
Abstract
The discovery of the endocannabinoid system (ECS) in the early 1990s of last century generated high expectations of new therapeutic opportunities. Its central role and pleiotropic character seemed to offer promising indications in the fields of pain, inflammation, CNS disorders, weight management and metabolic diseases. However, around 2007 the tide began to turn when several cannabinoid receptor type 1 (CB1) antagonists/inverse agonists failed as therapeutics against overweight and its complications. More recently, the development of FAAH (Fatty Acid Amide Hydrolase) inhibitors against pain has also faced serious setbacks. In retrospect the much greater complexity of the ECS than originally assumed has played a fundamental role in these difficulties. Although there is no doubt that endocannabinoids and their receptors are of great (patho-)physiological relevance, it has become clear that the ECS is intimately intertwined with other biological systems. Endocannabinoids exist in equilibrium with fatty acids and their metabolic derivatives, including eicosanoids and prostamides. Furthermore, there are several biologically active analogues of endocannabinoids, in particular fatty acid amides, with metabolic pathways overlapping those of the ECS. Finally, endocannabinoids per se and their congeners show “promiscuous” behaviour going beyond interactions with CB1 and CB2 receptors. It has become clear that the complexity of what may be called the “endocannabinoidome” demands for pharmacological approaches that take into account these dynamics. Targeting the “endocannabinoidome” continues to offer opportunities for prevention and therapy, in particular for chronic diseases. However, chances for success are more likely to come from “multiple-target” than from “single-target” approaches.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Fatty Acid Amide Hydrolase
- Transient Receptor Potential Ankyrin
- Fatty Amide
- Fatty Acid Amide Hydrolase Inhibitor
- GPR119 Agonist
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction: The Changing Views on the Endocannabinoid System
Endocannabinoids are signalling lipids playing important roles in a wide variety of biological processes. Together with their receptors and enzymes involved in their synthesis and breakdown they constitute the “endocannabinoid system” (ECS). By definition the term endocannabinoid is limited to those compounds displaying significant affinity to the cannabinoid receptors CB1 and CB2 [1, 2]. These receptors were discovered in the late 1980s [3, 4] and were shown to bind (−)-trans-Δ9-tetrahydrocannabinol (Δ9-THC) from the Cannabis plant. To date, nine “true” endocannabinoids are being distinguished (Fig. 9.1), which are all derived from long chain (C18 or longer) polyunsaturated fatty acids (LCPUFAs) [1]. The first two discovered and still the most studied are anandamide (N-arachidonoylethanolamine (AEA)), its name originating from the Sanskrit word “ananda” meaning “the bliss”, and 2-arachidonoylglycerol (2-AG).

Structural formulas of the classical endogenous cannabinoid receptor agonists, anandamide (N-arachidonoylethanolamine, AEA), 2-arachidonoyl glycerol (2-AG), N-arachidonoyldopamine (NADA), Noladin ether, O-arachidonoylethanolamine (Virodhamine), N-dihomo-γ-linolenoylethanolamine, N-oleoyldopamine (OLDA), N-docosatetraenoylethanolamine and oleamide
However, more than two decades of research have shown that the ECS per se is less specific and distinct than originally assumed. It is now widely accepted that it is tightly intertwined with other signalling mechanisms and that endocannabinoids are part of a larger class of structurally related amides, esters and ethers of fatty acids which exist in a continuous dynamic equilibrium with each other. The vast majority of these molecules belongs to the fatty (acid-) amides like AEA, although analogues of 2-AG including 2-oleoylglycerol and 2-linoleoylglycerol have also been found (see Sect. 2.3). Fatty acid amides (Lipid Maps class FA08; http://www.lipidmaps.org) are conjugates of different long chain fatty acids and amines including ethanolamine, neurotransmitters (serotonin, dopamine) or simple amino acids. They are abundantly present in nature and involved in various regulatory processes. In animals, their molecular targets go far beyond the classical CB receptors and include a wide range of receptors including GPR55, GPR18, GPR119, TRPA1 (transient receptor potential ankyrin 1), TRPV1 (transient receptor potential channel type V1), PPARs (peroxisome proliferator activated receptors) as well as several non-receptor targets [2, 5–7]
It has also become clear that some (if not all) of the “true” endocannabinoids themselves display “promiscuous” behaviour by activating or blocking other receptors besides CB1 or CB2 with potencies that differ little from those with which they interact with “true” cannabinoid receptors [2, 6]. In addition, anandamide, 2-AG and other CB ligands interact directly or indirectly with non-receptor targets [5]. Biochemical pathways for synthesis and degradation of endocannabinoids and their congeners show several crossroads with those of other lipid mediators, in particular eicosanoids. This not only creates a number of regulatory nodes but also leads to the formation of “hybrid” structures including prostamides and other oxidation products, often with bioactivity [8–11]. Taken together, an “expanded” view of the ECS is increasingly considered a better concept to comprehend its full dimensions [12]. In line with this, it has been suggested to apply the term “endocannabinoidome” to describe this family of molecules (Fig. 9.2). Mediators that are part of this endocannabinoidome are fluctuating in a time and tissue-specific way, modulated by various endogenous (e.g. energy status, inflammation) and environmental factors, including diet. These network dynamics have major consequences for drug development. Soon after its discovery it became clear that the ECS is involved in a number of important processes and (chronic) diseases including pain, anxiety/depression, GI/liver diseases, cancer, metabolic disease and eating behaviour. Several promising new pharmacological targets were suggested which often indeed showed encouraging effects in animal studies. In particular in relation to weight management and metabolic diseases expectations were high to develop CB1 antagonists or inverse agonists into a completely new drug class. Therefore, the failure of the first in class compound rimonabant because of severe anxiety and depression-related side-effects in predisposed persons [13] shocked the pharmaceutical industry. By the end of 2008 at least nine companies terminated active development projects with CB1 blockers. These included some with compounds in a well-advanced stage of development such as Taranabant (Merck) and CP-945,598 (Otanabant, Pfizer). In retrospect these failures illustrate that initial strategies to modulate a dynamic and pleiotropic like the ECS have been too narrow. The endocannabinoidome still holds many promises for both “food” and “pharmaceutical” applications. However, its complexity demands for more subtle multiple-target strategies instead of a classical one disease–one target–one drug approach. This chapter aims to illustrate some recent developments and activities in the field of the ECS, including some related receptors and mediators. Major therapeutic applications will be briefly illustrated. This will be elaborated in Sect. 4 by examples from two main domains, namely, “inflammation” and “weight management”.

Cartoon depicting the “expanding” view on the endocannabinoid system (ECS). The “classical” ECS (centre) is considered as part of an endocannabinoidome consisting of structurally related ligands, metabolites and enzymes involved. Endocannabinoids per se and their congeners interact with different non-cannabinoid receptors and other molecular targets
2 From Phytocannabinoids to Endocannabinoids: A classical Example of Reversed Pharmacology
2.1 Compounds with Pharmacological Activity from Cannabis spp.
Earliest written records on the physiological effects and medical use of Cannabis go back to about 2000 BC in the famous book Pe’n-ts’ao Ching attributed to the Chinese emperor Shen-nung [14]. This ancient pharmacopoeia describes a number of properties of Cannabis, including its capacity to “lighten one’s body”. Throughout history, medical use of Cannabis has been widely accepted and very common in different parts of the world until this began to decline around the beginning of the 1900s [15]. Since the last decades, there is a renewed interest in preparations and compounds prepared from the Cannabis plant. The term phytocannabinoids (phyto- used here to distinguish them from endocannabinoids) refers to a group of terpenophenolic compounds with 22 carbons (or 21 carbons for neutral form) of which more than 70 have been found so far and which can be categorised into ten main structural types [16, 17]. In general, Cannabis refers to the species Cannabis sativa, although there is ongoing discussion whether the genus Cannabis comprises more than one species, i.e. Cannabis sativa and C. indica [16]. Preparations from different Cannabis breeds show a great variety in absolute and relative concentrations of phytocannabinoids [18], of which only a few are ligands for CB1 or CB2 receptors. However, as the adjective “cannabinoid” predates the discovery of cannabinoid receptors by many years this term is still commonly used to describe also other compounds with structures similar to the phytocannabinoid Δ9-THC, irrespective of whether they are or are not cannabinoid receptor agonists or antagonists [2]. Recent breeding and selection of Cannabis for recreational purposes has primarily focussed on increasing the content of the psychotropic compound (−)-trans-Δ9-tetrahydrocannabinol (Δ9-THC, Fig. 9.3). At the same time, the renewed interest in Cannabis for medical use initiated the search for cultivars with completely different compositions and often much lower hallucinogenic activity. It is expected that the unravelling of the Cannabis sativa genome [19] will further stimulate these developments. In the plant, cannabinoids are produced as their carboxylic acid derivatives, known as cannabinoid acids. Their neutral counterparts can be formed through the action of heat (smoking), sunlight or during storage [20, 21]. Several cannabinoid acids themselves display biological activity, which are often distinct from those of their decarboxylated products [17, 20–22]. Chemical structures of some of the most studied phytocannabinoids are depicted in Fig. 9.3.

Examples of phytocannabinoids from Cannabis. The main psychoactive component is (−)-trans-Δ9-tetrahydrocannabinol (Δ9-THC). Two other compounds discussed in this chapter are (−)-cannabidiol (CBD) and (−)-trans-Δ9-tetrahydrocannabivarin (Δ9-THCV)
Although Δ9-THC (Dronabinol, Marinol®) has been available as medicinal preparation for oral use since the 1980s, its therapeutic use initially remained rather limited. Efficacy was reported to be variable, at least partly due to significant first-pass metabolism [23]. Since the beginning of this century there has been a slow but steady growth in the development and application of medicinal products based on herbal Cannabis or natural cannabinoids [23–25]. Differences between regions and therapeutic viewpoints are among the factors which determine whether the focus is more on the herb or on specific phytocannabinoids. Some countries and regions (The Netherlands, Canada, several US states) have official medicinal Cannabis policies, often referred to as “medical marijuana”. Herbal products are preferably taken by inhalation using special vaporizers, and there is an increasing trend towards “individualised” therapies using specially selected cultivars [18]. On the other hand, formulations with purified Δ9-THC, CBD and/or THCV for oral or oromucosal delivery are also being developed and implemented [26, 27]. Cannabis or phytocannabinoid-based preparations are used for a number of indications, including pain, the amelioration of chemotherapy-induced nausea and vomiting, stimulation of appetite and management of spasticity in multiple sclerosis [25, 27, 28]. An in-depth discussion on the role of Cannabis or cannabinoid-based preparations in medical therapy is considered outside the scope of this chapter. It is obvious though that the debate on this issue continues until today.
From a scientific and potentially therapeutic perspective it is of interest to note that increasing data are becoming available on the activity of phytocannabinoids other than Δ9-THC with only weak or no psychotropic effects. Compounds of interest include cannabidiol (CBD), cannabigerol (CBG), cannabichromene (CBC), Δ9-tetrahydrocannabivarin (THCV), cannabidivarin (CBDV) as well as (at least) some cannabinoid acids, Δ9-tetrahydrocannabinolic acid (Δ9-THCA) and cannabidiolic acid (CBDA). A detailed discussion of each of these molecules falls outside the scope of this chapter. For an excellent overview readers are referred to Izzo et al. [17]. However, two compounds are of specific interest and merit some extra attention here, namely, CBD and THCV.
Among the non-hallucinogenic phytocannabinoids, CBD (Fig. 9.3) is currently receiving the most attention. In dried Cannabis CBD contents range from very low (<1 %) to equal or even higher (up to around 8 %) compared to those of Δ9-THC, depending on the cultivar and preparation. The oromucosal spray Sativex®, prescribed for the treatment of spasticity due to multiple sclerosis contains CBD and Δ9-THC in a 1:1 ratio. Cannabidiol behaves like a typical multiple target compound. For reviews, see for example [17, 29]. It displays a highly diverse spectrum of activities including agonist activity for PPARγ, TRVP1 and TRPA1 receptors, antagonist of GPR55, a complex antagonistic behaviour towards CB1 and CB2, etc. (see also Sect. 3.2). Specifically in relation to neurodegenerative diseases and (neuropathic) pain the effects of CBD on glia cells are of interest to note. Several preclinical and an increasing number of clinical studies have suggested at least promising activities in chronic inflammatory and autoimmune diseases including IBD [30], MS [31], cancer [5, 32, 33] and different CNS disorders [34–36]. Remarkably, there is increasing evidence that CBD and Δ9-THC interact within the CNS thereby reducing the psychoactive effects of Δ9-THC and possibly even its psychogenic risks [37–39]. Future clinical studies, of which several are presently ongoing (source: ClinicalTrials.gov) should further demonstrate the full therapeutic potential of CBD, alone or combined with other cannabinoids or other compounds.
Δ9-tetrahydrocannabivarin (THCV, Fig. 9.3) occurs in Cannabis as a minor component in varying amounts [18]. Interestingly, this compound has been found to possess CB1 antagonist properties [40, 41]. Therefore, it is receiving attention as a natural alternative to the CB1 blockers/inverse agonists like rimonabant. Recently, THCV has been found to ameliorate insulin sensitivity in two mouse models of obesity [42].
2.2 Phytocannabinoids from Plants Other than Cannabis
Remarkably, structures with affinity to CB1 and CB2 have also been found in plants other than Cannabis [43]. These molecules may be divided into two categories. First, plants like most other organisms contain lipid-derived structures which are chemically related to the endocannabinoids as those found in mammals (see also Sect. 2.3), albeit shorter acyl chains (C12 or C14) appear to be more common in plant [43–46]. Next to this, an increasing number of other plant compounds with affinity for CB2 or CB1 have been characterised. Examples include (E)-ß-caryophyllene (present in many different spices and food plants including oregano, cinnamon and black pepper), falcarinol (found in carrots, parsley and celery), yangonin (present in Kava, Piper methysticum) and magnolol (from the medicinal plant Magnolia officinalis) (Fig. 9.4) [43, 44, 47–50].

Some plant-derived compounds with CB1 or (and) CB2 affinity present in plants other than Cannabis
Considering the wide abundance in nature and “promiscuity” of the ECS and related signalling systems, it does not seem unlikely that more natural compounds with similar properties will be found in common spices and herbs. It is tempting to speculate that such compounds may play a role in the culinary properties of some plants by inducing “hedonic” signals in the brain via CB1 receptor stimulation.
2.3 Endocannabinoids and Beyond
As mentioned in Sect. 1, the discovery of the prototypical endocannabinoids per se was followed by the finding that these molecules belong to much larger group of fatty acid-derived structures of which the biological effects go far beyond effects on CB1 and CB2.
The endocannabinoid anandamide (AEA) belongs to N-acylethanolamine (NAE) subclass of fatty acid amides. In addition to the NAEs, several other classes of fatty acid amides can be distinguished, including the primary fatty acid amides, the N-acylamino acids (=N-acylamines), N-acylarylalkylamines (N-acyldopamines, N-acylserotonins) (Fig. 9.5) [51, 52].

Examples of fatty acid amide structures not belonging to the endocannabinoids per se (see also Fig. 9.1)
It has been shown that cells are able to “combine” different fatty acids and biogenic amines to make several possible permutations of different fatty acid amides [7, 51]. Several studies have demonstrated that the local relative availability of fatty acid precursors, which in turn is modulated by dietary intake of lipids, plays an important role in determining the pattern of amide conjugates formed. For example, a number of studies in rodents and humans have shown that increasing the relative proportion of n-3 LC PUFAs in the diet can lead to a decrease in the formation of the “prototypic” endocannabinoids AEA and 2-AG, which are derived from the n-6 fatty acid arachidonic acid [53–56]. These changes are a direct consequence from a shift in n-3–n-6 balance of membrane lipids, resulting in compensatory increases in n-3 LC-PUFA-derived acyl conjugates. The same holds true for the local availability of amines. For example, we showed that serotonin conjugates with fatty acids are formed by gut tissue, where most of the body’s serotonin resides [57].
Compared to the many fatty acid amides, only little has been reported on 2-acylglycerol esters other than the endocannabinoid 2-AG. However, it is likely that several congeners will exist, for example formed out of triglycerides. In 1999, Ben-Shabat et al. reported the isolation of 2-linoleoyl-glycerol and 2-palmitoyl-glycerol (2-PG) from mouse spleen, brain and gut [58]. These two compounds did not directly bind to CB1 or CB2 receptors but apparently potentiated the binding of 2-AG and its capacity to inhibit adenylyl cyclase. Furthermore, both esters caused potentiation of some of the in vivo effects of 2-AG. Interestingly, 2-oleolyglycerol (2-OG) was found to stimulate GPR119 receptors (see Sect. 3.2.4) in vitro, and did this more potently than 2-AG, 2-PG and 2-linoleoyl-glycerol [59]. Subsequently 2-OG was given to human volunteers (2 g by jejunal delivery), which resulted in increased levels of plasma GLP-1 compared to administration of the precursor oleic acid. Triglycerides with oleic acid in the sn-2 position are very common in the diet and from these 2-OG can be formed in the gut in amounts larger than the dose given in the study of Hansen et al. [59]. Very recently, the presence of 2-linoleoyl-glycerol and 2-oleoylglycerol (2-OG) has also been demonstrated in Drosophila [60].
3 Biochemistry and Pharmacology of the ECS
3.1 Endocannabinoid Formation and Breakdown
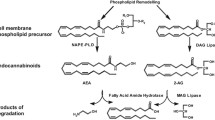
Synthesis and release of endocannabinoids and many related compounds are considered to take place “on demand” via different multi-step processes which are partly acting in parallel. For N-acyl-ethanolamines (NAEs) the most studied pathway is their formation from glycerophospholipids via N-acylphosphatidyl ethanolamines (NAPEs), present in phospholipid membranes. NAPEs function as stable precursors and source of their respective NAEs. Besides their role as precursor of NAEs, NAPEs have bioactive effects themselves [61]. NAPEs are formed by incorporation of fatty acids from the sn-1 position of a donor phospholipid like phosphatidylcholine and transfer to an ethanolamine phospholipid, e.g. phosphatidylethanolamine. This reaction is catalysed by a Ca2+-dependent N-acyltransferase [61, 62]. Next, free NAE can be generated by a NAPE-hydrolyzing phospholipase D (NAPE-PLD). In addition, other synthesis routes for NAEs have been found [61, 63]. The glycerol-ester 2-AG is synthesised from diacylglycerol (DAG), a very common second messenger, via the enzyme diacylglycerol lipase (DAGL), of which more than one form has been described [64]. Biosynthetic routes for other N-acylamides appear to be less well studied [52]. Huang et al. originally suggested that N-arachidonoyldopamine (NADA) may either be synthesised by condensation of dopamine with arachidonic acid (possibly via arachidonoyl-CoA) or via a pathway involving N-arachidonoyl-tyrosine [65]. Later, Hu et al. [66] reported that the latter may not be very likely. Instead they suggest a direct involvement of FAAH either as rate-limiting enzyme that liberates arachidonic acid from AEA, as a conjugation-enzyme, or both.
Conjugates of arachidonic acid (and possibly other fatty acids) with simple amino acids can be synthesised via the formation of the acyl-CoA thioesters, as was shown for N-arachidonoyl-glycine (NAGly) [67]. Interestingly, the arachidonic acid that conjugates with glycine appears to be a result of the hydrolysis of AEA [68]. An alternative pathway was proposed by Burstein et al. [69] who speculated that NAGly is produced by the oxidation of the ethanolamine in AEA, presumably through alcohol dehydrogenase. Evidence exists for both pathways [68].
Fatty amides and 2-acylglycerols are broken down by enzymatic hydrolysis. The primary NAE degrading enzyme is fatty acid amide hydrolase (FAAH, now also known as FAAH-1), localised on the endoplasmatic reticulum [70]. A second FAAH enzyme is found in humans located on cytoplasmic lipid droplets [70, 71]. Recently, a third NAE hydrolysing enzyme, N-acyl ethanolamine-hydrolysing acid amidase (NAAA) has been identified [72].
To reach their sites of catabolism within the cell, NEAs are bound to different proteins including fatty acid binding proteins 5 and 7, heat shock protein 70, albumin and fatty acid amide hydrolase-like AEA transporter protein [73, 74]. Intracellular trafficking of NAEs is also important to reach those receptors that are located intracellularly [55, 75]. Next to hydrolysis, NAEs are substrates for oxidative enzymes including cyclooxygenases (COXs), lipoxygenases (LOXs) and cytochrome P450 enzymes, yielding a range of prostaglandin-amides (prostamides), prostaglandin-glycerol esters and hydroperoxy-derivatives [5, 76, 77]. At least a number of these oxidation products show biological activity [76–78]. 2-AG is hydrolyzed via the enzyme monoacylglycerol lipase (MAGL), to a lesser extent by α/ß-hydrolase 12 (ABHD12) and α/ß-hydrolase 6 (ABHD6), and also by FAAH [79, 80]. Interestingly, AA in brain formed by hydrolysis of 2-AG via MAGL has been shown to serve as pool for pro-inflammatory eicosanoid synthesis, thus representing a connection between endocannabinoid and eicosanoid pathways [81]. 2-AG can also be oxygenated by COX-2 and LOX resulting in the formation of prostaglandin glycerol esters (PG-Gs) [76].
3.2 Cannabinoid and Related Receptors
According to the IUPHAR classification system the CB1 and CB2 receptors are the only bona fide cannabinoid receptors. They are phylogenetically restricted to the chordate branch of the animal kingdom [2]. Among other GPCRs, those structurally most closely related to CB1 and CB2 belong to the lysophospholipid receptors. These receptors for endocannabinoids or lysophospholipid-like molecules have evolved independently in different branches of the GPCR superfamily [1]. However, in terms of ligand binding characteristics the picture becomes more complicated. As mentioned before, endocannabinoids have a multitude of structural analogues. These compounds interact with different receptors and non-receptor targets. Several endocannabinoids per se, including anandamide, but also Δ9-THC and a number of synthetic CB1 or CB2 agonists and antagonists can activate or block different non-cannabinoid receptors with potencies that differ little from those with which they activate or block the “true” cannabinoid receptors [1]. According to nomenclature criteria of the NC-IUPHAR cannabinoid receptor subcommittee the TRPV1 channel might eventually come to be regarded as being either an “ionotropic cannabinoid CB3 receptor” or a dual TRPV1/CB3 receptor. In addition, some other receptors deserve further attention in this respect, namely, GPR18, GPR55, GPR119 and the peroxisome proliferator-activated receptors (PPARs) α and γ. Although these show little to no structural similarity to CB1 and CB2 they have been shown to respond to endocannabinoids, their endogenously present congeners and (or) plant-derived “phyto”-cannabinoids.
3.2.1 CB1 Receptors
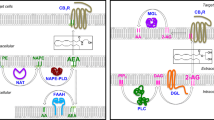
CB1 receptors are presynaptically located at central or peripheral nerve terminals and act as modulators of synaptic transmission by a process which has been called retrograde signalling {Wilson, 2002 #4848; Cachope, 2012 #3683; Vaughan, 2005 #4853}. Physiological stimulation of neurons induces the synthesis of endocannabinoids in the post-synaptic nerve terminal and this reduces synaptic inputs in a highly selective and restricted manner (Fig. 9.6).

Schematic representation of the mechanism of retrograde signalling by endocannabinoids at a synaptic cleft. Neuronal depolarization causes cleavage of membrane lipid precursors to induce de novo synthesis and release of endocannabinoids such as AEA, PEA, OEA and 2-AG into the synaptic cleft. These endocannabinoids activate cannabinoid CB1 receptors located on presynaptic terminals of neurons which reduces release of neurotransmitters (such as GABA or glutamate) onto the postsynaptic neuron. Endogenously released cannabinoids might also act via TRP ligand gated ion channels (e.g. TPRV1) and other GPCRs (e.g. GPR 119). Endocannabinoids are taken back up into neuronal and glial cells and then degraded by enzymes such as fatty acid amide hydrolase (FAAH) and MAG-lipase (MAGL)
The majority of CB1 receptors are coupled through Gi/o proteins. Their stimulation leads to inhibition of adenylate cyclase activity, effects on different Ca2+ and K+ channels, and stimulation of mitogen-activated protein (MAP) kinase. In some cases CB1 receptors signal through Gs proteins [1, 2, 80].
In contrast to what was originally assumed, the distribution of CB1 receptors is not limited to the CNS, and CB1 receptors are also found in the immune system, vascular endothelium, intestine, liver, skeletal muscles, peripheral nerve synapses and reproductive tissues. As a consequence of their localisation at the terminals of central and peripheral neurons where they mediate inhibition of neurotransmitter release, CB1 receptors are involved in a wide variety of biological processes [1] including learning and memory, anxiety, pain, eating behaviour, metabolism, reproduction and growth and development. As a result they have been associated with different disorders and diseases (Sect. 4). For example, their involvement in food intake regulation (Sect. 4.2) takes place at different levels, starting from receptors within the GI tract to the regulation of hedonic rewarding in the brain [55, 82–84]. Its presence in peripheral tissues also provides an explanation for the sustained effects of the CB1 inverse agonist rimonabant on body weight and the improvement of insulin resistance and blood lipids, in addition to its short-term appetite-decreasing effect. On Vagal afferents CB1 expression was found to be regulated by CCK [85] and high/low fat diets [86]. Remarkably, peripheral stimulation of CB1 receptors on Vagal afferents by anandamide was shown to reduce appetite, whereas central stimulation of CB1 receptors increased food intake [87]. In the brain, the CB1 is now regarded the most abundant G-protein coupled receptor [2]. A pioneering study on its distribution in brain was published in 1990 by Miles Herkenbaum et al. [88]. More recent reviews include the following references [89, 90]. As mentioned before, the central regulation of energy intake and metabolism is one of the major functions of the “classical” ECS. Within the brain, CB1 receptors have been linked to several both homeostatic and non-homeostatic regulation mechanisms, with endocannabinoids acting as modulators of orexigenic and anorexigenic neurotransmitters and neuropeptides by presynaptic regulation of their release. The brain ECS shows numerous anatomical and functional connections with other signalling pathways including dopaminergic, opioid and GABA-ergic systems involved in pleasure and reward, pain, anxiety, fear, etc. [55, 91–95].
3.2.2 CB2 Receptors
CB2 receptors are predominantly expressed on immune and haematopoietic cells, but functionally relevant expression has also been found in specific regions of the brain, other tissues and in various tumours. Like CB1 receptors they are coupled through Gi/o proteins, negatively to adenylate cyclase activity and positively to MAP kinase. Although several studies have suggested that CB2 activation is immunomodulatory and neuroprotective [96–98], some data remain inconclusive. This may be partly due to the fact that different components of the inflammatory cascade can be affected in a different direction [99]. Furthermore, discrepancies are caused by the use of different animal models, compounds and doses [100]. Disease-induced changes (usually increases) in CB2 receptor expression have been reported [101]. Furthermore, many synthetic CB2 receptor agonists have shown protective effects in a variety of preclinical disease models and pathological conditions (reviewed by ref. 101). Therefore, the application of selective CB2 agonists would be of interest for a number of disorders (Review: [28]). At the same time the wide abundance of CB2 receptors and the critical importance of retaining an adequate pro-inflammatory balance present challenges for their application as therapeutic targets [101]. Therefore, subtle and well-balanced approaches, including multiple targeted and/or localised therapies are likely to provide the best options [29].
3.2.3 Transient Receptor Potential (TRP) Cation Channels
Transient receptor potential (TRP) cation channels constitute a superfamily of receptors involved in the signal transduction of a wide range of stimuli, including effects elicited by endogenous lipids [2, 102–104]. Mammalian TRPs are subdivided into six protein families of which three are here considered of particular relevance because they bind endocannabinoids and related compounds and (or) phytocannabinoids. These are: the vanilloid receptor TRPs (TRPVs, in particular TRVP1), the melastatin or long TRPs (TRPMs, in particular TRPM8) and ankyrin transmembrane protein 1 (TRPA1).
The TRVP1 receptor is particularly known as the receptor for the vanilloid capsaicin present in red peppers. In addition it is perhaps the best established non-cannabinoid receptor for endocannabinoids, and for anandamide in particular [7, 95]. Several papers note the overlap between the ECS and what has been called “endovanilloid system” [95, 105–107]. Based on this it has been suggested to rename the TRVP1 receptor to “ionotropic cannabinoid CB3 receptor” or a dual TRPV1/CB3 receptor (see also Sect. 3.2). N-arachidonoyl-dopamine (NADA) was the first fatty acid amide shown to act as endogenous ligand of TRVP1 receptors [65]. Meanwhile several other N-acyl amides have also been demonstrated to activate TRPV1 [51]. TRVP1 is predominantly expressed in sensory neurons but also on non-neuronal cells including epithelial, endothelial and smooth muscle cells as well as in lymphocytes, hepatocytes and pancreatic cells [2, 5, 108]. Historically, TRPV1 has been considered a pro-inflammatory receptor in several conditions, including neuropathic pain, joint inflammation and inflammatory bowel disease. A number of TRVP1 antagonists have been developed as potential drugs against different forms of pain, but so far results in the clinic were not successful [108]. Recent evidence also demonstrates paradoxical, protective functions of TRVP1 in vivo [109]. The receptor also plays a role in energy metabolism and weight management as recently reviewed by Ahern [102]. For example, there is long-standing evidence that dietary consumption of chilli peppers can affect body weight. Treatment with capsaicin, or related “vanilloid” compounds, reduces weight gain and adiposity in animals consuming moderate to high-fat diets. An interesting finding was that the endogenous endocannabinoid congener N-arachidonoyl-serotonin (AA-5-HT) displays dual activity as both FAAH inhibitor and TRPV1 antagonist. The compound has shown marked effects against both acute and chronic peripheral pain in rodent models [110, 111]. Previous studies from our lab showed that this conjugate is particularly present in the gut, but so far its biological role has not been established [57]. In addition to TRPV1, other members of this family, including TRPV2-4 have been associated with, in particular effects of phytocannabinoids and (or) Cannabis extracts [2, 5, 112].
The TRPM8 receptor is involved in the detection of sensations such as cold. Activators include eucalyptol, menthol and icilin [113]. It is considered a therapeutic target for cold hypersensitivity and neuropathic pain [108]. Its expression was also found to be important for the survival of androgen receptor-dependent prostate cancer cells [5]. Both anandamide and N-arachidonoyl dopamine, but not 2-AG, were shown to antagonise the stimulatory effect of menthol and icilin on TRPM8 [114]. In addition, several phytocannabinoids show activity on TRPM8 [112, 114].
The TRPA1 receptor is receiving increasing attention as a key regulator of neuropeptide release, neurogenic inflammation and pain. See [108, 115] for reviews. TRPA1 was found to be activated by CBD [114]. Another phytocannabinoid, cannabichromene can also act as TRPA1 agonist. The receptor was shown to be involved in the inhibition of nitric oxide production in macrophages and the amelioration of murine colitis by cannabichromene [116].
3.2.4 GPR119
GPR119 ([117] for a recent review) has been described as a class A (rhodopsin-type) orphan GPCR but has no close primary sequence relative in the human genome. Two of its endogenous ligands discovered so far are the fatty acid amides oleoylethanolamide (OEA) and N-oleoyldopamine (OLDA). Furthermore the receptor can be activated, albeit with less potency, by PEA, EAE and linoleylethanolamine (LEA) [2, 118, 119]. As none of these compounds are ligands for CB1 or CB2 receptors, GPR119 is not considered a cannabinoid receptor per se [2]. Recently, GPR119 has also been found to respond to 2-oleoyl-glycerol, a compound formed out of common dietary triglycerides (described in Sect. 2.3). Following its de-orphanization in 2006 by Overton et al. [120] and the demonstration that small molecule agonists are able to reduce body weight gain in rodents, GPR119 has attracted considerable attention. The receptor is Gαs-protein coupled and predominantly expressed in pancreatic islets and gastrointestinal tract in humans and rodents. GPR119 agonists were found to increase intracellular cAMP, which in turn leads to increased GLP-1 secretion from entero-endocrine cells. Following the synthesis of the first ligands, including PSN632408 and AR-231,453 several pharmaceutical companies became active in developing GPR119 agonists. Many of these compounds have shown interesting activities in animal models of type 2 diabetes and obesity, including a reduction of blood glucose without causing hypoglycaemia, a reduction of food intake and body weight, and reduced diabetes progression. Presently, a number of GPR119 agonists are in advanced stages of development [121].
3.2.5 GPR 55
The discovery of the orphan GPCR GPR55 was first described in 2007 [122]. The receptor was shown to bind some CB1 and CB2 ligands. Therefore, it has been considered a “novel” or “third” cannabinoid receptor for some time, but this viewpoint has been abandoned. Structurally the receptor has no significant sequence similarity with the CB receptors, in particular not in the areas responsible for ligand binding [2]. GPR55 is expressed in the gut and found in cells of the immune system, including microglia in brain as well as in endothelial cells [123]. A recent study suggested that GPR55 regulates CB2 function in human neutrophils [124]. Following the report of Oka et al. [125] it is now assumed that its endogenous ligand is lysophosphatidylinositol (LPI). It was suggested by Ross [126] that LPI and GPR55 might play a role in driving cancer cell proliferation and migration. The phytocannabinoid CBD shows antagonist activity towards GPR55, which may of therapeutic relevance [127].
3.2.6 GPR18
The GPR18 gene was first cloned in 1997 [128] and at that time found to be highly expressed in human testis and spleen. In addition, its presence was shown in thymus, peripheral white blood cells and in the small intestine, whereas in many other tissues and organs it appeared to be absent. McHugh et al. [129] demonstrated that NaGly (N-arachidonoylglycine, see Sect. 3.1) serves as an endogenous ligand. The same group also reported that two cannabinoid agonists, AEA and THC, are full agonists at GPR18, whereas CBD displays low efficacy as agonist [130]. Considering its location on microglia cells [131] and on peripheral macrophages, GPR18 and its endogenous ligand(s) are receiving increasing attention in relation to inflammation.
3.2.7 Peroxisome Proliferator-Activated Receptors (PPARs)
PPARs can be activated by some non-cannabinoid NAEs including OEA and PEA. The same has been shown for some 2-AG derivatives of the COX/LOX/CYPP450 pathways, and to a lesser extent also for AEA and 2-AG itself. PPARs are ligand-activated transcription factors that play critical roles in very different biological pathways such as lipid, protein, glycerol, urea, glucose, glycogen, and lipoprotein metabolism, adipogenesis, trophoblast differentiation and cell migration. For recent reviews see for example [132, 133]. Their best known agonists are various fatty acids and their derivatives. Therefore, PPARs are commonly regarded as general—not very selective—lipid sensors monitoring local metabolic changes. The PPAR family consists of PPARα, PPARβ and PPARγ. The three PPAR iso-types are similar in homology, but show their own distribution pattern. In humans PPARα is localised in areas of high fatty acid catabolism (kidneys, liver, heart, brown adipose tissue and intestines). PPARγ is found as two isoforms: PPARγ1 (predominantly present in gut, brain, vascular cells and immune cells) and PPARγ2 (mainly in adipose tissue). PPARβ/δ has been found in many tissues and is particularly highly active in skeletal muscle, smooth muscle and skin [132].
The role of the PPARα receptor as a pivotal switch in different inflammatory and pain signalling pathways in the CNS and periphery is widely acknowledged [132, 134, 135]. Two well-known N-acylamides that are linked to this PPAR are PEA and OEA. For PEA (see also Sect. 4.3.2) it is assumed that its anti-inflammatory activity can largely be assigned to an agonist activity on PPARα [135–137]. PPARα is also playing a pivotal role in the satiety-inducing effects of OEA [138]. This NAE is formed form oleic acid in the epithelium of the proximal small intestine. PPARγ serves as the molecular target for the thiazolidinediones, an important class of anti-diabetic drugs. Its major natural ligands and activators are PUFAs and fatty acid-derived molecules. The beneficial action of PPARγ has typically been attributed to increased insulin sensitivity and reduced inflammation. Agonism of PPARγ is increasingly considered an important property of the phytocannabinoid CBD (Sect. 2.1). PPARγ and CBD are also receiving attention in relation to CNS diseases like Alzheimers’ disease because of the role of PPARγ in stimulating microglial function [139, 140].
3.3 Interactions of Endocannabinoids with Non-receptor Targets
Several studies suggest that the biological activities of at least some of the endocannabinoids and their congeners are not exclusively mediated through GPCRs or nuclear receptors. An example comes from the anti-inflammatory effects of N-docosahexaenoylethanolamine (DHEA, Fig. 9.5), the ethanolamine conjugate of DHA (docosahexaenoic; 22:6n-3). Its concentration in animal tissues and human plasma increases when diets rich in fish or krill oil are consumed. Comparing a series of NAEs from n-3 and n-6 LC-PUFAs, we found DHEA to be the most potent anti-inflammatory compound in LPS-stimulated RAW264.7 macrophages [141]. Later studies suggested that anti-inflammatory effects of DHEA are at least partly independent from CB1, CB2 or PPARɣ receptors and probably take place via inhibition of eicosanoids produced through COX-2 [56]. Interestingly, DHEA was also reported to inhibit growth of prostate and breast cancer cell lines which was at least partly independent from CB1 or CB2 interaction [142, 143]. Similarly, DHEA was shown to stimulate neurite growth, synaptogenesis and glutamatergic synaptic activity in developing hippocampal neurons via (at least) cannabinoid receptor-independent mechanisms [144]. Another example is N-arachidonoyl dopamine (NADA). Like DHEA, NADA was found to be potent inhibitor of PGE2 synthesis in lipopolysaccharide (LPS) stimulated primary glia cells [145, 146].
4 Endocannabinoids and Targets in Disease
4.1 General Aspects, Targets and Examples
The broad involvement of the endocannabinoidome in various biological processes and its many connections with other systems in terms of ligands, receptors and metabolic pathways explains why it is has been associated with so many disorders and diseases. However, it should be noted that associations with pathologies like those mentioned in Table 9.1 do not imply that suitable targets for prevention or intervention are at an easy reach. On the contrary, its wide abundance and high degree of pleiotropy present serious challenges to develop efficacious and specific drugs. It has also become clear that initial strategies to modulate the ECS have probably been too narrow and expectations too high. Two well-known examples in this respect are the experiences thus far with CB1 inverse agonists (Sect. 4.2) and FAAH blockers (Sect. 4.3). Furthermore, it is also clear that changes in the expression of certain receptors or ligands are often the result of other (patho-)physiological processes instead of being part of a modifiable cause of a disease. As can be seen from the list of disease areas of interest (Table 9.1) many of these are of a chronic and multifactorial character. It is increasingly acknowledged that such disorders are often better managed by multiple target strategies, instead of a “one disease–one target” approach. This involves the use of promiscuous drugs or targeted drug (of drug–food) combinations [147]. These developments stimulated by the evolution of “omics” technologies, system biology and bioinformatics and the endocannabinoidome lends itself well for such an approach [148]. Table 9.1 lists a non-exhaustive overview of disease areas of interest. In the next sections, two of these are further elaborated viz weight management (Sect. 4.2) and inflammation (Sect. 4.3). For other field readers are referred to the literature.
4.2 The Endocannabinoidome in Weight Management
The modulation of food intake and energy metabolism is generally considered one of the most pivotal roles of the ECS. It has also been the most intensively studied topic in this field, in particular until 2008 when the withdrawal of rimonabant caused a dramatic change. The ECS modulates food intake and energy metabolism at different levels, starting from CB1 receptors within the GI tract to the regulation of hedonic rewarding of foods in the brain [82–84, 190, 191]. From an evolutionary perspective it is thought that one of its main functions is as a pleiotropic regulator of energy uptake and storage and of non-homeostatic eating behaviour [192, 193]. In the past these mechanisms were biologically advantageous in order to survive periods of food shortage [194]. The discovery of the high abundance of CB1 in brain, and the observation that CB1 antagonists and reverse agonists induce a reduction of appetite and food-intake in animals fuelled an enormous activity of research in academia and industry, resulting in the market introduction of rimonabant 2006. Expectations, therapeutic and financial, were very high. The failure of rimonabant because of depression-related side-effects [13] shocked the research community and the pharmaceutical industry. By the end of 2008 at least nine companies terminated active development projects of CB1 blockers. Next to rimonabant, which has been on the market in Europe but not in the USA, several related compounds were in advanced stages of development, including taranabant (Merck), surinabant (Sanofi) and CP-945,598 (otanabant, Pfizer). In the meantime it has become clear that CB1 receptors are also abundantly present outside the CNS [12, 191]. In fact, it is now assumed that the central effects of rimonabant are responsible for the short-term reduction of food-intake, whereas the more sustained effects on body weight and the improvement of insulin resistance and blood lipids are largely due to its peripheral actions. In the gut, CB receptors show a specific distribution, being largely distributed in the enteric nervous system (ENS) [178]. Both CB1 and CB2 receptors are found on enteric neurons, nerve fibres and nerve terminals in the ENS. The CB1 receptor is found on nerve fibres throughout the wall of the gut, but with the highest density in the two ganglionated plexuses, the myenteric and submucosal plexus, of the ENS. CB1 expression on Vagal afferents was found to be regulated by CCK [85] and high/low fat diets [86]. Stimulation of central CB1 receptors, for example by anandamide has been shown to increase food-intake. Remarkably, stimulation of CB1 receptors on Vagal afferents seems to do the opposite [87].
Notwithstanding the failure of rimonabant and other CB1 blockers/inverse agonists, CB1 receptors remain of interest as a pharmacological target. The presence of CB1 receptors outside the CNS offers possibilities for treatment of type 2 diabetes and other complications of the metabolic syndrome. To improve tissue specific activity and reduce CNS side-effects so-called peripherally restricted CB1 antagonists are under investigation [12, 28, 121, 191, 195]. Furthermore, the use of CB1 neutral antagonists or partial agonists as opposed to inverse agonists such as rimonabant has been proposed as a strategy [191].
As described in Sect. 2.1 there exist also (at least) one natural weak CB1 antagonist, THCV from Cannabis which might offer possibilities in this respect [17, 41, 42].
In addition to CB receptors, related receptors may offer interesting targets in weight management, including TRVP1 (Sect. 3.2.3) and GPR119 (Sect. 3.2.4).
Although not discussed in further detail are the possibilities to target the ECS in order to increase appetite or food-intake in general. The use of Cannabis preparations in AIDS and cancer patients for this purpose has already been introduced in Sect. 2.1. The ECS is also receiving interest in relation to eating disorders like anorexia and bulimia nervosa [161, 196, 197].
4.3 Inflammatory Processes
Several receptors which are modulated by endocannabinoids or their structural analogues are involved in the regulation of inflammation, pain and immune-functions in a broad sense [100]. Of particular interest are CB2 (Sect. 3.2.2), TRVP1, TRPA1 and other TRP cation channels (Sect. 3.2.3), GPR18 (Sect. 3.2.6) and PPARs (Sect. 3.2.7), and this list is likely to increase. Furthermore, a number of endocannabinoids per se (Anandamide, 2-AG) and related compounds (PEA, SEA, OEA, DHEA, etc.) have shown anti-inflammatory and (or) immune modulating properties. Finally, the endocannabinoidome is deeply intertwined with other important lipid-based signalling systems including those regulated by COX and LOX. On the one hand, this broad involvement offers several potential targets for intervention. On the other hand, this complexity provides challenges in terms of specificity and side-effects. Some examples will be highlighted in this Section.
4.3.1 Modulators of Endocannabinoid Turnover
Inhibition of enzymes involved in the synthesis or breakdown of endocannabinoids, in particular DAGL, MAGL, FAAH or NAAA (N-acylethanolamine acid amidase) has been considered a manner to modulate inflammation and (or) pain. Diacylglycerol lipases (DAGLα and DAGLβ) are involved in the synthesis of 2-AG. Inhibition of DAGLβ has been found to lower 2-AG, as well as AA and eicosanoids, in mouse peritoneal macrophages in a manner that was distinct and complementary to disruption of cytosolic phospholipase-A2 [198]. Mono-acyl glycerol lipase (MAGL) catalyses the hydrolysis of 2-AG to arachidonic acids (AA). Inhibition of peripheral MAGL in rats using the selective MAGL inhibitor JZL184 was found suppressed LPS-induced circulating cytokines which in turn was suggested to modulate central cytokine expression [199]. In brain, AA formed by hydrolysis of 2-AG has been shown to serve as pool for pro-inflammatory eicosanoid synthesis, thus representing another crossroads between endocannabinoid and eicosanoid pathways [81]. MAGL-disrupted mice displayed neuroprotection in a model for Parkinson’s disease but showed no haemorrhaging in the gut as seen with COX inhibitors [200]. Inhibition of Fatty Acid Amide Hydrolase (FAAH) aiming to increase fatty amide levels has also been considered as intervention strategy in inflammation and (or) pain. A number of animal studies, for example with the inhibitor URB597, indeed showed reduction of inflammatory pain or modulation pro-inflammatory gene induction, although results were not always unambiguous [201, 202]. It has been suggested that inactivation of FAAH can modulate 2-AG tissue levels as well, either up or down, depending on the location [203]. Studies in human volunteers with FAAH inhibitors confirmed increased NAE levels, including that of AEA, OEA and PEA [204]. However, in a recent phase II clinical trial in patients with osteoarthritic knee pain the FAAH inhibitor PF-04457845 failed to show any effect [205, 206]. As FAAH activity was strongly inhibited and plasma NAE concentrations consistently elevated, it was suggested that alternative targets and pathways for breakdown might have counteracted the potentially beneficial effects of elevated anandamide levels on pain and inflammation [207]. Inhibition of NAAA provides an alternative approach to increase levels of for example PEA and OEA. Recently, the selective NAAA inhibitor ARN077 has been found to inhibit hyperalgesia and allodynia caused by inflammation or nerve damage [208]. Interestingly, the antinociceptive effects of ARN077 were prevented by the selective PPAR-α antagonist GW6471 and did not occur in PPAR-α knockout mice.
4.3.2 Endocannabinoid Congeners as Potential Anti-inflammatory Compounds
Several individual endocannabinoids, fatty amides and phytocannabinoids have been demonstrated to possess anti-inflammatory properties [100]. The n-3 LC-PUFA derived N-docosahexaenoylethanolamine (DHEA, Fig. 9.5) has already been described in Sect. 3.3. The same holds true for the Cannabis-derived compound CBD (Sect. 2.1). An interesting compound which is receiving increasingly attention is N-Palmitoylethanolamide (PEA, Fig. 9.5), an endogenous NAE originating from palmitic acid (C16:0), the most common saturated fatty acid found in animals [209]. Earliest reports on its anti-inflammatory properties date back to 1957. PEA shows a broad diversity of receptor affinities, including interactions with PPARα, GPR55 and TRVP1, as well as indirect activity via an “entourage” effect [137, 210]. The latter refers to a mechanism in which PEA reduces the enzymatic breakdown of AEA through competition for FAAH, resulting in higher AEA concentrations [211, 212]. The compound is presently receiving attention as potential drug or nutraceutical against chronic pain, (neuro-)inflammation and degenerative diseases of the central nervous system [137, 167, 209, 213, 214]. Increasing evidence indicates that non-neuronal cells within the CNS are crucially involved in mediating the effects of PEA [137, 215, 216]. These non-neuronal cells regulate inflammatory processes in the CNS and are key players in the communication between the immune system and the CNS during neurodegenerative disorders and in neuropathic pain. The C18 homologue of PEA, N-stearoyl ethanolamine (SEA) has also been associated with anti-inflammatory effects but this compound has been far less investigated [217].
5 Conclusions and Perspectives
More than two decades of research have changed our early view of the ECS. Initial expectations on the possibilities to develop new drug classes based on its key molecular targets have proven to be too high. It is now obvious that the “prototypical” ECS is deeply intertwined with other important signalling systems. Endocannabinoids have numerous bioactive congeners and metabolites, which often show “promiscuous” behaviour towards their receptors and other targets. This so-called endocannabinoidome is modulated by various endogenous (e.g. energy status, inflammation) and environmental factors in a time- and tissue-specific manner. The complexity and dynamics of the endocannabinoidome presents technical challenges and its understanding and modulation demands for a systems biology approach. At the same time the endocannabinoidome still holds many promises for both “food” and “pharmaceutical” applications as it is crucially involved in many disorders. Chronic diseases often involve tissue degeneration and remodelling, inflammation and pain, and are orchestrated by different interacting metabolic processes in which the “expanded” ECS is centrally involved.
Significant progress in their prevention and modulation is likely to come from a paradigm shift as it is currently taking place in the discovery and development process of drugs and nutritional products. These involve more subtle multiple-target strategies instead of a classical one disease–one target–one drug approach.
Abbreviations
- (LC-)PUFA:
-
(Long chain-)polyunsaturated fatty acid
- 2-AG:
-
2-Arachidonoylglycerol
- AA:
-
Arachidonic acid
- AEA:
-
N-arachidonoylethanolamine (anandamide)
- CB (receptor):
-
Cannabinoid (receptor)
- CBD:
-
Cannabidiol
- COX:
-
Cyclooxygenase
- DAGL:
-
Diacylglycerol lipase
- DHA:
-
Docosahexaenoic acid (22:6n-3)
- DHEA:
-
N-docosahexaenoylethanolamine
- ECS:
-
Endocannabinoid system
- FAAH:
-
Fatty acid amide hydrolase
- GPCR:
-
G-protein coupled receptor
- LOX:
-
Lipooxygenase
- MAGL:
-
Monoacylglycerol lipase
- NADA:
-
N-arachidonoyldopamine
- NAEs:
-
N-acylethanolamines
- OEA:
-
N-oleoylethanolamine
- PEA:
-
N-palmitoylethanolamine
- PPAR:
-
Peroxisome proliferator-activated receptor
- THCV:
-
Δ9-Tetrahydrocannabivarin
- TRVP1:
-
Transient receptor potential channel type V1
- Δ9-THC:
-
Δ9-Tetrahydrocannabinol
References
Pertwee RG, Howlett AC, Abood M, Barth F, Bonner TI, Cabral G et al (2011) IUPHAR/BPS guide to pharmacology. Cannabinoid receptors. http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=13. Accessed 7 May 2014
Pertwee RG, Howlett AC, Abood ME, Alexander SPH, di Marzo V, Elphick MR et al (2010) International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 62:588–631
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Brown I, Cascio MG, Rotondo D, Pertwee RG, Heys SD, Wahle KWJ (2013) Cannabinoids and omega-3/6 endocannabinoids as cell death and anticancer modulators. Prog Lipid Res 52:80–109
Alexander SPH, Kendall DA (2007) The complications of promiscuity: endocannabinoid action and metabolism. Br J Pharmacol 152:602–623
Di Marzo V, Bisogno T, de Petrocellis L (2007) Endocannabinoids and related compounds: walking back and forth between plant natural products and animal physiology. Chem Biol 14:741–756
Silvestri C, Martella A, Poloso NJ, Piscitelli F, Capasso R, Izzo A et al (2013) Anandamide-derived prostamide F2α negatively regulates adipogenesis. J Biol Chem 288:23307–23321
Woodward DF, Jones RL, Narumiya S (2011) International union of basic and clinical pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev 63:471–538
Woodward DF, Liang Y, Krauss AHP (2007) Prostamides (prostaglandin-ethanolamides) and their pharmacology. Br J Pharmacol 153:410–419
Woodward DF, Wang JW, Poloso NJ (2013) Recent progress in prostaglandin F2α ethanolamide (prostamide F2α) research and therapeutics. Pharmacol Rev 65:1135–1147
Silvestri C, di Marzo V (2013) The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab 17:475–490
Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A (2007) Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370:1706–1713
Li H-L (1974) The origin and use of cannabis in eastern asia linguistic-cultural implications. Econ Bot 28:293–301
Robson P (2005) Human studies of cannabinoids and medicinal cannabis cannabinoids. In: Pertwee RG (ed) Handbook of experimental pharmacology, vol 168. Springer, Berlin, pp 719–756
Flores-Sanchez IJ, Verpoorte R (2008) Secondary metabolism in cannabis. Phytochem Rev 7:615–639
Izzo AA, Borrelli F, Capasso R, di Marzo V, Mechoulam R (2009) Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci 30:515–527
Fischedick JT, Hazekamp A, Erkelens T, Choi YH, Verpoorte R (2010) Metabolic fingerprinting of Cannabis sativa L., cannabinoids and terpenoids for chemotaxonomic and drug standardization purposes. Phytochemistry 71:2058–2073
Van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR et al (2011) The draft genome and transcriptome of Cannabis sativa. Genome Biol 12:R102
Verhoeckx KCM, Korthout HAAJ, van Meeteren-Kreikamp AP, Ehlert KA, Wang M, van der Greef J et al (2006) Unheated Cannabis sativa extracts and its major compound THC-acid have potential immuno-modulating properties not mediated by CB1 and CB2 receptor coupled pathways. Int Immunopharmacol 6:656–665
Eichler M, Spinedi L, Unfer-Grauwiler S, Bodmer M, Surber C, Luedi M et al (2012) Heat exposure of Cannabis sativa extracts affects the pharmacokinetic and metabolic profile in healthy male subjects. Planta Med 78:686–691
Rock EM, Kopstick RL, Limebeer CL, Parker LA (2013) Tetrahydrocannabinolic acid reduces nausea-induced conditioned gaping in rats and vomiting in Suncus murinus. Br J Pharmacol 170:641–648
Grotenhermen F (2003) Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 42:327–360
Grotenhermen J (2004) Clinical pharmacodynamics of cannabinoids. J Cannabis Ther 2004(1):29–78
Robson PJ (2014) Therapeutic potential of cannabinoid medicines. Drug Test Anal 6:24–30
Stott CG, White L, Wright S, Wilbraham D, Guy GW (2013) A phase i study to assess the single and multiple dose pharmacokinetics of THC/CBD oromucosal spray. Eur J Clin Pharmacol 69:1135–1147
Hazekamp A, Heerdink ER (2013) The prevalence and incidence of medicinal cannabis on prescription in The Netherlands. Eur J Clin Pharmacol 69:1575–1580
Pertwee RG (2012) Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos Trans R Soc Lond B Biol Sci 367:3353–3363
Starowicz K, di Marzo V (2013) Non-psychotropic analgesic drugs from the endocannabinoid system: “magic bullet” or “multiple-target” strategies? Eur J Pharmacol 716:41–53
Esposito G, de Filippis D, Cirillo C, Iuvone T, Capoccia E, Scuderi C et al (2013) Cannabidiol in inflammatory bowel diseases: a brief overview. Phytother Res 27:633–636
Kozela E, Lev N, Kaushansky N, Eilam R, Rimmerman N, Levy R et al (2011) Cannabidiol inhibits pathogenic T cells, decreases spinal microglial activation and ameliorates multiple sclerosis-like disease in C57BL/6 mice. Br J Pharmacol 163:1507–1519
De Petrocellis L, Ligresti A, Schiano Moriello A, Iappelli M, Verde R, Stott CG et al (2013) Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: pro-apoptotic effects and underlying mechanisms. Br J Pharmacol 168:79–102
Massi P, Solinas M, Cinquina V, Parolaro D (2013) Cannabidiol as potential anticancer drug. Br J Clin Pharmacol 75:303–312
Campos AC, Moreira FA, Gomes FV, del Bel EA, Guimarães FS (2012) Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Philos Trans R Soc Lond B Biol Sci 367:3364–3378
Kowal MA, Hazekamp A, Colzato LS, van Steenbergen H, Hommel B (2013) Modulation of cognitive and emotional processing by cannabidiol: the role of the anterior cingulate cortex. Front Hum Neurosci 7(Art 147):1–4
Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C et al (2012) Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2:e94
Bhattacharyya S, Morrison PD, Fusar-Poli P, Martin-Santos R, Borgwardt S, Winton-Brown T et al (2009) Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology 35:764–774
Demirakca T, Sartorius A, Ende G, Meyer N, Welzel H, Skopp G et al (2011) Diminished gray matter in the hippocampus of cannabis users: possible protective effects of cannabidiol. Drug Alcohol Depend 114:242–245
Klein C, Karanges E, Spiro A, Wong A, Spencer J, Huynh T et al (2011) Cannabidiol potentiates Δ9-tetrahydrocannabinol (THC) behavioural effects and alters THC pharmacokinetics during acute and chronic treatment in adolescent rats. Psychopharmacology (Berl) 218:443–457
Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA et al (2005) Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol 146:917–926
Pertwee RG (2008) The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta-9-tetrahydrocannabinol, cannabidiol and delta-9-tetrahydrocannabivarin. Br J Pharmacol 153:199–215
Wargent ET, Zaibi MS, Silvestri C, Hislop DC, Stocker CJ, Stott CG et al (2013) The cannabinoid Δ9-tetrahydrocannabivarin (THCV) ameliorates insulin sensitivity in two mouse models of obesity. Nutr Diabetes 3:e68
Gertsch J, Pertwee RG, di Marzo V (2010) Phytocannabinoids beyond the Cannabis plant: do they exist? Br J Pharmacol 160:523–529
Gertsch J (2008) Immunomodulatory lipids in plants: plant fatty acid amides and the human endocannabinoid system. Planta Med 74:638–650
Coulon D, Faure L, Salmon M, Wattelet V, Bessoule J-J (2012) N-acylethanolamines and related compounds: aspects of metabolism and functions. Plant Sci 184:129–140
Kim S-C, Chapman KD, Blancaflor EB (2010) Fatty acid amide lipid mediators in plants. Plant Sci 178:411–419
Ligresti A, Villano R, Allarà M, Ujváry I, di Marzo V (2012) Kavalactones and the endocannabinoid system: the plant-derived yangonin is a novel CB 1 receptor ligand. Pharmacol Res 66:163–169
Gertsch J, Leonti M, Raduner S, Racz I, Chen J-Z, Xie X-Q et al (2008) Beta-caryophyllene is a dietary cannabinoid. Proc Natl Acad Sci U S A 105:9099–9104
Leonti M, Casu L, Raduner S, Cottiglia F, Floris C, Altmann KH et al (2010) Falcarinol is a covalent cannabinoid CB1 receptor antagonist and induces pro-allergic effects in skin. Biochem Pharmacol 79:1815–1826
Fuchs A, Rempel V, Müller CE (2013) The natural product magnolol as a lead structure for the development of potent cannabinoid receptor agonists. PLoS One 8:e77739
Bradshaw HB, Raboune S, Hollis JL (2013) Opportunistic activation of TRP receptors by endogenous lipids: exploiting lipidomics to understand TRP receptor cellular communication. Life Sci 92:404–409
Farrell EK, Merkler DJ (2008) Biosynthesis, degradation and pharmacological importance of the fatty acid amides. Drug Discov Today 13:558–568
Balvers MJ, Verhoeckx KM, Bijlsma S, Rubingh C, Meijerink J, Wortelboer H et al (2012) Fish oil and inflammatory status alter the n-3 to n-6 balance of the endocannabinoid and oxylipin metabolomes in mouse plasma and tissues. Metabolomics 8:1130–1147
Hansen HS (2013) Effect of diet on tissue levels of palmitoylethanolamide. CNS Neurol Disord Drug Targets 12:17–25
Maccarrone M, Gasperi V, Catani MV, Diep TA, Dainese E, Hansen HS et al (2010) The endocannabinoid system and its relevance for nutrition. Annu Rev Nutr 30:423–440
Meijerink J, Balvers M, Witkamp R (2013) N-acyl amines of docosahexaenoic acid and other n-3 polyunsatured fatty acids: from fishy endocannabinoids to potential leads. Br J Pharmacol 169:772–783
Verhoeckx KCM, Voortman T, Balvers MGJ, Hendriks HFJ, M wortelboer H, Witkamp RF (2011) Presence, formation and putative biological activities of N-acyl serotonins, a novel class of fatty-acid derived mediators, in the intestinal tract. Biochim Biophys Acta 1811:578–586
Ben-Shabat S, Fride E, Sheskin T, Tamiri T, Rhee MH, Vogel Z et al (1998) An entourage effect: inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur J Pharmacol 353:23–31
Hansen KB, Rosenkilde MM, Knop FK, Wellner N, Diep TA, Rehfeld JF et al (2011) 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J Clin Endocrinol Metab 96:E1409–E1417
Tortoriello G, Rhodes BP, Takacs SM, Stuart JM, Basnet A, Raboune S et al (2013) Targeted lipidomics in Drosophila melanogaster identifies novel 2-monoacylglycerols and N-acyl amides. PLoS One 8:e67865
Wellner N, Diep TA, Janfelt C, Hansen HS (2013) N-acylation of phosphatidylethanolamine and its biological functions in mammals. Biochim Biophys Acta 1831:652–662
Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D (1996) Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16:3934–3942
Tsuboi K, Ikematsu N, Uyama T, Deutsch DG, Tokumura A, Ueda N (2013) Biosynthetic pathways of bioactive N-acylethanolamines in brain. CNS Neurol Disord Drug Targets 12:7–16
Reisenberg M, Singh PK, Williams G, Doherty P (2012) The diacylglycerol lipases: structure, regulation and roles in and beyond endocannabinoid signalling. Philos Trans R Soc Lond B Biol Sci 367:3264–3275
Huang SM, Bisogno T, Trevisani M, Al-Hayani A, de Petrocellis L, Fezza F et al (2002) An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A 99:8400–8405
Hu SSJ, Bradshaw HB, Benton VM, Chen JSC, Huang SM, Minassi A et al (2009) The biosynthesis of N-arachidonoyl dopamine (NADA), a putative endocannabinoid and endovanilloid, via conjugation of arachidonic acid with dopamine. Prostaglandins Leukot Essent Fatty Acids 81:291–301
Huang SM, Bisogno T, Petros TJ, Chang SY, Zavitsanos PA, Zipkin RE et al (2001) Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem 276:42639–42644
Bradshaw HB, Rimmerman N, Hu SSJ, Burstein S, Walker JM (2009) Novel endogenous N-acyl glycines: identification and characterization. In: Gerald L (ed) Vitamins and hormones, vol 81, Chapter 8. Academic, Burlington, pp 191–205
Burstein SH, Rossetti RG, Yagen B, Zurier RB (2000) Oxidative metabolism of anandamide. Prostaglandins Other Lipid Mediat 61:29–41
Bisogno T (2008) Endogenous cannabinoids: structure and metabolism. J Neuroendocrinol 20:1–9
Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF (2006) A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem 281:36569–36578
Ueda N, Tsuboi K, Uyama T (2010) Enzymological studies on the biosynthesis of N-acylethanolamines. Biochim Biophys Acta 1801:1274–1285
Fowler CJ (2012) Anandamide uptake explained? Trends Pharmacol Sci 33:181–185
Kaczocha M, Glaser ST, Deutsch DG (2009) Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci U S A 106:6375–6380
Kaczocha M, Vivieca S, Sun J, Glaser ST, Deutsch DG (2012) Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem 287:3415–3424
Rouzer CA, Marnett LJ (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev 111:5899–5921
Vandevoorde S, Lambert DM (2007) The multiple pathways of endocannabinoid metabolism: a zoom out. Chem Biodivers 4:1858–1881
Yang R, Fredman G, Krishnamoorthy S, Agrawal N, Irimia D, Piomelli D et al (2011) Decoding functional metabolomics with docosahexaenoyl ethanolamide (DHEA) identifies novel bioactive signals. J Biol Chem 286:31532–31541
Blankman JL, Simon GM, Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356
Fonseca BM, Costa MA, Almada M, Correia-da-Silva G, Teixeira NA (2013) Endogenous cannabinoids revisited: a biochemistry perspective. Prostaglandins Other Lipid Mediat 102–103:13–30
Cao Z, Mulvihill MM, Mukhopadhyay P, Xu H, Erdélyi K, Hao E et al (2013) Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology 144:808–817, e15
Cota D, Tschop MH, Horvath TL, Levine AS (2006) Cannabinoids, opioids and eating behavior: the molecular face of hedonism? Brain Res Rev 51:85–107
Cota D (2008) The role of the endocannabinoid system in the regulation of hypothalamic-pituitary-adrenal axis activity. J Neurendocrinol 20:35–38
Quarta C, Mazza R, Obici S, Pasquali R, Pagotto U (2011) Energy balance regulation by endocannabinoids at central and peripheral levels. Trends Mol Med 17:518–526
Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ (2010) Expression of cannabinoid CB1 receptors by vagal afferent neurons: kinetics and role in influencing neurochemical phenotype. Am J Physiol Gastrointest Liver Physiol 299:G63–G69
Nefti W, Chaumontet C, Fromentin G, Tome D, Darcel N (2009) A high-fat diet attenuates the central response to within-meal satiation signals and modifies the receptor expression of vagal afferents in mice. Am J Physiol Regul Integr Comp Physiol 296:R1681–R1686
Hansen HS, Diep TA (2009) N-acylethanolamines, anandamide and food intake. Biochem Pharmacol 78:553–560
Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Coste BR et al (1990) Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A 87:1932–1936
Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066
Katona I, Freund TF (2012) Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 35:529–558
Fattore L, Melis M, Fadda P, Pistis M, Fratta W (2010) The endocannabinoid system and nondrug rewarding behaviours. Exp Neurol 224:23–36
Melis M, Pistis M (2012) Hub and switches: endocannabinoid signalling in midbrain dopamine neurons. Philos Trans R Soc Lond B Biol Sci 367:3276–3285
Solinas M, Goldberg SR, Piomelli D (2008) The endocannabinoid system in brain reward processes. Br J Pharmacol 154:369–383
Leishman E, Kokesh KJ, Bradshaw HB (2013) Lipids and addiction: how sex steroids, prostaglandins, and cannabinoids interact with drugs of abuse. Ann N Y Acad Sci 1282:25–38
Di Marzo V, de Petrocellis L (2012) Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R Soc Lond B Biol Sci 367:3216–3228
Basu S, Dittel B (2011) Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res 51:26–38
Basu S, Ray A, Dittel BN (2013) Cannabinoid receptor 2 (CB2) plays a role in the generation of germinal center and memory B cells, but not in the production of antigen-specific IgG and IgM, in response to T-dependent antigens. PLoS One 8:e67587
Pacher P, Mechoulam R (2011) Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50:193–211
Miller AM, Stella N (2008) CB2 receptor-mediated migration of immune cells: it can go either way. Br J Pharmacol 153:299–308
Witkamp R, Meijerink J (2014) The endocannabinoid system: an emerging key player in inflammation. Curr Opin Clin Nutr Metab Care 17:130–138
Pacher P, Kunos G (2013) Modulating the endocannabinoid system in human health and disease: successes and failures. FEBS J 280:1918–1943
Ahern GP (2013) Transient receptor potential channels and energy homeostasis. Trends Endocrinol Metab 24:554–560
Clapham D, Julius D, Montell C, Schultz G (2013) Transient receptor potential channels, introduction. Last modified 30 Aug 2013. Accessed 09 Nov 2013. IUPHAR/BPS guide to pharmacology
Ramsey IS, Delling M, Clapham DE (2006) An introduction to TRP channels. Annu Rev Physiol 68:619–647
Tsuji F, Murai M, Oki K, Seki I, Ueda K, Inoue H et al (2010) Transient receptor potential vanilloid 1 agonists as candidates for anti-inflammatory and immunomodulatory agents. Eur J Pharmacol 627:332–339
De Petrocellis L, Schiano Moriello A, Imperatore R, Cristino L, Starowicz K, di Marzo V (2012) A re-evaluation of 9-HODE activity at TRPV1 channels in comparison with anandamide: enantioselectivity and effects at other TRP channels and in sensory neurons. Br J Pharmacol 167:1643–1651
Starowicz K, Makuch W, Korostynski M, Malek N, Slezak M, ZychowskaM et al (2013) Full inhibition of spinal FAAH leads to TRPV1-mediated analgesic effects in neuropathic rats and possible lipoxygenase-mediated remodeling of anandamide metabolism. PLoS One 8, Article number e60040
Brederson J-D, Kym PR, Szallasi A (2013) Targeting TRP channels for pain relief. Eur J Pharmacol 716:61–76
Alawi K, Keeble J (2010) The paradoxical role of the transient receptor potential vanilloid 1 receptor in inflammation. Pharmacol Ther 125:181–195
Maione S, de Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E et al (2007) Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol 150:766–781
Costa B, Bettoni I, Petrosino S, Comelli F, Giagnoni G, di Marzo V (2010) The dual fatty acid amide hydrolase/TRPV1 blocker, N-arachidonoyl-serotonin, relieves carrageenan-induced inflammation and hyperalgesia in mice. Pharmacol Res 61:537–546
De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S et al (2011) Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol 163:1479–1494
Knowlton WM, McKemy DD (2011) TRPM8: from cold to cancer, peppermint to pain. Curr Pharm Biotechnol 12:68–77
De Petrocellis L, Vellani V, Schiano-Moriello A, Marini P, Magherini PC, Orlando P et al (2008) Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J Pharmacol Exp Ther 325:1007–1015
Bautista DM, Pellegrino M, Tsunozaki M (2013) TRPA1: a gatekeeper for inflammation. Annu Rev Physiol 75:181–200
Romano B, Borrelli F, Fasolino I, Capasso R, Piscitelli F, Cascio MG et al (2013) The cannabinoid TRPA1 agonist cannabichromene inhibits nitric oxide production in macrophages and ameliorates murine colitis. Br J Pharmacol 169:213–229
Zhu X Huang X, Qian X (2013) GPR119 agonists: a novel strategy for type 2 diabetes treatment, diabetes mellitus – insights and perspectives. In: Oluwafemi O (ed) InTech. ISBN:978-953-51-0939-6. doi: 10.5772/48444. http://www.intechopen.com/books/diabetes-mellitus-insights-and-perspectives/gpr119-agonists-a-novel-strategy-for-type-2-diabetes-treatment
Syed SK, Bui HH, Beavers LS, Farb TB, Ficorilli J, Chesterfield AK et al (2012) Regulation of GPR119 receptor activity with endocannabinoid: like lipids. Am J Physiol Endocrinol Metab 303:E1469–E1478
Overton HA, Fyfe MCT, Reynet C (2008) GPR119, a novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br J Pharmacol 153:S76–S81
Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G (2006) Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab 3:167–175
Witkamp RF (2011) Current and future drug targets in weight management. Pharm Res 28:1792–1818
Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J et al (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101
Henstridge CM, Balenga NAB, Kargl J, Andradas C, Brown AJ, Irving A et al (2011) Minireview: recent developments in the physiology and pathology of the lysophosphatidylinositol-sensitive receptor GPR55. Mol Endocrinol 25:1835–1848
Balenga NAB, Aflaki E, Kargl J, Platzer W, Schröder R, Blättermann S et al (2011) GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res 21:1452–1469
Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T (2007) Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun 362:928–934
Ross RA (2011) L-α-lysophosphatidylinositol meets GPR55: a deadly relationship. Trends Pharmacol Sci 32:265–269
Ross RA (2009) The enigmatic pharmacology of GPR55. Trends Pharmacol Sci 30:156–163
Gantz I, Muraoka A, Yang Y-K, Samuelson LC, Zimmerman EM, Cook H et al (1997) Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics 42:462–466
McHugh D, Hu SSJ, Rimmerman N, Juknat A, Vogel Z, Walker JM et al (2010) N-Arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci 11, Article number 44
McHugh D, Page J, Dunn E, Bradshaw HB (2012) Δ9-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br J Pharmacol 165:2414–2424
McHugh D (2012) GPR18 in microglia: implications for the CNS and endocannabinoid system signalling. Br J Pharmacol 167:1575–1582
Mandard S, Patsouris D (2013) Nuclear control of the inflammatory response in mammals by peroxisome proliferator-activated receptors. PPAR Res 2013, Article ID 613864
Wahli W, Michalik L (2012) PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab 23:351–363
Gervois P, Mansouri RM (2012) PPARα as a therapeutic target in inflammation-associated diseases. Expert Opin Ther Targets 16:1113–1125
LoVerme J, Fu J, Astarita G, la Rana G, Russo R, Calignano A et al (2005) The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 67:15–19
Skaper S, Facci L, Fusco M, Valle M, Zusso M, Costa B et al (2014) Palmitoylethanolamide, a naturally occurring disease-modifying agent in neuropathic pain. Inflammopharmacology 22:79–94
Skaper S, Facci L, Giusti P (2013) Glia and mast cells as targets for palmitoylethanolamide, an anti-inflammatory and neuroprotective lipid mediator. Mol Neurobiol 48:340–352
Piomelli D (2013) A fatty gut feeling. Trends Endocrinol Metab 24:332–341
Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT (2012) PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci 32:17321–17331
Lourenco MV, Ledo JH (2013) Targeting Alzheimer’s pathology through PPARγ signaling: modulation of microglial function. J Neurosci 33:5083–5084
Meijerink J, Plastina P, Vincken J-P, Poland M, Attya M, Balvers M et al (2011) The ethanolamide metabolite of DHA, docosahexaenoylethanolamine, shows immunomodulating effects in mouse peritoneal and RAW264.7 macrophages: evidence for a new link between fish oil and inflammation. Br J Nutr 105:1798–1807
Brown I, Cascio MG, Wahle KW, Smoum R, Mechoulam R, Ross RA et al (2010) Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines. Carcinogenesis 31:1584–1591
Brown I, Wahle KWJ, Cascio MG, Smoum-Jaouni R, Mechoulam R, Pertwee RG et al (2011) Omega-3 N-acylethanolamines are endogenously synthesised from omega-3 fatty acids in different human prostate and breast cancer cell lines. Prostaglandins Leukot Essent Fatty Acids 85:305–310
Kim HY, Spector AA (2013) Synaptamide, endocannabinoid-like derivative of docosahexaenoic acid with cannabinoid-independent function. Prostaglandins Leukot Essent Fatty Acids 88:121–125
Navarrete CM, Fiebich BL, de Vinuesa AG, Hess S, de Oliveira ACP, Candelario-Jalil E et al (2009) Opposite effects of anandamide and N-arachidonoyl dopamine in the regulation of prostaglandin E2 and 8-iso-PGF2α formation in primary glial cells. J Neurochem 109:452–464
Navarrete CM, Pérez M, de Vinuesa AG, Collado JA, Fiebich BL, Calzado MA et al (2010) Endogenous N-acyl-dopamines induce COX-2 expression in brain endothelial cells by stabilizing mRNA through a p38 dependent pathway. Biochem Pharmacol 79:1805–1814
Georgiou NA, Garssen J, Witkamp RF (2011) Pharma–nutrition interface: the gap is narrowing. Eur J Pharmacol 651:1–8
Maione S, Costa B, di Marzo V (2013) Endocannabinoids: a unique opportunity to develop multitarget analgesics. Pain 154:S87–S93
Wainwright CL, Michel L (2013) Endocannabinoid system as a potential mechanism for n-3 long-chain polyunsaturated fatty acid mediated cardiovascular protection. Proc Nutr Soc 72:460–469
Montecucco F, di Marzo V (2012) At the heart of the matter: the endocannabinoid system in cardiovascular function and dysfunction. Trends Pharmacol Sci 33:331–340
Bátkai S, Osei-Hyiaman D, Pan H, El-Assal O, Rajesh M, Mukhopadhyay P et al (2007) Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J 21:1788–1800
Pacher P, Mukhopadhyay P, Mohanraj R, Godlewski G, Batkai S, Kunos G (2008) Modulation of the endocannabinoid system in cardiovascular disease: therapeutic potential and limitations. Hypertension 52:601–607
Rajesh M, Mukhopadhyay P, Hasko G, Huffman JW, Mackie K, Pacher P (2008) CB2 cannabinoid receptor agonists attenuate TNF-alpha-induced human vascular smooth muscle cell proliferation and migration. Br J Pharmacol 153:347–357
Naccarato M, Pizzuti D, Petrosino S, Simonetto M, Ferigo L, Chiodo Grandi F et al (2010) Possible anandamide and palmitoylethanolamide involvement in human stroke. Lipids Health Dis 9:47
Hillard CJ, Weinlander KM, Stuhr KL (2012) Contributions of endocannabinoid signaling to psychiatric disorders in humans: genetic and biochemical evidence. Neuroscience 204:207–229
Micale V, di Marzo V, Sulcova A, Wotjak CT, Drago F (2013) Endocannabinoid system and mood disorders: priming a target for new therapies. Pharmacol Ther 138:18–37
Hill MN, Hillard CJ, Bambico FR, Patel S, Gorzalka BB, Gobbi G (2009) The therapeutic potential of the endocannabinoid system for the development of a novel class of antidepressants. Trends Pharmacol Sci 30:484–493
Ruehle S, Rey AA, Remmers F, Lutz B (2012) The endocannabinoid system in anxiety, fear memory and habituation. J Psychopharmacol (Oxf) 26:23–39
Finn DP (2010) Endocannabinoid-mediated modulation of stress responses: physiological and pathophysiological significance. Immunobiology 215:629–646
Finn DP, Viveros MP, Marco EM (2012) The endocannabinoid system and emotional processing: pathophysiology and therapeutic potential. J Psychopharmacol (Oxf) 26:3–6
Viveros MP, Bermúdez-Silva FJ, Lopez-Rodriguez A, Wagner EJ (2011) The endocannabinoid system as pharmacological target derived from its CNS role in energy homeostasis and reward. Applications in eating disorders and addiction. Pharmaceuticals 4:1101–1136
Hansen HS (2010) Palmitoylethanolamide and other anandamide congeners. Proposed role in the diseased brain. Exp Neurol 224:48–55
Tanveer R, McGuinness N, Daniel S, Gowran A, Campbell VA (2012) Cannabinoid receptors and neurodegenerative diseases. Wiley Interdiscip Rev Membr Transp Signal 1:633–639
Karl T, Cheng D, Garner B, Arnold JC (2012) The therapeutic potential of the endocannabinoid system for Alzheimer’s disease. Expert Opin Ther Targets 16:407–420
Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang Y-P et al (2012) Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Reports 2:1329–1339
Esposito E, Cuzzocrea S (2013) Palmitoylethanolamide is a new possible pharmacological treatment for the inflammation associated with trauma. Mini Rev Med Chem 13:237–255
Esposito E, Cuzzocrea S (2013) Palmitoylethanolamide in homeostatic and traumatic central nervous system injuries. CNS Neurol Disord Drug Targets 12:55–61
Maccarrone M (2013) Endocannabinoid signaling in cancer: a rather complex puzzle. Trends Pharmacol Sci 34:426–427
Guindon J, Hohmann AG (2011) The endocannabinoid system and cancer: therapeutic implication. Br J Pharmacol 163:1447–1463
Pisanti S, Picardi P, D’Alessandro A, Laezza C, Bifulco M (2013) The endocannabinoid signaling system in cancer. Trends Pharmacol Sci 34:273–282
Cridge BJ, Rosengren RJ (2013) Critical appraisal of the potential use of cannabinoids in cancer management. Cancer Manag Res 5:301–313
Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A et al (2013) Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut (in press), Published Online First 30 September 2013
Wright KL, Duncan M, Sharkey KA (2007) Cannabinoid CB2 receptors in the gastrointestinal tract: a regulatory system in states of inflammation. Br J Pharmacol 153:263–270
Izzo AA (2007) The cannabinoid CB(2) receptor: a good friend in the gut. Neurogastroenterol Motil 19:704–708
Di Marzo V, Capasso R, Matias I, Aviello G, Petrosino S, Borrelli F et al (2008) The role of endocannabinoids in the regulation of gastric emptying: alterations in mice fed a high-fat diet. Br J Pharmacol 153:1272–1280
Izzo AA, Camilleri M (2008) Emerging role of cannabinoids in gastrointestinal and liver diseases: basic and clinical aspects. Gut 57:1140–1155
Izzo AA, Camilleri M (2009) Cannabinoids in intestinal inflammation and cancer. Pharmacol Res 60:117–125
Izzo AA, Sharkey KA (2010) Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther 126:21–38
Rani Sagar D, Burston JJ, Woodhams SG, Chapman V (2012) Dynamic changes to the endocannabinoid system in models of chronic pain. Philos Trans R Soc Lond B Biol Sci 367:3300–3311
Desroches J, Charron S, Bouchard J-F, Beaulieu P (2014) Endocannabinoids decrease neuropathic pain-related behavior in mice through the activation of one or both peripheral CB1 and CB2 receptors. Neuropharmacology 77:441–452
Figueroa JD, Cordero K, Serrano-Illan M, Almeyda A, Baldeosingh K, Almaguel FG et al (2013) Metabolomics uncovers dietary omega-3 fatty acid-derived metabolites implicated in anti-nociceptive responses after experimental spinal cord injury. Neuroscience 255:1–18
Guindon J, Hohmann AG (2007) Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol 153:319–334
Kupczyk P, Reich A, Szepietowski JC (2009) Cannabinoid system in the skin: a possible target for future therapies in dermatology. Exp Dermatol 18:669–679
Bíró T, Tóth BI, Haskó G, Paus R, Pacher P (2009) The endocannabinoid system of the skin in health and disease: novel perspectives and therapeutic opportunities. Trends Pharmacol Sci 30:411–420
Petrosino S, Cristino L, Karsak M, Gaffal E, Ueda N, Tüting T et al (2010) Protective role of palmitoylethanolamide in contact allergic dermatitis. Allergy 65:698–711
Mallat A, Lotersztajn S (2008) Cannabinoid receptors as novel therapeutic targets for the management of non-alcoholic steatohepatitis. Diabetes Metab 34:680–684
Deveaux V, Cadoudal T, Ichigotani Y, Teixeira-Clerc F, Louvet A, Manin S et al (2009) Cannabinoid CB2 receptor potentiates obesity-associated inflammation, insulin resistance and hepatic steatosis. PLoS One 4:e5844
Mallat A, Teixeira-Clerc F, Deveaux V, Manin S, Lotersztajn S (2011) The endocannabinoid system as a key mediator during liver diseases: new insights and therapeutic openings. Br J Pharmacol 163:1432–1440
Mallat A, Teixeira-Clerc F, Lotersztajn S (2013) Cannabinoid signaling and liver therapeutics. J Hepatol 59:891–896
Di Marzo V, Ligresti A, Cristino L (2010) The endocannabinoid system as a link between homoeostatic and hedonic pathways involved in energy balance regulation. Int J Obes 33:S18–S24
Engeli S (2012) Central and peripheral cannabinoid receptors as therapeutic targets in the control of food intake and body weight appetite control. In: Joost H-G (ed) Handbook of experimental pharmacology, vol 209. Springer, Berlin, pp 357–381
Di Marzo V, Després JP (2009) CB1 antagonists for obesitywhat lessons have we learned from rimonabant? Nat Rev Endocrinol 5:633–638
Piazza PV, Lafontan M, Girard J (2007) Integrated physiology and pathophysiology of CB1-mediated effects of the endocannabinoid system. Diabetes Metab 33:97–107
DiPatrizio NV, Piomelli D (2012) The thrifty lipids: endocannabinoids and the neural control of energy conservation. Trends Neurosci 35:403–411
Klumpers LE, Fridberg M, de Kam ML, Little PB, Jensen NO, Kleinloog HD et al (2013) Peripheral selectivity of the novel cannabinoid receptor antagonist TM38837 in healthy subjects. Br J Clin Pharmacol 76:846–857
Gérard N, Pieters G, Goffin K, Bormans G, van Laere K (2011) Brain type 1 cannabinoid receptor availability in patients with anorexia and bulimia nervosa. Biol Psychiatry 70:777–784
Marco EM, Romero-Zerbo SY, Viveros MP, Bermudez-Silva FJ (2012) The role of the endocannabinoid system in eating disorders: pharmacological implications. Behav Pharmacol 23:526–536
Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF (2012) DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol 8:999–1007
Kerr DM, Harhen B, Okine BN, Egan LJ, Finn DP, Roche M (2013) The monoacylglycerol lipase inhibitor JZL184 attenuates LPS-induced increases in cytokine expression in the rat frontal cortex and plasma: differential mechanisms of action. Br J Pharmacol 169:808–819
Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG et al (2011) Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334:809–813
Okine BN, Norris LM, Woodhams S, Burston J, Patel A, Alexander SPH et al (2012) Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: evidence for plastic changes in the endocannabinoid system. Br J Pharmacol 167:627–640
Murphy N, Cowley TR, Blau CW, Dempsey CN, Noonan J, Gowran A et al (2012) The fatty acid amide hydrolase inhibitor URB597 exerts anti-inflammatory effects in hippocampus of aged rats and restores an age-related deficit in long-term potentiation. J Neuroinflammation 9, Article number 79
Di Marzo V, Maccarrone M (2008) FAAH and anandamide: is 2-AG really the odd one out? Trends Pharmacol Sci 29:229–233
Ozalp A, Barroso B (2009) Simultaneous quantitative analysis of N-acylethanolamides in clinical samples. Anal Biochem 395:68–76
Huggins JP, Smart TS, Langman S, Taylor L, Young T (2012) An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153:1837–1846
Li GL, Winter H, Arends R, Jay GW, Le V, Young T et al (2012) Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharmacol 73:706–716
Di Marzo V (2012) Inhibitors of endocannabinoid breakdown for pain: not so FA(AH)cile, after all. Pain 153:1785–1786
Sasso O, Moreno-Sanz G, Martucci C, Realini N, Dionisi M, Mengatto L et al (2013) Antinociceptive effects of the N-acylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain 154:350–360
Hesselink JMK, Kopsky DJ, Witkamp RF (2014) Palmitoylethanolamide (PEA) ‘promiscuous’ anti-inflammatory and analgesic molecule at the interface between nutrition and pharma. Pharma Nutr 2(1):19–25
Di Marzo V, Skaper SD (2013) Editorial: palmitoylethanolamide—biochemistry, pharmacology and therapeutic use of a pleiotropic anti-inflammatory lipid mediator. CNS Neurol Disord Drug Targets 12:4–6
Ho WSV, Barrett DA, Randall MD (2008) ‘Entourage’ effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol 155:837–846
Skaper SD, di Marzo V (2012) Endocannabinoids in nervous system health and disease: the big picture in a nutshell. Philos Trans R Soc Lond B Biol Sci 367:3193–3200
Bettoni I, Comelli F, Colombo A, Bonfanti P, Costa B (2013) Non-neuronal cell modulation relieves neuropathic pain: efficacy of the endogenous lipid palmitoylethanolamide. CNS Neurol Disord Drug Targets 12:34–44
Melis M, Carta G, Pistis M, Banni S (2013) Physiological role of peroxisome proliferator-activated receptors type alpha on dopamine systems. CNS Neurol Disord Drug Targets 12:70–77
Skaper SD, Facci L (2012) Mast cell-glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos Trans R Soc Lond B Biol Sci 367:3312–3325
Esposito E, Paterniti I, Mazzon E, Genovese T, di Paola R, Galuppo M et al (2011) Effects of palmitoylethanolamide on release of mast cell peptidases and neurotrophic factors after spinal cord injury. Brain Behav Immun 25:1099–1112
Dalle Carbonare M, del Giudice E, Stecca A, Colavito D, Fabris M, D’Arrigo A, Bernardini D, Dam M, Leon A (2008) A saturated N-acylethanolamine other than N-palmitoyl ethanolamine with anti-inflammatory properties: a neglected story…. J Neuroendocrinol 20:26–34
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Witkamp, R.F. (2014). The Endocannabinoid System: A Dynamic Signalling System at the Crossroads Between Metabolism and Disease. In: Folkerts, G., Garssen, J. (eds) Pharma-Nutrition. AAPS Advances in the Pharmaceutical Sciences Series, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-319-06151-1_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-06151-1_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-06150-4
Online ISBN: 978-3-319-06151-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)