
Abstract
Even if the presence of a cardiomyopathy involving the right ventricle (RV) usually identifies mainly a specific cardiac disease initially called “arrhythmogenic right ventricular cardiomyopathy” and recently renamed “arrhythmogenic cardiomyopathy,” myocardial insults of different etiologies may involve the RV and lead to structural and functional abnormalities. In this chapter, we describe in detail the pathologic basis, the clinical feature, and the genetic background of AC, which is an important cause of sudden death, especially in the young. Moreover, the recent identification of an AC form characterized by an early and predominant involvement of the left ventricle (arrhythmogenic left ventricular cardiomyopathy), that enters into differential diagnosis with dilated cardiomyopathy and chronic myocarditis, is discussed. Finally, we also briefly analyze RV cardiomyopathic involvement of other cardiac or systemic diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Arrhythmogenic cardiomyopathy
- Arrhythmias
- Sudden cardiac death
- Dilated cardiomyopathy
- Hypertrophic cardiomyopathy
- Cardiac sarcoidosis
Even if the presence of a cardiomyopathy involving the right ventricle (RV) usually identifies mainly a specific cardiac disease initially called “arrhythmogenic right ventricular cardiomyopathy (ARVC)” and recently renamed “arrhythmogenic cardiomyopathy,” it is important to emphasize that myocardial insults of various etiologies may involve the RV and lead to structural and functional abnormalities.
Thus, in this chapter besides describing in detail the pathologic basis and the clinical feature of AC, we also briefly analyze RV cardiomyopathic involvement of other cardiac or systemic diseases.
Arrhythmogenic Cardiomyopathy
Arrhythmogenic cardiomyopathy (AC) is an inherited form of heart disease characterized pathologically by myocardial necrosis with fibro-fatty replacement and clinically by ventricular arrhythmias and impairment of ventricular systolic function [1, 2].
Historical Notes and First Clinical Descriptions
The first historical description of the condition can be found in the book De Motu Cordis et Aneurysmatibus, published in 1736 by Giovanni Maria Lancisi, who described a large family with recurrence of heart failure and sudden death with presence on autopsy of RV aneurysms [3].
In 1961, Dalla Volta described a series of patients with a dilated right ventricle of nonischemic origin and in whom cardiac catheterization demonstrated the presence of auricularization (strong right atrial contraction) of the right ventricle pressure curve [4].
The first complete clinical description of the disease was done in 1982 by Marcus who reported a series of 24 adult patients with recurrent episodes of ventricular tachycardia with left bundle branch block morphology, inverted T waves on the right precordial leads at electrocardiogram (ECG), and RV dilatation [5]. Histology examination documented the presence of extensive substitution of the RV myocardium with fatty and fibrous tissue. Since that moment on, the disease has been called both “arrhythmogenic right ventricular dysplasia” or “arrhythmogenic right ventricular cardiomyopathy.”
Few years later Thiene reported detailed pathologic features of the disease, consisting of myocyte necrosis with fibro-fatty substitution, and identified this disease as an important cause of sudden cardiac death (SCD) in young subjects, with particular regard to athletes [6].
It is noteworthy that initially AC was hypothesized to be a result of a congenital defect of myocardial development, while in the following years the description of familial recurrence was in favor of a genetic origin [7].
Epidemiology
The AC prevalence is difficult to estimate due to the frequent misdiagnoses, but it reasonably ranges from 1:1000 to 1:5000 [2, 8]. While in the past AC was considered to be an endemic disease in North East Italy (“Venetian disease”), now its presence is well recognized in different ethnicities [9]. The disease usually becomes clinically overt in the second–fourth decades of life and males result to be more frequently affected compared to females (up to 3:1) [8, 9]. Nonetheless, the disease is rarely diagnosed before puberty.
Pathological Findings
The AC pathologic basis consists of myocardial ventricular atrophy followed by fibro-fatty tissue replacement; this process is progressive, starting from the epicardium and then extending to the endocardium, eventually becoming transmural [10, 11] (Fig. 15.1). The progression of the pathologic process can lead to wall thinning and aneurysms, typically located at the inferior, apical, and infundibular walls of the RV (the so-called triangle of dysplasia, the hallmark of AC). Although in the original description the disease was characterized by an exclusive or at least predominant RV involvement, in the last years the improvement of imaging techniques and in particular the introduction of cardiac magnetic resonance (CMR) with contrast agent injection have demonstrated that the left ventricle (LV) is frequently involved. For this reason the current phenotypic classification of the disease considers the presence of three variants: “right dominant,” characterized by the predominant RV involvement, with no or minor LV abnormalities; “biventricular” with a parallel involvement of the RV and LV; and “left dominant” (also referred to as “arrhythmogenic left ventricular cardiomyopathy: ALVC”) characterized by a predominant LV involvement, with no or minor RV abnormalities [12].

Sudden death in AC patient 30 years after his clinical presentation with chest pain. At autopsy, the heart in cross section reveals diffuse biventricular involvement (a) with transmural fibro-fatty replacement of the RV free wall (b, trichrome staining ×3) and subepicardial mid-mural involvement of the LV free wall (c, trichrome staining ×3). AC arrhythmogenic cardiomyopathy, LV left ventricle, RV right ventricle. (Reproduced form Bariani R et al., “‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy”. EP Eur 2020, under licence n. 5007620151917)
This fact has led over the last few years to use the broader term of “arrhythmogenic cardiomyopathy,” which includes all these phenotypic expressions [1, 13].
In AC patients histological examination reveals the presence of islands of surviving myocytes interspersed with fibrous and fatty tissue. Fatty infiltration, that in original descriptions was one of the milestones of the disease histologic feature, is now not considered anymore a sufficient morphologic hallmark of the disease, as a replacement-type fibrosis and myocyte degenerative changes should always be identified [2, 14].
Genetic Background
Since the first description, the presence of an inheritable pattern of the disease with familial recurrence has been demonstrated. In addition, it became evident that in the majority of cases the disease was inherited through an autosomal dominant transmission with incomplete penetrance and variable expressivity [1]. Notably, in 1986 a disease variant characterized by the association between AC and palmoplantar keratoderma/woolly hair (cardio-cutaneous syndrome) was described in Naxos Island of Greek and named Naxos syndrome [15]. Differently from isolated AC, Naxos syndrome is inherited in an autosomal recessive manner with full penetrance. In 2000 a deletion in desmosomal gene plakoglobin was identified as the underlying genetic cause of ACM [16].
Few years later, mutations in other genes encoding for main components of the desmosome were found to be linked to AC [17].
Thus, it has become evident that abnormalities in desmosome structure have a key role in AC pathogenesis and for this reason AC is now considered to be mainly a “disease of the desmosome.”
Desmosomes are complex structures consisting of proteins and are responsible for cell adhesion and signaling. One important function is to tether adjacent cells mechanically by joining their intermediate filaments to create a unified cytoskeletal network [1, 2] (Fig. 15.2).

Schematic representation of the complex integration of mechanical and electrochemical signaling at cardiac intercalated discs and it highlights the proposed remodeling of desmosomes, gap junctions, and ion channels in arrhythmogenic cardiomyopathy. (Reproduced under license (n. 5014280291051) from Elliott PM, Anastasakis A, Asimaki A, et al. Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. Eur J Heart Fail. 2019;21(8):955–964. doi:https://doi.org/10.1002/ejhf.1534)
Genetic studies demonstrated that approximately 30–50% of AC patients carry a pathogenic mutation in a desmosome gene.
The most frequent gene found in AC patients is PKP2 (19–46%) followed by DSP (1–16%), DSG2 (2.5–10%), DSC2 (1–8%), and JUP (1%). Moreover, approximately 10–25% of AC patients carry compound mutations.
Plakophilin 2 (PKP-2)
This is the most frequently mutated gene that is found in AC patients. The presence of heterozygous mutations in this gene is usually linked to the “classical” form of the disease with a predominant RV involvement. The majority of mutations have been identified in the C-terminal portion of the protein [18].
Desmoplakin (DSP)
Several mutations involving this gene have been identified so far, often leading to the synthesis of a truncated protein at the N-terminal or C-terminal side. Interestingly, these mutations can have different phenotypic expressions: if mutation involves N-terminal portion, the resulting phenotype is a classic form of AC with autosomal dominant transmission [19] while mutations on the C-terminal end (the one that interacts with intermediate filaments) are usually expressed with predominant LV involvement (ALVC) [19, 20]. Finally, the presence of a homozygous mutation can be characterized phenotypically by biventricular AC forms with almost exclusively fibrous infiltration associated with cutaneous involvement (Carvajal syndrome) [21]. The different clinical AC phenotypes linked to different protein domains of desmoplakin (N- or C-terminal) suggest the presence of distinct molecular mechanisms underlying the different disease variants. Thus, it has been speculated that the left-dominant variant may be secondary to an altered desmoplakin-desmin linkage, which compromises the integrity of the cytoskeleton in cardiomyocyte while an alteration in the relationship between desmoplakin and other components of desmosome would cause a classical disease phenotype [20]. Recently, a study compared clinical data of patients with truncating mutations in DSP and PKP-2 genes [22]. Authors concluded that LV involvement was exclusively present in patients with DSP mutations, which also had a preserved systolic function of both ventricles compared to PKP-2 patients. At CMR these patients frequently showed LGE on the LV, mainly located in lower and inferoseptal segments. Of note a frequent positive history of chest pain episodes in DSP patients was reported, both probands and family members; it has also been noted that acute episodes of myocardial damage can occur even in the presence of normal systolic function [22].
Desmoglein 2 (DSG-2)
Nine different mutations regarding this gene are currently known and are mainly located in the N-terminal region, responsible for a classical AC phenotype, even if in some cases a phenotypic overlap with dilated cardiomyopathy (DCM) has been reported [23].
Desmocollin 2 (DSC-2)
Mutations of this gene lead to premature truncation of desmocollin protein, with loss of its normal function, and they are associated with right-dominant forms. Both autosomal recessive and dominant transmission are reported [24].
Plakoglobin (JUP)
Deletion at the C-terminal end of this gene leads to formation of a truncated protein. Homozygous mutations are associated with Naxos cardio-cutaneous syndrome, with autosomal recessive transmission, while heterozygous mutations are expressed with only cardiac ventricular involvement [25].
Although less frequently found, mutations in non-desmosomal genes have also been linked to AC: desmin (DES), filamin C (FLNC), transmembrane protein 43 (TMEM-43), lamin A/C (LMNA), titin (TTN), phospholamban (PLN), α-T-catenin (CTNNA-3), cadherin-2 (CDH2), transforming growth factor-β3 (TGF-β3), ryanodine receptor 2 (RYR2), and Nav1.5 (SCN5A).
α-T-Catenin (CTNNA3)
This protein interacts with PKP-2 in the intercalated disc. Mutations in this gene lead to a decreased binding capacity to desmosomal components, resulting in impaired intercellular adhesion function. This form is usually characterized by incomplete penetrance [26].
Cadherin-2 (CDH2)
It is an integral glycoprotein that mediates cell adhesion in the presence of calcium. The intracellular domain is connected to actin filaments by catenins. Recently, in the worldwide cohort of patients affected by AC, previously negative at the genetic examination, mutations in CDH2 were detected. Moreover, these patients had an increased risk of ventricular arrhythmias, while evolution toward heart failure is rare [27].
Laminin (LMNA)
It is a nuclear matrix protein whose mutations are expressed with a wide phenotypic variety that may, although rarely, include cardiac involvement with an AC phenotype [28]. It is most frequently found in severe forms of the disease, with a dilated phenotype and high risk of sudden cardiac death [29].
Desmin (DES)
It is an intermediate filament protein that is essential for the organization of the cytoskeleton and structural maintenance of cardiomyocytes. Mutations in this gene, which often have complete penetrance, cause a group of skeletal myopathies associated with conduction blocks and cardiomyopathy, associated with a dilated or restrictive phenotype, and sometimes an AC forms [30]. It is interesting to underline that recently a mutation of this gene, compromising the binding between desmin and desmoplakin, has been described in AC subjects with a predominant and severe involvement of the LV [31].
Transmembrane Protein 43 (TMEM-43)
This is a nuclear protein that interacts with several other proteins in nucleus and with various transcription factors and it is reported to be responsible for a severe, full-penetrance phenotype of AC associated with a high risk of SCD [32].
Titin (TTN)
This giant protein is an important constituent of sarcomeres and one of the main pathogenic genes in cardiomyopathies with hypertrophic and dilated phenotype. A possible correlation between TTN and AC has been hypothesized considering its direct and close contact with intercalary discs. The AC phenotype linked to titin mutations is characterized by a biventricular involvement with high risk of heart failure, presence of arrhythmias (both supraventricular tachycardia and ventricular arrhythmias) and conduction blocks, configuring an “overlap syndrome” between different cardiomyopathies [33].
Filamin C (FLNC)
Filamins are a family of proteins that interconnect actin filaments, forming a network, and anchor membrane-associated proteins to the cytoskeleton, thus contributing both to structural stability and to signal transduction from cell membrane. FLNC is associated with sarcomere disc and mutations in the FLNC gene and mutations have been found in skeletal muscle myopathies and in dilated and restrictive cardiomyopathies. Truncated mutations of the FLNC gene have been associated with left-dominant forms of AC, with high prevalence of ventricular arrhythmias and signs of fibrosis on CMR and/or histological examination [34].
Ryanodine Receptor 2 (RYR2)
This was the first non-desmosomal gene to be described as associated with AC. RYR2 encodes for the cardiac ryanodine receptor, an important channel that allows calcium ions to pass from the sarcoplasmic reticulum to the cytoplasm during cardiac systole. This gene is usually associated with a primary nonstructural cardiomyopathy (catecholaminergic polymorphic ventricular tachycardia), but some mutations, probably due to a different molecular mechanism, are expressed with an AC phenotype characterized by frequent exercise-induced arrhythmias [35, 36].
Nav1.5 (SCNA5)
Voltage-dependent sodium channels play a central role in the creation and propagation of the action potential through cardiomyocytes. Mutations in SCNA5 gene, which encodes for the pore-forming subunit of the Na+ channel, have been associated with several arrhythmic diseases, including Brugada, long QT, and sick sinus node syndromes. They have also been found in a small percentage of AC patients, associated with an elongated QRS, but its role as disease gene has still to be elucidated [37].
Phospholamban (PLN)
This is a transmembrane protein of the sarcoplasmic reticulum involved in calcium transport by inhibiting the activity of the SERCA2 (sarcoplasmic/endoplasmic reticulum calcium ATPase) pump. Mutations in this gene are associated with restrictive, dilated, and arrhythmogenic cardiomyopathies; in the latter, a particularly severe phenotype is frequently found, with biventricular involvement and a peculiar electrocardiographic pattern (severe reduction of QRS voltages in limb leads) [38]. In a mouse model it was shown that a mutation in the PLN gene (Arg14del) leads to a high risk of developing DCM or AC with heart failure [39].
In the last 10 years, an increasing number of evidence and advances in molecular research have led to a change in the etiology of AC. From the initial idea of a monogenic disease, recent findings suggest rather a complex genetic condition, in which the phenotype is determined by the interaction of multiple genetic and environmental factors [1, 2]. The frequency of compound and digenic heterozygosity is reported to be 10–25% of cases depending on the study population. Noteworthy, genotype-phenotype correlation studies have shown that a higher mutational load correlates with an unfavorable clinical course, a higher risk of SCD, and frequent biventricular involvement [40,41,42].
Clinical Features and Natural History
In AC the presence of fibro-fatty tissue leads both to morphological ventricular abnormalities and circuits that constitute the anatomic basis of reentry ventricular arrhythmias. The diagnosis of the disease relies on the demonstration of anamnestic, clinical, morphological, and electrophysiological parameters which can be achieved from different instrumental tools. The phenotypic aspects of AC can variate in a considerable way, ranging from asymptomatic family members with mild forms of the disease to symptomatic patients who experienced life-threatening ventricular arrhythmias or refractory heart failure [1]. In affected families the presence of carriers of disease gene mutations has been demonstrated who do not show any signs of the disease (the so-called healthy carriers). The most common clinical presentation consists of arrhythmic symptoms such as palpitations, syncopal episodes, or cardiac arrest; unfortunately, SCD can be the first clinical manifestation of the disease in previously asymptomatic individuals, especially in the young and in competitive athletes [43, 44]. Classically, different clinical phases of the disease have been identified: (1) the initial “concealed phase” with absence of overt ventricular structural abnormalities, with or without minor ventricular arrhythmias; (2) the second phase of “clinically overt disease” characterized by the onset of ventricular arrhythmias and presence of ventricular functional and structural abnormalities; (3) the third phase that is due to progression of ventricular muscle disease leading to RV impairment with RV failure and relatively preserved LV function; and (4) the fourth phase “end stage” with biventricular pump failure. The prognosis in patients affected with AC is related to the degree of electric instability and of ventricular muscle disease. The overall mortality rate varies in literature, due to the different patients’ selection. In a study on 37 AC families with a mean follow-up of 8.5 years, a mortality of 0.08% per year was found [8], while in a series of 61 AC patients with a mean follow-up of 4.6 years the mortality rate was estimated to be of 4% per year [45]. This high variability is probably in relation to the different populations and reflects the wide spectrum of AC clinical phenotype [2].
Sports Activity and Arrhythmogenic Cardiomyopathy
James et al. first reported in humans that a history of intense exercise was more often associated with desmosomal gene mutation carriers developing the disease and patients with overt AC suffering from major ventricular arrhythmias [46]. From then on, many other studies [47,48,49,50,51,52,53] confirmed that sports activity, especially if prolonged, promotes the development of AC in genotype-positive/phenotype-negative patients, deteriorates ventricular function in patients with overt AC, triggers ventricular arrhythmias, and increases the likelihood of ICD interventions. Indeed, the physical activity generates a mechanical stress at the level of a previously genetically impaired cell-cell adhesion, thus promoting myocyte death. In particular, Ruwald et al. showed that participation in competitive sport was associated with an absolute risk of potentially lethal arrhythmic events of 61% at 40 years of age in AC patients [49]. Sawant et al. and Lie et al. reported more severe RV dysfunction, LV dysfunction, and heart failure when endurance training was carried on [47, 50]. Animal studies demonstrated that in heterozygous plakoglobin-deficient (JUP+/−) mice endurance training (daily swimming) promoted the development of RV abnormalities such as dilatation and dysfunction and ventricular arrhythmias [54]. Moreover, the effect of endurance training in enhancing RV abnormalities was demonstrated in a mouse model overexpressing a nonsense plakophilin-2 (PKP2) gene mutation after adeno-associated virus injection [55]. Conversely, the risk of VAs and mortality both in AC patients and genotype-positive relatives can be reduced by lowering exercise [8, 49, 50]. Thus, on the one hand, preclinical genetic testing among asymptomatic gene carriers with the aim to educate about lifestyle changes can prevent the development of the disease in this category. On the other hand, the identification of early stages of the disease by preparticipation screening and disqualification from competitive sports activity may prevent disease progression and fatal arrhythmias [56]. Accordingly, both European and American guidelines recommend restriction from competitive sports activity of AC patients and at-risk relatives as a measure aimed to reduce the risk of SCD [57, 58]. However, considering the general physical and mental health benefits related to exercise, ESC guidelines allow a maximum of 150 min of low-moderate-intensity (3–6 metabolic 292 equivalent) exercise per week in all affected and at-risk subjects [57].
Disease Diagnosis
AC diagnosis is multiparametric as a single specific diagnostic marker does not exist, due to the wide clinical phenotype and a clinical overlap with other cardiac diseases. In 1994, an international task force proposed a diagnostic scoring system with major and minor criteria with the aim to uniform the diagnosis [59]. Definite AC diagnosis included two major criteria or one major and two minor criteria or four minor criteria from different categories. In the following years, clinical studies demonstrated these criteria to be highly specific but lacked sensitivity for the diagnosis in mild forms of the disease. For this reason, diagnostic criteria were revised in 2010, with addition of quantitative measurement of imaging tools and of new ECG parameters. Moreover, genetic analysis results entered among diagnostic criteria [60]. In 2019 an International Expert Report provided an extensive critical review of the clinical performance of AC ongoing diagnostic criteria with the aim to identify potential areas of improvement [60]. Major limitations of the 2010 Task Force criteria were considered to be the incomplete understanding of the genetic background of the disease and the absence of specific criteria for the diagnosis of the broader spectrum of the disease phenotypes, including ALVC forms [61, 62]. Moreover, 2010 criteria did not consider tissue characterization findings provided by CMR which offer the possibility to identify myocardial fibrosis and play a key role in the accurate diagnosis of the LV phenotype. For these reasons in 2020 a modification of diagnostic criteria, called “Padua criteria” (Table 15.1) which included also CMR findings and presence of ALVC forms, was proposed [13].
Diagnostic Tools in AC
As stated above, current AC diagnostic criteria take into consideration different parameters regarding anamnesis, electrophysiological features, and findings coming from imaging techniques.
Personal and Familial Anamnesis
Patients could be completely asymptomatic or complain about palpitations, dizziness, or syncopal episodes. In the presence of a severely reduced ventricular function, heart failure symptoms and signs could be present. A careful family history investigation with particular regard to SCD cases and presence of relatives showing arrhythmic diseases or presenting with arrhythmic symptoms should be performed. Finally, a family pedigree should be created.
Twelve-Lead Electrocardiogram (ECG)
ECG pattern plays a central role in AC diagnosis, as loss of electrical forces secondary to myocardial atrophy, conduction abnormalities caused by fibrofatty replacement and/or right ventricular dilatation, and presence of a transmural voltage gradient between injured and healthy myocytes lead to ventricular depolarization/repolarization activities that can be appreciated on ECG [63]. Nonetheless, in a significant number of AC patients, ranging from 12% to 50% on different series, ECG can be normal [64,65,66]. The ECG patterns that can be found are the following (Figs. 15.3, 15.4, and 15.5):
-
Delay of terminal activation time: It is defined as a slurring of the S wave in leads V1–V3 with the longest value from the nadir of the S wave to the end of all QRS ≥55 ms in the absence of r′ in leads V1–V3.
-
Right ventricular conduction delay: Presence of conduction delay can be represented mainly by an incomplete right bundle branch block (RBBB) (rSr1 in lead V1 and QRS duration <120 ms) while a complete RBBB is less common.
-
QRS fragmentation: It is defined as the presence of additional spikes within the QRS complex due to a regional delay of the normal ventricular conduction linked to fibro-fatty infiltration.
-
Negative T waves: Inverted T waves on right precordial leads (V1, V2, and V3) in subjects older than 14 years are considered a diagnostic criterion for AC as they are related to the RV volumes. In addition, inverted T waves on inferior and lateral leads (V5–V6) can suggest a LV involvement.
-
Low QRS voltages: Defined as a p eak-to-peak QRS amplitude of less than 5 mm in the limb leads and/or less than 10 mm in the precordial leads [67]. The presence of low QRS voltages in limb leads has been demonstrated to be associated with the presence of LV late enhancement (LGE) and it is one of the criteria for ALVC diagnosis [13, 68].

ECG pattern of a patient affected by right-dominant arrhythmogenic cardiomyopathy. Presence of T-wave inversion right precordial leads (V1 through V3) secondary to the fibro-fatty replacement of the RV

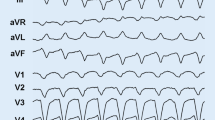
ECG pattern of a patient affected by arrhythmogenic cardiomyopathy with biventricular involvement. Negative or flattening T waves are present in both precordial and limb leads. Moreover, QRS complex voltages are reduced in all leads as a result of extensive fibro-fatty replacement in both ventricles

ECG of a patient affected by left-dominant arrhythmogenic cardiomyopathy. T-wave inversion in lateral and limb lead is suggestive of left ventricular involvement
Ventricular Arrhythmia (VA) Detection
VAs linked to AC usually have a left bundle branch block (LBBB) pattern. While a left-axis deviation can lead to suspecting the presence of an AC form, the main problem is the differentiation from idiopathic ventricular arrhythmias with right-axis deviations originating from the RV infundibulum. The most sensitive parameters are a QRS duration in lead I ≥120 ms, a QRS transition in V6, notching on any complex, and early QRS onset in V1 [69, 70]. Regarding their complexity, ventricular arrhythmias can be isolated or organized in runs of non-sustained ventricular tachycardia (NSVT) or sustained ventricular tachycardia (sVT). Ventricular fibrillation (VF) is mostly reported in young patients during the earlier phases of AC, whereas sustained VTs occur more commonly later in the disease course [71]. Bhonsale et al. [72] demonstrated that AC patients who experienced ventricular fibrillation and SCD were significantly younger (median age 23 years) than those presenting with sustained monomorphic VTs (median age 36 years). In addition, in patients with late presentation (>50 years) sustained VT was the predominant arrhythmic event, while in young population VF was more common [73]. This age-related behavior of arrhythmic pattern could be explained by the progressive nature of the disease, considering that monomorphic VT is usually linked to reentry circuits around stable fibro-fatty myocardial scars that are the result from a long pathologic process, while VF may be the result of acute electrical instability, particularly in the context of myocarditis-mediated bouts of acute myocyte necrosis [71, 74].
Two-Dimensional Echocardiography
Echocardiography is the first-line imaging modality in AC, since it is a noninvasive, widely available technique which can provide information about volumes and systolic function of both ventricles. However, echocardiographic diagnosis in AC requires a specific expertise due to the retrosternal position, the complex geometry, and the load dependency of the RV [75]. In addition to this, echocardiography shows a low sensitivity, especially in the early stages of the disease [76].
Regional wall motion abnormalities (RWMA), together with global RV dysfunction and dilation, represent the macroscopic results of the fibro-fatty changes at the histological level. The echocardiographic diagnosis of AC lies consequently in demonstrating their presence while performing the exam. Regional RV akinesia, dyskinesia, or aneurysm; right ventricle outflow tract (RVOT) diameter (measured from either parasternal long-axis [PLAX] view or parasternal short-axis [PSAX] view); and RV-fractional area change (FAC) are the only standard echocardiographic measures included in the 2010 Task Force criteria [60] (Table 15.2).
Moreover, even if not proved to raise the diagnostic sensibility, other parameters such as the RV basal diameter and those obtained by advanced echocardiographic methods have been proposed to strengthen the suspicion of AC in dubious cases [77].
Advanced Echo Modalities
Contrast echocardiography, Doppler tissue imaging, tissue deformation imaging, and 3D echocardiography are emerging tools in the echocardiographic assessment in AC.
Contrast echocardiography can enhance the detection of RWMAs if the image quality is not satisfying [78]. Doppler tissue imaging for the measurement of the peak systolic annular velocity (s′) of the RV can provide additional information about the RV longitudinal systolic function. The latter can also be assessed by tissue deformation imaging that typically shows a reduced RV strain and an increased mechanical dispersion in AC patients [48, 79, 80]. Since these parameters are altered early before other macroscopic changes become evident, they appear particularly useful when early stages are suspected or when looking for the disease in relatives. 3D echocardiography allows a precise assessment of RV volumes and function, despite losing accuracy when end-stage forms with extremely enlarged ventricles are addressed [81].
Cardiac Magnetic Resonance
Cardiac magnetic resonance can allow a comprehensive evaluation of volumes, function, and tissue characterization in a single investigation. A standardized protocol is recommended when approaching AC patients, with cine images performed using steady-state free precession (SSFP) sequences and LGE images using phase-sensitive inversion recovery (PSIR) sequences [82]. Most of our knowledge about common findings as evidenced by CMR comes from studies carried out in patients affected by the classic right phenotype. Recognized CMR features associated with AC are RV wall thinning, RVOT enlargement, trabecular disarray, fibro-fatty replacement, ventricular dilatation, focal bulges, microaneurysms, and global or regional systolic dysfunction [82] (Fig. 15.6).

CMR images of a patient affected by right-dominant arrhythmogenic cardiomyopathy. (a, b) Apical four-chamber view and mid short-axis view of cine images showing severe dilatation of the RV. (c, d) Apical four-chamber view and mid short-axis view of post-contrast sequences showing extensive LGE of the RV
The CMR criteria in 2010 TF [60] included quantitative metrics, such as RV dilatation or global dysfunction, and qualitative findings, like akinesia, dyskinesia, and dyssynchronous contraction (i.e., RWMAs) (Table 15.3).
Besides these morpho-functional anomalies, CMR can show also structural alterations, such as fat infiltration by T1-weighted spin-echo images, and LGE by post-contrast sequences at the RV level. However, given the low reproducibility and inconsistency of these measures, neither of them was included into the 2010 TF criteria. As stated above in 2020 Padua criteria included also LGE among the criteria for diagnosis. Notably, detections of LGE at the RV level are hampered by the thin RV wall, which makes the LGE analysis less consistent than for the LV [83]. Moreover, LGE cannot distinguish between fat and fibrosis [82]. Also, LGE is a nonspecific finding that can be found in other diseases that mimic AC, such as myocarditis, sarcoidosis, and dilated cardiomyopathy. Likewise, intramyocardial fat has been demonstrated in older, obese patients and it is a common finding in autopsy cases dying for noncardiac causes [84,85,86]. Despite these limitations, contrast-enhanced CMR is currently the ideal technique to address the emerging biventricular and the left-dominant variants, thanks to its tissue characterization capability that allows the detection of both fibrosis and fibro-fatty infiltration at the LV level (Fig. 15.7).

CMR images of a patient affected by arrhythmogenic cardiomyopathy with biventricular involvement. (a) Apical four-chamber view of cine images showing dilatation of both ventricles with thinning of the LV lateral wall. (b) T1-weighted black blood sequences with fat suppression demonstrating fatty infiltration of septal and lateral walls. (c) Post-contrast sequences showing LGE in the same locations as in (b)
Endomyocardial Biopsy
It is an invasive procedure that is performed via venous access and catheterization of right heart and which allows the sampling of myocardium from free wall of the RV, which is then subjected to histological analysis. Although it is a part of the diagnostic 2010 Task Force criteria, this examination, due to its invasiveness, is reserved for selected AC patients, in which phenocopies (dilative cardiomyopathy, myocarditis, sarcoidosis) should be excluded [87]. As mentioned above, the sample is taken preferably from the free wall of the ventricle (the septum is rarely involved in classic variants of AC), and, in order to increase sensitivity and reduce risk of wall perforation, it should be guided by electro-anatomical mapping or CMR [14, 87]. Endomyocardial biopsy offers an in vivo characterization of the distinctive element of the disease, like fibro-fatty replacement and loss of myocytes. In details, according to the parameters of the International Task Force [88] if on morphometric analysis the residual myocytes are <60%, the major criterion is considered, and if between 65% and 70%, the minor criterion is considered. This method can help in the differential diagnosis between AC and so-called phenocopies of disease, such as DCM, myocarditis, sarcoidosis, or other conditions leading to myocardial tissue replacement. However, although endomyocardial biopsy can unequivocally detect the presence of fibrous or fibroadipose replacement and quantify the proportion of residual myocytes, it is severely limited by its poor sensitivity. Indeed, since pathological process affects the heart muscle focally and proceeds from epicardium through the endocardium, a negative histological sample does not necessarily imply the absence of disease. Immunohistochemical analytical tests have recently been revised to evaluate on the sample the possible alteration in the distribution of desmosomal proteins [89]; unfortunately, specific findings for AC have not been yet identified.
Arrhythmogenic Left Ventricular Cardiomyopathy
Arrhythmogenic left ventricular cardiomyopathy (ALVC) is an AC form characterized by an early and predominant involvement of the left ventricle [12]. In contrast to the biventricular variant, where the degree of ventricular dysfunction is similar in the two ventricles, in this case right ventricular involvement, if present, has a minor significance. First evidence of ALVC came from autopsy reports and soon after from screening of families with mutations in the DSP gene [19, 90]. The first clinical description of the disease was made in 2008 [12] and unfortunately no validated diagnostic criteria for this AC form have been provided so far. In addition, 2010 TFC criteria have proved to be insensitive in the ALVC diagnosis [22]. Commonly, ECG shows T-wave inversion typically located in lower and/or lateral leads and presence of low QRS voltages in peripheral leads. It has been speculated that this may be secondary to fibro-fatty replacement of the left ventricle, but conclusive studies are not yet available. Arrhythmias are characterized by a RBBB morphology and variable axis, frequently originating from the lateral wall of the LV. Notably, the degree of electrical instability seems not to correlate with the degree of LV, this being a distinguishing feature from DCM. CMR plays a pivotal role in diagnosis mostly through the possibility to highlight the presence of fibrous tissue by means of LGE detection. The peculiar pattern of LGE distribution is represented by LV subepicardial stria, most frequently located on inferolateral basal segments with variable extension and in some cases leading to a circumferential involvement of the entire ventricle (Fig. 15.8). Unfortunately, this pattern is not exclusive of AC and enters into differential diagnosis with other diseases such as myocarditis. Recent studies demonstrated that in patients with acute myocarditis LGE can be found in 41% of cases on subepicardial layers of LV inferior and lateral walls [91] with nonsignificant changes only in 30% of cases at 6 months’ follow-up [92]. Conversely in ALVC, because of its progressive pathogenesis, LGE is unchanged or increased in almost all patients. For these reasons, the diagnosis cannot be based solely on instrumental examinations, but must take account of family history, genetic findings, and, in sporadic cases, endomyocardial biopsy. Similarly, DCM forms with clinical and instrumental features similar to those of ALVC have been described (so-called dilated cardiomyopathies with an arrhythmogenic phenotype) [93]. However, studies comparing clinical and CMR characteristics of a group of DCM and AC patients demonstrated that the amount of LGE and its distribution are significantly different between the two groups. Interestingly, in AC gadolinium on the LV showed a peculiar pattern, as described above, while in DCM a common finding was an intramural stria at septal level. In addition, the amount of LGE was significantly higher in AC group. The explanation for these findings can be obtained by analyzing the different pathophysiology of the two cardiomyopathies. In DCM, fibrosis is a secondary phenomenon due to ventricular enlargement, while in AC it is a primary phenomenon resulting from the death of cardiomyocytes through necrosis and apoptosis [94]. Patients with ALVC may experience “hot phases” characterized by chest pain and enzymatic release. Unfortunately, differential diagnosis with acute myocarditis can be quite difficult. It is estimated that “hot phase” phenomenon has an incidence that ranges from 5% to 25% of patients in different series [22, 95]. In a recent paper our group analyzed clinical and instrumental findings of a series of 23 patients affected by AC, mainly with a ALVC or a biventricular phenotype, who experienced one or more episodes of myocardial injury [95]. From those data, myocarditis-like picture seems to be a rather uncommon clinical presentation of AC, often occurring in the pediatric age, and CMR is the first-choice examination for the differential diagnosis between AC and acute myocarditis. Moreover, in patients with this clinical presentation EMB can have a pivotal role in differential diagnosis as well as family screening and genetic test. As stated above, signs of myocardial injury that precede systolic dysfunction were found in a significant number of subjects carrying DSP truncating mutations [22]. To date, it is unclear why some AC patients develop episodes of myocardial injury and it has been speculated that this is in relation with the wall thickness considering that these episodes are more intense and symptomatic when involving the LV as in ALVC forms. Finally, their role in disease progression and arrhythmic risk remains to be elucidated. In contrast to classical variants, no conclusive data exist for ALVC as far as prognosis and arrhythmic risk stratification are concerned. While parameters such as degree of RV dilatation and dysfunction, extension of T-wave inversion, and degree of electrical instability exist for right and biventricular variants [96], no validated predictors are present for ALVC forms to date. Recently, a risk score has been developed to help the clinician in the decision to implant an ICD in primary prevention [97]. However, it seems to lack sensitivity for left-dominant forms [98].

CMR images of a patient affected by left-dominant arrhythmogenic cardiomyopathy. (a) Apical four-chamber view of cine images showing a mild dilated left ventricle with a thinned lateral wall compared to the septum. (b, c) Four-chamber view and short-axis view of post-contrast sequences showing subepicardial LGE in the form of stria of the anterolateral and inferolateral walls
Therapeutic Strategies in AC
Physical Restriction
Sports activity enhances AC progression and worsens the disease arrhythmic substrate [44, 47, 49, 50, 54]. Conversely, the risk of ventricular arrhythmias (VAs) and mortality can be lowered by reducing exercise [8, 49, 50, 99].
Different categories of AC patients show a dose-dependent association between exercise exposure and disease penetrance. Genotype-positive relatives undergoing competitive sports and high-intensity physical exercise are affected by an increased risk of VAs and heart failure as documented by clinical studies [46, 100]. With this regard, pre-symptomatic genetic testing has a role because it can detect those individuals in whom a lifestyle change can reduce the risk of developing AC. Likewise, in patients with an overt phenotype, preparticipation screening and disqualification may prevent SCD [56].
Accordingly, both European and American guidelines recommend restriction from competitive sports activity of AC patients and at-risk relatives as a measure aimed to reduce the risk of SCD [57, 58].
Drug Therapy
Beta-Blockers
Ventricular arrhythmias and cardiac arrest in AC are usually promoted by adrenergic stimulation and occur typically during or early after a physical effort. Thus, beta-blockers are recommended in AC patients symptomatic for frequent premature ventricular complex (PVCs) and non-sustained ventricular tachycardias (NSVT), patients with recurrent VT, appropriate ICD therapies, or inappropriate ICD interventions resulting from sinus tachycardia, supraventricular tachycardia, or atrial fibrillation/flutter with high ventricular rate.
In addition, since reducing the ventricular wall stress lowers the myocardial disease progression and is considered as a first-line medication in the management of heart failure, beta-blockers should be offered in all patients with a definite diagnosis of AC, irrespective of arrhythmias.
So far, in phenotype-negative gene carriers prophylactic use of these drugs is not justified [101, 102].
Antiarrhythmic Drugs
When beta-blockers alone are not sufficient to control the arrhythmic burden, anti-arrhythmic drug therapy is indicated for symptomatic patients with frequent PVCs and/or NSVT. In particular, sotalol and amiodarone (alone or associated with beta-blockers) are the most effective drugs with a relatively low proarrhythmic risk [101, 102].
Heart Failure Drugs
The standard pharmacological treatment for heart failure (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, beta-blockers, and diuretics) is recommended in patients who develop right, left, or biventricular heart failure [102].
New Drugs
Therapeutic strategies targeting the Wtn/β and NFκB pathways appear to lower the disease in animal models and thus may be promising options in the future [103].
Catheter Ablation
Catheter ablation should be considered as a therapeutic option for patients symptomatic for PVCs or VT or frequent appropriate ICD interventions on VT despite optimal medical therapy, in order to improve symptoms and prevent ICD shocks, respectively [101]. The initial experience with this technique reported high acute success rates followed by high rates of recurrences due to the progressive nature of the disease leading to the development of multiple arrhythmogenic foci over time [104,105,106]. Moreover, regions of fibro-fatty replacement—that are regarded as arrhythmogenic substrate for VT—are mostly located in the subepicardial RV layers, thus partially explaining the failure of the traditional endocardial approach. Epicardial catheter ablation appears to be a feasible and more effective approach for patients in whom one or more endocardial procedures have been unsuccessful [102, 107]. Importantly, neither antiarrhythmic drugs nor catheter ablation proved to reduce the risk of SCD. Thus, they should be considered as measures to reduce the frequency of arrhythmic episodes rather than to improve prognosis. The only effective therapy for the prevention of SCD in such patients is ICD implantation [102].
ICD Implantation
Regarding AC recommendation of ICD implantation in AC patients, three risk categories (“high,” “moderate,” and “low”) have been defined. Those who have a history of cardiac arrest or hemodynamically unstable VT or who have severe ventricular dysfunction (either right or left or both ventricles) are considered “high-risk” subjects and receive a class I recommendation for ICD implantation.
Patients with major risk factors, such as syncope, non-sustained ventricular tachycardias, or moderate dysfunction of the right or left or both ventricles, are classified as “intermediate-risk” subjects, and receive a class IIa recommendation for ICD implantation. Recently, a score system including ECG, CMR, and degree of electrical instability has been proposed [97]. Since the presence of scars in AC may not affect the LV performance, but can still trigger adverse arrhythmic events, ICD implant for primary prevention should be considered in the presence of extensive LGE/fibrosis even if the LV systolic function is not severely depressed [102].
Heart Transplant
Heart transplant still represents the final therapeutic option for AC patients with advanced stages of the disease who suffer from refractory congestive heart failure and/or uncontrollable arrhythmic storms, despite previous attempts with catheter ablation and ICD therapy [102].
Right Ventricular Myocardial Changes in Specific Diseases
Different systemic or cardiac diseases can directly affect the right ventricle. A right ventricular involvement can be present in different cardiomyopathies having both genetic and non-inherited origin as hypertrophic cardiomyopathy, Fabry cardiomyopathy, DCM, or peripartum cardiomyopathy. Moreover, a RV involvement can be demonstrated in patients with systemic diseases as amyloidosis, sarcoidosis, or systemic sclerosis. Finally, RV physiologic changes can be detected in highly trained athletes.
Cardiac Sarcoidosis
The differential diagnosis between cardiac sarcoidosis and AC is often challenging because of both clinical and imaging features common to the two entities. In cardiac sarcoidosis life-threatening arrhythmias and heart failure can occur as a consequence of granulomatous infiltrates and fibrosis. The septum and the LV free wall are the most common locations at the LV level, while the RV free wall is involved in up to 40% of cases. Nevertheless, some peculiar features distinguish sarcoidosis from AC, and thus can help the diagnostic assessment. First, differently from AC, AV conduction delays are frequent because of the granulomatous infiltration of the interventricular septum. In addition to this, sarcoidosis is usually a systemic disease involving different organs such as lungs, skin, liver, and eyes. Conversely, cardiac isolated forms are less frequently found. Finally, advanced imaging techniques can offer some useful tips for the diagnosis. Extracardiac findings can be evidenced during the exam. At post-contrast sequences, LGE shows an intramural or patchy appearance, localizes mostly at the basal lateral wall, and is responsive to immunosuppressive therapy. When combined with positron-emission tomography (PET), the fluorodeoxyglucose uptake can reveal active inflammatory lesions [108,109,110].
Dilated Cardiomyopathy
DCM is currently defined by the presence of left ventricular or biventricular systolic dysfunction and dilatation that are not explained by abnormal loading conditions or coronary artery disease [111]. In DCM patients RV function may be reduced due to the same process leading to LV cardiomyopathy or hemodynamic consequences of LV dilation, dysfunction, or increased filling pressure. At the same time, the reduced RV function may worsen LV preload. The prevalence of RV dysfunction in DCM ranges from 34% to 65% [112]. In DCM patients RV function has a relevant prognostic value and CMR studies confirmed that RV function strongly predicts cardiac mortality in patients with HF. Thus assessment of RV size and function in DCM patients appears to be crucial for providing relevant prognostic and therapeutic information [112].
Hypertrophic Cardiomyopathy
Even if in patients with hypertrophic cardiomyopathy (HCM) myocardial hypertrophy mainly involves the LV and RV hypertrophy and dysfunction can also be present. The degree of RV wall thickness has been found to correlate significantly with LV wall thickness, even if RV hypertrophy is not associated with a particular pattern of LV hypertrophy [113]. Of note, HCM patients with severe RV hypertrophy have a poor clinical outcome and CMR studies demonstrated that RV hypertrophy is associated with RV LGE and that it is an independent predictor of cardiovascular event occurrence [114, 115]. Even in the presence of RV hypertrophy, detection of RV outflow tract obstruction in HCM patients is quite uncommon, not dynamic, and mainly due to RV hypertrophy. Regarding non-sarcomeric HCM, RV hypertrophy has been described also in Anderson-Fabry disease and has been proved to correlate with disease severity and LV hypertrophy [112].
References
Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res. 2017;121:784–802.
Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–300.
Lancisi GM. Arrhythmogenic right ventricular cardiomyopathy, De Motu Cordis et Aneurysmatibus. Naples, 1728.
Dalla-Volta S, Battaglia G, Zerbini E. ‘Auricularization’ of right ventricular pressure curve. Am Heart J. 1961;61:25–33.
Marcus FI, et al. Right ventricular dysplasia: a report of 24 adult cases. Ann Noninvasive Electrocardiol. 1999;4:97–111.
Thiene G, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–33.
Nava A, et al. Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol. 1988;12:1222–8.
Nava A, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–33.
Pilichou K, et al. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis. 2016;11:33.
Basso C, et al. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–91.
Thiene G, Basso C. Arrhythmogenic right ventricular cardiomyopathy: an update. Cardiovasc Pathol. 2001;10:109–17.
Sen-Chowdhry S, et al. Left-dominant arrhythmogenic cardiomyopathy. An under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–87.
Corrado D, et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int J Cardiol. 2020;319:106–14.
Basso C, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29:2760–71.
Protonotarios N, et al. Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J. 1986;56:321–6.
McKoy G, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355:2119–24.
Karmouch J, Protonotarios A, Syrris P. Genetic basis of arrhythmogenic cardiomyopathy. Curr Opin Cardiol. 2018;33:276–81.
Petros S, et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:356–64.
Bauce B, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005;26:1666–75.
Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16:927–35.
Norgett EE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–6.
Smith ED, et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation. 2020;141:1872–84.
Syrris P, et al. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype–phenotype characterization of familial disease. Eur Heart J. 2007;28:581–8.
Heuser A, et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79:1081–8.
Tsatsopoulou AA, Protonotarios NI, McKenna WJ. Arrhythmogenic right ventricular dysplasia, a cell adhesion cardiomyopathy: insights into disease pathogenesis from preliminary genotype-phenotype assessment. Heart. 2006;92:1720–3.
Van Hengel J, et al. Mutations in the area composita protein at-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–10.
Alice G, et al. Cadherin 2-related arrhythmogenic cardiomyopathy: prevalence and clinical features. Circ Genomic Precis Med. 2021;14:e003097.
Quarta G, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33:1128–36.
Glöcklhofer CR, et al. A novel LMNA nonsense mutation causes two distinct phenotypes of cardiomyopathy with high risk of sudden cardiac death in a large five-generation family. Europace. 2018;20:2003–13.
Augusto JB, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging. 2020;21:326–36.
Bermúdez-Jiménez FJ, et al. Novel Desmin mutation p.Glu401Asp impairs filament formation, disrupts cell membrane integrity, and causes severe arrhythmogenic left ventricular cardiomyopathy/dysplasia. Circulation. 2018;137:1595–610.
Merner ND, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–21.
Matthew T, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy–overlap syndromes. Circulation. 2011;124:876–85.
Ortiz-Genga MF, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–51.
Tiso N, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001;10:189–94.
Austin KM, et al. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2019; https://doi.org/10.1038/s41569-019-0200-7.
Te Riele ASJM, et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017;113:102–11.
Van Rijsingen IAW, et al. Outcome in phospholamban R14del carriers results of a large multicentre cohort study. Circ Cardiovasc Genet. 2014;7:455–65.
Eijgenraam TR, et al. The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unresponsive to standard heart failure therapy. Sci Rep. 2020;10:9819.
Xu T, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–97.
Rigato I, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:533–42.
Bauce B, et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010;7:22–9.
Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990;89:588–96.
Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003;42:1959–63.
Lemola K, et al. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: long term experience of a tertiary care centre. Heart. 2005;91:1167–72.
James CA, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy–associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–7.
Sawant AC, et al. Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J Am Heart Assoc. 2014;3:e001471.
Saberniak J, et al. Comparison of patients with early-phase arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract ventricular tachycardia. Eur Heart J Cardiovasc Imaging. 2017;18:62–9.
Ruwald A-C, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36:1735–43.
Lie ØH, et al. Harmful effects of exercise intensity and exercise duration in patients with arrhythmogenic cardiomyopathy. JACC Clin Electrophysiol. 2018;4:744–53.
Ruiz Salas A, et al. Impact of dynamic physical exercise on high-risk definite arrhythmogenic right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2018;29:1523–9.
Müssigbrodt A, et al. Effect of exercise on outcome after ventricular tachycardia ablation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Int J Sports Med. 2019;40:657–62.
Paulin FL, et al. Exercise and arrhythmic risk in TMEM43 p.S358L arrhythmogenic right ventricular cardiomyopathy. Hear. Rhythm. 2020;17:1159–66.
Kirchhof P, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–806.
Cruz FM, et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J Am Coll Cardiol. 2015;65:1438–50.
Corrado D, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a Preparticipation screening program. JAMA. 2006;296:1593–601.
Pelliccia A, et al. 2020 ESC guidelines on sports cardiology and exercise in patients with cardiovascular disease: the task force on sports cardiology and exercise in patients with cardiovascular disease of the European Society of Cardiology (ESC). Eur Heart J. 2021;42:17–96.
Maron BJ, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 3: hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e273–80.
McKenna WJ, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society. Br Heart J. 1994;71:215–8.
Marcus FI, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–41.
Elliott PM, et al. Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. Eur J Heart Fail. 2019;21:955–64.
Corrado D, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41:1414–29.
di Gioia CRT, et al. Nonischemic left ventricular scar and cardiac sudden death in the young. Hum Pathol. 2016;58:78–89.
Nunes de Alencar Neto J, Baranchuk A, Bayés-Genís A, Bayés de Luna A. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: an electrocardiogram-based review. Europace. 2018;20:f3–f12.
Te Riele ASJM, et al. Malignant arrhythmogenic right ventricular dysplasia/cardiomyopathy with a normal 12-lead electrocardiogram: a rare but underrecognized clinical entity. Heart Rhythm. 2013;10:1484–91.
Steriotis AK, et al. Electrocardiographic pattern in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2009;103:1302–8.
Borys Surawicz TK. Chou’s electrocardiography in clinical practice: adult and pediatric—Borys Surawicz. Timothy Knilans—Google Libri.
De Lazzari M, et al. Relationship between electrocardiographic findings and cardiac magnetic resonance phenotypes in arrhythmogenic cardiomyopathy. J Am Heart Assoc. 2018; https://doi.org/10.1161/JAHA.118.009855.
Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J Am Coll Cardiol. 2018;72:784–804.
Hoffmayer KS, et al. Electrocardiographic comparison of ventricular arrhythmias in patients with arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2011;58:831–8.
Mattesi G, Zorzi A, Corrado D, Cipriani A. Natural history of arrhythmogenic cardiomyopathy. J Clin Med. 2020;9:878.
Bhonsale A, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36:847–55.
Bhonsale A, et al. Cardiac phenotype and long-term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm. 2017;14:883–91.
Migliore F, et al. Prognostic value of endocardial voltage mapping in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Arrhythmia Electrophysiol. 2013;6:167–76.
Yoerger DM, et al. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia. J Am Coll Cardiol. 2005;45:860–5.
Borgquist R, et al. The diagnostic performance of imaging methods in ARVC using the 2010 task force criteria. Eur Heart J Cardiovasc Imaging. 2014; https://doi.org/10.1093/ehjci/jeu109.
Haugaa KH, et al. Comprehensive multi-modality imaging approach in arrhythmogenic cardiomyopathy—an expert consensus document of the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2017;18:237–53.
López-Fernández T, Ángel Garcı́a-Fernández M, Pérez David E, Moreno Yangüela M. Usefulness of contrast echocardiography in arrhythmogenic right ventricular dysplasia. J Am Soc Echocardiogr. 2004;17:391–3.
Sarvari SI, et al. Right ventricular mechanical dispersion is related to malignant arrhythmias: a study of patients with arrhythmogenic right ventricular cardiomyopathy and subclinical right ventricular dysfunction. Eur Heart J. 2011;32:1089–96.
Teske AJ, et al. Early detection of regional functional abnormalities in asymptomatic ARVD/C gene carriers. J Am Soc Echocardiogr. 2012;25:997–1006.
Prakasa KR, et al. Feasibility and variability of three dimensional echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2006;97:703–9.
te Riele ASJM, Tandri H, Bluemke DA. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J Cardiovasc Magn Reson. 2014;16:50.
Tandri H, et al. Magnetic resonance imaging findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2003;14:476–82.
Basso C, Thiene G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovasc Pathol. 2005;14:37–41.
Tandri H, et al. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia. Sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006;48:2277–84.
Tansey DK, Aly Z, Sheppard MN. Fat in the right ventricle of the normal heart. Histopathology. 2005;46:98–104.
Angelini A, Basso C, Nava A, Thiene G. Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J. 1996;132:203–6.
Marcus FI, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2010;121:1533–41.
Asimaki A, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–84.
Collett BA, Davis GJ, Rohr WB. Extensive fibrofatty infiltration of the left ventricle in two cases of sudden cardiac death. J Forensic Sci. 1994;39:1182–7.
Aquaro GD, et al. Cardiac MR with late gadolinium enhancement in acute myocarditis with preserved systolic function: ITAMY study. J Am Coll Cardiol. 2017;70:1977–87.
Aquaro GD, et al. Prognostic value of repeating cardiac magnetic resonance in patients with acute myocarditis. J Am Coll Cardiol. 2019;74:2439–48.
Spezzacatene A, et al. Arrhythmogenic phenotype in dilated cardiomyopathy: natural history and predictors of life-threatening arrhythmias. J Am Heart Assoc. 2015;4:e002149.
Cipriani A, et al. Arrhythmogenic right ventricular cardiomyopathy: characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. J Am Heart Assoc. 2020; https://doi.org/10.1161/JAHA.119.014628.
Bariani R, et al. ‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy. Europace. 2020; https://doi.org/10.1093/europace/euaa343.
Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376:61–72.
Cadrin-Tourigny J, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2019;40:1850–8.
Aquaro GD, et al. Prognostic value of magnetic resonance phenotype in patients with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2020;75:2753–65.
Wang W, et al. Arrhythmic outcome of arrhythmogenic right ventricular cardiomyopathy patients without implantable defibrillators. J Cardiovasc Electrophysiol. 2018;29:1396–402.
Saberniak J, et al. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur J Heart Fail. 2014;16:1337–44.
Priori SG, et al. ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–867.
Corrado D, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Circulation. 2015;132:441–53.
Van Der Voorn SM, et al. Arrhythmogenic cardiomyopathy: pathogenesis, pro-arrhythmic remodelling, and novel approaches for risk stratification and therapy. Cardiovasc Res. 2020;116:1571–84.
Marchlinski FE, et al. Electroanatomic substrate and outcome of catheter ablative therapy for ventricular tachycardia in setting of right ventricular cardiomyopathy. Circulation. 2004;110:2293–8.
Verma A, et al. Short- and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation. 2005;111:3209–16.
Dalal D, et al. Morphologic variants of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. A genetics-magnetic resonance imaging correlation study. J Am Coll Cardiol. 2009;53:1289–99.
Assis FR, Tandri H. Epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy. Card Electrophysiol Clin. 2020;12:329–43.
Vita T, et al. Complementary value of cardiac magnetic resonance imaging and positron emission tomography/computed tomography in the assessment of cardiac sarcoidosis. Circ Cardiovasc Imaging. 2018;11:e007030.
Philips B, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy and cardiac sarcoidosis. Circ Arrhythmia Electrophysiol. 2014;7:230–6.
Yatsynovich Y, Dittoe N, Petrov M, Maroz N. Cardiac sarcoidosis: a review of contemporary challenges in diagnosis and treatment. Am J Med Sci. 2018;355:113–25.
Pinto YM, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37:1850–8.
Cavigli L, et al. The right ventricle in “left-sided” cardiomyopathies: the dark side of the moon. Trends Cardiovasc Med. 2020; https://doi.org/10.1016/j.tcm.2020.10.003.
Maron MS, et al. Right ventricular involvement in hypertrophic cardiomyopathy. Am J Cardiol. 2007;100:1293–8.
Guo X, et al. The clinical features, outcomes and genetic characteristics of hypertrophic cardiomyopathy patients with severe right ventricular hypertrophy. PLoS One. 2017;12:e0174118.
Nagata Y, et al. Right ventricular hypertrophy is associated with cardiovascular events in hypertrophic cardiomyopathy: evidence from study with magnetic resonance imaging. Can J Cardiol. 2015;31: 702–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bariani, R., Mattesi, G., Cipriani, A., Bauce, B. (2021). Right Ventricular Cardiomyopathies. In: Gaine, S.P., Naeije, R., Peacock, A.J. (eds) The Right Heart. Springer, Cham. https://doi.org/10.1007/978-3-030-78255-9_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-78255-9_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-78254-2
Online ISBN: 978-3-030-78255-9
eBook Packages: MedicineMedicine (R0)