
Abstract
Clonal heterogeneity in cell populations with respect to properties such as growth rate, motility, metabolism or signaling, has been observed for some time. Unraveling the dynamics and the mechanisms giving rise to such variability has been the goal of recent work, largely aided by quantitative/ mathematical tools. Quantitative evaluation of cell-to-cell variability (heterogeneity) poses technical challenges that only recently are being overcome. Clearly, a mathematical theory of cellular heterogeneity could have fundamental implications. For instance, a theory of cell population growth variability, coupled with experimental measurements, may in the long term be crucial for an in-depth understanding of physiological processes such as stem cell expansion, embryonic development, tissue regeneration, or of pathological ones (e.g., cancer, fibrosis, tissue degeneration). We focus on recent advances, both theoretical and experimental, in quantification and modeling of the clonal variability of proliferation rates within cell populations. Our aim is to highlight a few stimulating examples from this fledgling and exciting field, in order to frame the issue and point to challenges and opportunities that lie ahead. Furthermore, we emphasize work carried out in cancer-related systems.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Heterogeneity and Growth Variability
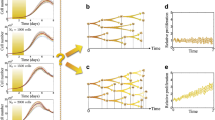
Clonal heterogeneity in cell populations with respect to properties such as growth (Fig. 1), motility (Fig. 2), metabolism or signalling (Fig. 3), has been observed for some time (see, e.g., Altschuler [1], Quaranta [12] and references therein). However, quantitative evaluation of this cell-to-cell variability (heterogeneity) poses technical challenges that only recently are being overcome [1, 12]. Furthermore, mathematical foundations for interpreting these quantitative experimental observations of heterogeneity are in need of development. Far from being exclusively academic, a mathematical theory of cellular heterogeneity could have fundamental implications, similar to a theory on population biology or ecology [8]. For instance, a theory of cell population growth variability, coupled to experimental measurements, may in the long term be crucial for an in-depth understanding of physiological processes such as stem cell expansion, embryonic development, tissue regeneration, or of pathological ones (e.g., cancer, fibrosis, tissue degeneration).

Cell-to-cell variability of intermitotic times within human cultured cell lines. Note that the heterogeneity of intermitotic times within seemingly homogeneous isogenic cell lines (populations) is quite broad, and distributed in non-Gaussian fashion. Intermitotic time encompasses hours from the end of one cell division to the start of the successive one. Single cells were tracked by automated confocal microscopy collecting images at regular intervals by automated microscopy as described [19]. Intermitotic times were calculated as described [19] and fitted to an exponentially modified Gaussian (EMG) distribution http://en.wikipedia.org/wiki/Exponentially_modified_Gaussian_distribution. Human cultured cell lines are as follows: A375, melanoma; PC9, non-small cell lung carcinoma; MCF10A, immortalized non-tumorigenic breast epithelium; CA1D, H-Ras transformed MCF10A. \(n\) = number of cells tracked; \({\mu }, {\sigma }\) and \({\kappa }\) are parameters for the EMG distribution; ks p-value was calculated by the Kolmogorov-Smirnoff statistic test.

Cell-to-cell speed variation within mammary gland human cell lines. Spontaneous, non-directed motility was tracked in over 1,500 individual cells from one immortalized (MCF10A) and two transformed MCF10A-derived (AT1 and CA1d) breast epithelial cell lines. Cell-to-cell variability of motility was evaluated with respect to speed under two culture conditions, full-supplement or serum/EGF-depleted media, respectively. a Box-and-whisker plot of individual cell speed (color-coded by individual experiment). b Population histogram of frequency (the number of cells) and the normal (Gaussian) fit for each set of data (based around the average). Shapiro-Wilks W tests confirmed that distributions are non-normal and positively skewed (more cells are likely to move at lower speeds) with long tails (at higher speeds).

Single cell variability in metabolic and signaling activity. a Single-cell measurements of glucose uptake using 2- deoxy- 2- [(7- nitro- 2, 1, 3- benzoxadiazol- 4- yl)amino]- D- glucose (2-NBDG). Fluorescent representative images of CA1d (right, higher magnification) after 10 min incubation with 300\({\mu }\)M of 2-NBDG as described in [7]. The variability in subcellular distribution of the probe was apparent in CA1d cells (right panel). b Staining patters of BT-474 lapatinib resistant cell line reveals variability (heterogeneity). BT-475LR cell lines were plated overnight and treated with 1 \({\mu }\)M lapatinib for 1 h at 37 C. Cells were fixed and stained with fluorescent probes (DNA/pAKT(pS473)/Ac-Histone3) and imaged with a Zeiss confocal microscope (LSM 510).
Here, we focus on recent advances, both theoretical and experimental, in quantification and modeling of the clonal variability of proliferation rates within cell populations. Our aim is to highlight a few stimulating examples from this fledgling and exciting field, in order to frame the issue and point to challenges and opportunities that lie ahead. Furthermore, we emphasize work carried out in cancer-related systems. As our aim is not an exhaustive review, we apologize in advance for inevitable omissions.
Variability of growth rates, among other indicators of heterogeneity in growth kinetics of individual tumours, has long been detected, but precision in quantification may have been made possible only in the past few years by methods developed by, among others, Quaranta and his group (see [12, 19]). For instance, a team from Verona, Italy, quantified growth variability of tumour cell clones from a human leukaemia cell line, by cloning Molt3 cells, and measuring the growth of 201 clonal populations by microplate spectrophotometry. Growth rate of each clonal population was estimated by fitting data with the logistic equation for population growth [18]. Their results indicated that growth rates vary between clones. Six clones with growth rates above or below the mean growth rate of the parent population were further cloned, and the growth rates of their offspring were measured. Researchers noted that distribution of subclone growth rates did not significantly differ from that of the parent population, supporting the conjecture that growth variability has an epigenetic origin [18]. Such variability in growth rates may be amenable to further quantitative analysis of population dynamics with analytic tools developed in Tyson et al. [19].
In the paper “Characterizing heterogeneous cellular responses to perturbations” [14], Slack et al. approached the challenge of heterogeneity with a mathematically-appealing assumption that cell populations may be described as mixtures of a limited number of phenotypically distinct subpopulations. Methods for characterizing spatial heterogeneity observed within cell populations are developed, starting from the extraction of phenotypic measurements of the activation and colocalization patterns of cellular readouts from large numbers of cells in diverse conditions. Phenotypic stereotypes are identified within the total population, and probabilities assigned to cells belonging to subpopulations modeled on these stereotypes. Each population or condition may then be characterized by a probability vector—its subpopulation profile—estimating the number of cells in each subpopulation. Responses of heterogeneous cellular populations to perturbations (e.g., anti-cancer drugs) are summarized as probabilistic redistributions of these mixtures. In the study by Slack et al., this computational method was applied to heterogeneous responses of cancer cells to a panel of drugs. The finding is that cells treated with drugs of similar mechanism exhibited the same pattern of heterogeneity redistribution.
In subsequent work from this group, Singh et al. [13] employed the same computational framework to investigate whether patterns of basal signaling heterogeneity in untreated cell populations could distinguish cellular populations with different drug sensitivities. As in the earlier study, cellular heterogeneity in populations was modeled as a mixture of stereotyped signaling states. Interestingly, the researchers found that patterns of heterogeneity could be used to separate the most sensitive and most resistant populations to the drug paclitaxel within a set of H460 lung cancer clones and within the NCI-60 panel of cancer cell lines, but not for a set of less heterogeneous immortalized noncancer HBEC (human bronchial epithelial cell) clones. Stockholm et al. [17] used both computer simulation and experimental analysis to address the issue of the origin of phenotypic differentiation in clonal populations. Two models—referred to as the “extrinsic” and “intrinsic” models—explaining the generation of diverse cell types in a homogeneous population, were tested using simple multi-agent computer modeling. The approach takes each cell as an autonomous “agent”, and following defined rules governing the action of individual agents, the behavior of the system emerges as an outcome of the agents’ collective action.
As the term suggests, the “extrinsic” model attributes the occurrence of a phenotypic switch to extrinsic factors. Identical cells may become different because they encounter different local environments that induce alternative adaptive responses. Changing its phenotype, the cell contributes to changes in the local environment, inducing responses in surrounding cells, and ultimately influencing the dynamics of the cell population. The second model assumes that the phenotype switch is intrinsic to the cells. Phenotypic changes could occur even in a homogenous environment and may result from asymmetric segregation of intrinsic fate determinants during cell division that lead to the change in gene expression patterns, [17].
The Stockholm study cites an experiment where two subpopulations appear spontaneously in C2C12 mouse myogenic cells—the main population (MP), and a side population (SP). The two cell types are phenotypically distinct, and researchers take off from the lab experiment to perform agent-based modeling computer simulation on two cell types subject to two sets of hypotheses (the extrinsic and intrinsic models). The models are built on a limited number of simplified assumptions about how individual cells migrate, interact with each other, divide and die. The agent-based model assumes that each cell divides at each iteration step but survival of daughter cells depends on local cell density. In the intrinsic model, the phenotypic switch occurs under the assumption of cell autonomy, with the environment playing no ostensible role in the switching; rather, switching from one cell type to the other occurs at fixed probabilities. In the extrinsic model, local cell density determines phenotypic switching, hence local density is surrogate for the complex of factors affecting cell survival, such as gradient of nutrients, oxygen, secreted factors, etc, and cell types represent two forms of adaptation to high and low density environments. The extrinsic and intrinsic hypotheses were implemented by varying the parameters (assuming cell migration velocities within experimentally guided limits of values). Simulations for the intrinsic model result in the two cell types being distributed randomly both during growth and equilibrium, suggesting that the randomness of cell type spatial distribution is characteristic of the intrinsic model. On the other hand, the spatial distribution of cells resulting from simulations of the extrinsic model is different from that in the intrinsic model, with cluster formation as an observed feature. Moreover, this feature is robust in the range of parameter values considered.
Both intrinsic and extrinsic models generate in the simulations heterogeneous cell populations with a stable proportion of the two cell types. Experimental verification of model predictions, using the C2C12 myogenic cell line, indicated that neither one of the models can fully account for the spatial distribution of the cell types at equilibrium, as some clustering of the rare SP cell was observed in low density regions, while distribution in high density regions was generally uniform. A hybrid model combining both intrinsic and extrinsic hypotheses was in better agreement with the clustering behavior of the rare SP cells. In the end, it is not solely the local environment, nor, on the other hand, merely a cell-autonomous propensity for differentiation that activates the phenotype switch. Rather, it may be a combination of the two.
A similar “agent” model framework is utilized in mathematical models of cancer invasion, with emphasis on tumor microenvironment, compared in [11]. In that review, three independent computational models for cancer progression are discussed, all pointing to an essential role of the tumor microenvironment (mE) “in eliciting invasive patterns of tumor growth and enabling dominance of aggressive cell phenotypes.” Both the evolutionary hybrid cellular automata (EHCA) and the Hybrid Discrete Continuum (HDC) models treat cells as points on a lattice. In the case of the EHCA, the grid itself represents the mE, and the only variable on the grid, apart from cells, is the concentration of oxygen, with a partial differential equation controlling the oxygen dynamics in space and time. In the HDC model, the mE consists of a two-dimensional lattice of extracellular matrix upon which oxygen diffuses and is produced/consumed, and matrix degrading proteases are produced/used. The HDC model has the mE variables controlled by reaction-diffusion equations with tumor cells occupying discrete lattice points. Notably, a key feature of the HDC model is that the tumor cell population is heterogeneous, each cell phenotype being defined from a pool of 100 pre-defined phenotypes within a biologically relevant range of cell-specific traits. Mutation is incorporated into the model by assigning to cells a small probability of changing some traits at cell division. If a change occurs, the cell is randomly assigned a new phenotype from the pool of about 100. Taken together with a third model—the Immersed Boundary method (IBCell)—the models describe the process of cancer invasion on multiple scales: The EHCA at the molecular (gene expression) scale, the IBCell at the cell scale, the HDC at the tissue scale. Though not highlighted, heterogeneity is an issue addressed in the models, with the microenvironment driving cancer progression in a major way, and on multiple scales. From representative simulations of the models (see [11] for details), analysis of the effect of mE variables on tumor growth point to “competitive adaptation to mE conditions as a determining factor for invasion: both invasive tumor morphology (“fingering”) and evolution of dominant aggressive clonal phenotypes appear to occur by a process of progressive cell adaptation to mE’s that support sustained competition between distinct cancer cell phenotypes.”
In their 2011 paper [6] on models of heterogeneous cell populations, Hasenauer et al. discuss a framework for modeling genetic and epigenetic differences among cells. With the approach to intracellular biochemical reaction networks modeled by systems of differential equations (which may characterize metabolic networks and signal transduction pathways), heterogeneity in populations is accounted for by differences in parameter values and initial conditions. Using population snapshot data, a Bayesian approach is used to infer parameter density of the model describing single cell dynamics. Using maximum likelihood methods, single cell measurement data is processed for parameter density estimation; the proposed framework includes a noise model, as well as methods for determining uncertainty of the parameter density. For computational tractability, the population model is converted into a density-based model, where the variables are not states of single cells but density of the output (see [6] for details). Towards verifying efficacy of the proposed modeling framework, the model of TNF (tumor necrosis factor) signaling pathway was studied under a hypothetical experimental set-up with artificial data involving a cell population responding to the TNF stimulus. The model, introduced in [3], is based on known inhibitory and activating interactions among key signaling proteins of the TNF pathway. Cellular response to the TNF stimulus has been observed to be highly heterogeneous within a clonal population. Heterogeneity at the cell level is modeled by differences in two parameter values, one quantifying the inhibitory effect of NF-êB via the C3a inhibitor XIAP onto the C3 activity, and the other the activation of I-kB via NF-kB. The authors conclude that the method yields good estimation results.
In the abovementioned framework, the assumption was that network structure was identical in all cells and spatial effects and stochasticity of the biochemical reactions are negligible. Moreover, the mechanisms for cell-to-cell interactions typically characterized by differential equations, are reasonably well-understood and formulated, from actual experiment.
In an effort to uncover sources of cell-to-cell variation, Colman-Lerner et al. [4] looked into cell-to-cell variability of a prototypical eukaryotic cell fate decision system, the mating pheromone response pathway in yeast. Cell-to-cell variation was quantified by the output in the cell-fate decision system—the pheromone response pathway in the yeast Saccharomyces cerevisiae. The fate decision to switch from the normal vegetative growth to mating events including gene transcription, cell cycle arrest, etc. is induced by the alpha-factor, a pheromone secreted by cells of the mating type. Pheromone-induced expression of fluorescent protein reporter genes was used as a readout. To dis-aggregate differences due to the operation of the signal transduction pathway from cell-to-cell differences in gene expression from the reporters, yeast strains containing genes for the yellow and cyan fluorescent protein were generated. The analytical framework used considered the alpha-factor response pathway and the reporter gene expression mechanism to measure its activity as a single system, with two connected subsystems—pathway and expression. In each of the two subsystems, two sources of variation are considered—stochastic fluctuations and cell-to-cell differences in “capacity”, depending on number, localization and activity of proteins that transmit the signal (pathway capacity) or express genes into proteins (expression capacity). About half of the observed variation was attributed to pre-existing differences in cell cycle position at the time of pathway induction, while another large component of the variation in system output is due to differences in cell capacity to express proteins from genes. Very little variation is due to noise in gene expression. Although the study did not specifically refer to molecular mechanisms underlying cell-to-cell variation, it does provide a basis for further investigation into these mechanisms, including, as mentioned elsewhere, network architecture.
Heterogeneous cell populations have been the subject of mathematical modeling since about the 1960s, with the cell population balance (CPB) approach by Frederickson and a few others (see [16] for references). The models use partial integro-differential equations for the dynamics of the distribution of the physiological state of cells and ordinary integro- differential equations to describe substrate availability. For CPB models, heterogeneity arises from physiological functions leading to different growth and division rates of the cells, as well as for unequal partitioning effects. When the physiological state vector (whose components include intracellular content, morphometric characteristics like size) has two or more components, the approach leads to multidimensional models that are highly unwieldy computationally. Stamatakis notes that CPB models cannot account for the inherent stochasticity of chemical reactions occurring in cellular control volumes or stochastic DNA-duplication. To account for this stochasticity, refinements were considered by Gillespie and others (see [16] for references) using the chemical master equation. A relatively recent approach, referred to as the Langevin approach, uses stochastic differential equations in modeling stochasticity in intracellular reactions. In recent work Stamakis and Zygourakis (2010) [16] propose a mathematical framework to account for all the various sources of cell population heterogeneity, namely growth rate variability, stochasticity in DNA duplication and cell division, and stochastic reaction occurrences for the genetic network, through the cell population master equation (CPME) that governs the temporal dynamics of the probability of finding the cell population at a specific state, together with a Monte Carlo algorithm that enables simulation of exact stochastic paths of this master equation. Employing the population balance framework, each cell is described by a state vector containing information about its chemical content and morphometric characteristics such as length, etc (Stamatakis uses volume only). The state of the overall population is given by a vector w, which reflects the number v of individual cells and the state of each vector. The master equation is derived as a probability balance describing the evolution of a probability distribution for the cell population, using submodels of probability inflows and probability outflows accounting for chemical reactions, DNA duplication, cell growth (here using exponential growth), a propensity function (for cells to divide).
In an earlier study, Mantzaris [9] also looked into models of cell population heterogeneity, incorporating into a prior deterministic single-cell model, two extra parameters (one, a rate of operator fluctuations) to quantify two main sources of stochasticity at the single cell level for the reaction network, namely small number of molecules and slow operator fluctuations. Starting from a deterministic cell population balance model (DCPB), Mantzaris used stochastic differential equations to refine the CPB model (to account for extrinsic and intrinsic sources of population heterogeneity—respectively, the unequal partitioning between daughter cells of intracellular components on division, and random fluctuations in reaction rates regulated by a small number of regulatory molecules) through the Stochastic Variable Number Monte Carlo method/model. Simulations on a genetic network with positive feedback revealed differences arising from different sources of stochasticity on regions of the parameter space where the system is bistable.
Although much of the modeling of heterogeneity has not specifically investigated implications on cancer treatment, a 2012 study (see [10]) looks into cell-cycle heterogeneity and its effects on solid tumor response to chemotherapy. In their paper, Powathil et al. raise the difficulty of treating cancer with chemotherapeutic drugs due to the development of cell-cycle mediated drug resistance. Elsewhere (see references in [10]) it has been suggested that this may be due to the presence of functionally heterogeneous cells and can be addressed to some extent by using combinations of chemotherapy drugs that target different phases of the cell-cycle kinetics. Hence, it is important to study and analyze the underlying heterogeneity within a cell and within a solid tumour due to the presence of the unfavourable microenvironment and the cellcycle position. A hybrid multi-scale cellular automaton model is used to simulate the spatio-temporal dynamics at the cell level, incorporating feedbacks between these cell level dynamics and molecular variations of intercellular signalling and macroscopic behaviour of tissue oxygen dynamics. Each cell has its own cell-cycle dynamics and this is incorporated into the CA model for cellular proliferation using a set of ordinary differential equations, from an early model by Tyson and Novak [20]. Chemical processes within the cell are quantified using concentration of key chemical components, considered as functions of time, and a 6-variable system of differential equations describe the processes of production, destruction and interactions. These kinetic relations are then used to explain transitions between two steady states—the G1 and the S-G2-M state, assumed to be controlled by cell mass. With cells located spatially in the dynamic microenvironment, depending on variations in oxygen concentration and with drug distribution dynamics in the growing tumor also affecting the state of individual cells, partial differential equations (for oxygen, a reaction diffusion equation) model changes in oxygen and drug concentration. In simulating the model, parameters were chosen based on earlier work (mainly from Tyson and Novak); notably, to account for the “natural” variability between cell growth rates, and to have a non-synchronous cell population, a multiple of the value from a probability density function with uniform distribution between -1 and 1 is added to an identified value for growth rate, effectively incorporating cell cycle heterogeneity. Computational simulations were run first on cell-cycle and oxygen tumor growth, assuming zero drug concentration, and subsequently on tumours treated with cell-cycle specific drugs. The results revealed that cytotoxic effect of combination therapy depends on timing of drug delivery, time-delay between doses of chemotherapeutic drugs, and cell-cycle heterogeneity. Not surprisingly, drug effectiveness also depends on distribution of tumor cell mass as it affects the tumor microenvironment and drug distribution. The current direction towards patient specific optimal treatment strategies seems to be supported by the model simulations. It is worth noting that non-synchronous cell population can be parameterized from experimental data [5] due to recent automated microscopy advances, making it possible to validate models such as the one described by Powathil et al. [10].
In a recent review by Bendall and Nolan [2], the authors assert that “stem cell hierarchies, transcription start sites, cell signaling pathways (and more) all function against a backdrop that assumes that carefully orchestrated single-cell stochastics, in concert with mass action, is what determines outcome.” Since all kinds of heterogeneity may drive treatment decisions, it is crucial to develop better technologies to study heterogeneity in single-cells. Notably, the statement is made that recent research indicate that the biology of single cells “is rarely deterministic.” Snijder and Pelkmans [15] take the view instead that “a large part of phenotypic cell-to-cell variability is the result of deterministic regulatory processes.” Although not necessarily in conflict, these seemingly opposing views point to the necessity to further investigate various and diverse aspects and mechanisms driving phenotypic heterogeneity in cells and cell populations. As Snijder points out, population context has been shown to contribute in major ways to cellular behavior, including sporulation, genetic competence and motility, giving rise to adaptation in gene transcription, protein translation, cellular growth, rate of proliferation, sensitivity to apoptosis, metabolic activity, cell shape and/or cell polarization. These adaptations cause cells themselves to alter population context, eventually determining single-cell distribution of phenotype properties in a population. Such complex feedback/ regulatory mechanisms may involve many entities and interactions, in the absence of a full understanding of which, a stochastic distribution may somewhat account for the variability [21].
2 Conclusions
What emerges from the models so far developed is that apparently “stochastic/variable behavior” in single cells and populations can be reasonably quantified, if not fully understood. In many of the above-mentioned mathematical models for population heterogeneity, the key to characterization of population behavior is a fairly holistic understanding of the key “players” (cells), their environment, and reactions and feedback mechanisms among components. Integration of these theoretical and quantitative tools will be paramount for distinguishing between relevant and noisy heterogeneity [1]. While this field of investigation is still in its infancy, it is not difficult to imagine the impact it will have on our understanding of cellular response to perturbations, including drugs.
References
S.J. Altschuler, L.F. Wu, Cellular heterogeneity: do differences make a difference? Cell 141 (4), 63–559 (2010)
S.C. Bendall, G.P. Nolan, From single cells to deep phenotypes in cancer. Nat. Biotechnol. 30(7), 47–639 (2012)
M. Chaves, T. Eissing, F. Allgöwer, Bistable biological systems: a characterization through local compact input-to-state stability. IEEE Trans. Autom. Control. (8ADNov.) 13(53), 87–100 (2008)
A. Colman-Lerner, A. Gordon, E. Serra, T. Chin, O. Resnekov, D. Endy, et al. Regulated cell-to-cell variation in a cell-fate decision system. Nature 2005th ed. 437(7059), 699–706 (2005)
P. Gabriel, S.P. Garbett, V. Quaranta, D.R. Tyson, G.F. Webb, The contribution of age structure to cell population responses to targeted therapeutics. J. Theor. Biol. 311, 19–27 (2012)
J. Hasenauer, S. Waldherr, M. Doszczak, N. Radde, P. Scheurich, F. Allgöwer, Identification of models of heterogeneous cell populations from population snapshot data. BMC Bioinform. 12, 125 (2011)
M. Hassanein, B. Weidow, E. Koehler, N. Bakane, S. Garbett, Y. Shyr et al. Development of high-throughput quantitative assays for glucose uptake in cancer cell lines. Mol. Imaging. Biol. 13(5), 52–840 (2011)
C.J. Krebs, in The problem of abundance: Populations, ed. by B Cummings. Ecology: The Experimental Analysis of Distribution And Abundance, 6th edn. Benjamin/Cummings, Menlo Park, 2008), p. 111
N.V. Mantzaris, From single-cell genetic architecture to cell population dynamics: quantitatively decomposing the effects of different population heterogeneity sources for a genetic network with positive feedback architecture. Biophys. J. 92(12), 88–4271 (2007)
G.G. Powathil, K.E. Gordon, L.A. Hill, M.A.J. Chaplain, Modelling the effects of cellcycle heterogeneity on the response of a solid tumour to chemotherapy: biological insights from a hybrid multiscale cellular automaton model. J. Theor. Biol. 308, 1–19 (2012)
V. Quaranta, K.A. Rejniak, P. Gerlee, Anderson AR. Invasion emerges from cancer cell adaptation to competitive microenvironments: quantitative predictions from multiscale mathematical models. Semin. Cancer Biol. 2008th ed. 18(5), 338–48 (2008)
V. Quaranta, D.R. Tyson, S.P. Garbett, B. Weidow, M.P. Harris, W. Georgescu, Trait variability of cancer cells quantified by high-content automated microscopy of single cells. Meth. Enzymol. 467, 23–57 (2009)
D.K. Singh, C.-J. Ku, C. Wichaidit, R.J. Steininger, L.F. Wu, S.J. Altschuler, Patterns of basal signaling heterogeneity can distinguish cellular populations with different drug sensitivities. Mol. Syst. Biol. 6, 369 (2010)
M.D. Slack, E.D. Martinez, L.F. Wu, S.J. Altschuler, Characterizing heterogeneous cellular responses to perturbations. Proc. Natl. Acad. Sci. USA 105(49), 11–19306 (2008)
B. Snijder, L. Pelkmans, Origins of regulated cell-to-cell variability. Nat. Rev. Mol. Cell Biol. 12(2), 25–119 (2011)
M. Stamatakis, K. Zygourakis, A mathematical and computational approach for integrating the major sources of cell population heterogeneity. J. Theor. Biol. 266(1), 41–61 (2010)
D. Stockholm, R. Benchaouir, J. Picot, P. Rameau, T.M.A. Neildez, G. Landini et al., The origin of phenotypic heterogeneity in a clonal cell population in vitro. PLoS ONE 2(4), e394 (2007)
C. Tomelleri, E. Milotti, C. Dalla Pellegrina, O. Perbellini, A. Del Fabbro, M.T. Scupoli, A quantitative study of growth variability of tumour cell clones in vitro. Cell Prolif. 2008th ed. 41(1), 177–191 (2008)
D.R. Tyson, S.P. Garbett, P.L. Frick, V. Quaranta, Fractional proliferation: a method to deconvolve cell population dynamics from single-cell data. Nat. Methods 2012th ed. 9, 923–928 (2012)
J.J. Tyson, B. Novak, Regulation of the eukaryotic cell cycle: molecular antagonism, hysteresis, and irreversible transitions. J. Theor. Biol. 210(2), 63–249 (2001)
H. Youssefpour, X. Li, A.D. Lander, J.S. Lowengrub, Multispecies model of cell lineages and feedback control in solid tumors. J. Theor. Biol. 304, 39–59 (2012)
Acknowledgments
We would like to thank the contributors of data included: Fig. 1—Darren R. Tyson (Center for Cancer Systems Biology Center and Department of Cancer Biology, Vanderbilt School of Medicine) and Peter L. Frick (Center for Cancer Systems Biology Center and Chemical and Physical Biology Program, Vanderbilt School of Medicine); Fig. 2—Mark P. Harris (NextGxDx, Nashville, TN) and Shawn P. Garbett (Center for Cancer System Biology and Department of Cancer Biology, Vanderbilt School of Medicine); and Fig. 3—Mohamed Hassanein (Pulmonary Medicine, Vanderbilt School of Medicine) and Hironobu Yamashita (Department of Gynecology and Obstetrics, Tohoku university, Sendai, Japan). We would also like to thank all the members of the Quaranta laboratory for useful discussions and Lourdes Estrada for discussing key aspects of the manuscript. Furthermore, we would like to acknowledge the support from the Integrative Cancer Biology Program (ICBP) U54CA113007 (V.Q.) and the Philippines’ Commission on Higher Education for funding Dr. Priscilla Macansantos’ visit to Vanderbilt School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this paper
Cite this paper
Macansantos, P., Quaranta, V. (2014). Quantitative Approaches to Heterogeneity and Growth Variability in Cell Populations. In: Delitala, M., Ajmone Marsan, G. (eds) Managing Complexity, Reducing Perplexity. Springer Proceedings in Mathematics & Statistics, vol 67. Springer, Cham. https://doi.org/10.1007/978-3-319-03759-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-03759-2_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-03758-5
Online ISBN: 978-3-319-03759-2
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)