
Abstract
Azospirillum and other related plant growth-promoting rhizobacteria produce several phytohormones and signaling molecules. Among them, nitric oxide is now recognized as a key second messenger in plants and in beneficial and pathological plant-microorganism interactions switching on and off different processes. Nitric oxide has been associated to the signaling cascades leading to lateral and adventitious root development induced by Azospirillum in tomato, in the Azospirillum biofilm formation, and in the nodule development in rhizobia-legume symbiosis. As a central component of N cycle, nitric oxide is produced and consumed in different metabolic pathways such as denitrification and nitrification, and it is closely related to other N compound like nitrate and nitrite. The emerging importance of nitric oxide in the biology of bacteria-plant relation is a challenge for understanding the molecular and chemical basis underpinning the nitric oxide actions in the association of plant growth-promoting bacteria with roots. In this chapter we describe several techniques that allow detecting and quantifying endogenously produced and exogenously applied nitric oxide in bacteria cultures and inoculated plants, including real-time and/or in situ nitric oxide production. The most used methods—Griess assay, electron paramagnetic resonance, fluorescent probes, and electrochemical sensors—are described with detailed protocols, discussing their advantages and drawbacks. In addition, we remark factors affecting nitric oxide production like growth conditions, metabolic inhibitors, and others. Finally, pharmacological, genetic, and histochemical strategies to study the role of nitric oxide in the association of Azospirillum with plant roots are presented with examples and methodological procedures.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Nitric Oxide
- Electron Paramagnetic Resonance
- Nitrate Reductase
- Paracoccus Denitrificans
- Alcaligenes Faecalis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Rhizobacteria secrete metabolites into the rhizosphere which can act as signaling compounds perceived by neighboring cells within the same microcolony, by other bacterial species present in the rhizosphere, or by root cells of the host plant. Signals derived from changes in the soil environment trigger selective root and shoot responses. The mechanisms by which rhizobacteria elicit plant growth promotion, from the viewpoint of signal transduction pathways is a matter of debate. In this scenario, the interrelationships established between roots and the biotic components of the rhizosphere would have a strong impact not only on plant growth, but also on microorganisms. Nitric oxide (NO) is a small bioactive molecule produced either by eukaryotic and prokaryotic cells which in plants has a central role in the signaling pathway for growth and development of roots. The plant growth-promoting rhizobacteria (PGPR) Azospirillum brasilense has been proved to produce NO that participates in the signaling cascades inducing lateral and adventitious root formation in tomato (Creus et al. 2005; Molina-Favero et al. 2008) and also in the induction of bacterial biofilm formation (Arruebarrena Di Palma et al. 2013). Therefore, it would be useful and desirable to handle different approaches and techniques that deal with NO metabolism in PGPR and their interaction with plants.
The chemical properties of NO make it a versatile signal molecule that functions through interactions with cellular targets via either redox or additive chemistry. Its small Stokes’ radius and neutral charge allows rapid membrane diffusion and, together with its short half-life, makes NO ideal as a near real-time signal between cells (Lamattina et al. 2003). In cells, NO can exist in the form of three interconverting compounds: a free radical nitric oxide (NO•), a nitrosonium cation (NO+), and a nitroxyl anion (NO−), each one with different chemical reactivities and properties. Furthermore, the existence of an unpaired electron makes NO highly reactive with oxygen (O2), superoxide (O2 −), N derivatives, and transition metals (Lamattina et al. 2003).
Nitric oxide can be produced by several pathways in bacteria: (1) Denitrification: is the stepwise dissimilative reduction of nitrate (NO3 −) to nitrite (NO2 −), NO, nitrous oxide (N2O), and dinitrogen (N2) by the corresponding N-oxide reductases. This pathway allows denitrifiers to grow under low-oxygen or anaerobic conditions, since NO3 − is used instead of O2 as a final electron acceptor in respiration (Cutruzzolá 1999). In addition to anaerobic denitrification, it is accepted that denitrification can also occur under fully aerobic conditions (Jetten et al. 1997); (2) Nitrification: is an aerobic ammonium (NH4 +) oxidation pathway yielding hydroxylamine (NH2OH), NO2 −, and finally, NO3 − (Wrage et al. 2001) in autotrophic conditions. NO is generated as an intermediary in the reduction of NO2 − to N2. This process can also be performed by heterotrophic microorganisms but without energy generation; (3) NO synthase: NO is produced by the oxidation of L-arginine to L-citrulline, in the presence of O2 by a bacterial NOS-like enzyme (bNOS) harbored mainly by Gram-positive microorganisms (Stuehr 1997).
Since its discovery as an endogenous-free radical, NO has been proposed to be either cytoprotective or cytotoxic (Stamler 1994; Beligni and Lamattina 1999). The cytoprotection is based on NO’s ability to regulate the level and toxicity of reactive oxygen species. NO-mediated toxicity is mainly generated by reaction with O2 −, leading to the formation of the strong oxidant peroxinitrite, which can oxidize thiols and nitrate peptides and proteins at the phenyl group of tyrosine (Lamattina et al. 2003). These same reactions, along with the ability of NO to transiently bind to numerous sites of proteins—including heme, iron–sulfur clusters, and thiols—enable NO to impact cell activities from the transcriptional to posttranscriptional levels (Cohen et al. 2010). This dual behavior of NO reinforces the necessity of a proper knowledge in the designing, measurement, and results interpretation of experimental approaches to reveal NO actions in the study of plant–microbe interaction.
2 Production of NO by Beneficial Rhizospheric Microorganisms: Detection and Quantification
2.1 Methods for NO Measuring
2.1.1 Griess Assay
Nitric oxide production is often measured indirectly by quantifying the stable end products of its metabolism since NO is not stable and persistent in stored biological samples. Two of the more stable oxidation products of NO metabolism are inorganic nitrite and nitrate (Archer 1993). In 1879, the German organic chemist Johan Peter Griess described a colorimetric protocol for nitrite measurement, which is to date one of the most widely used assays for NO measurement because of its simplicity and commercial availability. In this method, nitrite is first treated with a diazotizing reagent, e.g., sulfanilamide (SA), in acidic media to form a transient diazonium salt. This intermediate is then allowed to react with a coupling reagent, N-naphthyl-ethylenediamine (NED), to form a stable azo compound. The intense purple color of the product allows assaying nitrite with high sensitivity and can be used to measure nitrite concentration as low as ~0.5 μM. The absorbance at 540 nm of the formed adduct is linearly proportional to the nitrite concentration in the sample (Xu et al. 2000). Through the years, many variations on the original reaction have been described. The most popular version seems to be the sequential method in which nitrite is mixed with SA first, immediately followed by the addition of NED. This method seems to give highest yield of the chromophore, and therefore, it is the most sensitive way to perform Griess Reaction assay (Verdon et al. 1995; Guevara et al. 1998). This approach is adopted in most commercial kits.
For a more accurate measurement of NO produced in a sample, the nitrate formed via oxidation of nitrite must also be measured. This is often accomplished by reducing nitrate to nitrite immediately prior to the addition of the Griess reagents to the initial sample (Arita et al. 2007). The available kits use either cadmium or nitrate reductase to reduce nitrate into nitrite (Sun et al. 2003).
This approach was widely employed to identify denitrifying rhizobacteria in soil communities (Braker et al. 2010; Mora-Ravelo et al. 2013; Vercellino and Gómez 2013).
Materials and Reagents
-
Reagent A: Sulfanilamide Solution (1 % (w/v) sulfanilamide in 5 % (v/v) phosphoric acid).
-
Reagent B: NED Solution (0.1 % (w/v) N-1-napthylethylenediamine dihydrochloride).
-
Standard Nitrite Solution: 0.1 M sodium nitrite.
-
Polystyrene 96-well microtiter plate.
-
Microtiter plate reader with 520–550 nm filter.
Griess Assay
-
Add 100 μL of the cell-free culture or standard nitrite solution to a well of a microtiter plate in triplicate.
-
Transfer 50 μL of reagent A to each well.
-
Incubate 5–10 min at room temperature, protected from light.
-
Add 50 μL of reagent B to the wells.
-
Incubate at room temperature for 5–10 min, protected from light. A purple/magenta color will begin to form immediately.
-
Record the absorbance at 540 nm with the microtiter plate reader.
-
Compare the obtained values with the standard curve prepared with sodium nitrite (linear between 0 and 100 μM).
Considerations and Recommendations
-
Sulfanilamide and NED compete for nitrite in the Griess reaction (Fiddler 1977), thus greater sensitivity is achieved when the two components are added sequentially.
-
It is recommended to perform two-times reading to decrease the background variations between wells or samples (Xu et al. 2000).
-
The final pH of a sample after addition of reagent A is critical for the Griess reaction. Lower final pH results in higher absorbance at 540 nm. When the pH is lower than 1.8, the absorbance at 540 nm is stable (Xu et al. 2000). For samples with a high buffer capacity, more acid should be added to reagent A.
-
In a condition where there is low level of NO production, high amount of nitrate (or nitrite) in the media will make the measurement difficult due to the high background (Arita et al. 2007), thus it is important to know minutely nitrite and nitrate concentration in the media broth used and also to minimize as much as possible the content of these compounds from water in the solutions.
2.1.2 Electron Paramagnetic Resonance
Electron paramagnetic resonance (EPR), also known as electron spin resonance (ESR), is a versatile technique specific for atoms, molecules, and complexes with unpaired electrons (free radicals). Among its several biological applications (for examples see García Rubio 2004), this method allows to identify and quantify a paramagnetic molecule as NO with high specificity and sensibility (detection limit ~10−9 M). The advantages of EPR are: (1) it is the only method that unequivocally discriminate NO from other related no free radicals, as nitrate and nitrite; (2) it can report NO levels in vitro and in vivo; and (3) it is possible to use non-transparent (i.e., turbid and/or opaque) and non-purified samples (Kleschyov et al. 2007). The main drawback of EPR is that specialized equipment and technical expertise are needed.
Unpaired electrons, as spinning charges, generate a magnetic field becoming a magnetic dipole. Similar to a compass, these dipoles align themselves in external magnetic fields. EPR is based in the property of these dipoles to take two possible orientations that show “resonance” as the electrons are flipped when energy in the microwave range is applied in presence of an external magnetic field. The amount of energy absorbed is proportional to the amount of unpaired electrons in the sample, and the value of the external magnetic field at which resonance occur, in a constant microwave frequency, is related to g, that is a spectroscopic factor characteristic of a given paramagnetic center. The shape and the hyperfine structure of the spectral line are also characteristic of a paramagnetic substance (Hogg 2010).
The transient nature and concentration of NO in biological samples prevent its direct detection with EPR. Instead, NO must be trapped to form stable paramagnetic adducts before being quantified. Several diamagnetic compounds have been used as “spin trap” of NO, among them are DETC (diethyldithiocarbamate), MGD (N-methyl-d-glucamine dithiocarbamate), and DTCS (dithiocarboxy sarcosine) (Venkataraman et al. 2002; Puntarulo et al. 2009).
EPR has been used to study NO metabolism in several microorganisms. As examples, the mechanisms for NO production by nitrite reductase (Nir) in Pseudomonas spp. (Radoul et al. 2012) and Geobacillus stearothermophilus (Davydov et al. 2009), and for NO consumption by respiratory NO reductase in Paracoccus denitrificans (Field et al. 2008) were assessed by this technique. In PGPR, EPR was employed to show the formation of nitrosylleghaemoglobin (LbNO) within soybean nodules of Bradyrhizobium japonicum (Meakin et al. 2007), the synthesis of NO by a Nir in Sinorhizobium meliloti (Ferroni et al. 2012), and to quantify the NO production by A. brasilense Sp245 wt and its IAA- and periplasmic nitrate reductase (Nap−) mutants (Creus et al. 2005; Molina-Favero et al. 2008). EPR methodology has also been used for assaying enzyme activity in A. brasilense (Vanoni et al. 1992).
Materials and Reagents
-
Bacterial sample (a minimum of approximately 0.15 g is needed). Samples do not need to be purified and can simply consist in cells from cultures grown in the condition desired and harvested by centrifugation.
-
Solution of 10 mM sodium N-methyl-d-glucamine dithiocarbamate (MGD) in 1 mM FeSO4.
-
Aqueous solution of 4-hydroxy-2,2,6,6-tetramethyl piperidine 1-oxyl, TEMPOL (a stable free radical used as standard to obtain the concentration of other free radical adducts).
-
Bottom-sealed Pasteur pipettes.
-
EPR spectrometer.
EPR Assay
-
Add 220 μL MGD solution to 150 mg of bacterial sample.
-
Transfer the sample with the spin trap to a bottom-sealed Pasteur pipette and introduce in the spectrometer cavity for measurements.
-
Record the spectra at room temperature (~18 °C) in the EPR spectrometer operating at 9.5 GHz (X-band). Set the instrument at 200 G field scan, 83.886 s sweep time, 327.68 ms time constant, 5.983 G modulation amplitude, 50 kHz modulation frequency, and 20 mW microwave power. Different modulation amplitude and/or microwave power can be selected to improve the resolution of spectra depending on the spectrometer used.
-
Record the spectra of the TEMPOL solution with the same spectrometer setting.
-
NO concentration is quantified by double integration of the three-line spectra (Fig. 9.1a) and is referenced to the TEMPOL spectra, which have a known area to concentration ratio.
Fig. 9.1 
Nitric oxide detection in A. brasilense cultures. (a) Three-line EPR spectra characteristic of the NO-MGD adduct recorded in pellets of A. brasilense Sp245 grown in liquid OAB medium with NO3 − or NH4 + and in cultures boiled for 5 min. The readout resulting from the detector is a first derivative of the absorption spectrum, whose area is proportional to the concentration of unpaired electrons (in this case NO) and the intensity is measured in an arbitrary scale, so the y axis is omitted. (b) Kinetics of NO production by A. brasilense Sp245 determined with the fluorescent probe DAF-2 DA. Kinetics of controls without probe, non-inoculated OAB medium, and culture plus the scavenger cPTIO (0.5 mM) are indicated. The fluorescence is showed as arbitrary units (AU)

2.1.3 Fluorescent Probes: Diaminofluorescein Fluorescent Dyes
Diamine derivatives of fluorescein (DAF) are the most widely used NO fluorescent probes (Namin et al. 2013) and are one of the best options for NO detection due to their high sensitivity and simple procedure. Diaminofluorescein-2 (DAF-2) was the first product in the DAF series to become commercially available. It reacts with NO, in presence of oxygen, resulting in the formation of a triazolo-fluorescein analogue (DAF-2T) that exhibits a strong green fluorescence (Kojima et al. 1998a, b). DAF-2 DA, a diacetate analogue of DAF-2, was designed in particular for imaging of NO produced in living cells (Kojima et al. 1998a). The diacetate group is removed by cellular esterases, allowing the DAF-2 form to react with intracellular NO (Planchet and Kaiser 2006; Mur et al. 2011). Because of the membrane permeability characteristic of DAF-2 DA, it is possible to monitor endogenous NO production within the cells in a real-time fashion, conferring an obvious advantage in efficient uptake of the fluorescent probe (Arita et al. 2007). DAF-FM has been developed as a more sensitive NO sensor than DAF-2 DA (~3 nM and ~5 nM, respectively; Murad 1999). Contrary to DAF-2 DA, it has also been suggested that the fluorescent signal of DAF-FM is not affected by pH above 5.
DAF-2 DA was employed to detect NO production by A. brasilense Sp245 growing under aerobic conditions (Fig. 9.2; Creus et al. 2005; Molina-Favero et al. 2008) and to establish the involvement of NO in the formation of lateral roots induced by Azospirillum (Creus et al. 2005). Schreiber (2006) made use of DAF-FM DA to image NO production by Bacillus subtilis and by a nitrifying biofilm. Using these probes, several authors have detected NO production by confocal microscopy in functional nodules during legume-rhizobia symbiosis (see Sect. 9.3.3. below).

A. brasilense Sp245 cells showing NO production examined by epifluorescence microscopy. The bacteria was resuspended in HEPES-NaOH with or without the scavenger cPTIO for 30 min. Bacteria was further incubated for 2 h with DAF-2 DA and examined at 1,000× magnification (Creus et al. 2005; with kind permission from Springer Science and Business Media)
Materials and Reagents
-
NO-specific probes DAF-2 DA or DAF-FM DA (e.g., Calbiochem, La Jolla, CA, USA).
-
DAF-2T or DAF-FMT as standard (e.g., Calbiochem, La Jolla, CA, USA).
-
Polystyrene 96-well microtiter plate.
-
Fluorescence plate reader (e.g., Fluoroskan Ascent; Thermo Electron) with the appropriate filters (excitation 480 nm; emission 515 nm).
Reagent Preparation
Prepare a 1× working solution of DAF-probe immediately prior to use; a 1:500 dilution in water or in 0.1 M phosphate buffer pH 7.4 is generally required. Do not store 1× solution for later use.
Observations: DAF-probes are very light-sensitive; protect the reagent and the stained samples from direct light. Bovine Serum Albumin (BSA) and phenol red in culture media may affect fluorescence.
Probe Assay
-
Transfer 100 μL of the Azospirillum culture of each treatment to a 96-well microtiter plate. Do at least three replicates per measurement.
-
Add 1× working solution of DAF-probe at 10 μM final concentration.
-
Incubate 20 min at room temperature, in dark and with gentle agitation.
-
Place the 96-well microtiter plate into a fluorescence plate reader. The fluorescence intensity must be measured every 2–4 min over a period of at least 2 h.
-
Determinate NO concentration by comparison with the DAF-2T (or DAF-FMT) reference curve (linear between 0 and 1,000 nmol).
Considerations and Recommendations
-
Three types of controls must be done in order to establish if DAF-fluorescence is actually due to NO detection and not caused by methodological artifacts: (1) culture media without the addition of DAF-probe as background fluorescence, (2) non-inoculated media with DAF-probe to establish if the media cause any interference with the reaction, and (3) inoculated media with DAF-probe plus an NO scavenger (e.g., cPTIO, see Sect. 9.3.1. below) for NO specificity. Figure 9.1b shows kinetics of NO production by A. brasilense Sp245 in different conditions using DAF-2 DA and the controls described above.
-
It is now established that simultaneous presence of a NO source and hydrogen peroxide (H2O2) augments the fluorescence (Balcerczyk et al. 2005), therefore it is important to determine if the treatment applied produces H2O2 by cellular lysis. Check this possibility using an inverted fluorescence microscope to visualize potential increases in background fluorescence.
-
Only NO production kinetics can be measured with these probes because the reaction is cumulative. For NO consumption, other method must be used.
2.1.4 Electrochemical Sensors
Electrochemical methods are the most practical in measuring NO in biological samples due to small electrode size, in vivo capability, nondestructive properties, minimal or no reagents requirements, high sensitivity, simplicity, and the possibility to be operated with limited electrochemistry knowledge (Taha 2003).
Nitric oxide can be detected amperometrically using an NO-specific electrode, typically carbon- or platinum-coated, where the voltage of the electrode is held constant above the oxidation potential of NO (+900 mV against an AgCl reference electrode). The current generated is a linear function of NO concentration at the electrode surface. The available sensors are integrated, which means that there is no need for additional reference or counter electrode. They employ a gas permeable membrane for selectivity assurance, coupled with a controlled instrumental parameter. Membranes can be made from different compounds such as chloroprene rubber, cellulose acetate, collodion/polystyrene, polytetrafluoroethylene (PTFE), and phenylenediamine (Davies and Zhang 2008). These sensors are very sensitive and specific to NO because the gas permeable membrane eliminates all ions and other compounds except gases, and the applied electrical potential and electrode material eliminate interferences from other gases such as oxygen, carbon monoxide, carbon dioxide, etc. Detection levels are as low as 0.3–0.5 nM and a linear range can be achieved up to 25 μM NO (Taha 2003; Arita et al. 2007). Being electrochemical sensors, NO sensors are sensitive to temperature fluctuations (Taha 2003), but newer models are available with a temperature compensation option.
Electrochemical sensors have been extensively used in Escherichia coli (Gardner et al. 1998; Pathania et al. 2002; Bang et al. 2006) and in many other pathogenic bacteria (Ouellet et al. 2002; Arai et al. 2005; Lama et al. 2006; Avila-Ramirez et al. 2013) to study real-time NO consumption related to hemoglobin activity. In rhizobacteria, Arruebarrena Di Palma et al. (2013) demonstrated NO production in A. brasilense Sp245 biofilm formation employing this method.
Materials and Reagents
-
Nitric Oxide Measuring System (e.g., inNO-T-II System, Innovative Instruments, Inc., Tampa, FL. USA).
-
NO-specific sensor (e.g., amiNO-2000, Innovative Instruments, Inc., Tampa, FL. USA).
-
1 M sulfuric acid.
-
Solid potassium iodide.
-
100 μM potassium nitrite as standard solution.
Sensor Calibration
-
Before calibrating and using the sensor, it should have been polarized for few hours, preferably overnight and immersed in calibration solution or water.
-
Prepare calibration solution adding 18 mL of water, 2 mL of 1 M sulfuric acid, and approximately 20 mg of potassium iodide to a 20 mL vial. A small stirring bar should be used for mixing and obtaining a uniform solution. When this solution becomes light yellow, due to the formation of iodine, prepare a new solution.
-
Immerse the tip of the sensor in calibration solution. For the amiNO-2000, submerge 2–3 mm from the tip.
-
Wait for the background to decrease to stable value and then zero the background.
-
The in situ generation of NO is achieved by the addition of standard nitrite solution to an acidified solution in the presence of a reducing agent such as iodide ion. In this reaction the molar ratio of nitrite to nitric oxide is 1:1 and consequently, the amount of NO generated equals the amount of nitrite added.
-
Add 10 μL of 100 μM nitrite standard solution to a 20 mL stirred calibration solution; this result in 50 nM NO.
-
Before the second addition, wait until the current reaches its maximum potential and begins to decline. This normally takes a few seconds.
-
Add 20 μL of nitrite standard solution. A final NO concentration of 100 nM will be obtained. Include more additions if you will. It is recommendable using calibration concentrations in the range of the test solution.
-
Measure the peak height of the additions. Plot current vs concentration to make a reference curve.
Notes
-
Calibration should be done at the same temperature of the sample to be tested.
-
Other procedures can be chosen for calibration. For example, NO gas dissolved in aqueous solution or a NO-producing agent like S-nitroso-N-acetylpenicillamine (SNAP) can be used.
Real-Time Azospirillum NO Production
-
Immediately previous to use, stabilize the microelectrode 15 min running in PBS buffer pH 7.2, followed by 15 min in fresh NFb-malic medium. If another medium will be used previously, make sure it does not generate interference in the measurement.
-
Zero the background.
-
Immerse microelectrode 3–4 mm in the bacterial culture and start recording changes on current potential. Usually, 30–40 min of recording time is needed per sample to measure NO production in Azospirillum cultures.
-
Enter the obtained current value in the standard curve to establish NO concentration in the samples.
Considerations and Recommendations
-
If you fail to observe the release of NO, you can also analyze for nitrite in free cell supernatants using the same solution employed for calibration. It is recommendable to make dilutions of the supernatant to avoid electrode and solution saturation. For A. brasilense Sp245 growing in NFb-NO3 − media, 20 μL of a 1:25 dilution is sufficient for nitrite detection in 20 mL of calibration solution.
2.2 Factors Affecting NO Production in Azospirillum Culture
2.2.1 Culture Media and Growth Conditions
In order to set the conditions for different experiments, it is necessary to determine those circumstances in which NO production by rhizobacteria is favored.
One important issue to take into account is the bacterial growth phase as this represents a particular physiological stage in which some metabolic pathways are activated and other repressed. Therefore, NO production must be investigated in each phase to acquire a complete picture of the NO metabolism in the studied bacteria. For example, A. brasilense Sp245 produces NO throughout the exponential growth phase, with the highest concentration at the end of it (~16 h in OAB media).
On the other hand, the composition of the growth media is determinant of NO production and, depending on the pathway the bacteria use to biosynthesize NO, the media must provide the precursor for its synthesis. In denitrifying bacteria, media with NO3 − and low O2 tension must be supplied. If bacteria, as in the case of A. brasilense, harbors a periplasmic nitrate reductase not inhibited nor repressed by O2, the agitation can be high. We have characterized aerobic NO production by A. brasilense Sp245 in OAB medium with NO3 − as N source, in which bacteria produced ~120 nmol NO per gram, a concentration 25-fold higher than that observed for NH4 + containing media (4.2 nmol NO per gram of bacteria) (Molina-Favero et al. 2008).
If one suspects that the major source of NO production in the studied bacteria is heterotrophic nitrification, that possibility must be tested by incubating in media with NH4 + as N source. In this media, the addition of hydroxylamine or nitrite should enhance NO production. A four-time increase in the NO production rate was observed when A. brasilense Sp245 was incubated in the presence of 0.1 mM hydroxylamine (concentrations up to 0.5 mM had no effect or were inhibitory; Molina-Favero 2014).
If the bacteria produce NO by the action of a bNOS, L-arginine, the substrate of this enzyme, must be provided in the media in concentrations about 10–15 mM.
2.2.2 NO Production Pathways: Inhibitors
The inhibition of key enzymes from metabolic pathways leading to NO production could help in the study of the biogenic origin of NO when specific deficient mutants are not available. In these cases, simple and cheap pharmacological application of inhibitors substances or analogues of substrates result in blocking the activity of key specific enzymes in a given pathway.
The denitrification pathway that accounts for the majority of the NO produced by A. brasilense when grown in nitrate is driven by nitrate reductase (NR), nitrite reductase (Nir), nitric oxide reductase (Nor), and nitrous oxide reductase (Nos) (Zumft 1997).
Two different dissimilatory NR are present in some strains of denitrifiers. The dissimilatory membrane bound nitrate reductase (Nar) generates ATP by proton motive force across plasma membrane, and the periplasmic nitrate reductase (Nap) which is not repressed nor inactivated by O2 and is not involved in ATP generation.
The catalytic centre of NR joins a guanine molibdopterin dinucleotide cofactor. Tungstate can compete with molybdate for incorporation into the enzyme complex and results in the inactivation of molybdate-dependent enzymes (Harper and Nicholas 1978; Hille 2002). Tungstate is widely used as a NR inhibitor in plant NO research (Mendel 2007) and also in bacteria culture (Betlach and Tiedje 1981). In nitrogen fixing nodules of Medicago truncatula, the use of tungstate could inhibit NO production, showing that the NR is involved in the production of NO by the nodule, but not in roots and leaves as the inhibition by tungstate was not achieved in that tissues (Horchani et al. 2011).
However, evidence indicates that tungstate not only inactivates NR, but also inhibits other molybdate-dependent enzymes at least in plants (Xiong et al. 2012). In addition, a number of investigations have shown that tungstate also inhibits root growth, affects cortical microtubule formation, and induces programmed cell death in plants, just like other heavy metals do (Adamakis et al. 2008). Therefore, this inhibitor must be used with caution in analyzing plant microbe interaction experiments, keeping in mind that it is not completely specific (Xiong et al. 2012). The majority of the literature revised uses 1 mM of tungstate as final concentration in liquid bacteria broth (Kletzin and Adams 1996).
On the other hand, dissimilatory Nir is considered the major source of NO in bacteria. Its localization is periplasmic and two different types of Nir can be encountered in denitrifiers even though, so far, there is no report on bacteria harboring the two types (Zumft 1997). One of these types is cytochrome cd1-d Nir that contains heme c and heme d1 as prosthetic group. The other is a Cu-containing -d Nir (Ye et al. 1994).
A Cu-containing Nir from Alcaligenes faecalis strain S-6 was purified and crystallized. The purified enzyme was strongly inhibited by KCN, but only slightly by sulfhydryl reagents such as p-chloromercuribenzoate and N-ethylmaleimide (Kakutani et al. 1981). The use of this type of inhibitors is appropriate when testing purified enzymes, but not when pharmacological experiments are done with living bacteria that would result damaged by blocking other vital processes yielding confusing results and leading to misinterpretation. A combination of pharmacological and biochemical analysis with a genetic approach will be necessary in order to investigate the roles of NO in plants (Xiong et al. 2012), bacteria, and their interaction.
Many Gram-positive bacteria also harbor a specific bNOS which catalyzes the conversion of L-arginine to L-citrulline and NO (Stuehr 1997). Sequence analysis of the genes encoding bNOS reveals great similarity with the N-terminal NOSoxy domain of mammalian NOS, whereas the NOSred domain is completely absent in bNOS. Nevertheless, bNOS are functional and effectively synthesize NO from L-arginine, also showing inhibition by the mammalian NOS inhibitors (Chen and Rosazza 1994; Choi et al. 1997; Sari et al. 1998; Adak et al. 2002). The main used inhibitors for this enzyme are NG-nitro-L-arginine methyl ester hydrochloride (L-NAME) and L-N5-(1-iminoethyl)-ornithine dihydrochlorid (L-NIO). Both of them are selective competitors with L-arginine. They are used in a concentration range between 50 and 100 μM in liquid bacteria broth (Creus et al. 2005).
3 Study of the NO Functions in the Plant–Azospirillum Association
3.1 Pharmacological Approach: NO Donors and Scavengers
In order to fully understand the diverse bioregulatory functions of NO, several experimental strategies have been developed. The methodological approach mostly employed in NO research involves the application of exogenous NO. Such a relatively simple methodology has yielded numerous evidences for specific physiological functions of NO, although some of them seemed rather ambiguous and controversial (Floryszak-Wieczorek et al. 2006).
Nitrogen oxide donors are compounds which generate NO or related N-oxide species in a controlled manner. By definition, all NO donors release NO producing NO-related activity when applied to biological systems and thus are principally suited to either mimic an endogenous NO-related response or substitute an endogenous NO deficiency (Feelisch 1998). The pathways leading to enzymatic and/or non-enzymatic formation of NO differ significantly among individual compound classes, as well as their chemical reactivities and kinetics of NO release.
Several chemical NO donors are currently being used in experimental studies. The most commonly used donor is sodium nitroprusside (SNP), a transition metal NO complex that generates mainly NO+. Other donors frequently employed in NO research include S-nitrosothiols, being the main members within this group S-nitrosoglutathione (GSNO) and SNAP. The utilization of these compounds, along with other NO donors, allowed investigators to make a remarkable progress in the field of NO physiology. In bacteria, SNP and GSNO have been extensively used to produce nitrossative stress (Poole and Hughes 2000; Brandes et al. 2007; Avila-Ramirez et al. 2013), to mimic NO activity as signal molecule in biofilm formation (Barraud et al. 2006; Arruebarrena Di Palma et al. 2013; Barnes et al. 2013) and in plant biotic interactions (Creus et al. 2005; Molina-Favero et al. 2007; Scheler et al. 2013; Puppo et al. 2013), and also to complement mutations in NO production pathways (Arruebarrena Di Palma et al. 2013).
On the other hand, the use of different types, concentrations, and ways of application of the NO-releasing compounds has turned out into discrepancies between experimental results obtained with different NO donors. Hence, in order to make the obtained data reliable, it is necessary to complement with experiments using different inhibitors of NO synthesis or NO-scavengers and to monitor precisely the current concentration of NO in the biological sample (Neill et al. 2003).
There are several compounds available that can act as NO scavengers which differ in specificity and in their ability to gain access to the site of NO action. One of the first compounds used as NO scavenger is oxygenated hemoglobin, which in reaction with NO produces methemoglobin and nitrate (Doyle and Hoekstra 1984). This reaction is fast and stoichiometric, but with low specificity for NO (Arita et al. 2007) and non-suitable for intracellular scavenging (Feelisch 1998). Another class of compounds used as NO scavengers are the nitronyl nitroxides, which were introduced earlier for the quantification of NO by EPR (Yoshida et al. 1994). Among them, 2-(4-carboxyphenyl)-4,5-dihydro-4,4,5,5-tetramethyl-1H-imidazolyl-1-oxy-3-oxide (cPTIO) developed by Akaike and coworkers has proved to be very effective for NO scavenging because of its high specificity and stoichiometric reaction with NO (Akaike and Maeda 1996). The combination of DAF and cPTIO, although arguably (see Arita et al. 2007 for more information), has been frequently applied for proving in vivo NO production in animals (Pittner et al. 2003; Uruno et al. 2005; Negri et al. 2013), plants (Arita et al. 2007; Corpas et al. 2011; Tossi et al. 2012; Verma et al. 2014), and bacteria (Creus et al. 2005; Baudouin et al. 2006; Horchani et al. 2011). Additionally, the combined action of SNP and cPTIO treatments has been used to set up the participation of NO in several biological systems including the Azospirillum-promoted lateral root formation in tomato seedlings (Creus et al. 2005; Molina-Favero et al. 2007).
In order to make a proper experimental design using this methodology, the investigator should be aware of the key properties and differences between NO donor classes, including the mechanism and kinetics of NO release from the donor, dependence on the external factors, toxicity of accumulated products, etc. Here are some considerations to take into account when using either SNP or GSNO as NO donors.
-
In a biological system, the redox form/s of NO (NO+, NO• or NO−) that is/are actually released makes a substantial difference to the NO donor’s reactivity towards other biomolecules, the profile of by-products, and the bioresponse. For example, depending on the reaction conditions, GSNO can act as donors of NO, NO−, or NO+ (Singh et al. 1996).
-
It is important to know the appropriate information about the susceptibility of a given NO donor to, e.g., oxygen, light, temperature, and changes in pH, in order to set appropriate conditions of the experiment and also to prevent decomposition occurring in the stock solution. In this regard, the release of NO from SNP has been shown to be caused by photochemical reactions (Leeuwenkamp et al. 1984). In contrast, GSNO was shown to release NO in the dark, even though light seems to be necessary to initiate the decomposition process (Floryszak-Wieczorek et al. 2006). In general, stock solutions should be made up fresh before use, kept on ice, and protected from light (SNP solution is prepared in water; for GSNO preparation see Hart (1985)). Final dilutions should preferably be prepared in assay buffer immediately before application and checked for pH.
-
Formation of compound-specific by-products may arise during decomposition or metabolism, sometimes in amounts far exceeding those of NO released and even toxic. In the case of SNP, the reduction and subsequent decomposition of the nitrosyl complex is accompanied by cyanide release (Feelisch 1998), therefore controls with this anion should be included. Another negative control to check reactivity of the SNP degradation by-products can be made using an old SNP solution. On the other hand, photolytic and transition metal ion-mediated decomposition of GSNO can lead to GSH generation (Singh et al. 1996), a molecule with an important activity as antioxidant. Controls with this molecule should be included.
-
Because of the duality nature of the NO (signal molecule vs. stressing agent), the effect of its action will depend to a large extent on its concentration. Hence, at too high NO donor concentration—instead of stimulation—an inhibition of the process may be observed. For instance, in Azospirillum Sp245 growing with nitrate as N-source, the addition of GSNO induces biofilm formation in a dose-dependent manner up to 50 μM GSNO, but higher concentrations produce in turn biofilm disaggregation (see Chap. 11 in this book; Arruebarrena Di Palma et al. 2013). Concentrations above 2 mM GSNO affect Azospirillum viability (own data not published). In addition, the efficiency of NO release by SNP is better than GSNO (Ederli et al. 2009) and consequently lower concentrations of SNP than GSNO are required to obtain similar biological effect.
-
The temporal signatures of NO release differ significantly among NO donor classes. It is rapid and transient (between 30 and 60 min) for GSNO, while for SNP, a slow and sustained release of NO was registered, with a maximum peak at 24 h (Simontacchi et al. 2012; Grossi and D’Angelo 2005).
-
The product of exposure time and concentration of the NO donor determines the quality and magnitude of the biological response to exogenously applied NO. Thus, short-lived NO donors may have to be administered as continuous infusions rather than in a bolus form in order to avoid the delivery of only a short burst of NO. In most cases, NO will have to be delivered continuously over the entire period of incubation in order to best mimic its biological activity.
3.2 Genetic Approach
Previously, we introduce the pharmacological approach to study NO, which is the most widely used strategy for plants and plant–microbe interactions (Delledonne 2005). However, it is highly recommended that the use of NO donors, scavengers, and inhibitors is complemented with a genetic approach to substantiate pharmacological findings and to overcome side effects of chemicals.
The genetic approach offers important tools to elucidate the biochemical pathways synthesizing NO and/or its signaling mechanism. The use of mutants produced by insertion of Tn5 transposon and lacking one or more activities has been used as strategy by several authors. Baudouin et al. (2006) inoculated M. truncatula seedlings with different S. meliloti 1,021 derivatives in order to identify the source of NO in fixating nodules. These mutants, impaired in N fixation (NifH−) or denitrification (NirK− and NorD−), produced nodules with an unaffected NO level, indicating that these genes are not involved in NO production. Going further, Horchani et al. (2011) used a nodule-targeted RNA interference (RNAi) strategy to specifically knockdown M. truncatula nitrate reductases NR1 and NR2 (MtNr1/2 mutant), thus decreasing NR activity in the N2-fixation zone without affecting nitrogen metabolism in the whole plant. By inoculating MtNR1/2 with S. meliloti 2011 wt and denitrification mutants affected in napA and nirK genes, these authors showed that both plant and bacterial NR contribute to NO production in functional nodules. Similarly, we characterized NO production in A. brasilense Sp245 wt and two Tn5 derivative mutants impaired in IAA synthesis (ipdC −, Faj009 strain, Costacurta et al. 1994) and in Nap activity (napA −, Faj164 strain, Steenhoudt et al. 2001a), both in ammonia- and nitrate-containing medium. Subsequently, tomato seedlings were inoculated with these mutated Azospirillum strains and results demonstrated that bacterial root-growth-promoting activity is highly dependent on NO production, particularly by Nap (Molina-Favero et al. 2008). The use of both pharmacological and genetic approaches allows us to establish NO as a signaling molecule in the Azospirillum–root interaction and the cross talk of NO with auxin in the promoting effects (Creus et al. 2005; Molina-Favero et al. 2007, 2008).
Another genetic approach, successfully adopted by Pothier et al. (2007), involved the use of promoter traps technique. In this methodology short DNA fragments, harboring promoter sequences, are cloned upstream of reporter genes. Screening a random promoter library of Azospirillum, these authors found that the NO-associated nirK gene is up-regulated by wheat seed extracts.
The post-genetic “omics” strategies can also be useful to understand NO function in PGPR–plant interactions. Boscari et al. (2013) showed the transcriptomic profiles during nodule development in M. truncatula-S. meliloti symbiosis and how the expression pattern is modulated by NO. In a similar manner, Van Puyvelde et al. (2011) studied the transcriptomic response of A. brasilense wt and Faj009 to exogenous and endogenous auxin. They found evidence of a putative connection between the IAA and NO biosynthetic pathways since the Nap operon was up-regulated in the ipdC − mutant.
Considering that genomes of A. brasilense Sp245 (Wisniewski-Dyé et al. 2011) and Azospirillum sp. B510 (Kaneko et al. 2010), and in a close future also the Az39 genome (Cassán 2014), are published and annotated, bioinformatics tools are now available. Using these tools and a PCR-based strategy, Arruebarrena Di Palma (2008) failed to show the presence of a bacterial NOS-like gene in A. brasilense Sp245, but he was able to show the presence of a putative ammonium monooxigenase gene possibly involved in NO synthesis. This gene is expressed during the growth of A. brasilense Sp245 wt and the mutant Faj164 (Molina-Favero 2014).
The following considerations should be kept in mind when a genetic approach is used:
-
The conditions needed for a mutation to be expressed. As examples, NO production in M. truncatula-S. meliloti nodules rapidly increases in hypoxia and it is in this condition when the differences in NO levels between wt and mutants (both rhizobia and plant) are noticed (Horchani et al. 2011). Also, the knockout mutation of A. brasilense Sp245 Faj164 in the napA gene is only relevant when nitrate is present. Therefore, differences in NO synthesis and root growth-promoting effects due to inoculation are only recorded in incubation media with 2–10 mM nitrate (Molina-Favero et al. 2008).
-
Mutations can have pleiotropic effects. As a case, some A. brasilense mutated in NR are reported to have a reduced colonization capacity (Steenhoudt et al. 2001b). For this reason, in inoculated tomato seedlings an obligated control is to check that the number of wt and Nap mutant Faj164 cells in roots are not significantly different (Molina-Favero et al. 2008). Specific controls must be performer in each case.
-
The degree in which a single mutation affects a metabolic pathway and/or the number of genes which must to be mutated to completely knock out an activity. Analogous to IAA, several different NO biosynthetic pathways exist in bacteria (Schreiber et al. 2012) and in plants (Gupta et al. 2011). In the latter, NO null mutants has not been obtained so far (Gupta et al. 2011). In A. brasilense Sp245, there are also redundancies in metabolic pathways leading to NO production (Molina-Favero et al. 2007). As well, some compensation or a complementation can exist between plant and microorganism to produce NO (Horchani et al. 2011).
3.3 Histochemical Approach
So far in this chapter, we have introduced different methodologies to study NO production in Azospirillum and related bacteria, and diverse strategies to analyze the role of NO in bacteria and during beneficial plant biotic interaction by using pharmacological and genetic approaches.
To deeply understand NO functions in plant-rhizobacteria associations, the researcher should also analyze whether NO is actually produced differentially during the interaction and, if possible, identify its source, timing, and localization. These goals can be accomplished monitoring real-time NO production in root tissues of inoculated plants by using fluorescent probes under a microscope.
This technique allowed several authors to observe transiently NO production in Lotus japonicus and Medicago sativa roots 4 h after inoculation with their cognate symbionts, suggesting that NO production could result from the specific recognition of the plant and bacterial partners (Shimoda et al. 2005; Nagata et al. 2008). Furthermore, in M. truncatula roots, using DAF-2 DA, del Giudice et al. (2011) demonstrated that NO is produced at different sites during the infection process. Using DAF-FM DA, Baudouin et al. (2006) detected NO production by confocal microscopy in functional nodules during M. truncatula-S. meliloti symbiosis. Nitric oxide was specifically localized in the bacteroid-containing cells of the nodule fixation zone.
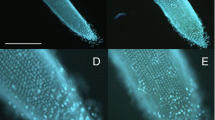
This approach was also employed by Creus et al. (2005) to analyze NO accumulation in Azospirillum-inoculated and control tomato roots grown in the presence or absence of 1 mM cPTIO (Fig. 9.3). Using DAF-2 DA, they demonstrated that: (1) roots from inoculated seeds displayed higher fluorescence intensity compared to non-inoculated ones and that this fluorescence could be partially blocked by cPTIO (Fig. 9.3a); (2) the fluorescence was located mainly at the vascular tissues and subepidermal cells (Fig. 9.3b). The protocol employed in this work is detailed below.

Nitric oxide detection by fluorescence with DAF-2 DA in Azospirillum-inoculated tomato roots. Tomato seeds were inoculated with A. brasilense Sp245 and incubated in distilled water (H2O) or cPTIO for 7 days. Control seeds were not inoculated (−). Root segments were incubated with DAF-2 DA and observed by epifluorescence (upper panels) or bright-field (lower panels) microscopy. (a) Longitudinal view of squashed roots. Bar = 0.3 mm. (b) Transversal sections of the primary root showing the green fluorescence corresponding to NO. Bar = 0.05 mm (Creus et al. 2005; with kind permission from Springer Science and Business Media)
Materials
-
1× dye solution: 15 μM DAF-2 DA in 20 mM HEPES-NaOH pH 7.5.
-
20 mM HEPES-NaOH buffer pH 7.5.
-
Bright-field and epi-fluorescence microscope with appropriate filters (e.g., Eclipse E 200, Nikon, Tokyo).
Tissue Sections Stain
-
Choose the tissue section to analyze. Only fresh tissue sections should be used. Do not fix sections with organic solvents or formalin as this will denature enzymes and give false readings from fixatives.
-
Wash the sections with 20 mM HEPES-NaOH pH 7.5 or other buffers free of serum, BSA, and phenol.
-
Flood the sections with the 1× dye solution and incubate for 2 h in dark.
-
Wash sections three times for 15 min each in the dark with fresh buffer to remove excess dye.
-
Prepare the stained tissue sections to be examined by epifluorescence (excitation 490 nm; emission 525 nm) and bright-field microscopy (e.g., the stained root tomato segments shown in Fig. 9.3 were squashed (a) or transversally cut with a razor blade (b)).
-
For relative fluorescence quantification, analyze the images using suitable software (e.g., IMAGE J).
Note
-
Since increased NO accumulation in inoculated plants could be the consequence of NO production by PGPR itself and/or by the plant (Creus et al. 2005), it is recommended to use this methodology with genetically modified plants or PGPR strains displaying diminished NO generation in order to discriminate the origin of NO between both organisms.
4 Concluding Remarks and Perspectives
With the beginning of the twenty-first century, we have faced an impressive advancement of the knowledge in the field of the diversity, biochemistry, and influence of the rhizosphere in plant growth determination.
The results of improving our understanding of how the “interactome” works in the diversity of species inhabiting the microbiota in rhizosphere should help us in learning about the consequences of greatly disturbing the soil homeostasis. It is a challenge nowadays identifying significant plant–microbe interactions at the rhizosphere at individual level. Knowing the genomes composition of the association could lead to the incorporation of plant–microbiome interaction as a trait to be considered into plant breeding programmes (Lundberg et al. 2012; Peiffer et al. 2013; Wagner et al. 2014).
Soils contribute with almost 20 % of the global atmospheric NO budget (Conrad 1995). Nitric oxide is a product of denitrification, nitrification, and reduction of nitrate to ammonia through microbial activity in soils (Zumft 1997). On the other hand, there is a significant increase in the emission of NO from soils fertilized with either biological or inorganic products (IPCC 1995; Ruser et al. 1998), but, remarkably, no studies have been conducted to distinguish between the effects of NO emission from soils and the effect of N fertilization per se on the crops’ yield. Besides, NO emission appears to be also dependent on crop species (Roelle et al. 2001), soil temperature, and water content, in addition to the level of total extractable nitrogen from soils (Davidson and Kingerlee 1997).
As a result, intriguing aspects concerning the significance of the NO produced by the biological activity in soils remains to be elucidated: (1) Is the contribution of the microbial activity in rhizosphere a source of NO important for the root perception of the rhizosphere composition and biodiversity, and for keeping the homeostasis required for the normal root growth and developmental processes?; (2) Is it possible to distinguish, among the increased NO fluxes emitted from the microbial activity in soils, between the NO that is acting as a signal for specific growth and developmental processes, from the NO derived as a by-product of N metabolism? (3) Is it possible to find a reliable method to distinguish the contribution of NO from microbiota in rhizosphere from the NO produced by plants?
These topics represent strong challenges to focus the attention of researchers working on plant–microbe interactions in soils in the coming decades.
References
Adak S, Bilwes KS, Panda K et al (2002) Cloning, expression, and characterization of a nitric oxide synthase protein from Deinococcus radiodurans. Proc Natl Acad Sci U S A 99:107–112
Adamakis I-DS, Eleftheriou EP, Rost TL (2008) Effects of sodium tungstate on the ultrastructure and growth of pea (Pisum sativum) and cotton (Gossypium hirsutum) seedlings. Environ Exp Bot 63:416–425
Akaike T, Maeda H (1996) Quantitation of nitric oxide using 2-phenyl-4,4,5,5- tetramethylimidazoline-1-oxyl 3-oxide (PTIO). Method Enzymol 268:211–221
Arai H, Hayashi M, Kuroi A et al (2005) Transcriptional regulation of the flavohemoglobin gene for aerobic nitric oxide detoxification by the second nitric oxide-responsive regulator of Pseudomonas aeruginosa. J Bacteriol 187(12):3960–3968
Archer S (1993) Measurement of nitric oxide in biological models. FASEB J 7(2):349–360
Arita NO, Cohen MF, Tokuda G et al (2007) Fluorometric detection of nitric oxide with diaminofluoresceins (DAFs): applications and limitations for plant NO research. In: Polaco JC, Lamattina L (eds) Nitric oxide in plant growth, development and stress physiology. Springer, Berlin, pp 269–280
Arruebarrena Di Palma A (2008) Producción de óxido nítrico por nitrificación heterotrófica en Azospirillum. Graduate Thesis Dissertation, Universidad Nacional de Mar del Plata
Arruebarrena Di Palma A, Pereyra CM, Moreno Ramirez L et al (2013) Denitrification-derived nitric oxide modulates biofilm formation in Azospirillum brasilense. FEMS Microbiol Lett 338(1):77–85
Avila-Ramirez C, Tinajero-Trejo M, Davidge KS et al (2013) Do globins in microaerophilic Campylobacter jejuni confer nitrosative stress tolerance under oxygen limitation? Antioxid Redox Signal 18(4):424–431
Balcerczyk A, Soszynski M, Bartosz G (2005) On the specificity of 4-amino-5-methylamino-2′, 7′-difluorofluorescein as a probe for nitric oxide. Free Radic Bio Med 39(3):327–335
Bang IS, Liu L, Vazquez-Torres A, Crouch ML et al (2006) Maintenance of nitric oxide and redox homeostasis by the Salmonella flavohemoglobin Hmp. J Biol Chem 281(38):28039–28047
Barnes RJ, Bandi RR, Wong WS et al (2013) Optimal dosing regimen of nitric oxide donor compounds for the reduction of Pseudomonas aeruginosa biofilm and isolates from wastewater membranes. Biofouling 29(2):203–212
Barraud N, Hassett DJ, Hwang SH et al (2006) Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J Bacteriol 188(21):7344–7353
Baudouin E, Pieuchot L, Engler G et al (2006) Nitric oxide is formed in Medicago truncatula- Sinorhizobium meliloti functional nodules. Mol Plant Microbe Interact 19:970–975
Beligni MV, Lamattina L (1999) Is nitric oxide toxic or protective? Trends Plant Sci 4:299–300
Betlach M, Tiedje J (1981) Kinetic explanation for accumulation of nitrite, nitric oxide, and nitrous oxide during bacterial denitrification. Appl Environ Microbiol 42(6):1074–1084
Boscari A, del Giudice J, Ferrarini A et al (2013) Expression dynamics of the Medicago truncatula transcriptome during the symbiotic interaction with Sinorhizobium meliloti: which role for nitric oxide? Plant Physiol 161:425–439
Braker G, Schwarz J, Conrad R (2010) Influence of temperature on the composition and activity of denitrifying soil communities. FEMS Microbiol Ecol 73(1):134–148
Brandes N, Rinck A, Leichert L et al (2007) Nitrosative stress treatment of E. coli targets distinct set of thiol-containing proteins. Mol Microbiol 66(4):901–914
Cassán F (2014) Mecanismos de promoción del crecimiento en rizobacterias de interés agronómico. El género Azospirillum. Actas de las IV Jornadas Bonaerenses de Microbiología de Suelos para una Agricultura Sustentable. Argentina. pp 12–13
Chen Y, Rosazza PN (1994) A bacterial nitric oxide synthase from a Nocardia species. Biochem Biophys Res Commun 203:1251–1258
Choi W, Chang M, Han J et al (1997) Identification of nitric oxide synthase in Staphylococcus aureus. Biochem Biophys Res Commun 237:554–558
IPCC Intergovernmental Panel on Climate Change (1995) Climate change 1995. The science of climate change. Contribution of Working Group I to the Second Assessment Report of the Intergovernmental Panel on Climate Change. London: Cambridge University Press, pp. 588
Cohen MF, Lamattina L, Yamasaki H (2010) Nitric oxide signaling by plant-associated bacteria. In: Hayat S, Mori M, Pichtel J, Ahmad A (eds) Nitric oxide in plant physiology. Wiley, Weinheim, pp 161–172
Conrad R (1995) Soil microbial processes involved in production and consumption of atmospheric trace gases. Adv Microb Ecol 14:207–250
Corpas FJ, Leterrier M, Valderrama R et al (2011) Nitric oxide imbalance provokes a nitrosative response in plants under abiotic stress. Plant Sci 181(5):604–611
Costacurta A, Keijers V, Vanderleyden J (1994) Molecular cloning and sequence analysis of an Azospirillum brasilense indole-3-pyruvate decarboxylase gene. Mol Gen Genet 243:463–472
Creus CM, Graziano M, Casanovas EM et al (2005) Nitric oxide is involved in the Azospirillum brasilense-induced lateral root formation in tomato. Planta 221:297–303
Cutruzzolá F (1999) Bacterial nitric oxide synthesis. Biochem Biophys Acta 1411:231–249
Davidson EA, Kingerlee W (1997) A global inventory of nitric oxide emissions from soils. Nutr Cycl Agroecosyst 48:37–50
Davies IR, Zhang X (2008) Nitric oxide selective electrodes. Method Enzymol 436:63–95
Davydov R, Sudhamsu J, Lees NS et al (2009) EPR and ENDOR characterization of the reactive intermediates in the generation of NO by cryoreduced oxy-nitric oxide synthase from Geobacillus stearothermophilus. J Am Chem Soc 131:14493–14507
del Giudice J, Cam Y, Damiani I et al (2011) Nitric oxide is required for an optimal establishment of the Medicago truncatula-Sinorhizobium meliloti symbiosis. New Phytol 191:405–417
Delledonne M (2005) NO news is good news for plants. Curr Opin Plant Biol 8:390–396
Doyle MP, Hoekstra JW (1984) Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J Inorg Chem 14:351–358
Ederli L, Reale L, Madeo L et al (2009) NO release by nitric oxide donors in vitro and in planta. Plant Physiol Biochem 47(1):42–48
Feelisch M (1998) The use of nitric oxide donors in pharmacological studies. Naunyn Schmiedebergs Arch Pharmacol 358(1):113–122
Ferroni FM, Guerrero SA, Rizzi AC et al (2012) Overexpression, purification, and biochemical and spectroscopic characterization of copper-containing nitrite reductase from Sinorhizobium meliloti, 2011. Study of the interaction of the catalytic copper center with nitrite and NO. J Inorg Biochem 114:8–14
Fiddler RM (1977) Collaborative study of modified AOAC method of analysis for nitrite in meat and meat products. J Assoc Off Anal Chem 60:594–599
Field SJ, Thorndycroft FH, Matorin AD et al (2008) The respiratory nitric oxide reductase (norBC) from Paracoccus denitrificans. Method Enzymol 437:79–101
Floryszak-Wieczorek J, Milczarek G, Arasimowicz M et al (2006) Do nitric oxide donors mimic endogenous NO-related response in plants? Planta 224(6):1363–1372
García Rubio I (2004) Aplicaciones Biológicas de la EPR. Cuarta Escuela de Resonancia Paramagnética Electrónica. Universidad de Alicante, Alicante, pp 151–183
Gardner PR, Gardner AM, Martin LA et al (1998) Nitric oxide dioxygenase: an enzymic function for flavohemoglobin. Proc Natl Acad Sci U S A 95(18):10378–10383
Griess PJ (1879) Bemerkungen zu der abhandlung der H.H. Weselsky und Benedikt “Ueber einige azoverbindungen”. Ber Dtsch Chem Ges 12:426–428
Grossi L, D’Angelo S (2005) Sodium nitroprusside: mechanism of NO release mediated by sulfhydryl-containing molecules. J Med Chem 48:2622–2626
Guevara I, Iwanejko J, Dembińska-Kieć A et al (1998) Determination of nitrite/nitrate in human biological material by the simple Griess reaction. Clin Chim Acta 274(2):177–188
Gupta KJ, Fernie AR, Kaiser WM et al (2011) On the origins of nitric oxide. Trends Plant Sci 16:160–168
Harper JE, Nicholas JC (1978) Nitrogen metabolism of soybeans I. Effect of tungstate on nitrate utilization, nodulation, and growth. Plant Physiol 62:662–664
Hart TW (1985) Some observations concerning the S-nitroso and S-phenylsulphonyl derivatives of L-cysteine and glutathione. Tetrahedron Lett 26:2013–2016
Hille R (2002) Molybdenum and tungsten in biology. Trends Biochem Sci 27:360–367
Hogg N (2010) Detection of nitric oxide by electron paramagnetic resonance spectroscopy. Free Radic Bio Med 49:122–129
Horchani F, Prévot M, Boscari A et al (2011) Both plant and bacterial nitrate reductases contribute to nitric oxide production in Medicago truncatula nitrogen-fixing nodules. Plant Physiol 155:1023–1036
Jetten MSM, Logemann S, Muyzer G et al (1997) Novel principles in the microbial conversion of nitrogen compounds. Anton Leeuw 71:75–93
Kakutani T, Watanabe H, Arima K et al (1981) Purification and properties of a copper-containing nitrite reductase from a denitrifying bacterium, Alcaligenes faecalis strain S-6. J Biochem 89(2):453–461
Kaneko T, Minamisawa K, Isawa T et al (2010) Complete genomic structure of the cultivated rice endophyte Azospirillum sp. B510. DNA Res 17:37–50
Kleschyov AL, Wenzel P, Munzel T (2007) Electron paramagnetic resonance (EPR) spin trapping of biological nitric oxide. J Chromatogr B 851:12–20
Kletzin A, Adams M (1996) Tungsten in biological systems. FEMS Microbiol Lett 18(1):5–63
Kojima H, Nakatsubo N, Kikuchi K et al (1998a) Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal Chem 70:2446–2453
Kojima H, Sakurai K, Kikuchi K et al (1998b) Development of a fluorescent indicator for nitric oxide based on the fluorescein chromophore. Chem Pharm Bull 46:373–375
Lama A, Pawaria S, Dikshit KL (2006) Oxygen binding and NO scavenging properties of truncated hemoglobin, HbN, of Mycobacterium smegmatis. FEBS Lett 580(17):4031–4041
Lamattina L, García-Mata C, Graziano M et al (2003) Nitric oxide: the versatility of an extensive signal molecule. Annu Rev Plant Biol 54:109–136
Leeuwenkamp OR, Van Bennekom WP, Van der Mark EJ et al (1984) Nitroprusside, antihypertensive drug and analytical reagent. Review of (photo)stability, pharmacology and analytical properties. Pharm Weekbl Sci 6:129–140
Lundberg DS, Lebeis SL, Paredes SH et al (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90
Meakin GE, Bueno E, Jepson B et al (2007) The contribution of bacteroidal nitrate and nitrite reduction to the formation of nitrosylleghaemoglobin complexes in soybean root nodules. Microbiology 153:411–419
Mendel RR (2007) Biology of the molybdenum cofactor. J Exp Bot 58:2289–2296
Molina-Favero C (2014) Rol del óxido nítrico (NO) en la asociación de Azospirillum con raíces de plantas superiores. Doctoral dissertation, Universidad Nacional de Mar del Plata
Molina-Favero C, Creus MC, Lanteri ML et al (2007) Nitric oxide and plant growth promoting rhizobacteria: common features influencing root growth and development. Adv Bot Res 46:1–33
Molina-Favero C, Creus CM, Simontacchi M et al (2008) Aerobic nitric oxide production by Azospirillum brasilense Sp245 and its influence on root architecture in tomato. Mol Plant Microbe Interact 21:1001–1009
Mora-Ravelo SG, Reyes FG, Moreno JP et al (2013) Dinamic of bacteria desnitrificants and nitrificants in the rizospheric of wheat with slow release of fertilizer, irrigated with waste or well water. Adv Microbiol 3(04):343
Mur LAJ, Mandon J, Cristescu SM et al (2011) Methods of nitric oxide detection in plants: a commentary. Plant Sci 181:509–519
Murad F (1999) Discovery of some of the biological effects of nitric oxide and its role in cell signaling (Nobel lecture). Biosci Rep 38(13–14):1856–1868
Nagata M, Murakami E, Shimoda Y et al (2008) Expression of a class 1 hemoglobin gene and production of nitric oxide in response to symbiotic and pathogenic bacteria in Lotus japonicus. Mol Plant Microbe Interact 21:1175–1183
Namin SM, Nofallah S, Joshi MS et al (2013) Kinetic analysis of DAF-FM activation by NO: toward calibration of a NO-sensitive fluorescent dye. Nitric Oxide 28:39–46
Negri P, Maggi M, Correa-Aragunde N et al (2013) Nitric oxide participates at the first steps of Apis mellifera cellular immune activation in response to non-self recognition. Apidologie 44(5):575–585
Neill SJ, Desikan R, Hancock JT (2003) Nitric oxide signalling in plants. New Phytol 159:11–35
Ouellet H, Ouellet Y, Richard C et al (2002) Truncated hemoglobin HbN protects Mycobacterium bovis from nitric oxide. Proc Natl Acad Sci U S A 99(9):5902–5907
Pathania R, Navani NK, Gardner AM et al (2002) Nitric oxide scavenging and detoxification by the Mycobacterium tuberculosis haemoglobin, HbN in Escherichia coli. Mol Microbiol 45(5):1303–1314
Peiffer JA, Spor A, Koren O, Jin Z et al (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci U S A 110:6548–6553
Pittner J, Liu R, Brown R et al (2003) Visualization of nitric oxide production and intracellular calcium in juxtamedullary afferent arteriolar endothelial cells. Acta Physiol Scand 179(3): 309–317
Planchet E, Kaiser WM (2006) Nitric oxide (NO) detection by DAF fluorescence and chemiluminescence: a comparison using abiotic and biotic NO sources. J Exp Bot 57:3043–3055
Poole RK, Hughes MN (2000) New functions for the ancient globin family: bacterial responses to nitric oxide and nitrosative stress. Mol Microbiol 36:775–783
Pothier JF, Wisniewski-Dyé F, Weiss-Gayet M et al (2007) Promoter-trap identification of wheat seed extract-induced genes in the plant-growth-promoting rhizobacterium Azospirillum brasilense Sp245. Microbiology 153:3608–3622
Puntarulo S, Jasid S, Boveris AD et al (2009) Electron paramagnetic resonance as a tool to study nitric oxide generation in plants. In: Hayat S, Mori M, Pichtel J, Ahmad A (eds) Nitric oxide in plant physiology. Wiley, Weinheim, pp 17–30
Puppo A, Pauly N, Boscari A et al (2013) Hydrogen peroxide and nitric oxide: key regulators of the legume-Rhizobium and mycorrhizal symbioses. Antioxid Redox Signal 18(16): 2202–2219
Radoul M, Barak Y, Rinaldo S et al (2012) Solvent accessibility in the distal heme pocket of the nitrosyl d1-heme complex of Pseudomonas stutzeri cd1 nitrite reductase. Biochemistry 51:9192–9201
Roelle PA, Aneja VP, Gay B, Geron C, Pierce T (2001) Biogenic nitric oxide emissions from cropland soils. Atmos Environ 35:115–124
Ruser R, Flessa H, Schilling R, Steindl H, Beese F (1998) Soil compactation and fertilization effects on nitrous oxide and methane fluxes in potato fields. Soil Sci Soc Am J 62:1587–1595
Sari MA, Moali C, Boucher JL et al (1998) Detection of a nitric oxide synthase possibly involved in the regulation of the Rhodococcus sp R312 nitrite hydratase. Biochem Biophys Res Commun 250:364–368
Scheler C, Durner J, Astier J (2013) Nitric oxide and reactive oxygen species in plant biotic interactions. Curr Opin Plant Biol 16(4):534–539
Schreiber F (2006) Detection and function of nitric oxide in microbial communities. Doctoral dissertation, University of Bremen
Schreiber F, Wunderlin P, Udert KM et al (2012) Nitric oxide and nitrous oxide turnover in natural and engineered microbial communities: biological pathways, chemical reactions, and novel technologies. Front Microbiol 23:372. doi:10.3389/fmicb.2012.00372
Shimoda Y, Nagata M, Suzuki A et al (2005) Symbiotic rhizobium and nitric oxide induce gene expression of non-symbiotic hemoglobin in Lotus japonicus. Plant Cell Physiol 46:99–107
Simontacchi M, Buet A, Lamattina L, Puntarulo S (2012) Exposure to nitric oxide increases the nitrosyl-iron complexes content in sorghum embryonic axes. Plant Sci 183:159–166
Singh RJ, Hogg N, Joseph J et al (1996) Mechanism of nitric oxide release from S-nitrosothiols. J Biol Chem 271(31):18596–18603
Stamler JS (1994) Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell 78:931–936
Steenhoudt O, Keijers V, Okon Y et al (2001a) Identification and characterization of a periplasmic nitrate reductase in Azospirillum brasilense Sp245. Arch Microbiol 175:344–352
Steenhoudt O, Ping Z, Vande Broek A et al (2001b) A spontaneous chlorate-resistant mutant of Azospirillum brasilense Sp245 displays defects in nitrate reduction and plant root colonization. Biol Fertil Soils 33:317–322
Stuehr DJ (1997) Structure-function aspects in the nitric oxide synthases. Annu Rev Pharmacol 37:339–359
Sun J, Zhang X, Broderick M et al (2003) Measurement of nitric oxide production in biological systems by using Griess reaction assay. Sensors 3(8):276–284
Taha ZH (2003) Nitric oxide measurements in biological samples. Talanta 61(1):3–10
Tossi V, Lombardo C, Cassia R et al (2012) Nitric oxide and flavonoids are systemically induced by UV-B in maize leaves. Plant Sci 193:103–109
Uruno A, Sugawara A, Kanatsuka H et al (2005) Upregulation of nitric oxide production in vascular endothelial cells by all-trans retinoic acid through the phosphoinositide 3-kinase/Akt pathway. Circulation 112(5):727–736
Van Puyvelde S, Cloots L, Engelen K et al (2011) Transcriptome analysis of the rhizosphere bacterium Azospirillum brasilense reveals an extensive auxin response. Microb Ecol 61:723–728
Vanoni MA, Edmondson DE, Zanetti G et al (1992) Characterization of the flavins and the iron-sulfur centers of glutamate synthase from Azospirillum brasilense by absorption, circular dichroism, and electron paramagnetic resonance spectroscopies. Biochemistry 31:4613–4623
Venkataraman S, Martin SM, Schafer FQ et al (2002) Detailed methods for the quantification of nitric oxide in aqueous solutions using either an oxygen monitor or EPR. Free Radic Bio Med 29:580–585
Vercellino M, Gómez MA (2013) Denitrifying capacity of rhizobial strains of Argentine soils and herbicide sensitivity. Ann Microbiol 63(4):1563–1570
Verdon CP, Burton BA, Prior RL (1995) Sample pretreatment with nitrate reductase and glucose-6-phosphate dehydrogenase quantitatively reduces nitrate while avoiding interference by NADP+ when the Griess reaction is used to assay for nitrite. Anal Biochem 224:502–508
Verma A, Malik CP, Gupta VK (2014) Sodium nitroprusside-mediated modulation of growth and antioxidant defense in the in vitro raised plantlets of peanut genotypes. Peanut Sci 41(1): 25–31
Wagner MR, Lundberg DS, Coleman-Derr D et al (2014) Natural soil microbes alter flowering phenology and the intensity of selection on flowering time in a wild Arabidopsis relative. Ecol Lett 17:717–726
Wisniewski-Dyé F, Borziak K, Khalsa-Moyers G et al (2011) Azospirillum genomes reveal transition of bacteria from aquatic to terrestrial environments. PLoS Genet 7(12):e1002430
Wrage N, Velthof GL, Van Beusichem ML et al (2001) Role of nitrifier denitrification in the production of nitrous oxide. Soil Biol Biochem 33:1723–1732
Xiong J, Fu G, Yang Y et al (2012) Tungstate: is it really a specific nitrate reductase inhibitor in plant nitric oxide research? J Exp Bot 63(1):33–41
Xu J, Xu X, Verstraete W (2000) Adaptation of E. coli cell method for micro-scale nitrate measurement with the Griess reaction in culture media. J Microbiol Method 41(1):23–33
Ye RW, Averill BA, Tiedje J (1994) Denitrification: production and consumption of nitric oxide. Appl Environ Microbiol 60:1053–1058
Yoshida M, Akaike T, Wada Y et al (1994) Therapeutic effects of imidazolineoxyl N-oxide against endotoxin shock through its direct nitric oxide-scavenging activity. Biochem Biophys Res Commun 202:923–930
Zumft WG (1997) Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61:533–616
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Amenta, M., Molina-Favero, C., Creus, C.M., Lamattina, L. (2015). Nitric Oxide in Azospirillum and Related Bacteria: Production and Effects. In: Cassán, F., Okon, Y., Creus, C. (eds) Handbook for Azospirillum. Springer, Cham. https://doi.org/10.1007/978-3-319-06542-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-06542-7_9
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-06541-0
Online ISBN: 978-3-319-06542-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)