by direct reaction with the photogenerated holes.

Active oxygen species generated in the photocatalytic process
Because of high energy of carbon–fluorine bonds (C–F, 116 kcal/mol) and high reduction potential of 3.6 V (Eq. (3.1)), the PFCs are chemically stable and resistant to oxidation, reduction, and biodegradation. Hence, the cleavage of C–F bonds was the most important criteria for PFCs detoxification and persistence elimination [29].
Hydroxyl radicals normally react with saturated organics through an H-atom abstraction to form water (Eq. (3.2)) and react with unsaturated organics primarily via an addition reaction. The hydroxyl radical reacts with most aliphatic and aromatic organics at near diffusion-controlled rates at environmentally relevant pH values. PFOA contains no hydrogen atoms for abstraction by OH• and perfluorination reduces electron density of –COO− group in PFOA, thus the direct electron transfer between OH• and –COO– group in PFOA is not favorable. Therefore, the hydroxyl radical must act through a direct electron transfer to form the less thermodynamically favored hydroxyl ion (Eq. (3.3)) [20].
Thus the oxygen-containing radicals generated in the photocatalytic process cannot decompose PFOA. It is reported that the first step of the decomposition of PFCA in a TiO2-mediated heterogeneous photocatalysis is initiated by an electron transfer from the adsorbed perfluorocarboxylate to the holes in the valence band (Eq. (3.4)).
In advanced oxidation processes, hydroxyl radicals generated through hydrogen peroxide photolysis, ozonation, photo-Fenton process, sonolysis, and peroxone chemistry. Therefore, some conventional advanced oxidation methods based on active oxygen radicals are not effective for PFOA decomposition.
3.2.1 The Photocatalytic Performance of In2O3 and TiO2
It was reported that the most common photocatalyst, TiO2, is ineffective in degrading PFOA. Thus, we investigated the activity of other semiconductors for PFOA decomposition. Here we report the photocatalytic activity of commercial In2O3 relative to TiO2. The commercial In2O3 nanoparticles with Brunauer–Emmett–Teller (BET) surface area of 12.6 m2/g and Degussa P25 TiO2 with BET surface area ~50 m2/g were used as photocatalysts for degradation of PFOA.
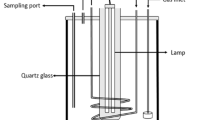
A tubular quartz vessel reactor was used for photocatalytic decomposition of PFOA (Fig. 3.2). As shown in Fig. 3.2, a low-pressure mercury lamp (23 W) emitting 254 nm was placed in the center of the reactor with a quartz tube protection. The reaction temperature was kept at ~25 °C with a cooling water jacket around the reactor. Oxygen gas was continuously bubbled into the reactor through a porous glass plate during the whole reaction. The photocatalyst was suspended in the PFOA aqueous solution by the bubbling of oxygen gas. The initial concentration of PFOA aqueous solution was ~100 μmol/L (41.4 mg/L) and the dosage of photocatalysts in suspension was ~0.5 g/L.

Schematic illustration of the photocatalytic system
Concentrations of PFOA and degradation intermediates formed were measured on a Waters Acquity UPLC system, coupled with a Micromass Quattro Premier tandem quadrupole mass spectrometric system (Waters, Milford, USA), i.e., UPLC-MS/MS system. PFOA weakly absorbs UV light with wavelengths longer than 220 nm, thus the direct photolysis by 254 nm UV light is very slow and considered negligible in these experiments.
Indium oxide (In2O3) possesses much higher photocatalytic activity than TiO2 [30]. The photocatalytic decomposition of PFOA followed pseudo-first-order kinetics. The rate constant by In2O3 is about 0.378 h−1, and it is 0.045 h−1 by TiO2 and 0.026 h−1 for the direct photolysis, respectively, which means the decomposition rate constant of PFOA by In2O3 is 8.4 times higher than that by TiO2.
The main degradation intermediates were shorter-chain PFCAs, including perfluoroheptanoic acid (PFHpA, C6F13COOH), perfluorohexanoic acid (PFHxA, C5F11COOH), perfluoropentanoic acid (PFPeA, C4F9COOH), perfluorobutanoic acid (PFBA, C3F7COOH), pentafluoropropionic acid (PFPrA, C2F5COOH), and trifluoroacetic acid (TFA, CF3COOH).
Formations of fluoride ions (i.e., F−) during PFOA decomposition were monitored by ion chromatography (Dionex ICS-2000, USA). TOC (total organic carbon) measurements are usually used to assess mineralization of the substrate. However, in the study of photocatalytic decomposition of PFOA, TOC data from the commercial TOC analyzer could not be obtained reliably. PFOA and its degradation intermediates are fully fluorinated hydrocarbons, which are resistant to most conventional treatment processes, and they are very stable under conditions under which their hydrocarbon analogues are degraded. To thermally decompose them, high temperatures (~1,200 °C) are required. And two commercial methods to measure TOC (i.e., catalytic oxidation at 680 ºC or UV-persulfate oxidation) cannot completely oxidize PFOA and its degradation intermediates. Other indices, i.e., defluorination ratio (formed fluoride ion/total amount of fluorine contained in initial PFOA), can be used to reflect the mineralization of PFOA.
During the photocatalysis, most fluorine atoms (F) stay in aqueous solution, although others may be transformed into gaseous products. Total F content in aqueous solution consists of four parts; remaining PFOA, shorter-chain PFCAs, F− and PFCAs adsorbed on catalyst surface. The F content on the catalyst surface could be estimated in terms of the adsorption amount and the XPS quantitative result. The gaseous products were analyzed by ATD/GC-MS. A small amount of HCOOH and C4–C6 alkane such as C n F2n+2 and C n HF2n+1 were detected in the gas phase.
3.2.2 Coordination of PFOA to In2O3 and TiO2
To elucidate why In2O3 is a better photocatalyst than TiO2, we investigated the adsorption status of PFOA on In2O3 and TiO2. Adsorption of reactants to the photocatalyst surface is a critical step in the photocatalytic process. The diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) is a highly sensitive method to characterize structural changes of adsorbed species. Figure 3.3 shows the spectra of PFOA/In2O3, PFOA/TiO2, and PFOA/KBr. An absorbance at 1,769 cm−1 in IR spectrum of PFOA/KBr is typical of C = O vibration for carboxylic acid. The strong bands in the range of 1,300–1,100 cm−1 are assigned to C–F stretching. The C–OH vibration is overlaid with an absorbance peak at 1,210 cm−1 [31]. The broadbands at 1,640–1,600 cm−1 are assigned to adsorbed water molecules. As for potassium perfluorooctanoate (PFOK), the ν(C=O) vibration peak is replaced with the asymmetric (ν as(COO−)) and symmetric stretches (ν s(COO−)) of carboxylate, appearing at 1,660 and 1457 cm−1 respectively. Upon adsorption on In2O3, ν(C = O) stretch of PFOA vanishes, and two new peaks appear at 1,641 and 1,443 cm−1, which are assigned to the asymmetric and symmetric stretching modes of the –COO− group [31–34]. Similarly, upon adsorption on TiO2, ν(C = O) vibration peak disappears, and ν as(COO−) and ν s(COO−) appear at 1,686 and 1,408 cm−1 respectively. These results indicate that PFOA coordinates with both In2O3 and TiO2 via its carboxylate group.

DRIFT spectra of PFOA mixed with KBr and adsorption-equilibrium on In2O3 and TiO2 photocatalysts at room temperature. The samples of PFOA/photocatalyst were filtered and dried at room temperature for 24 h before measurement. Reproduced with permission from [30]
According to the stretching frequencies of the carboxylate, Deacon et al. concluded an empirical relationship between the frequency difference [35], Δν = ν as(COO−) − ν s(COO−), and the types of bonding of carboxylate to cations. The Δν value which is substantially greater than the ionic Δν indicates a monodentate coordination, while the Δν value which is significantly less than the ionic Δν indicates a bidentate or bridging coordination. When the Δν value is close to the ionic Δν, chelating and/or bridging carboxylates cannot be excluded. The Δν value observed for PFOA/TiO2 (278 cm−1) is substantially greater than that of K+ salt (203 cm−1), which indicates that PFOA coordinates to TiO2 in a monodentate mode. While the Δν value for PFOA/In2O3 (198 cm−1) is somewhat smaller than the ionic Δν, considering the stronger adsorption of PFOA on In2O3 (In2O3, 30 μmol/g; TiO2, 14.2 μmol/g), it can be concluded that PFOA is bound to In2O3 in a bidentate or bridging configuration.
Figure 3.4 shows the 19F MAS NMR spectra of PFOA and samples adsorbed on In2O3 or TiO2. For the sample of PFOA/KBr, peaks at −80.1, −116.9, and −124.6 ppm are assigned to the terminal CF3, the CF2 group next to the carboxylic group, and the CF2 group adjacent to the terminal CF3 respectively. Peaks in the range from −119.6 to −120.9 ppm are assigned to other CF2 groups. The 19F chemical shifts of PFOA in solution was also confirmed by Buchanan et al. [36, 37], who used 19F–19F correlation spectrometry technique. After PFOA adsorbed, changes of the chemical shifts and line widths were observed. The terminal CF3 group showed a significant shift to the high field about 1.5 and 1.8 ppm for In2O3 and TiO2 respectively, and the CF2 group adjacent to CF3 group also shifted upfield. The shift of the CF3 and its adjacent CF2 group can be attributed to these groups located at the air/monolayer interface as Pawsey et al. [38] explained for PFCAs adsorbed on ZrO2. In addition, the line width of the CF2 group next to the carboxylic group broadened, which is attributed to the deprotonation of carboxylic acid group and its coordination to the photocatalyst. Notably, there is a great difference in 19F line width of other inner CF2 groups between In2O3 and TiO2. For the PFOA/TiO2, the peaks heavily overlapped and lumped into a big peak, while those for PFOA/In2O3 were less changed and remained a shape similar to those of the bulk acid. These observations indicate that the inner CF2 groups of PFOA may interact with TiO2 surfaces, but have little interaction with In2O3.

19F MAS NMR spectra of PFOA mixed with KBr and adsorption-equilibrium on In2O3 and TiO2 photocatalysts at room temperature. The samples of PFOA/photocatalyst were filtered and dried at room temperature for 24 h before measurement. Reproduced with permission from [30]
According to the above results, the schematic configurations of PFOA adsorbed on In2O3 and TiO2 are shown in Fig. 3.5. As for PFOA/In2O3, PFOA closely coordinates to In2O3 in a bidentate or bridging mode, resulting in a vertical and ordered configuration of PFOA chain on In2O3 surface. While in the case of PFOA/TiO2, PFOA binds on TiO2 surface in a monodentate mode with its carboxylate group, resulting in a tilted configuration of PFOA on TiO2 surface. Consequently, the inner CF2 group of PFOA may interact with TiO2 surface OH group via hydrogen bonds. The formation of hydrogen bonds at the organic–inorganic interface have been widely reported and observed by solid-state NMR.

Schematic diagram of PFOA configurations adsorbed on In2O3 and TiO2. Reproduced with permission from [30]
3.2.3 Mechanism for Photocatalytic Degradation of PFOA
Photocatalytic oxidation can proceed via direct hole oxidation or via indirect •OH radicals, with the tight adsorption of the electron donor an essential requisite for direct hole oxidation [39]. PFOA closely coordinates to In2O3 in a bidentate or bridging configuration, and it is reasonable that the PFOA decomposition may be induced by direct hole oxidation. We detected •OH radicals formed in the presence of different photocatalysts by ESR using DMPO as the spin-trap reagent. The DMPO-•OH generation was characterized by four peaks with intensity 1:2:2:1 appearing in the ESR spectra. Figure 3.6 shows that the intensities of •OH generated in TiO2 suspension were much greater than those in In2O3 suspension. This indicates that the photogenerated holes in the valence band of TiO2 are largely transformed into •OH radicals, while those of In2O3 react slowly with surface-bound water or hydroxyl group (–OH). After addition of PFOA, the signals of •OH peaks increased greatly in the TiO2 system. This indicates that the production of •OH radicals was enhanced by PFOA. It was reported that F− and CF3COO− adsorbed on TiO2 have strong electron-withdrawing ability to reduce the recombination of electrons and holes [40–42], PFOA may also have this ability. As photogenerated holes of TiO2 are quickly and mostly transformed into •OH radicals after addition of PFOA, and •OH radicals are not effective to degrade PFOA [43, 44], it is understandable that TiO2 shows a low activity for the PFOA decomposition. However, intensities of •OH radicals in the In2O3 system were slightly lowered after addition of PFOA, which implied that photogenerated holes could directly react with PFOA and thus fewer holes were transformed into OH radicals. The possible mechanisms of PFOA decomposition on In2O3 and TiO2 are summarized in Fig. 3.7. Compared with TiO2 material, In2O3 has a higher adsorption capacity and tightly coordinates with PFOA in a bidentate or bridging configuration, which is beneficial for PFOA to be decomposed via the direct hole oxidation. Thus, In2O3 possesses much higher activity than TiO2 to decompose PFOA under UV irradiation.

DMPO spin-trapping ESR spectra under UV irradiation for 4 min at room temperature of water and PFOA solution in the presence of In2O3 or TiO2. Reproduced with permission from [30]

Possible mechanisms of the photocatalytic decomposition of PFOA by In2O3 and TiO2. Reproduced with permission from [30]
3.2.4 Potential Applications in Wastewater Treatment
In real wastewater, PFOA generally coexists with other chemical compounds, such as organic pollutants, natural organic matter, and bicarbonate, which may reduce the PFOA decomposition efficiency [45, 46]. The characteristics of the secondary-treated effluent were shown in Table 3.1. The decomposition of PFOA by In2O3 photocatalysis in the original secondary effluent was almost inhibited.
Moreover, the adsorption of PFOA on In2O3 became insignificant, which can be attributed to the competitive adsorption of bicarbonate anion on In2O3 carrying positive charges at pH 7.8 (the pHzpc of In2O3 is 8.7). Bicarbonate (HOCOO−) has the same carboxyl group as PFOA, and its concentration usually much higher than PFOA concentration added in the secondary-treated effluent, the surface of In2O3 was mostly occupied by bicarbonate. As a result, little PFOA was adsorbed on In2O3 and its decomposition was accordingly inhibited. The adsorption of PFOA on In2O3 increased, to some extent, when the secondary-treated effluent was adjusted to acidic conditions. However, this has little influence on the PFOA decomposition in secondary effluent, which means it is necessary to degrade coexisting organic matters to recover the photocatalytic activity of In2O3 for PFOA decomposition.
Ozone addition can greatly accelerate the photocatalytic removal of organic matter. When the pH value of the secondary-treated effluent was adjusted to 4, and ozone gas was simultaneously added, PFOA decomposed almost as fast as in pure water. This means that the impacts of bicarbonate and organic matter in wastewater can be mostly avoided via pH adjustment and ozone addition.
3.2.5 The Enhancement of TiO2 Photocatalysis of PFOA
The photocatalytic degradation of PFOA by using TiO2 is very slow. And some efforts also have been made to explore the effectiveness of TiO2 composites in degrading PFOA. TiO2 photocatalytic degradation of PFOA is slightly enhanced by the addition of phosphotungstic (H3PW12O40) acid, which acts as an electron shuttle from TiO2 to O2 [43]. Photoelectrocatalysis utilizing TiO2 with a submonolayer Ni–Cu coating was also developed to enhance the rate of PFOA decomposition [47]. A rough analysis yields a power density-normalized absolute rate of TiO2/Ni–Cu to be 7.4 times faster than TiO2 alone. A reductive degradation process was proposed.
A combination of treatment techniques have been studied largely for the treatment of wastewater with organic pollutants. The efficiency of PFOA photocatalysis by TiO2 is enhanced when coupled with sonolysis. The sonication enhances the photocatalytic decomposition of PFOA from 22 to 45 % after 7 h with TiO2 as photocatalyst [48]. Sonication improves the photocatalytic performance through physical dispersion of TiO2 and easing the mass transfer which keeps regenerating the TiO2 surface.
The photocatalytic process is influenced by pH and surface chemistry. TiO2 has a pH of zero point of charge (pHzpc) of 6.25, which indicates that TiO2 has a positive surface charge at pH < 6.25 and a negative charge at pH > 6.25. This charge-specific behavior of TiO2 favors anionic electron donors at pH < pHzpc in photocatalysis. Therefore positive holes are considered to be the predominant oxidizing species at lower pHs while hydroxyl radicals play an important role at higher pH values. Owning to the involvement of electron transfer from the adsorbed perfluorocarboxylate to the valence hole, it was thought that an acidic solution might favor the decomposition of PFCAs more effectively. Perchloric acid (HClO4) has been reportedly used as an additive to enhance the TiO2 photocatalysis [49], the decomposition of 99 % and defluorination of 38 % were achieved in 7 h under highly acidic conditions (pH = 0.9–1.2).
Reducing the combination of e−–h− pairs is an important approach to improve photocatalytic efficiency. Carbon nanotubes (CNTs) are an effective support for TiO2 due to their electron accepting and transport capacity, and the possibility to modify their porosity and surface. They provide a convenient way to direct the flow of photogenerated charges and increase the lifetime of e−–h− pairs generated by semiconductor photocatalysts. The porosity of CNTs favors adsorption of pollutants on the catalyst surface. The composite TiO2 with multiple wall CNTs (TiO2–MWCNT) has been used as a photocatalyst to degrade PFOA in water [50]. TiO2–MWCNT displayed much higher photocatalytic ability towards PFOA relative to pure TiO2 under UV irradiation.
It has been reported that the photocatalytic decomposition of PFOA in aqueous solution using Fe and Nb co-doped TiO2 (Fe:Nb–TiO2) [51] prepared by the sol-gel method showed higher activity compared to both undoped and commercially available TiO2. The enhanced activity was attributed to the effects of co-doping on both the physicochemical properties and surface interfacial charge transfer.
Although some advancement has been made, the photocatalytic activity of TiO2 on the decomposition of PFOA is still lower than that of In2O3.
3.3 In2O3 Nanomaterials for Photocatalytic Degradation of PFOA
In2O3 is an indirect band semiconductor with a direct band gap of 3.6 eV and an indirect bandgap of 2.8 eV [52]. As a wide bandgap semiconductor, In2O3 has been applied to improve the photocatalytic efficiency of other semiconductors. In2O3 and its composite oxides have also been investigated as water-splitting photocatalysts. As mentioned above, In2O3 showed higher photocatalytic activity for PFOA decomposition than TiO2. We have found that a high adsorption capacity of In2O3 and tight coordination to PFOA is beneficial for PFOA degradation via direct hole oxidation. The following discussion introduces the preparation of In2O3 nanomaterials and photocatalytic performance of nanostructured In2O3 in detail. The photocatalytic decomposition of PFOA was conducted in a tubular quartz reactor under 254 UV light irradiation as described above. The initial concentration of PFOA was ~30 mg/L and the dosage of photocatalyst was ~0.5 g/L.
3.3.1 In2O3-Graphene Nanocomposites
Graphene, a monolayer of two-dimensional (2D) carbon atomic sheets, exhibits excellent mobility of charge carriers (200,000 cm2/V s), large surface area (calculated value is as high as 2,630 m2/g), optical transparency, and chemical stability [53]. The decoration of photocatalysts such as TiO2, ZnO, CdS, and Sr2Ta2O7 with graphene can enhance their activity because the introduction of graphene can reduce the recombination of photogenerated electron–hole pairs. Because the decomposition of PFOA is initiated by an electron transfer from adsorbed PFOA to a valence band hole, the coverage with graphene on In2O3 nanoparticles influences the photocatalytic activity of In2O3.
The In2O3-graphene composites were synthesized via a sonication-assisted solution route [54]. In2O3 nanoparticles and graphene oxide (GO) were dispersed in water by ultrasonication to form two uniform suspensions, which were then mixed and continuously sonicated to form the GO–In2O3 nanocomposite. The GO–In2O3 was reduced by hydrazine and ammonia at 95 °C. The In2O3-graphene composite (In2O3–G) obtained was calcined under nitrogen atmosphere. In2O3-graphene composite treated at 100, 350, and 400 °C were named as In2O3-G100, In2O3-G350, and In2O3-G400, respectively.
The coverage of graphene on the surface of In2O3 nanoparticles was identified by Raman spectrum. Figure 3.8 shows the Raman spectra of graphene oxide and In2O3-graphene composite. The Raman-active E 2g mode at about 1,598 cm−1 is characteristic of the presence of sp2 carbon-type structures within the In2O3-graphene composites. The D band around 1,351 cm−1 is associated with the presence of defects in the hexagonal graphical layers [55]. The 2D band around 2,679 cm−1 provides evidence for the formation of graphene. The shape of Raman spectrum changes depending on graphene structure and layer.

Raman spectra of graphene oxide and In2O3-graphene composites. Reproduced with permission from [54]
The coverage of graphene on In2O3 was influenced by the calcination temperature, which was confirmed by HRTEM. Figure 3.9 shows the HRTEM images of In2O3 nanoparticles and In2O3-graphene composite. It can be seen that the carbon layers surrounding In2O3 particles cracked to some extent with increased heat. Therefore, the graphene-wrapped area of In2O3-graphene composite decreases with increases in heat treatment temperature. It has been reported that the absorption intensity of the graphene-semiconductor composites in the visible-light region is enhanced by increased graphene amounts [56, 57]. We found that the absorption intensity of In2O3-graphene composites in the visible-light region also decreases with the decrease of graphene-wrapped area, which was caused by heat treatment.

HRTEM images of samples. (a) In2O3 nanoparticles, (b) graphene-wrapped In2O3 nanocomposite (In2O3-G100), (c) In2O3-graphene nanocomposite with heat treatment at 350 °C (In2O3-G350), and (d) In2O3-graphene nanocomposite with heat treatment at 400 °C (In2O3-G400). The white arrow in (b) indicates the graphene. Reproduced with permission from [54]
The photocatalytic performance of PFOA decomposition depends on the coverage of graphene on In2O3. Compared with In2O3 nanoparticles and other composite photocatalysts, In2O3-G400 exhibits the highest photocatalytic activity. The PFOA decomposition appears to follow pseudo-first-order kinetics. The reaction rate constants (k) and half-life of PFOA (τ 1/2) by different photocatalysts are listed in Table 3.2. The defluorination ratio of PFOA by In2O3-G400, In2O3-G350, In2O3-G100, In2O3 nanoparticles reached 60.9 %, 37.7 %, 12.9 %, and 29.7 % within 3 h, respectively.
The photocatalytic activity of In2O3-graphene nanocomposites depends on two factors: exposed surface area of In2O3 as active adsorption sites, and supported graphene for separation of photogenerated carriers. The In2O3-graphene nanocomposites have the following advantages: (1) the exposed In2O3 surface can provide more active sites for the adsorption of PFOA, which favors the electron transfer from the adsorbed perfluorocarboxylate to valence band holes; (2) the supported graphene can act as an electron shuttle to carry excited electrons from the semiconductor to the electron acceptor, which is beneficial for efficient separation of photogenerated hole–electron pairs.
3.3.2 In2O3 Porous Nanostructures
Porous materials offer many advantages. Large surface area and porous structures are conducive to mass transfer, which are helpful for the photocatalytic reaction. Moreover, for photocatalysts with a porous structure, recombination of photogenerated carriers (e−/h+) in the bulk is reduced and a fast surface e−/h+ separation is achieved. As mentioned above, high adsorption of In2O3 is beneficial for PFOA decomposition via direct hole oxidation. Accordingly, In2O3 photocatalysts with porous nanostructure and high specific surface area are expected to show high photocatalytic activity for PFOA decomposition.
In2O3 nanospheres with nanoporous structures and a uniform size at ~100 nm were synthesized by a solvothermal method followed by calcinations [58]. For preparation of precursor of In2O3 nanospheres, i.e., In(OH)3 porous nanospheres, ethylenediamine—ethanol was used as a mixed solvent. In(NO3)·4.5H2O was dissolved in ethanol, and the obtained solution was mixed with ethylenediamine. The mixture was transferred into a Teflon-lined stainless steel autoclave, and maintained at 180 °C for 16 h. The resulting white precipitate (precursor) was thoroughly washed with deionized water and ethanol. The dried precursor was calcined for 2 h in air to form In2O3 nanospheres.
In2O3 has two crystal phases, i.e., cubic In2O3 (c–In2O3) and hexagonal In2O3 (h–In2O3). The In2O3 nanosphere that we prepared is cubic phase, as confirmed by X-ray diffraction.
Figure 3.10a–c show the TEM and SEM images of In2O3 nanospheres. The nanospheres were built of numerous nanoplates, which connected each other to form a nanoporous structure. The ring-like SAED pattern (inset of Fig. 3.10a) indicates a polycrystalline structure of as-synthesized In2O3 nanospheres, and the presence of obvious discrete spots suggests that the materials were well crystallized. The high-magnification TEM image in Fig. 3.10c shows In2O3 nanospheres were constructed by the interlocking stack of ultrathin nanoplates and confirms the single crystalline property of each nanoplate. The TEM image in Fig. 3.10c also reveals that the plate-like nanoparticles are irregular polygonal in shape, with size of about 5–12 nm. Commercial In2O3 nanoparticles (Beijing Chemical Co., Ltd., >99.99 %) and TiO2 (Degussa P25) were adopted as the reference with which to compare the photocatalytic activity under the same experimental conditions. The diameters of most commercial In2O3 nanoparticles were in the range of 40–90 nm (Fig. 3.10d).

(a) Low-magnification TEM images, (b) FESEM image, and (c) high-magnification TEM image of In2O3 nanospheres; (d) FESEM image of commercial In2O3 nanoparticles. The inset of (a) is SAED pattern. Reproduced with permission from [58]
In2O3 nanospheres have characteristics of mesoporous materials, as confirmed by BET gas sorptometry measurements. The existence of a number of ~6 nm pores was identified, and those pores presumably arose from the stacks of nanoplates. In2O3 nanospheres had a high BET surface area about 39.0 m2/g (Table 3.3), while that of commercial In2O3 nanoparticles was only 12.7 m2/g. The BET-specific surface area of TiO2 (P25) was ~50 m2/g. The difference of the BET surface area between In2O3 nanospheres and In2O3 nanoparticles can be attributed to the fact that the former have nanoporous architecture.
The absorption of In2O3 nanospheres showed a slight blue-shift compared with those of In2O3 nanoparticles (Fig. 3.11). In2O3 has a small Bohr exciton radius of 2.14 nm. The slight blue-shift of UV-vis absorption may be resulted from the quantum confinement effect.

UV-vis absorption spectra of In2O3 nanoparticles, In2O3 nanospheres and TiO2. Reproduced with permission from [58]
Comparative experiments were conducted to investigate the photocatalytic activity of In2O3 nanospheres, In2O3 nanoparticles and TiO2 for PFOA decomposition. The PFOA decomposition appears to follow pseudo-first-order kinetics. The reaction rate constants (k) and half-life of PFOA (τ 1/2) by different photocatalysts are listed in Table 3.3. The rate constant of In2O3 nanospheres was 5.89 h−1, which was nearly 9 and 54.6 times higher than that of In2O3 nanoparticles and TiO2 respectively. In2O3 nanospheres also showed a higher defluorination ratio. The defluorination ratio of PFOA by In2O3 nanospheres, In2O3 nanoparticles and TiO2 reached 71.0 %, 29.7 %, and 5.1 % after 3 h, respectively.
The much higher activity of In2O3 nanospheres can be attributed to larger specific surface area and nanoporous structure. Large surface area can offer more adsorption and reaction sites, and accordingly, has a beneficial effect on the photocatalytic activity. The adsorption percentages of PFOA in the dark on In2O3 nanospheres, In2O3 nanoparticles, and TiO2 were 27.4 %, 19.4 %, and 2.23 %, respectively. The nanoporous structure is beneficial for the diffusion of reactants and intermediates during reaction, which is one of the important steps determining the reaction rate. Besides, the nano-size of ultrathin plates constructing In2O3 nanospheres shortens the diffusion distance of photogenerated carriers and reduces recombination of electron–hole pairs [59], which accordingly favors the photocatalysis of PFOA.
3.3.3 Effects of In2O3 Morphology on Photocatalytic Degradation of PFOA
The catalytic performance of nanomaterials is determined either by the composition in terms of the atomic structure [60], or by the morphologies that affect surface atomic arrangements, coordination, and specific surface area [61, 62]. Earlier discussions have demonstrated that the photocatalytic decomposition of PFOA involves electron transfer from PFOA to photocatalyst [30]. The morphology of photocatalysts is thus thought to play an important role in improving the decomposition rate. In2O3 nanostructures with different morphologies have been developed and exhibit novel shape-dependent properties, such as improved optical properties and excellent gas-sensing characteristics. In the case of photocatalytic decomposition of PFOA, it is very important to study the relationship of photocatalytic performance and morphologies of In2O3 [63]. Thus three In2O3 nanostructures with different morphologies were synthesized to study the correlation between activity and morphology.
In2O3 microspheres were synthesized as follows. The preparation of precursors was carried out in a 100-mL Teflon-lined stainless steel autoclave. In(NO3)·4.5H2O was used as the source material. In(OH)3 microspheres were synthesized by a solvothermal process and a mixed solvent of ethanol/1,2-propane-diamine at 180 °C for 16 h. In2O3 porous microspheres were obtained by calcination of the precursor In(OH)3 microsphere at 500 °C for 2 h. In2O3 nanocubes and nanoplates were synthesized as follows. In2O3 nanoplates and nanocubes were synthesized by a similar procedure, except that the solvents were changed to H2O/1,2-propane-diamine for nanocubes, and H2O/1,3-propane-diamine for nanoplates, respectively.
The crystal structure and the phase purity of the as-synthesized samples with different solvents were verified by XRD. All three synthesized products, microspheres, nanocubes, and nanoplates, are pure body-centered cubic (bcc) phase of In(OH)3 (JCPDS No. 01-073-1810) and no other impurities were detected.
Figure 3.12 shows the FESEM images of the calcined samples, i.e., as-obtained In2O3 products. Figure 3.12a shows the In2O3 microspheres have a diameter of about 180 nm and a porous structure. The porous microsphere is composed of several nanoparticles with diameters of 5–15 nm. As shown in Fig. 3.12b, the synthesized In2O3 nanocubes have a side length of about 40–150 nm. Although some In2O3 samples agglomerated and their morphology distorted, many particles still had a cube-like shape as indicated by white arrows in Fig. 3.12b. Figure 3.12c shows that the surface of nanoplates displayed some defects. The morphologies and structures of In(OH)3 precursors usually play an important role in the transformation from indium hydroxide to indium oxide during calcination.

FESEM images of as-obtained In2O3 products: (a) microspheres, (b) nanocubes and (c) nanoplates. Reproduced with permission from [63]
In2O3 microspheres have a characteristic of mesoporous materials, as confirmed by BET gas sorptometry measurements. In2O3 microspheres have a narrow pore-size distribution centered at 6 nm. The BET surface area values of In2O3 microspheres, In2O3 nanoplates and In2O3 nanocubes were 42.3, 18.9, and 13.6 m2/g, respectively (Table 3.4). The relatively higher BET surface area of the microspheres confirms that the In2O3 microspheres have a porous structure.
Comparative experiments were carried out to investigate the photocatalytic activities of In2O3 products with different morphologies for PFOA decomposition. In2O3 microspheres showed the highest activity for PFOA among the three samples. The photocatalytic decomposition of PFOA by different photocatalysts followed pseudo-first-order kinetics. The rate constants of PFOA decomposition by In2O3 microspheres, nanoplates, and nanocubes were 7.94, 4.45, and 1.83 h−1, respectively (Table 3.4). The corresponding half-life of PFOA decomposition with In2O3 microspheres was only 5.3 min, while 9.4 and 22.7 min were the experimentally-determined half-lives for In2O3 nanoplates and In2O3 nanocubes, respectively.
Differing photocatalytic activities possessed by In2O3 in various nanostructures can be attributed to differences in their surface area and oxygen vacancies as discussed below. Since the photocatalytic reaction takes place on the surface of a photocatalyst, normally a high specific surface area has a beneficial effect on the activity of catalysts. As shown in Table 3.4, the values of BET surface area of In2O3 microspheres, In2O3 nanoplates, and In2O3 nanocubes correspond linearly with their corresponding reaction rates.
Due to PFOA decomposition involving the direct charge transfer between In2O3 and PFOA, the chemical states of surface In and O elements may have significant effects on PFOA adsorption and subsequent decomposition. Figure 3.13 shows the high-resolution XPS spectra of three In2O3 photocatalysts with different morphologies. As indicated in Fig. 3.13, all the O 1s core-level spectra are asymmetrical with a hump on the higher binding energy (BE) side, which can be fitted to two components with peaks around 529.7 and 531.1 eV, respectively. The two peaks of O 1s are distinguished as Oa and Ob [64–66]. The Ob has a BE about 1.4 eV higher than that of the Oa. Fan [64] and Donley [66] suggested that the Oa peak is assigned to O2− ions surrounded by In atoms with their full complement of six nearest-neighbor O2− ions and the Ob peak is assigned to the oxygen atom adjacent to oxygen deficiency sites (i.e., they do not have neighbor In atoms with their full complement of six nearest-neighbor O2− ions). It is hypothesized that oxygen atoms adjacent oxygen defect sites donate some of their electron density towards In atoms that are no longer fully coordinated, which appears to cause the O 1s peak to shift towards the higher binding energy [60]. The Ob/Oa ratio therefore magnifies and provides a sensitive indicator of the level of oxygen vacancy in the material. The Ob/Oa ratios of three as-obtained In2O3 materials, i.e., microspheres, nanoplates, and nanocubes were determined as 0.80, 0.69, and 0.56, respectively. The high Ob/Oa ratio of In2O3 microspheres may be ascribed to the ethanol solvent used during their solvothermal synthesis. It is well known that ethanol is a weak reducing agent under high temperature and pressure. When the precursor is treated solvothermally (ethanol as the solvent) at 180 °C for 16 h, the oxygen vacancy should be generated on the surface of In2O3 nanocrystals.

XPS spectra of O 1s of In2O3 samples with different morphologies. Reproduced with permission from [63]
The Ob/Oa ratio of three In2O3 materials linearly corresponds to their photocatalytic activity towards PFOA, as shown in Fig. 3.14, which is similar to those reported in the literature [67–69]. It was reported that the presence of oxygen vacancy on the surface of ZnO nanorods promotes the separation of photogenerated electron–hole pairs, thus enhancing photocatalytic activity [67]. Meng et al. [68] reported that organic dye can bind around the O vacancy by inserting an O atom from –COOH group into the vacant position on TiO2 surfaces; the oxygen vacancy defects stabilize dye adsorption and facilitate charge injection. Furthermore, STM observations revealed that formic acid can also bond to TiO2 (110) surface by inserting an O atom of formate into an oxygen vacancy site to form bridge and monodentate configurations, and oxygen vacancies are essential for the catalytic dehydration reaction process of formic acid on the TiO2 surface [69]. Considering that PFOA molecules own a terminal –COOH group, it is suggested that PFOA molecules can also insert an O atom from its –COOH group into an oxygen vacancy site on the In2O3 surface to form tight and close contact with In2O3, which is beneficial for direct charge transfer and subsequent photocatalytic decomposition under UV irradiation. The In2O3 nanomaterials, especially porous microspheres, have high oxygen vacancy defects, thus demonstrating efficient photocatalytic activity to degrade PFOA.

Photocatalytic activity of three nanostructured In2O3 materials vs. their Ob/Oa ratio. Reproduced with permission from [63]
3.3.4 Quantum Efficiency of Photocatalytic Degradation of PFOA
The quantum efficiency (QE) of PFOA decomposition was estimated as follows (Eq. (3.5)):
The quantum efficiency (QE) of PFOA decomposition using different photocatalysts is shown in Table 3.5. The incident UV intensity (the main wavelength is 254 nm) was ca. 3.2 W/cm2, and the UV radiation area was ca. 113 cm2, thus the estimated UV power was 0.36 W.
The photocatalytic decomposition of PFOA proceeds via sequential steps, i.e., one CF2 unit is removed, and shorter-chain PFCAs are generated in each step. Shorter-chain PFCAs can also be photocatalytically decomposed by In2O3 photocatalysts. Moreover, the •OH generated cannot decompose PFCAs including PFOA, so it is difficult to calculate the QE of the reaction accurately. Equation (3.5) only estimates QE of the PFOA decomposition reaction. The QE of the entire reaction using In2O3 photocatalysts is actually higher than estimated here.
3.4 Ga2O3 Nanomaterials for Photocatalytic Decomposition of PFOA
Gallium oxide (Ga2O3) is an important semiconductor with a wide bandgap (E g = 4.9 eV) and excellent chemical and thermal stability, unique conduction and luminescence properties. Ga2O3 has five polymorphs (α, β, δ, γ, and ε phases). Among these polymorphs, β-Ga2O3 is a thermodynamically stable phase with monoclinic structure, while the others are metastable and the ε-Ga2O3 exhibits the lowest symmetry. All the phases can be obtained from orthorhombic gallium oxide hydroxide (α-GaOOH) by annealing at a suitable temperature with the transformation dependent on the type of gallium precursors and the methodology used. For photocatalytic applications, β-Ga2O3 has been reported to show high activity and stability towards benzene degradation under UV irradiation [70]. As reported in the literature [70], the high activity and long-term stability of β-Ga2O3 is ascribed to its stronger oxidative capability and higher specific surface area.
We prepared the needle-like β-Ga2O3 and sheaf-like β-Ga2O3 nanostructures, and investigated their photocatalytic activity towards the decomposition of PFOA in pure water and wastewater.
3.4.1 Sheaf-Like β-Ga2O3 and Needle-Like β-Ga2O3
The sheaf-like β-Ga2O3 was synthesized by a hydrothermal method followed by calcination. In a typical procedure, Ga(NO3)3 · xH2O and PVA (MW = 22,000) were dissolved in pure water [71]. The mixture was transferred into a Teflon-lined stainless steel autoclave and maintained at 200 °C for 8 h. The white precipitates (precursor of Ga2O3) were collected by centrifugation, and then washed with pure water and ethanol. β-Ga2O3 powder was obtained from the precursor via calcination at 700 °C for 2 h under a nitrogen atmosphere. The needle-like β-Ga2O3 was synthesized with a similar procedure as above, except that the pH value of the aqueous solution of Ga(NO3)3 · xH2O and PVA was adjusted to 6.4 using NaOH solution [72]. The sheaf-like and needle-like products were all monoclinic phases of β-Ga2O3 and no other impurity was found, as confirmed by X-ray diffraction.
Figure 3.15 shows the FESEM images of β-GaOOH precursor and β-Ga2O3 sheaf-like nanostructure. Figure 3.15a, b indicates that the individual sheaf (β-GaOOH) has a length in the range of 2–3 μm and an average diameter in the range of 0.5–1 μm. The individual nanoplates have average widths of 100 nm and thicknesses of 10 nm. Upon calcination at 700 °C for 2 h under a nitrogen atmosphere, as-obtained Ga2O3 product largely retains the morphology and architecture of its precursor, as show in Fig. 3.15c. Figure 3.15d shows the surface of material has become rough due to the dehydration shrinkage during calcination.

(a) Low- and (b) high-magnification FESEM images of as-synthesized β-GaOOH and (c, d) β-Ga2O3. Reproduced with permission from [71]
Figure 3.16 shows the FESEM images of the β-Ga2O3 needle-like nanostructure. The low-magnification image of Fig. 3.16a shows a needle-like morphology with a fairly uniform shape and size. Fig. 3.16b shows that individual needles have a length of 3–6 μm, and a width of 100–200 nm. The surfaces of the needles were relatively rough due to dehydration shrinkage during calcination.

(a) Low- and (b) high-magnification SEM images of the β-Ga2O3 nanostructure. Reproduced with permission from [72]
The sheaf-like β-Ga2O3 and needle-like β-Ga2O3 contained mesopores in the structure, which were identified by BET gas sorptometry measurements. The pore-size distributions of these two products were both in the range of 2–4 nm. These small pores may arise from the loss of adsorbed PVA molecules, and dehydration shrinkage during the conversion process from β-GaOOH to β-Ga2O3. The BET surface area values of the sheaf-like β-Ga2O3 and needle-like β-Ga2O3 were 36.1 and 26.0 m2/g, respectively (Table 3.6). The BET-specific surface area of commercial β-Ga2O3 was 11.5 m2/g. The commercial β-Ga2O3 was adopted as the reference with which to compare the photocatalytic activity under the same experimental conditions.
3.4.2 UV Photocatalysis of PFOA in Pure Water by β-Ga2O3
The photocatalytic decomposition of PFOA was conducted in a tubular quartz reactor vessel under ultraviolet irradiation (254 nm) as described above. The initial concentration of aqueous PFOA solution was ~500 μg/L, and the dosage of photocatalyst was ~0.5 g/L.
The reaction rate constants and half-lives are shown in Table 3.6. The sheaf-like β-Ga2O3 showed remarkably high activity in comparison to the needle-like β-Ga2O3 and commercial β-Ga2O3 as the larger surface area of the sheaf-like β-Ga2O3 provides more adsorption and reaction centers. Meanwhile, compared to TiO2, all three β-Ga2O3 samples exhibited better photocatalytic activity for PFOA decomposition. The excellent performance of the synthesized β-Ga2O3 for PFOA decomposition can be attributed to their unique bonding to PFOA, which is similar to that between In2O3 and PFOA. This is beneficial for PFOA decomposition by holes of the photocatalyst generated under UV irradiation.
3.4.3 UV Photocatalysis of PFOA in Sewage Water
To validate the feasibility of the sheaf-like Ga2O3 photocatalysis to decompose PFOA in wastewater in which coexisting compounds may reduce decomposition efficiency, we investigated the decomposition of PFOA added to a secondarily-treated effluent (its composition is listed in Table 3.1) taken from a municipal wastewater plant in Beijing, China. The experiment conditions were the same as those described for pure water.
In the original secondarily-treated effluent with pH of 7.8, the decomposition of PFOA was obviously retarded, the rate constant was reduced to 1.00 h−1 (Table 3.7). Similarly, the decomposition rate of PFOA in the presence of commercial Ga2O3 or P25 TiO2 was also reduced. The lower decomposition rate of PFOA in the secondarily-treated effluent can be attributed to the influence of bicarbonate and organic material. Bicarbonate (HCO3 −) has the same carboxyl group as PFOA, and its concentration (4.76 mmol/L) was nearly 3,000 times higher than that of PFOA added to the effluent, the competitive adsorption of bicarbonate on the surface of Ga2O3 inhibited the adsorption of PFOA, thus reducing its decomposition efficiency. After the pH value of the secondary effluent was adjusted to 4.3, transforming bicarbonate into carbonic acid, the rate constant increased from 1.00 to 1.43 h−1. However, PFOA decomposition was still inhibited compared to pure water, reflecting the impact of organic materials in the effluent, which may also competitively adsorb and attenuate UV penetration in water.
3.4.4 Vacuum Ultraviolet Photocatalysis of PFOA in Wastewater
Considering the synergistic effects of vacuum ultraviolet (VUV) irradiation on a photocatalyst and its ability to degrade natural organic materials [73, 74], the combination of Ga2O3 with VUV irradiation was evaluated for degradation of PFOA in wastewater. The solution of PFOA (500 μg/L) in wastewater was irradiated with a 185 nm VUV lamp in the presence of the sheaf-like Ga2O3, commercial Ga2O3, or P25 TiO2. To avoid the competitive influence of bicarbonate, pH was adjusted to 4.3 with HCl. A negative control (i.e., direct VUV photolysis), was carried out under the same conditions but without a photocatalyst.
Since PFOA strongly absorbs from the deep UV region to 200 nm [13], VUV irradiation itself can degrade PFOA. As shown in Table 3.7, the degradation rate constants of PFOA in wastewater for sheaf-like Ga2O3/VUV was 4.29 h−1, which was nearly as effective as sheaf-like Ga2O3/UV in pure water (4.85 h−1). While that was 1.21 h−1 for direct VUV irradiation. The high efficiency and stability of the sheaf-like Ga2O3/VUV process for PFOA removal from wastewater is not only enhanced by VUV irradiation by eliminating adverse impacts of coexisting organic materials, but also attributed to the unique role of nanostructured Ga2O3 to decompose PFOA. When P25 TiO2 or commercial Ga2O3 was used to replace sheaf-like Ga2O3 under VUV irradiation, as shown in Table 3.7, no significant improvement occurred compared with the VUV direct photolysis.
3.5 Summary
Heterogeneous photocatalysis based In2O3 or Ga2O3 is a promising technique for degradation of environmentally persistent PFOA. We first demonstrated that In2O3 and β-Ga2O3 exhibit higher photocatalytic activity for PFOA degradation relative to TiO2. The terminal carboxylate group of PFOA molecule tightly coordinates to the In2O3 (β-Ga2O3) surface in a bidentate or bridging configuration, which is beneficial for PFOA to be directly decomposed by photogenerated holes of In2O3 under UV irradiation. While PFOA coordinates to TiO2 in a monodentate mode, and photogenerated holes of TiO2 preferentially transform into hydroxyl radicals, which are inert to react with PFOA. The surface modification of In2O3 by graphene can enhance its photocatalytic activity for PFOA degradation, which was influenced by the coverage ratio of graphene on the surface of In2O3 nanoparticles.
In addition, several nanostructured In2O3 including nanospheres, nanoplates, nanocubes, and microspheres have been synthesized to obtain more efficient photocatalysts for PFOA degradation. The In2O3 nanospheres show excellent activity under mild conditions due to its porous nanostructures, with the first-order rate constant ~9 and 54.6 times higher than that by In2O3 nanoparticles and TiO2 respectively. In2O3 microspheres, In2O3 nanoplates, and In2O3 nanocubes exhibit different photocatalytic activities, the corresponding first-order rate constants were of 7.94, 4.45, and 1.83 h−1. The magnitude of oxygen vacancies owned by the different nanostructured In2O3 was found to be linear to their photocatalytic activity. Similarly, two kinds of nanostructured β-Ga2O3, i.e., sheaf-like β-Ga2O3 and needle-like β-Ga2O3, were also synthesized for PFOA degradation. The morphology of β-Ga2O3 also influences its activity for PFOA degradation.
Finally, the feasibility of nanostructured In2O3 and β-Ga2O3 for photocatalytic degradation of PFOA from real wastewater was investigated. The adverse impacts of bicarbonate and coexisting organic matters in wastewater can be mostly eliminated via pH adjustment and 185 nm VUV irradiation. For the real application of In2O3 and β-Ga2O3, further studies are necessary, such as coating of these nanomaterials and their activity for other pollutants.
References
J.P. Benskin, L.W.Y. Yeung, N. Yamashita, S. Taniyasu, P.K.S. Lam, J.W. Martin, Perfluorinated acid isomer profiling in water and quantitative assessment of manufacturing source. Environ. Sci. Technol. 44, 9049–9054 (2010)
H.M. Shin, V.M. Vieira, P.B. Ryan, R. Detwiler, B. Sanders, K. Steenland, S.M. Bartell, Environmental fate and transport modeling for perfluorooctanoic acid emitted from the Washington roliferat works facility in west Virginia. Environ. Sci. Technol. 45, 1435–1442 (2011)
G.B. Post, J.B. Louis, K.R. Cooper, B.J. Boros-Russo, R.L. Lippincott, Occurrence and potential significance of perfluorooctanoic acid (PFOA) detected in New Jersey public drinking water systems. Environ. Sci. Technol. 43, 4547–4554 (2009)
T. Zhang, Q. Wu, H.W. Sun, X.Z. Zhang, S.H. Yun, K. Kannan, Perfluorinated compounds in whole blood samples from infants, children, and adults in China. Environ. Sci. Technol. 44, 4341–4347 (2010)
B.G. Loganathan, K.S. Sajwan, E. Sinclair, K.S. Kumar, K. Kannan, Perfluoroalkyl sulfonates and perfluorocarboxylates in two wastewater treatment facilities in Kentucky and Georgia. Water Res. 41, 4611–4620 (2007)
R. Guo, W.J. Sim, E.S. Lee, J.H. Lee, J.E. Oh, Evaluation of the fate of perfluoroalkyl compounds in wastewater treatment plants. Water Res. 44, 3476–3486 (2010)
R. Loos, G. Locoro, S. Comero, S. Contini, D. Schwesig, F. Werres, P. Balsaa, O. Gans, S. Weiss, L. Blaha, M. Bolchi, B.M. Gawlik, Pan-European survey on the occurrence of selected polar organic persistent pollutants in ground water. Water Res. 44, 4115–4126 (2010)
J.Y. Hu, J. Yu, S.H. Tanaka, S.G. Fujii, Perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) in water environment of Singapore. Water Air Soil Pollut. 216, 179–191 (2011)
H.W. Sun, F.S. Li, T. Zhang, X.Z. Zhang, N. He, Q. Song, L.J. Zhao, L.N. Sun, T.H. Sun, Perfluorinated compounds in surface waters and WWTPs in Shenyang, China: mass flows and source analysis. Water Res. 45, 4483–4490 (2011)
G.W. Olsen, T.R. Church, E.B. Larson, G. van Belle, J.K. Lundberg, K.J. Hansen, J.M. Burris, J.H. Mandel, L.R. Zobel, Serum concentrations of perfluorooctanesulfonate and other fluorochemicals in an elderly population from Seattle, Washington. Chemosphere 54, 1599–1611 (2004)
L.W. Yeung, M.K. So, G. Jiang, S. Taniyasu, N. Yamashita, M. Song, Y. Wu, J. Li, J.P. Giesy, K.S. Guruge, P.K. Lam, Perfluorooctanesulfonate and related fluorochemicals in human blood samples from China. Environ. Sci. Technol. 40, 715–720 (2006)
K. Harada, A. Koizumi, N. Saito, K. Inoue, T. Yoshinaga, C. Date, S. Fujii, N. Hachiya, I. Hirosawa, S. Koda et al., Historical and geographical aspects of the increasing perfluorooctanoate and perfluorooctane sulfonate contamination in human serum in Japan. Chemosphere 66, 293–301 (2007)
A.M. Calafat, L.Y. Wong, Z. Kuklenyik, J.A. Reidy, L.L. Needham, Polyfluoroalkyl chemicals in the US population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ. Health Perspect. 115, 1596–1602 (2007)
K.S. Betts, Perfluoroalkyl acids: what is the evidence telling us? Environ. Health Perspect. 115, A250–A256 (2007)
C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens, J. Seed, Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol. Sci. 99, 366–394 (2007)
I.G. Shabalina, T. Panaretakis, A. Bergstrand, J.W. DePierre, Effects of the rodent peroxisome proliferator and hepatocarcinogen, perfluorooctanoic acid, on apoptosis in human hepatoma HepG2 cells. Carcinogenesis 20, 2237–2246 (1999)
X.M. Fang, Y.X. Feng, Z.M. Shi, J.Y. Dai, Alterations of cytokines and MAPK signaling pathways are related to the immunotoxic effect of perfluorononanoic acid. Toxicol. Sci. 108, 367–376 (2009)
X. Yao, L. Zhong, Genotoxic risk and oxidative DNA damage in HepG2 cells exposed to perfluorooctanoic acid. Mutat. Res. 587, 38–44 (2005)
A. Takagi, K. Sai, T. Umemura, R. Hasegawa, Y. Kurokawa, Short-term exposure to the peroxisome proliferators, perfluorooctanoic acid and perfluorodecanoic acid, causes significant increase of 8-hydroxydeoxyguanosine in liver DNA of rats. Cancer Lett. 57, 55–60 (1991)
C.D. Vecitis, H. Park, J. Cheng, B.T. Mader, M.R. Hoffmann, Treatment technologies for aqueous perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA). Front. Environ. Sci. Eng. China 3, 129–151 (2009)
J.W. Martin, D.M. Whittle, D.C.G. Muir, S.A. Mabury, Perfluoroalkyl contaminants in a food web from Lake Ontario. Environ. Sci. Technol. 38, 5379–5385 (2004)
U.S. Environmental Protection Agency, Provisional Health Advisories for perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS), (2009), http://www.epa.gov/waterscience/criteria/drinking/pha-PFOA_PFOS.pdf
H. Hori, E. Hayakawa, H. Einaga, S. Kutsuna, K. Koike, T. Ibusuki, H. Kiatagawa, R. Arakawa, Decomposition of environmentally persistent perfluorooctanoic acid in water by photochemical approaches. Environ. Sci. Technol. 38, 6118–6124 (2004)
H. Hori, A. Yamamoto, E. Hayakawa, S. Taniyasu, N. Yamashita, S. Kutsuna, Efficient decomposition of environmentally persistent perfluorocarboxylic acids by use of persulfate as a photochemical oxidant. Environ. Sci. Technol. 39, 2383–2388 (2005)
S. Kutsuna, H. Hori, Rate constants for aqueous-phase reactions of SO4 − with C2F5C(O)O− and C3F7C(O)O− at 298 K. Int. J. Chem. Kinet. 39, 276–288 (2007)
H. Hori, A. Yamamoto, K. Koike, S. Kutsuna, I. Osaka, R. Arakawa, Photochemical decomposition of environmentally persistent short-chain perfluorocarboxylic acids in water mediated by iron(II)/(III) redox reactions. Chemosphere 68, 572–578 (2007)
Y. Wang, P. Zhang, G. Pan, H. Chen, Ferric ion mediated photochemical decomposition of perfluorooctanoic acid (PFOA) by 254 nm UV light. J. Hazard. Mater. 160, 181–186 (2008)
H. Park, C.D. Vecitis, J. Cheng, W. Choi, B.T. Mader, M.R. Hoffmann, Reductive defluorination of aqueous perfluorinated alkyl surfactants: effects of ionic headgroup and chain length. J. Phys. Chem. A 113, 690–696 (2009)
K. Zhang, J. Huang, G. Yu, Q. Zhang, S. Deng, B. Wang, Destruction of perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) by ball milling. Environ. Sci. Technol. 47, 6471–6477 (2013)
X. Li, P. Zhang, L. Jin, T. Shao, Z. Li, J. Cao, Efficient photocatalytic decomposition of perfluorooctanoic acid (PFOA) by indium oxide and its mechanism. Environ. Sci. Technol. 46, 5528–5534 (2012)
F.P. Rotzinger, J.M. Kesselman-Truttmann, S.J. Hug, V. Shklover, M. Gratzel, Structure and vibrational spectrum of formate and acetate adsorbed from aqueous solution onto the TiO2 rutile (110) surface. J. Phys. Chem. B 108, 5004–5017 (2004)
M.I. Franch, J.A. Ayllon, J. Peral, X. Domenech, Enhanced photocatalytic degradation of maleic acid by Fe(III) adsorption onto the TiO2 surface. Catal. Today 101, 245–252 (2005)
T. Meulen, A. Mattson, L. Osterlund, A comparative study of the photocatalytic oxidation of propane on anatase, rutile, and mixed-phase anatase-rutile TiO2 nanoparticles: role of surface intermediates. J. Catal. 251, 131–144 (2007)
K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, 4th edn. (Wiley, New York, 2009)
G.B. Deacon, R.J. Phillips, Relationships between the carbon-oxygen stretching frequencies of carboxylato complexes and the type of carboxylate coordination. Coord. Chem. Rev. 33, 227–250 (1980)
G.W. Buchanan, E. Munteanu, B.A. Dawson, D. Hodgson, Concerning the origin of 19F-19F NMR COSY and NOESY connections in the spectra of perfluorooctanoic acid, RF-palmitic acid-F13 and diethyl perfluorosuberate. Magn. Reson. Chem. 43, 528–534 (2005)
W.R. Dolbier, Guide to fluorine NMR for organic chemists (Wiley, New Jersey, 2009)
S. Pawsey, L. Reven, 19F fast magic-angle spinning NMR studies of perfluoroalkanoic acid self-assembled monolayers. Langmuir 22, 1055–1062 (2006)
M.R. Hoffmann, S.T. Martin, W. Choi, D.W. Bahnemann, Environmental applications of semiconductor photocatalysis. Chem. Rev. 95, 69–96 (1995)
J.C. Yu, W.K. Ho, J.G. Yu, S.K. Hark, K. Iu, Effects of trifluoroacetic acid modification on the surface microstructures and photocatalytic activity of mesoporous TiO2 thin films. Langmuir 19, 3889–3896 (2003)
J.G. Yu, W.G. Wang, B. Cheng, B.L. Su, Enhancement of photocatalytic activity of mesoporous TiO2 powders by hydrothermal surface fluorination treatment. J. Phys. Chem. C 113, 6743–6750 (2009)
S. Yang, C. Sun, X. Li, Z. Gong, X. Quan, Enhanced photocatalytic activity for titanium dioxide by co-modifying with silica and fluorine. J. Hazard. Mater. 175, 258–266 (2010)
S. Kutsuna, Y. Nagaoka, K. Takeuchi, H. Hori, TiO2-induced heterogeneous photodegradation of a fluorotelomer alcohol in air. Environ. Sci. Technol. 40, 6824–6829 (2006)
H.F. Schroder, R.J.W. Meesters, Stability of fluorinated surfactants in advanced oxidation processes—a follow up of degradation products using flow injection-mass spectrometry, liquid chromatography-mass spectrometry and liquid chromatography-multiple stage mass spectrometry. J. Chromatogr. A 1082, 110–119 (2005)
J. Cheng, C.D. Vecitis, H. Park, B.T. Mader, M.R. Hoffmann, Sonochemical degradation of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in landfill groundwater: environmental matrix effects. Environ. Sci. Technol. 42, 8057–8063 (2008)
J. Cheng, C.D. Vecitis, H. Park, B.T. Mader, M.R. Hoffmann, Sonochemical degradation of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in groundwater: kinetic effects of matrix inorganics. Environ. Sci. Technol. 44, 445–450 (2010)
J. Chen, P. Zhang, L. Zhang, Photocatalytic decomposition of environmentally persistent perfluorooctanoic acid. Chem. Lett. 35, 230–231 (2006)
S.C. Panchangam, A.Y. Lin, J. Tsai, C. Lin, Sonication-assisted photocatalytic decomposition of perfluorooctanoic acid. Chemosphere 75, 654–660 (2009)
S.C. Panchangam, A.Y. Lin, K.L. Shaik, C. Lin, Decomposition of perfluorocarboxylic acids (PFCAs) by heterogeneous photocatalysis in acidic aqueous medium. Chemosphere 77, 242–248 (2009)
C. Song, P. Chen, C. Wang, L. Zhu, Photodegradation of perfluorooctanoic acid by synthesized TiO2-MWCNT composites under 365 nm UV irradiation. Chemosphere 86, 853–859 (2012)
C.R. Estrellan, C. Salim, H. Hinode, Photocatalytic decomposition of perfluorooctanoic acid by iron and niobium co-doped titanium dioxide. J. Hazard. Mater. 179, 79–83 (2010)
F. Quarto, C. Sunseri, S. Piazza, M. Romano, Semiempirical correlation between optical band gap values of oxides and the difference of electronegativity of the elements. Its importance for a quantitative use of photocurrent spectroscopy in corrosion studies. J. Phys. Chem. B 101, 2519–2525 (1997)
W. Wang, D. Wang, W. Qu, L. Lu, A. Xu, Large ultrathin anatase TiO2 nanosheets with exposed {001}facets on graphene for enhanced visible light photocatalytic activity. J. Phys. Chem. C 116, 19893–19901 (2012)
Z. Li, P. Zhang, J. Li, T. Shao, L. Jin, Synthesis of In2O3-graphene composites and their photocatalytic performance towards perfluorooctanoic acid decomposition. J. Photochem. Photobiol. A 271, 111–116 (2013)
O. Akhavan, M. Abdolahad, A. Esfandiar, M. Mohatashamifar, Photodegradation of graphene oxide sheets by TiO2 nanoparticles after a photocatalytic reduction. J. Phys. Chem. C 114, 12955–12959 (2010)
Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan, J.R. Gong, Highly efficient visible-light-driven photocatalytic hydrogen production of CdS-cluster-decorated graphene nanosheets. J. Am. Chem. Soc. 133, 10878–10884 (2011)
P. Wang, Y. Ao, C. Wang, J. Hou, J. Qian, A one-pot method for the preparation of graphene-Bi2MoO6 hybrid photocatalysts that are responsive to visible-light and have excellent phtotocatalytic activity in the degradation of organic pollutants. Carbon 50, 5256–5264 (2012)
Z. Li, P. Zhang, T. Shao, X. Li, In2O3 nanoporous nanosphere: a highly efficient photocatalyst for decomposition of perfluorooctanoic acid. Appl. Catal. B Environ. 125, 350–357 (2012)
Z. Zhang, C.C. Wang, R. Zakaria, J.Y. Ying, Role of particle size in nanocrystalline TiO2-based photocatalysts. J. Phys. Chem. B 102, 10871–10878 (1998)
J. Zhang, K. Sasaki, E. Sutter, R.R. Adzic, Stabilization of platinum oxygen-reduction electrocatalysts using gold clusters. Science 315, 220–222 (2007)
R. Narayanan, M.A. El-Sayed, Shape-dependent catalytic activity of platinum nanoparticles in colloidal solution. Nano Lett. 4, 1343–1348 (2004)
J.C. Yu, J. Yu, J. Zhao, Enhanced photocatalytic activity of mesoporous and ordinary TiO2 thin film by sulfuric acid treatment. Appl. Catal. B 36, 31–43 (2002)
Z. Li, P. Zhang, T. Shao, J. Wang, L. Jin, X. Li, Different nanostructured In2O3 for photocatalytic decomposition of perfluorooctanoic acid (PFOA). J. Hazard. Mater. 260, 40–46 (2013)
J.C.C. Fan, J.B. Goodenough, X-ray photoemission spectroscopy studies of Sn-doped indium-oxide films. J. Appl. Phys. 48, 3524–3531 (1977)
J. Li, H. Fu, L. Fu, J. Hao, Complete combustion of methane over indium tin oxides catalysts. Environ. Sci. Technol. 40, 6455–6459 (2006)
C. Donley, D. Dunphy, D. Paine, C. Carter, K. Nebesny, P. Lee, D. Alloway, N.R. Armstrong, Characterization of indium-tin oxide interfaces using X-ray photoelectron spectroscopy and redox processes of a chemisorbed probe molecule: effect of surface pretreatment conditions. Langmuir 18, 450–457 (2002)
Y. Zheng, L. Zheng, Y. Zhan, X. Lin, Q. Zheng, K. Wei, Ag/ZnO heterostructure nanocrystals: synthesis, characterization, and photocatalysis. Inorg. Chem. 46, 6980–6986 (2007)
S. Meng, E. Kaxiras, Electron and hole dynamics in dye-sensitized solar cells: influencing factors and systematic trends. Nano Lett. 10, 1238–1247 (2010)
M. Aizawa, Y. Morikawa, Y. Namai, H. Morikawa, Y. Iwasawa, Oxygen vacancy promoting catalytic dehydration of formic acid on TiO2(110) by in situ scanning tunneling microscopic observation. J. Phys. Chem. B 109, 18831–18838 (2005)
Y.D. Hou, X.C. Wang, L. Wu, Z.X. Ding, X.Z. Fu, Efficient decomposition of benzene over a β-Ga2O3 photocatalysts under ambient conditions. Environ. Sci. Technol. 40, 5799–5803 (2006)
T. Shao, P. Zhang, L. Jin, Z. Li, Photocatalytic decomposition of perfluorooctanoic acid in pure water and sewage water by nanostructured gallium oxide. Appl. Catal. B Environ. 142–143, 654–661 (2013)
T. Shao, P. Zhang, Z. Li, L. Jin, Photocatalytic decomposition of perfluorooctanoic acid in pure water and wastewater by needle-like nanostructured gallium oxide. Chin. J. Catal. 34, 1551–1559 (2013)
G. Imoberdorf, M. Mohseni, Degradation of natural organic matter in surface water using vacuum-UV irradiation. J. Hazard. Mater. 186, 240–246 (2011)
W.Y. Han, P.Y. Zhang, W.P. Zhu, J.J. Yin, L.S. Li, Photocatalysis of p-chlorobenzoic acid in aqueous solution under irradiation of 254 nm and 185 nm UV light. Water Res. 38, 4197–4203 (2004)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Zhang, P., Li, Z. (2014). Photocatalytic Degradation of Perfluorooctanoic Acid. In: Hu, A., Apblett, A. (eds) Nanotechnology for Water Treatment and Purification. Lecture Notes in Nanoscale Science and Technology, vol 22. Springer, Cham. https://doi.org/10.1007/978-3-319-06578-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-06578-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-06577-9
Online ISBN: 978-3-319-06578-6
eBook Packages: EngineeringEngineering (R0)