
Abstract
The beneficial effects of nanostructured material systems have provided a significant momentum to accomplish high-efficiency thermoelectric materials for power generation and cooling applications. The quantum size effects have been widely explored in order to shrink the contribution of lattice thermal conductivity of the thermoelectric systems, thereby enhancing the overall figure-of-merit. Modifying the nanoscale level structural features and the creation of additional phonon scattering sites in the form of grain boundary interfaces became the basis for fabrication of nanostructured materials. The requirement of specific physical features in nanostructured thermoelectrics also brought a variety of changes to the fabrication processes. In this chapter, we review some of the prominent techniques for fabrication of such nanostructured material systems. An overview of the concepts and techniques for theoretical modeling of the charge carrier and phonon transport mechanisms in the interfacial regions is presented. Further, the constructive effects of nanostructuring in thermoelectric materials are discussed based on a theoretical approach via Boltzmann transport equation under the relaxation time approximation. The calculations are used to demonstrate the advantages and disadvantages of nanoscale effects in the well-known material systems of Si x Ge1−x and Mg2Si.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Grain Boundary
- Spark Plasma Sinter
- Seebeck Coefficient
- Thermoelectric Material
- Lattice Thermal Conductivity
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
The unique technique of harvesting waste heat energy for power generation by means of thermoelectric (TE) materials was originally discovered by T. J. Seebeck in 1821 [1]. This effect was later enriched by J. C. A. Peltier and Lord Kelvin via their discoveries of the Peltier and Thomson effects, respectively [2, 3]. The Seebeck effect promoted the power generation characteristics of the thermoelectric materials whereas the Peltier effect stimulated the development of the materials and devices for cooling applications. The Thomson effect introduced a comprehensive interrelationship between the Seebeck and the Peltier effects via thermodynamically related coefficients [3]. The thermocouple may have been the most widely used thermoelectric device where a voltage difference generated across the terminals of an open circuit made of two dissimilar metals is used to measure the temperature.
A quantitative characterization of the Seebeck coefficient and the electrical conductivity also paved a path for the identification of many suitable materials for thermoelectric device fabrication [4]. Material systems such as Bi2Te3, PbTe, Si1−x Ge x , etc. were originally found to have favorable characteristics for developing high-efficiency thermoelectric materials. However, the initially identified material systems were not adequately efficient for major application areas such as heat engines and refrigeration systems. The pursuit of developing high-efficiency thermoelectric materials was boosted greatly by the emergence of the concept of the dimensionless figure-of-merit (ZT), introduced by A. F. Ioffe in 1949 [5, 6]. The figure-of-merit mathematically relates the fundamental properties such as the electrical conductivity (σ), Seebeck coefficient (S), the thermal conductivity due to carriers (κ e) and the lattice (κ l) of the TE material, and is defined as [5],
The introduction of the figure-of-merit led to the design and development of numerous optimization techniques for thermoelectric materials that primarily focused on maximizing the (σ/k) ratio while maintaining a substantial Seebeck coefficient. Figure 4.1 illustrates the recently reported ZT versus corresponding temperature ranges for various high-performance bulk TE materials [6, 7]. The state-of-the-art bulk TE material systems include many compounds that are suitable for low and medium temperature ranges. However, as indicated by the vertical line in Fig. 4.1, a dearth of high-performance TE materials systems is markedly observed at high temperatures.

4.1.1 Methods to Enhance Efficiency of Thermoelectric Materials
Material systems based on alloys were largely promoted to reduce the lattice thermal conductivity (κ l) via enhanced scattering of phonons at point defects [5, 8, 9]. Therefore, alloying was employed as the initial technique for thermoelectric material optimization. Later, Slack and Mahan independently defined that in the optimal form the chemical characteristics of a good thermoelectric material essentially include phonon–glass/electron–crystal type features [10, 11]. These features include optimum band gap, high mobility, multiple valleys near the Fermi surface, and low thermal conductivity. Such discoveries led to the evolvement of numerous other techniques for optimization of thermoelectric material systems.
The technique of alloying for reducing thermal conductivity was originally proposed in the 1950s [8]. Alloying of compounds with similar band structure was found to reduce the thermal conductivity significantly without disrupting the electrical properties of the compounds. Other methods of introducing point defects through nanoinclusions were also found to have a significantly positive impact on the thermoelectric figure-of-merit. In addition to alloying, further optimization techniques based on material characteristics were also instigated. Techniques such as microstructure modifications [12], bulk nanostructuring [13, 14], energy filtering techniques [15, 16], resonant energy levels [15], quantum wells and superlattices [17], complex material systems [18], etc. have been subsequently introduced.
The nanostructured systems primarily rely on the reduction of lattice and electronic thermal conductivity by enhanced scattering of phonons at the grain boundaries of the same material system. Nanocomposites fundamentally implement the same principle of scattering of phonons; however, the scattering locations are created by nano-sized grains of more than one material compacted to the locations next to each other. Well-known material systems like nanostructured Si x Ge1−x and Bi x Sb2−x Te3 were reported to benefit from very low thermal conductivities ensuing in high figure-of-merit over a wide range of temperatures [13, 19, 20]. However, recent theoretical estimations also revealed that the same nanostructuring techniques could prove to be detrimental to the thermoelectric characteristics of some materials systems such as Mg2Si owing to the losses incurred in electrical conductivity [21–23].
The techniques of nanostructure modifications were also successfully implemented in material systems like p-type Bi0.52Sb1.48Te3 by means of embedding nanocrystalline domains in an amorphous matrix of the host material [13]. The nanostructure, also comprised nanocrystals with fine and coherent grain boundaries, and resulted in enhanced electrical conductivity via promoting of selective phonon scattering. The unique structural properties lead to a figure-of-merit of 1.56 at 300 K [13]. Energy filtering methods were also found to enhance the Seebeck coefficient by means of increasing the average energy of the carriers as a result of scattering of low energy carriers at grain boundaries. This type of preferential low energy carrier scattering via energy filtering techniques was employed on materials systems like InGaAs/InGaAlAs superlattices and bulk nanostructured PbTe [16, 24]. A significant enhancement in the power factor (S 2 σ) of the superlattices was attained; however, nanostructured PbTe suffered loss in mobility due to scattering of carriers at grain boundaries.
Modifications in electronic properties such as band structure and density of states (DOS) for optimization of the thermoelectric properties were also reported. It was theoretically demonstrated that sharp features in the DOS close to the Fermi energy result in superior thermoelectric power factor [17]. An impurity-activated resonant level in the DOS is an analogous technique by which additional energy states are generated near to the conduction or valence band edges. The additional states are expected to contribute to the carrier transport and boost the magnitude of the Seebeck coefficient when the Fermi level is considerably close to these states. The enhancement in the Seebeck coefficient by means of impurity-activated resonant levels was reported by Heremans et al. for Tl-doped PbTe [25]. A figure-of-merit of twice that of the undoped bulk PbTe was achieved in Tl-doped PbTe as a direct result of the enhancement of the Seebeck coefficient due to the resonant states created by Tl near to the valence band. Combining the two concepts of bulk nanostructuring and resonant levels can potentially result in both low thermal conductivity and enhanced power factor to make more promising TE materials systems.
Quantum confinement techniques were also explored in the form of low-dimensional quantum wells and superlattices. The methods predominantly aimed at dramatically improving the figure-of-merit by means of controlling the electron and phonon energy spectra via structural alterations. Significant reduction in thermal conductivity and a subsequent enhancement in the figure-of-merit were reported in superlattice structures such as Bi2Te3/Sb2Se3 and PbTe/PbTeSe quantum-dot superlattices [26, 27]. Other complex material systems such as quantum wires, clathrates, and skutterudites have also been reported to help achieve better thermoelectric properties.
In the recent years, the nanoscale effects in techniques such as nanostructuring and quantum confinement have significantly contributed to the advancement of thermoelectric material systems. Therefore, a thorough understanding of the nanoscale effects pertaining to the structural modifications is required. The emphasis of this chapter is to provide a methodical insight into the nanoscale effects in state-of-the-art thermoelectric materials. In this chapter, the bulk nanostructured material systems will be referred with a new terminology, namely, Nano Bulk.
4.2 Comparison of Bulk and Thin Film Thermoelectrics
With the new stimuli of the research community in the thermoelectric (TE) area, TE research has continued in two general directions: (1) theoretical models that predict TE properties and offer superior materials and structural designs and (2) experimental efforts in finding new materials or structures that show enhanced TE properties. The follow-ups to these efforts have been in two main directions. One direction has been in finding new bulk materials and their advanced engineering to achieve a higher ZT [28, 29]. Complex crystal structures that yield low lattice thermal conductivity such as FeCo3Sb12 [30], Yb14MnSb11 [31], Ba8Ga16Ge30 [32], Zn4Sb3 [33], or Ag9TlTe5 [34] are several examples of this direction. Other examples of this direction include engineered energy band structures with increased number of energy band minima close to the Fermi surface such as PbTe0.85Se0.15:Na or with sharp features in the density of states close to the band edge such as Tl0.02Pb0.98Te [25] that result in the enhancement of the TE power factor [35].
The second direction has been in the use of nanostructures to reduce the thermal conductivity more than the electrical conductivity through the scattering of phonons at interfaces [26, 36]. It has also been possible to maintain or improve the TE power factor through quantum size effects [27, 36, 37] or interface energy filtering [38, 39] while the thermal conductivity was reduced. Some examples of this group are Bi2Te3/Sb2Te3 superlattices (SL) [27], and PbTe/PbSeTe quantum dot superlattices (QDSL) [26]. The main reason for the improvement in ZT in these structures comes from the reduction of the phonon thermal conductivity. For some other cases such as ErAs:InGaAs/InGaAlAs SLs [40], the enhancement in ZT was due to the increase of the Seebeck coefficient due to the hot carrier energy filtering [16, 39] and the reduction of the thermal conductivity. Bulk nanostructured Bi0.5Sb1.5Te3 [41], Si0.8Ge0.2 [42, 43], and PbTe-SrTe [44] materials have also shown high ZT resulting from the reduction of the thermal conductivity due to the increase of the phonon interface scattering.
With the rising demand on new applications of TE materials both for power generation and cooling functions, there is an increasing interest in material structures that are synthesized with cost effective approaches from earth-abundant and environmentally friendly elements. For this reason, superlattice structures made with thin film deposition techniques are usually too expensive for large scale applications and their use is limited to niche applications such as thermal management of telecommunication lasers or micro-power generators for medical or military applications. However, nano bulk materials made with the powder processing and sintering approaches are potentially inexpensive to synthesize and are appropriate for batch processing [20, 39]. They increase the figure-of-merit of the bulk materials typically based on the same principle as in superlattices, that the scattering of phonons should be enhanced relative to electrons. Hence, prior investigations of low-dimensional superlattices have now evolved into research and development of bulk materials containing nanostructured features.
Furthermore, the irreversible losses due to the electrical and thermal contact resistances become predominant factors when the thickness of the TE device is usually less than 100 μm. Thin film superlattices grown by vacuum deposition techniques are typically 5–20 μm thick, versus 200 μm for the thinnest thermoelectric devices based on bulk materials [45]. As a result, TE devices made from bulk materials, even when sliced to a few hundred microns, have shown larger efficiency than the ones made by thin film deposition.
The fabrication of TE modules from thin films also still suffers from a low yield due to the sensitivity to the height variation for making the electrical contacts [46]. Nevertheless, thin film TE devices can address certain thermal management problems in microelectronics that are not often possible through bulk devices. Thermal stabilization of laser diodes, infra-red detectors, charge-coupled devices (CCD), light-emitting diodes (LED), and other optoelectronic devices are among applications that can benefit from a faster response time and cooling power density of on-chip micro-thermoelectric refrigerators.
4.2.1 Significance of the Nano Bulk TE Materials
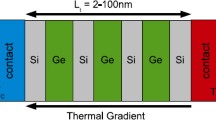
Bulk material systems such as Bi2Te3, Sb3Te3, PbTe, Si1−x Ge x , etc. profited majorly in their nano bulk structured forms. For the case of Bi0.5Sb1.5Te, the improvement in TE performance was possible owing to the significantly beneficial effect of nanostructuring in this material as shown in Fig. 4.2 [14]. It can be observed from Fig. 4.2a that nano bulk Bi0.5Sb1.5Te demonstrates an approximate 50 % drop in lattice thermal conductivity compared to the bulk crystalline form over a wide range of temperature.

Nano Bulk Si x Ge1−x is also well known for its superior TE properties compared to its bulk crystalline form [47, 48]. The ZT of p-type bulk alloy of Si0.8Ge0.2 was enhanced from 0.65 (bulk) to 0.95 at 800 °C with the help of nanostructuring. A significant drop in thermal conductivity was demonstrated by nano bulk phosphorous-doped n-type Si0.8Ge0.2 resulting in a ZT of 1.5 at 900 °C [48]. The thermal and electrical performance of nano bulk Si0.8Ge0.2 was recently reported to show significant dependence on the average crystallite size [42]. Theoretical calculations performed on bulk and nano bulk Si0.8Ge0.2 demonstrated that the lattice part of the thermal conductivity makes up for 80 % of the total thermal conductivity as shown in Fig. 4.2b. Images from the scanning electron microscopy of the fractured surface of the as-pressed sample and the transmission electron microscopy of the powder are shown in Fig. 4.3a, b, respectively, depicting the crystallite sizes in the nano bulk Si0.8Ge0.2 which lead to the low thermal conductivities of less than 3.0 W/mK. Similarly, other highly efficient techniques such as nanoparticle-in-alloy were explored theoretically and a figure-of-merit that is nearly 2.5 times the value of the bulk system of SiGe alloys was predicted [49]. The nanoscale effects owing to the crystallite sizes have been theoretically and experimentally substantiated in many recent reports [19, 42].

(a) Scanning Electron Microscopy (SEM) and (b) Transmission Electron Microscopy (TEM) images of nanostructured Si0.8Ge0.2 [42]
For a superior synopsis of the recent advances in TE materials through nanostructuring, the readers could refer to the CRC Handbook of Thermoelectrics [50] and published review articles [6, 15]. However, within the scope of this chapter, the primary focus will be on the effects of bulk nanostructuring comprehended by means of a theoretical approach as will be discussed in further sections. In order to thoroughly provide insight into the effects of bulk nanostructuring on the TE properties of materials, a simulation based assessment of the TE characteristics of Si x Ge1−x and Mg2Si follows. In the upcoming sub-sections of this chapter a systematic discussion of the experimental methods for making nano bulk materials, electronic properties of these materials, theoretical approaches to nanostructuring mechanisms, modeling strategies, carrier transport properties, phonon scattering mechanisms and the effect of grain sizes on the TE properties in Si x Ge1−x and Mg2Si is presented. As the end, the simulation results are critically discussed in order to conclusively show that in certain material systems detrimental effects due to nanostructuring may diminish the overall benefits.
4.3 Fabrication of Nano Bulk Thermoelectrics
Fabrication of nanostructured materials through a bulk process is challenging, but modification of bulk process conditions so that the ZT is enhanced is even more difficult. The modification process does not always lead to improved thermoelectric efficiency. Thermal stability of the devices made by nanostructured materials at high temperatures is another influencing factor as the nanoscale features may be removed due to the diffusion and energies of the interfaces. There are two major approaches via top-down and bottom-up methods to fabricate nano bulk thermoelectrics and each of them can be divided in to several branches. These methods are introduced in the following sections and their advantages and disadvantages are briefly discussed.
4.3.1 Top-Down Approach
The top-down methods start from a bulk material and the corresponding nanostructured features are obtained by structural deformations. There are two commonly used techniques which are based on sever plastic deformation (SPD) or thermal processing.
4.3.1.1 Sever Plastic Deformation (SPD)
The basis of the SPD method is to increase the free energy of the polycrystals and generate many more defects and grain boundaries in various nonequilibrium processes so that the microstructure is transformed into nano-sized crystallites while the structure of the coarse-grained material remains unchanged without any associated changes in the cross sectional dimensions of the samples [51]. Key principles of SPD are increasing dislocation density by heavily deforming materials, forming dislocation walls, and making transformation of dislocation walls into grain boundaries. Some known methods in this category are listed below:
-
High pressure torsion: A small thin disk is placed between two massive anvils under high pressure and intense shear strain is introduced by rotating the two anvils with respect to each other. In this method, the applied hydrostatic pressure is high; therefore, the fracture is suppressed and thus the method is applicable to hard and less ductile materials.
-
Equal channel angular pressing (extrusion) [52, 53]: In equal channel angular pressing (ECAP) the billet is placed in a die and a high pressure is exerted to it through a punch. It is one of the SPD methods for producing ultra-fine grain structures in nanoscale which applies a large amount of shear strain into the materials without changing the shape or dimensions of the billets.
-
Accumulated roll bonding [54, 55]: This is another SPD process to produce ultra-fine grain structures in nanoscale which is applicable for continuous production of the nanostructured materials. In this method 50 % rolled material is cut in two, stacked to make the initial dimension, and then rolled again. By repeating the deformation and bonding processes, the nanostructured materials can be produced.
4.3.1.2 Thermal Processing
Thermal processing techniques have widely been applied to induce nanoscale precipitates [56–58]. In order to fabricate such nanostructures in a metastable solid-solution the starting material is subjected to a thermal processing procedure like spinodal decomposition, nucleation, or the growth mechanism [56]. In the thermal process of matrix encapsulation the liquid mixture is cooled rapidly so that the insoluble minority phase is precipitated, and the embedded nanoparticles in the host material are created [57]. This technique has been applied for n-type AgPbTe2/PbTe [58] and Pb1−x Sn x Te/PbS [56] and p-type Na1−x Pb m Sb y Te m+2 [59] and NaPbTe-SrTe [60].
4.3.2 Bottom-Up Processes
This type of method starts from atoms and molecules as building blocks and the bulk materials are subsequently assembled from elemental constituents. Bottom-up methods are used to assemble nanoparticles into dense solid phases to attain a nanostructured material. The main technique frequently applied for thermoelectric materials consists of powder processing and subsequent consolidation.
4.3.2.1 Powder Processing
The powder characteristics facilitate low temperature sintering, which is a prerequisite for mitigating grain coarsening during the sintering process. The power can be synthesized by a chemical or physics method. The nanoparticles should be preferably non-agglomerating and uniform in size and shape or preferably monodispersed to result in a dense nano bulk material after consolidation. Non-agglomerated nanoparticles are made by wet chemical methods which are usually expensive procedures. The physical method is mainly based on mechanical milling which often results in agglomerated nanoparticles of different sizes in an inexpensive way.
-
Chemical methods: Several chemical methods have been developed to fabricate metal nanostructured materials with different shapes such as silver (Ag) nanoplates [61], silver nanodisks [62, 63], gold(Au) nanoplates [64], and gold nanodisks [65]. It has been shown that a high-temperature organic solution approach and a hydrothermal process can be used to prepare hexagonal nanoplates of Bi2Te3 and Bi2Se3 respectively [66]. High-yield two-dimensional micrometer-sized hexagonal nanoplates of Sb2Te3 using a solution-based synthesis method have been also produced [67].
-
Mechanical milling: In this process the powder is milled with different techniques during which the particles are cold welded and fractured many times resulting in ultimate fineness. The most commonly used milling method has been ball milling where the powder mixture is placed in the ball mill and is subjected to high energy collision from the balls. Among these, vibratory ball mills, planetary ball mills, and attritor mills have been common equipment for ball milling.
4.3.2.2 Consolidation
-
Hot pressing [41, 47, 68, 69]: It is a pressure assisted method for compacting nanopowders to obtain bulk nanostructured materials, which is usually accompanied by sintering at various times, temperatures, and atmospheres. The method of ball milling and hot pressing is cost effective, applicable for many material systems, and can produce nano bulk materials which are thermally stable. Several research groups have reported the implementation of this technique for n-type and p-type Si0.8Ge0.2 and some types of Bi x Sb2−x Te3 [41, 42, 70]. Spark plasma sintering [71–73] is a newer technique compared with conventional sintering approach where the heat is provided by an external source. In this method the heat is generated by passing a pulsed DC current through a die and conductive powder. This method creates a very high heating rate (100–1,000 K/min) which leads to a fast sintering process. In a similar approach, a DC current is used for heating the material [41]. The high heating rate allows sintering the material with reducing the grain growth therefore attaining smaller gains in the bulk material.
4.3.3 Theory of Charge Carrier and Phonon Transport at Interfaces
Theoretical studies of phonon transport in superlattices showed that reduction in lattice thermal conductivity does not need a well-defined geometry or a perfect interface at atomistic level [74]. The only requirement is having a high density of interfaces which is present in nano bulk structures. To enhance the ZT, the phonons have to be scattered more efficiently than charge carriers. Therefore, as a rule of thumb, the size of nanostructures should be smaller than the phonon mean free path and larger than the charge carrier mean free path. In practice, charge carriers have narrower spectrum than that of phonons. The interface spacing below which the electrical conductivity is reduced considerably can be estimated through the spectrum of the mean free path. If the interface size is larger than a single scattering site, it will have deteriorative impact on charge transport and decreases the ZT. Continued reduction in characteristic lengths in nanostructures has led to increasing importance of interfaces in transport properties of materials. In fact, a large portion of the advances in enhancing ZT for nanostructured materials comes from the beneficial effects of the interfaces.
4.3.3.1 Interface Definition and Types
The region in a crystalline or polycrystalline material that separates two crystallites is called interphase boundary or simply interface. The interface represents a discontinuity with a minimum thickness of two atoms. The neighboring grains may have different orientations. A random connection of two different crystallites may lead to dangling bonds, large crystallographic discrepancies, or voids in the interface region. If the crystallites’ chemical composition and lattice parameters are the same, the interface is called phase boundary; and if the crystallites belong to different materials, the interface is called heterophase boundary. In an interface, the atoms are shifted from their regular positions as compared to a perfect crystal and introduce defects into crystal structure. Interfaces play an important role in determining the properties of the nano bulk structure due to the large relative fraction of atoms that are present in the interphase boundary. Interfaces decrease the electrical and thermal conductivities of the material and provide preferred places for the precipitation of the new phases from the solid.
To get a sense of the atomic arrangement of the neighboring crystals at an interface, the interfaces can be divided into four classes:
-
1.
Coherent interface: There is a perfect match between two nanocrystals across the interface.
-
2.
Strained coherent interface: Imperfect matching between two nanocrystals causes some strain at the interface, which increases with the size of the crystallites.
-
3.
Semi-coherent interface: Although dislocations contribute to the energy of the system, they reduce the strain energy. The coherent interface with strain is likely to be transformed to a semi-coherent interface with dislocations.
-
4.
Incoherent interface: There is no lattice match between the crystallites across the interface.
As the matching between the crystallites improves, the interfacial free energy becomes smaller. In incoherent interfaces atoms that have more freedom to move are considered mobile, so they have higher energy. Liu et al. [75] established that the coherent interfaces generally exist in nanocrystalline composites, and according to the experimental results, they are largely unaffected by the fabrication process. It is rational that the coherent interfaces play a key role in assembling the composite nanostructures because the coherent interfaces require that the crystal planes from the two adjacent crystallites to have similar basal spacing and that the crystallites bind tightly to form interfaces which are thermodynamically stable. There is currently very little known about the effect of nanoscale coherent interface on energy transport. Nonetheless, some researchers have reported excellent ZT for Ag1 − x Pb m SbTe2 + m with m = 10 and 18 (2.1 at 800 K) which consist coherent interfaces [58]. The observation of coherent nanointerface in a PbTe/GeTe nanocomposite was also reported at the atomic level and atomistic modeling was applied to characterize the local chemical composition and crystalline structure and the coherency of interfaces was confirmed [76]. It was found that the large value of ZT was attributed to the nanoscale coherent interfaces that improved phonon scattering while having minimal effect on the charge carrier transport [76]. The coherent nanointerfaces between the matrix and nanoscale inclusions offer three advantages in thermal design for ZT improvement:
-
(a)
The nanoscale inclusions significantly reduce the mean free path of the phonons, thereby reducing the lattice thermal conductivity.
-
(b)
The strain at the coherent interface considerably enhances phonon scattering and causes a large thermal resistance at the interface, which helps to further reduce the overall thermal conductivity.
-
(c)
Electron scattering remains largely unaffected by the coherent interfacial structure, which leads to a very little decrease in the electrical conductivity. Such a combined effect is very favorable for enhancing ZT.
Charge carriers and phonons encounter different resistances across an interfacial region. The charge carrier boundary resistance originates from the impact of the charge carriers to a coulomb potential at the interface. This potential depends on the band structure and doping concentration of the interfacial materials. Both a potential barrier and a well can scatter charge carriers due to the disruption of the periodic potential. Charge carriers can tunnel through the barrier or be thermally excited over it depending on the width and height of the barrier [77]. Phonons lose their memory after encountering the interface. The reflectivity or transmission of phonons is determined by density of states and phonon velocities of different modes. The reflected phonons form the phonon boundary resistance which depends on the available energy states at both sides of the interface [78]. The charge carrier and phonon resistances at interfaces lead to nonequilibrium transport properties near the interface and at a distance from the interface, named energy relaxation length, they reach to a new thermal equilibrium.
4.3.3.2 Nano Bulk Structures
In a nano bulk material, grains and interfacial structures may have three distinct types as depicted in Fig. 4.4. Type 1 (Fig. 4.4a) is a nanocrystalline structure made of a single material composition. Type 2 (Fig. 4.4b) consists of embedded nanoparticles inside the lattice. Figure 4.4c is a nanocrystalline structure made from mixture of two or more of different materials or phases. In this structure the grains and/or the grain boundary regions are made from different materials. Experimentally, a nano bulk thermoelectric material may have a structure similar to any of these types or a combination of them. For example, a peak ZT of 1.4 at 100 °C was achieved in a p-type nanocrystalline Bi0.5Sb1.5Te3 bulk alloy using the type 1 interface, which separates single phase crystallites [41]. Also, the p-type PbTe nanostructured bulk with SrTe precipitates, which demonstrated a ZT value of 2.2 at 615 °C, took advantage of the combination of Type 1 and 2 structures to more effectively scatter heat carrying phonons across the integrated length scales [44]. A combination of Type 3 with 1 and/or 2 can make various other structures such as core–shell nanoparticle doped structures, or nanocrystalline structures with embedded nanoparticles. There is yet no report of enhanced ZT through Type 3 nano bulk structure.

Three types of nano bulk structures: (a) single component nanocrystalline material, (b) bulk material with embedded nanoparticles, (c) multicomponent nanocrystalline material
4.3.3.3 Interfacial Effects on Charge and Phonon Transport
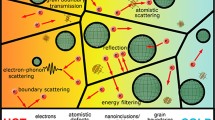
The interface in a nano bulk material is characterized by mismatch in bulk properties and irregularities in the region near the interface. Different carriers of charge and heat encounter different resistances across this interfacial region. Charge carriers encounter a coulomb potential at the interface resulting in the carrier boundary resistance, and phonons hit the interface and lose their memory. The probability of phonon being scattered to one side of the interface or the other is related to the density of states and the phonon group velocity of each vibrational mode. These interfacial resistances at an interface region result in electron and phonon thermal nonequilibrium near the interface, which can affect the thermoelectric properties significantly. The schematic diagram of Fig. 4.5 lists the interfacial effects on charge and heat carriers transport parameters and their connections with the thermoelectric properties.

Schematic illustration of the mechanisms that influence charge and thermal transport properties in interfacial regions
4.3.3.4 Charge Transport at Interface
There are three major challenges in analyzing the charge transport in nano bulk materials. First, the characteristic lengths in a nano bulk material can become smaller than the charge carrier wavelength as the grains become too small. This will reduce the accuracy of the Boltzmann Transport Equation results as the BTE is valid only if the charge carrier mean free path is much larger than the wavelength [79, 80]. The second difficulty arises from the unknown properties and the composition of the interfaces. As the density of the interfaces increases in the nano bulk material, their volume fractions become comparable with that of the host material; therefore, the transport properties of the material should be derived not just from the scattering processes, but from a new phase of charge and phonon transport making the determination of the interface properties a cumbersome problem.
Interfaces can influence the Seebeck coefficient, electrical conductivity, and the electronic thermal conductivity. In fact, interfaces have negative and positive effects on carrier transport. Although the interface scattering reduces the charge mobility, it can be exploited to improve the Seebeck coefficient through energy filtering and reduce the ambipolar thermal diffusion through preferential scattering of the minority carriers. The following is a brief description of how different concepts and quantities can change the contributions of the interfaces in charge transport properties.
4.3.3.4.1 Seebeck Coefficient
As can be seen from the Mott’s relation,
in which g(E) is the electronic density of states and v is the charge group velocity, the interfaces can be beneficial to Seebeck coefficient by three effects as listed below.
-
Quantum size effects can cause sharp changes in the density of states g(E) which in turn improves S and enhances ZT [37].
-
Interfaces can introduce potential barriers that can scatter carriers and affect the thermoelectric efficiency. In the presence of such potential barriers, carriers that are passed over the barriers have higher average energy than carriers far from the barriers. Carrier energy distribution function becomes that of the bulk material in a length equal to the energy relaxation length \( {\mathcal{l}}_{\mathrm{E}} \) from the barrier. If the grain sizes does not exceed \( {\mathcal{l}}_{\mathrm{E}} \), the carrier distribution function does not return to that of the bulk between GBs, and thus the effective Seebeck coefficient of the nano bulk material is higher than that of the parent single crystalline bulk material. Also, if the momentum relaxation length is much shorter than the distance between the GBs, the behavior of the carriers with energies greater than that of the barrier may be similar to that of carriers in the bulk. In other words, for carriers with energies above those of barriers, the differential conductivity σ(ε) of a material with potential barriers can be approximated by that of the material without potential barriers if \( {\mathcal{l}}_{\mathrm{m}}<<\mathcal{l} \). Therefore, under the condition of \( {\mathcal{l}}_{\mathrm{m}}<<\mathcal{l}<{\mathcal{l}}_{\mathrm{E}} \), the energy filtering mechanism can increase the effective material Seebeck coefficient. It is also understood that since such defects can reduce the electron mobility, the overall effect appears in the power factor S 2 σ.
Figure 4.6a shows the asymmetric contribution of electrons with different energies around the Fermi energy to the conduction process (differential conductivity). A more asymmetric differential conductivity would result in higher Seebeck coefficient. While the Seebeck coefficient is relatively high, the number of conduction electrons is small as the Fermi energy is close to the band edge. Figure 4.6b shows that when the Fermi energy is deep inside the conduction band, the differential conductivity is more symmetric with respect to the Fermi energy resulting in a small Seebeck coefficient. Removing low energy carriers by a potential barrier can lead to an improvement in Seebeck coefficient as shown in Fig. 4.6c. The new scattering mechanism introduced by the interface potential scatters low energy carriers and reduces their contribution to the transport properties so the Seebeck coefficient is increased. The successful implementation of this strategy has been reported for InGaAs/InGaAlAs superlattice [17] and PbTe-based nanostructured materials [18, 81].

Schematic representation of electron filtering. (a) Low doped (b) highly doped (c) energy filtered highly doped
Energy filtering improves the Seebeck coefficient through the reduction of bipolar effect too. To this end, the minority carriers which have lower energy are affected by fabricating a nanostructure which scatters them more strongly than the majority carriers. Such a reduced bipolar effect has been reported for nanostructured Bi0.5Sb1.5Te3 [41].
-
Any change in the dielectric constant caused by the strain or the change of the lattice constant at the interface can appear in ionized impurity, polar optical-mode phonon, and nonpolar optical-mode phonon scattering times [82]. Changes in these scattering times directly affect the Seebeck coefficient according to the Mott’s relation.
4.3.3.4.2 Electrical Conductivity
Interfaces form two-dimensional networks or line charges in juxtaposition. Interfaces change the electrical conductivity through several possibilities such as:
-
Interface roughness scattering at the interface decreases the electron mobility in the material [83]. However, the roughness scattering can lead to a significant increase in the thermionic current. A rough interface scattering conserves the energy but not the transverse momentum of the charge carriers. Therefore, the scattering at a rough interface mixes the planar motion of the charge carriers with the longitudinal motion perpendicular to the interface. In this case, the transmission probability depends on the total kinetic energy of the electrons rather than only the perpendicular component to the barrier. This will increase the number of electrons that are transmitted over the barrier, which can significantly increase the electrical conductivity. Non-conservation of transverse momentum does not significantly change the Seebeck coefficient; therefore, optimizing the material parameters (such as doping and barrier height) to gain an overall benefit from the interface roughness scattering is possible. For an accurate treatment of the interface scattering, one needs to develop a comprehensive model that includes all the important effects on the charge mobility and the transmission probability at the interface.
-
There are several mechanisms for the existence of an interfacial potential. One is the accumulation of charges at the interface, namely, the space charge electric potential. The broken bounds at the interfaces attract free carriers and form a space charge. The high density of states at interfaces and a space charge on either side of the interface represents a double Schottky barrier at the boundary thereby a high resistance to the lateral current flow. Early works on modeling of the interfaces in polycrystalline materials used a double diode or a two back-to-back diode model [84]. Since then there have been many different models for the effect of the interface space charge on electrical conductivity [85].
-
Another mechanism is due to the different orientations of the adjacent grains or simply crystallite rotation. Apart from the space charge potential barrier, electrons may experience a potential barrier (or well) due to the rotation of the adjacent grains. For example, an electron moving in L valley in one grain has to enter the Σ valley in the next grain. In a nano bulk material, the grains are randomly distributed with a random orientation. Electrons with sufficiently large energies can pass over the barrier and enter a different valley in the adjacent grain. Electrons with smaller energies may encounter an intervalley scattering and enter an equivalent valley in the adjacent grain (Fig. 4.7). This can happen through electron scattering by phonons or the interface roughness potential. Such scatterings reduce the boundary resistance as they provide alternate means for electron transport between the grains.
Fig. 4.7 
Pictorial depiction of the grain boundary interfaces and intervalley transitions. L–L and X–X lines demonstrate the equivalent intervalley scattering between adjacent grains. L–X line shows transition to a different valley that would disturb the energy spectra of the charge carrier
-
Dielectric constant can be changed due to the permanent dipoles which are induced by the electric field in the interfacial regions. The change in the dielectric constant leads to the change in polar optical-mode phonon and nonpolar optical-mode phonon scattering times which in turn alters the electrical conductivity [82].
-
Deformation potential theory is a band model to describe the charge transport and works well if the electron–phonon coupling is weak [86]. Deformation potential impacts on the electrical conductivity as well as the total phonon relaxation time. Local deformations in the crystal lattice caused by the interfaces can form an effective electric potential due to the strain and its effects on the band structure. Therefore, the changes in the lattice constant in the interfacial region can also affect the electrical conductivity through the change in the deformation potential [87].

4.3.3.4.3 Electronic Thermal Conductivity
The charge carriers also transport heat and add to the thermal conductivity. At first glance it seems that reducing the thermal conductivity through the reduction in the charge mobility or concentration is not favorable as it would adversely affect the ZT through electrical conductivity. However, the key is the different value of the Lorenz number in many thermoelectrics. Moreover, the ambipolar diffusion mechanisms can favorably behave differently at the presence of the interfaces. Wiedemann–Franz law states that the electronic thermal conductivity k e and electrical conductivity σ in a metal or degenerate semiconductor are directly proportional as:
where L is the Lorenz number with the value L = 2.45 × 10− 8 WΩ/K2, σ is the electrical conductivity, and T is the absolute temperature. Therefore, Wiedemann–Franz law affects the electronic thermal conductivity in two ways. First, k e is decreased due to the deteriorative effect of the interfaces on the charge carrier mobility. Second, k e is a function of the Lorenz number which itself is a function of the relaxation time. Therefore, it is possible to reduce the contribution of carriers in thermal conductivity by reducing the Lorenz number.
If a temperature difference is maintained across a thermoelectric material, a positive concentration gradient of thermally excited electron–hole pair in the direction of the temperature increment occurs. Diffusion causes a net flux of these electron–hole pairs towards the cold side of the material and their subsequent recombination that would heat up the lattice. This process is known as ambipolar thermal diffusion whose strength depends on the band gap and the balance of the electron and hole conductivities. Interfaces can preferentially scatter minority carriers more than majority ones due to the difference in their energy. For example, since the minority carriers have less energy than the majority ones in a bulk material, the former ones are scattered more strongly by the interface potential barriers resulting in an imbalance between the electron and hole electrical conductivities; hence, reducing the ambipolar diffusion.
4.3.3.5 Phonon Transport at Interface
According to the following relation, which is derived from a simple kinetic theory and neglects the normal phonon–phonon interactions, the lattice thermal conductivity can be calculated from [88, 89]:
where C is the heat capacity, v g is the phonon group velocity, τ is the total relaxation time, and ω c is the cutoff frequency. It is expected that the interfacial resistance of the grains in a nano bulk material should reduce the lattice thermal conductivity either through the reduction of the group velocity of the phonons or the reduction of the relaxation time. The reduction in relaxation time corresponds to the reduction in phonon mean free path. In nano bulk materials, the reduction of the phonon relaxation time can be achieved through point defects and/or grain boundary scatterings. Due to the complicated interplay of phonon scattering, group velocity, and heat capacity, a frequency dependent analysis of k latt is crucial to design low thermal conductivity thermoelectrics. Some of the main mechanisms affecting the lattice thermal conductivity are introduced briefly in the following.
4.3.3.5.1 Group Velocity
The one-dimensional Born–von Karman model, as a chain of atoms connected by springs with linear restoring forces to investigate the effect of the reduction in phonon group velocity on the spectral lattice thermal conductivity, indicates that the decrease in v g which decreases the k latt(ω) occurs in three ways: [90]
-
As the mass contrast increases, the optical mode flattens, and if Umklapp scattering dominates, k latt(ω) is proportional to v g(ω)2; therefore, the k latt(ω) is reduced significantly [91].
-
As the number of atoms in the primitive unit cell increases, due to their low velocity of optical modes, the v g(ω)2 is decreased considerably; hence, the k latt(ω) is decreased [92, 93].
-
In open framework compounds (such as clathrates and skutterudites) the avoided crossing effect in the vicinity of the guest atom vibrational modes reduces v g(ω)2 through the reduction of the acoustic contribution to k latt(ω); hence, k latt(ω) is decreased [94, 95].
4.3.3.5.2 Relaxation Time
Interfaces can scatter phonons via several different possibilities:
-
Regular reflection and refraction, arising from the difference of the phonon group velocities in the adjacent grains.
-
Diffusive scattering due to the corrugation of the interface, caused by impurities or interface roughness.
-
Diffraction of waves when the wavelength is comparable to the particle size. If the interface region is much smaller than the wavelengths of the excited phonons, the problem should be treated as a diffraction process (Rayleigh scattering).
-
Anharmonic effects are also able to extremely decrease the lattice thermal conductivity as reported by Morelli et al. [96]. Anharmonic bands are characterized by Grüneisen parameter γ. Good thermoelectric materials require large γ. Hopkins et al. [97] suggested a new model for thermal conductivity that takes into account the anharmonic coupling, or inelastic scattering events, at the interface of two grains. This is achieved by considering specific ranges of phonon frequency interactions and the conservation of the phonon number density. Their suggested model considered the contributions of the inelastically scattered phonons to the thermal conductivity and provided a good agreement with the experimental results at the Pb–diamond and Au–diamond interfaces.
-
Strain can be caused by the change of the lattice constant at the interface. The strain affects the vibrational modes and changes the relaxation time [98].
-
Deformation potential: This potential represents the interaction of charge carriers with phonons. The deformation potential theory has been developed and applied by many researchers [87, 99, 100]. Lattice mismatch at the interfaces can cause a lattice strain that would alter the deformation potential. Therefore, interfaces can affect the lattice thermal (and electrical) conductivity indirectly through the deformation potential.
Figure 4.8 depicts the schematic representation of the scattering mechanisms for phonons. For regular reflection and refraction, the relaxation time has no dependency on angular frequency. If the wavelength is comparable with the particle or the interface size, the relaxation time will be a function of ω 2(λ ~ d, τ(ω 2)) and if the wavelength is much larger than the particle or the interface size, the relaxation time will be proportional to ω 4 (λ ≫ d, τ(ω 4)).

Schematic representation of the scattering mechanisms for phonons from interfaces
As the characteristic lengths of the nano bulk materials approach the phonon mean free paths, the effect of scattering mechanisms at interfaces becomes much more important than inside the bulk material. Phonon reflection at a boundary leads to the Kapitza resistance, a phenomenon that has been known for a long time but has escaped quantitative description except at very low temperatures when the phonon wavelength is long so that the reflection can be treated based on acoustic wave theory. At room temperature, it is difficult to calculate the phonon reflectivity mainly because of the diffuse phonon reflection as well as the phonon spectrum mismatch between the two sides of the interface.
Phonons have a large range of frequency and relaxation time. Phonons with different frequencies have different effects. Phonons may behave like a plane wave if the interfacial roughness is small compared with the phonon wavelength. They may behave like a particle if their wavelengths are much smaller than the interfacial roughness. Microstructural obstacles like inclusions and precipitates have great effect on the lattice thermal conductivity. Until now, no model has been able to predict the interface thermal conductance except at very low temperatures. The Fourier heat conduction theory, as a common approach to model the effect of interfaces in thermal conductivity, underpredicts the effect of the interfacial scattering. This is mainly because this approach is based on the diffusion picture while the phonon mean free path is longer than the characteristic lengths of the interface region. Phonon transmission and reflectivity are crucial parameters to model the heat conduction in thermoelectric materials. In the case of the nano bulk materials, two major theoretical problems are known for understanding of the phonon transport. First, nano bulks show a lattice thermal conductivity lower than that of their alloy limit. Second, it is not clearly understood how the interface affects the phonon transport. The first problem can be explained by determining the amount of contribution of each wavelength of the phonons to the thermal conductivity. Phonons with short wavelength are strongly scattered by point defects but phonons with long wavelength have a long mean free path. Nevertheless, the contribution of the long wavelength phonons in thermal conductivity is small [101]. If the involved structures have larger characteristic length than that of alloy point defects, interfaces scatter long wavelength phonons that would further reduce the lattice thermal conductivity beyond the alloy limit. Moreover, some researchers believe that the low limit of thermal conductivity in nano bulk materials is smaller than that of bulk materials modeled by isotropic scattering mechanisms due to the highly anisotropic interfacial scattering in the nanostructures.
Nanoparticles in a host material have multiple interfaces and knowing the amount of the reflected or transmitted phonons from a single interface is crucial for designing nano bulks with reduced thermal conductivity. Available theories do not predict phonon reflectivity and transmission at an interface accurately as they do not clarify the contribution of the phonon scattering inside a nanoparticle or the role of the interface roughness.
4.4 Methods for Modeling Charge Carrier and Phonon Transport at Interfaces
4.4.1 Modeling by BTE
The BTE has been used extensively to model the thermoelectric properties of bulk materials. As long as all the scattering mechanisms are elastic and the relaxation time approximation is valid, electrical and thermal properties can be expressed as integrals of the relaxation times [79, 80]. To model the effect of interfaces, one can add an interface scattering time to the scattering times existent in the bulk material using Matthiessen’s rule. Therefore, the only thing that one needs is an expression for the interface or the grain boundary scattering, and the general algorithm to solve the BTE is unchanged. Modeling of the interface scattering, however, is not accurate at this stage.
A particular implementation of the BTE is the acoustic mismatch model (AMM) which can be considered as the most simplified and widely used method to treat the phonon transport at the interfaces. AMM compares the impedance ratios at an interface of two crystals. Since this model has been designed for specular elastic scattering of phonons at abrupt interfaces, it is proper for low temperature. The AMM, like other BTE-based models, does not account for the wave nature of the phonon transport that is driven by the underlying lattice dispersion and the boundary conditions on either side. Moreover, it does not take into account the nonequilibrium status of the region between the two thermal reservoirs. In addition, extending this model to cover multiple scattering phenomena in nanostructured materials has not been formulated yet.
4.4.2 Modeling by Molecular Dynamics (MD)
MD simulation can provide a deep insight of the mechanisms responsible for phonon transport in nanostructured materials. This method has been extensively developed and has found applications in research on thermal transport in nanoscale interfaces [102, 103]. After the pioneering work of Maiti et al. [104] using the nonequilibrium molecular dynamics (NEMD) for studying the Kapitza resistance across an interphase boundary, several studies on the interfaces between different phases [105] and nanostructures [106] have been performed. Additionally some researchers have used MD to calculate the thermal conductivity of some thermoelectric materials such as Bi2Te3 [107], Pb1−x Sn x Te [108], Pb1−x Ge x Te [108], and Si/Ge nanocomposites [109]. Although MD provides some atomistic details, since it is mainly based on empirical potentials, it gives poor estimates of the thermal transport properties.
4.4.3 Modeling by Nonequilibrium Green’s Functions (NEGF)
The Nonequilibrium Green’s Functions (NEGF) formalism has been extensively used to study the electron transport in nanostructured materials [110–112]. It has been used mainly for structures where characteristic wavelengths of the charge carriers become comparable to, or larger than, the sample size. The method can be readily developed to include the incoherent scattering and the electron–phonon correlation effects. In this method, a two probe system is defined and divided into three regions, left electrode, central region, and right electrode. It is assumed that the electrodes have bulk properties. First, the properties of the electrodes using standard density functional theory (DFT) techniques for periodic systems are calculated. The solution for the electrodes determines boundary conditions for the central region, and then in the second step, DFT equations for the central region are solved self-consistently. The charge carrier transport properties are calculated by using the Kohn–Sham Hamiltonian for propagating each electron.
The NEGF method has been also applied to treat phonon transport across the interfaces in nanostructures. First, some ab initio methods like tight binding, DFT, or MD are used to relax the crystal structures across the interface and to determine the interatomic force constants between the atoms. The computational unit cell should be large enough to get a sufficient number of nearest neighbor interactions. Then the phonon Green’s function is computed based on the extracted force constants. Moreover, the converged semiclassical Poisson electrostatic potential is used to calculate the transport properties. For example, the method has been applied to investigate the phonon transport in carbon nanotubes with defects and isotope scattering in carbon and boron-nitride nanotubes [113–115]. It is notable that the strength of the NEGF is its ability to include more complicated incoherent scatterings such as anharmonic phonon mechanisms.
4.5 Nanostructuring in Si x Ge1−x and Mg2Si
As discussed earlier in Sect. 4.1, nanostructuring techniques have steered the performance of many thermoelectric (TE) compounds towards attaining a ZT > 1 in the room to medium temperature ranges. Optimization of device performance was widely achieved through additional scattering mechanisms in the form of grain boundaries and interfaces created via nanostructuring techniques. Nanostructuring techniques were principally designed to scatter mid to long wavelength phonons, a feat that could not be effectively achieved through alloy or point-defect scattering [116]. Consequently, the alloy limit to the reduction of thermal conductivity was overcome by means of restricting the grain sizes to those comparable with the characteristic lengths of phonons.
4.5.1 Properties and Modeling of Silicon Germanium and Magnesium Silicide Nano Bulks
The binary compounds of Si x Ge1−x and Mg2Si hold favorable thermoelectric (TE) properties for power generation in the high (900–1,100 K) and medium temperature ranges (500–800 K), respectively [43, 117–120]. In addition to being cost efficient and allowing for scalable synthesis methods, both compounds were characterized for having high mobilities, electrical conductivities, Seebeck coefficients, and low lattice thermal conductivities [43, 50, 120]. Table 4.1 shows the important electronic and material properties of silicon germanium (Si x Ge1−x ) and magnesium silicide (Mg2Si).
A favorable feature of Si1−x Ge x is its ability to facilitate band structure engineering through variation in the alloy composition (x) of the elements. The alloy composition and the temperature affect both the electrical and thermal properties. Additionally, the similarities between the chemical and the physical properties of Si and Ge make the processing of Si1−x Ge x relatively simple. Si1−x Ge x crystallizes with a diamond lattice structure under atmospheric pressure. The type of the conduction and the carrier concentration in Si1−x Ge x could be controlled by doping with phosphorous (n-type) or boron (p-type). Si1−x Ge x is an indirect band-gap material with the primary conduction energy gap between the X and L valleys being ~0.8 eV [43, 50].
Among the materials suitable for thermoelectric power generation in the medium temperature range, Mg2Si stands apart due to the distinctive features of its elemental constituents. The elements of Mg and Si are nontoxic, widely available in nature, and are suitable for making light-weight and recyclable compounds. Additional related intermetallic compounds such as Mg2Ge and Mg2Sn are also suitable for thermoelectric power generation in the medium temperature range. Mg2Ge and Mg2Si possess semiconducting properties due to the valence electrons in these compounds being equal to the number of states in the Brillouin zone [120]. Mg2Sn has a very low energy gap and is considered as a semimetal.
Similar to Si1−x Ge x , the bulk crystalline form of Mg2Si possesses a high melting point, low thermal conductivity, and high electrical conductivity as shown in Table 4.1. Numerous experimental techniques such as mechanical alloying, hot-pressing, and spark plasma sintering have been reported for the preparation and synthesis of Mg2Si-based thermoelectric materials [121–125]. Recent reports also demonstrate novel methods such as microwave processing to fabricate nano bulk structures of Si x Ge1−x and Mg2Si [126, 127].
Elements such as antimony, aluminum, and bismuth are typical dopants for synthesis of n-type Mg2Si. However, the synthesis of p-type Mg2Si requires dopants such as silver or copper [125, 128]. As cited in the previous subsections, well-known techniques such as mechanical alloying, hot-pressing, and spark plasma sintering have been used to fabricate nano bulk compounds of Si x Ge1−x and Mg2Si. In some cases, the thermal processing methods such as solid-state reactions were also implemented. Methods to fabricate nano bulk compounds of Mg2Si were also widely implemented recently [129–132]. In these TE compounds, although a reduction in the thermal conductivity was obtained as a result of nanostructuring, the ZT remained lower than one. Unlike compounds such as Si1−x Ge x , the performance trend of nano bulk Mg2Si illustrated only meager enhancements. In order to understand the physical reasoning behind this unexpected trend, a detailed examination of the electron and phonon transport in these nanostructures is necessary. The primary parameters that are affected by bulk nanostructuring are the phonon mean free path (PMFP) and charge carrier mean free path (CMFP). The mean free path is defined as the distance travelled by the phonon or charge carrier between consecutive scattering events. The PMFP effectively determines the maximum distance of heat transport for each phonon. It is mathematically represented as the product of the phonon relaxation time (τ) and group velocity (v g). On the other hand, the lattice thermal conductivity (κ l) is the magnitude of thermal transport that occurs via the crystal lattice vibrations of the TE material and depends on τ, v g, and the phonon wavelength. The lattice thermal conductivity is affected by various physical characteristics of the material such as point defects in the lattice, ionized impurity sites, grain boundaries, etc. In particular, as the crystallite size reduces to feature sizes of the electrons and phonons in the nanocrystalline materials, interfacial sites and barrier potentials at the grain boundaries modify the typical behavior of the transport mechanisms by affecting the phonon and electron mean free paths. It should also be noted that the electrical transport properties are also a function of the defect density, impurity concentration, and the size of the crystallite in the TE material systems. Therefore, the effects of nanostructuring could be perceived by a reduction in both the electrical and the lattice thermal conductivities due to enhanced interface scatterings. Additional effects of nanostructuring may include selective carrier filtering which increases the Seebeck coefficient. In order to quantitatively comprehend the electrical and thermal transport properties of the nanostructured TE materials, semiclassical theoretical models have been implemented under the relaxation time approximation [43].
An effective calculation methodology to describe the nano bulk effects in Mg2Si and Si1−x Ge x is based on solving the Boltzmann transport equation coupled with the relaxation time approximation [21, 22]. The Debye model can further be applied simultaneously in order to determine the thermal conduction properties of the two materials being discussed. Such a rigorously simulation-tested modeling methodology was presented in detail in [22] and [43] for Mg2Si and Si1−x Ge x , respectively.
4.5.2 Modeling Methodology
For the material systems in discussion, a multiband transport mechanism is taken into account in order to efficiently estimate the thermoelectric properties [22, 120]. The sequence of the steps included in the modeling and simulation strategy are shown in Fig. 4.9. Under the assumption that the band structure-related and electronic properties of the material are supplied, the calculation methodology primarily includes the following stages:

Flow diagram of the methodology of modeling of TE properties
-
1.
Under the assumption of the charge conservation, calculation of the Fermi level, E F, based on (4.5a and 4.5b).
-
2.
Calculation of the charge carrier and phonon relaxation times based on scattering mechanisms.
-
3.
Estimation of the charge carrier mobility, Seebeck coefficient, and electrical conductivity.
-
4.
Estimation of the electronic and bipolar thermal conductivities.
-
5.
Determination of the lattice thermal conductivity based on the Debye model.
Where n o is the charge carrier concentration. For a given temperature and doping concentration, the Fermi level is determined by solving (4.5a and 4.5b). The Matthiessen’s rule shown in (4.6) determines the total relaxation times for phonon and carriers where i in represents the type of scattering mechanism [120].
The model is strongly established on the material parameters related to the physical properties such as the lattice properties and the band structure parameters.
Mg2 X-based compounds, where X = Si, Ge, or Sn, are known to have face centered cubic lattice structure. The forbidden energy gap for Mg2Si is indirect (Г 15–X 3) with E ing = 0.77 eV and forms the primary energy gap [133]. The multiband assumption for Mg2Si is justified from the fact that Mg2Si has three conduction band minima located at X 3, X 1, and L 1 points [80]. However, there is only one valence band maximum which is located at Г 15. The inter-band distances such as the X 1 and X 3 gap (0.4 eV) and the X 3–L 1 separation of 0.5 eV are also provided as the input based on the band structure [133]. Band structural features such as the non-parabolicity are considered for all bands and are implemented under a modified E(k) relation shown in (4.7) [43],
where k l and k t are the longitudinal and transverse components of the electron wavevector, respectively; m l and m t are the longitudinal and transverse components of the effective mass of each valley, and α is the non-parabolicity coefficient.
Analogous to the parameters for Mg2Si, the calculation model for Si0.8Ge0.2 was built based on the band structure properties, physical and structural characteristics, and electronic and lattice parameters [43]. The simulation parameters for both materials are summarized in Table 4.2.
4.5.2.1 Scattering Mechanisms
The doped compound of bulk Si0.8Ge0.2 is known to accommodate phonon scatterings due to acoustic phonon, intravalley phonon, ionized impurities, and point defects. In the nano bulk structures, the calculations account for an additional scattering mechanism in the form of grain boundary scattering. Similarly, in the crystalline form of doped bulk Mg2Si, scattering mechanisms are dominated by acoustic phonons and ionized impurities in the lattice. The energy dependent relaxation times can be estimated based on the Brooks–Herring formula via Thomas–Fermi approximation defined in (4.8) [80],
where λ s is the Debye screening length and is defined as,
where n I is the number of ions, E is the energy relative to the band edge, z is ionization number, m* is the effective mass, e is the charge of electron, f o is the Fermi–Dirac distribution obtained from (4.5b), ε is the low frequency dielectric constant, ε ∞ is the high frequency dielectric constant, and ℏ is the Planck’s constant. Likewise, the intravalley acoustic and optical phonon deformation potential scattering rates can be estimated using the equations [80],
where E is the energy relative to the band edge, D A and D v are acoustic phonon deformation potentials for the scattering of electrons and holes, respectively. T is the temperature, α is the non-parabolicity parameter, D(E) is the density of states, ρ is the density, and v s is the speed of sound.
For the calculations of TE properties of nano bulk systems, a potential barrier due to the presence of the grain boundary (GB) locations and/or due to the presence of the nanoparticles can be considered as shown in Fig. 4.10. The shaded circles schematically indicate the situations where nanoparticle doping such as nanoparticles dispersed in the host matrix is applicable. The randomly oriented lines in Fig. 4.10 specify the additional interfaces created due to the crystallite boundaries. These boundaries act as scattering regions for phonons and charge carriers. The potential barrier in these two cases can be modeled as shown in Fig. 4.10. The grain boundary region can be modeled as a location consisting of a space charge region with a cylindrical symmetry and width indicated by the height of the cylinder (z-axis). The barrier potential can be modeled to be exponentially decaying towards the edges of the space charge region. The mathematical functions that define the variation of the potential barriers in the direction perpendicular to the grain boundary and/or the nanoparticle are shown in (4.12) and (4.13), respectively. In (4.12) and (4.13), U c and U s is the heights of the grain boundary potential and r o is a constant in the order of the screening length. The Fermi’s Golden rule can be applied in order to estimate the effective scattering rate for a chosen grain boundary potential. The grain boundary potentials typically vary in the range of tens of meV to eV. As shown in Table 4.2, a constant value of 100 meV was assumed in these calculations for both material systems to emulate the effect of nanostructuring.

The schematic of the potential barriers due to the grain boundaries (cylindrical) and nanoparticle (circular) scattering zones
An effective implementation of this model accounts for the assumption of coherent carrier scattering at the grain boundaries. It is known that in typical nano bulk TE material systems, the crystallite sizes vary in a wide range. In order to estimate the TE characteristics efficiently, a Gaussian distribution of grain sizes with a certain average crystallite size can be considered. In the calculation results to be discussed in the next sub-sections, a Gaussian distribution of grain sizes with an average size of 20 nm was implemented.
4.5.2.2 Relaxation Time
In order to account for the effect of phonon and carrier energies in conjunction to the type of scattering mechanisms, the energy dependent scattering rates for each scattering mechanism were individually estimated. The scattering rates were then combined using the Matthiessen’s rule. Figure 4.11 shows the variation of the relaxation times for various scattering mechanisms in Mg2Si. As indicated by Fig. 4.11, the acoustic phonons dominate in the scattering of electrons with energies higher than 0.1 eV. The ionized impurity scattering (IIS) is only dominant at lower energy levels.

Calculated relaxation time versus energy for various scattering mechanisms in n-type Mg2Si [21]
It is indicative from Fig. 4.11 that the grain boundary scattering mechanism introduces additional scattering and is relatively dominant for carriers with lower (<0.1 eV) energies. Analogous to this situation, for carriers with energies greater than 0.1 eV, the ionized impurity and acoustic phonon scattering are dominant. It can also be inferred from Fig. 4.11 that in highly doped systems the mechanism of grain boundary scattering may be ineffective for a majority of carriers due to their high energy ranges.
4.5.2.3 Estimation of Lattice Thermal Conductivity
The thermal conductivity characteristics for both Mg2Si and Si0.8Ge0.2 can be calculated accurately under the contributions of the phonon scattering mechanisms such as 3-phonon, phonon–electron, alloy scattering, and grain boundary scattering. The relaxation times can be estimated for each scattering mechanism and the lattice thermal conductivity, κ l, the electronic contribution to thermal conductivity, κ e, and bipolar thermal conductivity can be estimated using (4.14)–(4.19) [21, 22, 43, 120]
in which x = ω/ω D, where ω D is the Debye frequency, v = (θ D/T)η, θ D is the Debye temperature, η is a fitting parameter to incorporate the effect of higher order phonon scattering (see Table 4.2), β is the ratio of Umklapp to normal-mode scattering, τ and τ U are the total and Umklapp relaxation times, respectively, T is absolute temperature, i, j are the valleys indices, σ is the conductivity, and S is the Seebeck coefficient. L is the carrier concentration dependent Lorentz number that can be numerically calculated to estimate the electronic part of the thermal conductivity as discussed earlier via (4.3). Lastly, the total thermal conductivity k total can be estimated as the sum of all three components κ total = κ l + κ c + κ b.
Figure 4.12 shows the variation of the calculated phonon mean free path (PMFP) in n-type Mg2Si plotted versus the inverse of the phonon wavelength that is proportional to the phonon energy. It is indicative from the plot that the slope of PMFP varies with phonon energy. It is also known that PMFP is a function of the scattering type [21]. At very low energies of phonons, it is known that electron–phonon (e–p) scattering is dominant and PMFP has a frequency dependency of 1/ω. With the increasing energy of phonons, the slope sharply increases with 1/ω 2 dependency as the 3-phonon (3-p) scattering dominates. At higher energies, the PMFP has a 1/ω 4 dependency due to the dominancy of the point defect scattering [21]. Figure 4.12 also shows the dependency of the accumulated lattice thermal conductivity, κ l, on the phonon energy. The κ l remains at a very low magnitude for low energy phonons, but increases rapidly with phonon energy after a certain energy. The effect of nanostructuring on the PMFP and κ l is comprehensively shown in Fig. 4.12. The calculations were performed after assuming an average crystallite size of 20 nm. It can be observed in Fig. 4.12 that at very low phonon energies, the PMFP saturates to approximately the chosen size of the grains. The line labeled GB PMFP also suggests the dominance of the grain boundary scattering resulting in such saturation. The resultant drop in κ l in the nano bulk Mg2Si is also presented in comparison with the bulk crystalline system.

4.5.2.4 Estimation of the Electrical Conductivity and the Seebeck Coefficient
In the multiband transport model for both Mg2Si and Si0.8Ge0.2, the electrical conductivity and Seebeck coefficient assume the forms shown in (4.20–4.22).
In Equations (4.20–4.22), i and j represent the valleys; q is the charge of the carrier; μ n and μ p are the carrier mobilities for electrons and holes, respectively; n and p indicate the electron and hole concentrations, respectively.
4.5.3 The Effect of the Grain Size on the TE Properties of Mg2Si and Si0.8Ge0.2
To better understand the benefits and secondary effects of nanostructuring, the thermoelectric characteristics of bulk (solid lines) and nano bulk (dashed lines) n-type Si0.8Ge0.2 and n-type Mg2Si are calculated with the best fitting models shown in Table 4.2. The predicted accumulated electrical conductivity (σ) and lattice thermal conductivity (κ l) plotted as a function of wavelength in n-type Si0.8Ge0.2 (calculated at T = 1,300 K) and n-type Mg2Si (calculated at 850 K) are shown in Fig. 4.13a, b. A grain size of 20 nm and a GB potential of 100 meV were assumed for both the material systems.

Comparison of (a) accumulated electrical conductivity and (b) accumulated thermal conductivity in bulk (solid lines) and nano bulk (dashed lines) n-type Si0.8Ge0.2 (calculated at 1,300 K) and n-type Mg2Si (calculated at 850 K)
Figure 4.13a, b shows a comparison of the effects of nanostructuring in both the material systems. As can be observed from Fig. 4.13a, the electrical conductivity (σ) in Mg2Si suffers significantly with a drop of nearly 40 % in the nano bulk system as compared to the bulk crystalline system. Such an unfavorable effect in nano bulk Mg2Si is a result of the loss in carrier mobility as will be discussed later in this section. In the corresponding calculations for Si0.8Ge0.2 the deterioration in electrical conductivity (σ) is a mere ~10 % of the bulk crystalline system.
The lattice thermal conductivity depicted in Fig. 4.13b shows that the short wavelength phonons carry most of the heat. It can be observed that phonons with wavelengths in the range of 0.5–10 nm contribute significantly to κ l in both Si0.8Ge0.2 and Mg2Si. The thermal conductivity increases rapidly with cumulative contribution from the short wavelength phonons and remains almost unchanged with adding the contribution of the long wavelength phonons. However, the limitation of grain size to 20 nm leads to ~60 % reduction in κ l of Si0.8Ge0.2, whereas the same in Mg2Si is only ~35 %. Considering the loss in σ for the same range of grain size in Mg2Si, the corresponding loss in power factor could nearly cancel out the effect of the reduction in κ l, eventually leading to little or no improvement in the ZT.
The detrimental effect of the nanostructuring in Mg2Si is primarily due to the reduction of charge carrier mobility due to the enhanced scattering at the grain boundaries. Such an effect is the result of the PMFP and CMFP falling in the same range for Mg2Si [21]. In order to carefully perceive this effect, the differential electrical conductivity and differential lattice thermal conductivity plotted versus energy for both the material systems are shown in Fig. 4.14a, b. The area under each curve is indicative of the corresponding total contribution of each component.

Comparison of the differential electrical and thermal conductivities in bulk (solid lines) and nano bulk (dashed lines) systems of (a) n-type Si0.8Ge0.2 at 1,300 K and (b) n-type Mg2Si at 850 K [22]
It can be observed from Fig. 4.14a that more than 50 % drop in differential electrical conductivity is resulted from restricting the grain size to 20 nm. This magnitude of drop is less than that of Si0.8Ge0.2 shown in Fig. 4.14b. Similarly, the magnitude of the reduction in differential thermal conductivity is not comparable to Si0.8Ge0.2 as indicted by the green lines in Fig. 4.14a, b.
Carrier filtering effects, which result in the increase of the average energy of the carriers, were seen from the predicted electrical conductivity plots. A slight increase in the Seebeck coefficient was estimated using (4.23) where ΔE is the change in the energy of the carrier and T is the temperature. The estimated increase of the Seebeck coefficients was 15 μV/K and 13 μV/K for Mg2Si and Si0.8Ge0.2, respectively [22].
Figure 4.15a, b shows a comparison of the calculated Hall mobility versus temperature in bulk and nano bulk Mg2Si and Si0.8Ge0.2 at selected optimum doping concentrations for n-type and p-type systems, respectively. The carrier mobility linearly decreases with temperature pertaining to the carrier scattering by acoustic phonons which increases with temperature. The introduction of the grain boundaries worsens the magnitude of the mobility, particularly in both n-type and p-type Mg2Si. In comparison, the n-type and p-type Si0.8Ge0.2 suffer a lower magnitude loss in mobility and the loss is much lower at high temperatures where the acoustic phonons are dominant carrier scatterers. Subsequently, an Mg2Si system gets adversely affected over the entire temperature range.

Figure 4.16 shows the electrical and thermal conductivity of Mg2Si plotted against the grain size. It is estimated from Fig. 4.16 by means of a sudden descent at grain sizes less than 20 nm that the electrical conductivity is affected more severely compared to the reduction in thermal conductivity in nano bulk Mg2Si. An unfavorable 35 % drop in electrical conductivity was predicted as a result of the decrease in the grain size from 20 to 5 nm. Such deterioration in the electrical conductivity is expected to affect the power factor negatively given the fact that the carrier filtering effect also leads to a very slight increase in the Seebeck coefficient as discussed earlier via Fig. 4.14.

Comparison of calculated electrical and thermal conductivity versus grain size in bulk and nanostructured n-Mg2Si at 850 K [22]
Figure 4.17a, b depicts the predicted ZT versus doping concentration for bulk and nano bulk systems in n-type and p-type Mg2Si. The results are shown in comparison to n-type and p-type Si0.8Ge0.2. It is evident from Fig. 4.17a that the nanostructuring does not show any significant effect on the maximum attainable ZT of Mg2Si over the chosen doping concentration range. Similar calculation for the p-type materials shows that nanostructuring of p-type Mg2Si enhances the ZT to a maximum value of 0.9 at an optimum carrier concentration of 4.3 × 1020 cm−3.

Comparison of the calculated figure-of-merit versus temperature for (a) n-type and (b) p-type bulk and nanostructured Mg2Si at 850 K
4.6 Future Directions
A fundamental understanding of the transport properties of the charge carriers and phonons in nanoscale can lead to more practical developments and better material designs. A proper theory should be able to explain how the nanoscale structure or structural dependencies at atomistic level such as the crystallite size, crystallite orientation, energy band discontinuity, and the randomness associated with these parameters in nanocrystalline structures affect the bulk properties. Moreover, the calculations of the dispersion curves, momentum, and energy relaxation times are necessary. The enhancement of ZT in nano bulk materials has been achieved through the reduction in thermal conductivity below that of the bulk material, so the next improved materials will feature both the reduction in thermal conductivity and improvement in power factor. It is also necessary to quantify the contribution of different geometries of the nanostructures in the reduction of the lattice thermal conductivity and the improvement of the power factor. This effort should address the role of nanostructures, grain and nanoparticle sizes, grain and nanoparticle size distributions, and finally grain and nanoparticle orientations.
It is expected that the theoretical calculations have a major role in providing techniques for the design and optimization of the new materials and their synthesis processing parameters. Material synthesis parameters should allow for prevention of the grain growth during processing and maintaining the properties of the materials at their working temperature or other required working conditions.
References
Seebeck, T.J.: Magnetische Polarisation der Metalle und Erze Durch Temperatur-Differenz. Ostwald’s Klassiker der Exakten Wissenshaften Nr. 70, 1822–1823 (1895)
Peltier, J.: Nouvelles Experiences sur la Caloriecete des Courans Electrique. Ann. Chim. LVI, 371–387 (1834)
Thomson, W.: On the dynamical theory of heat. Trans. R. Soc. Edinb. 3, 91–98 (1851)
Haken, W.: Beitrag zur Kenntnis der thermoelektrischen Eigenschaften der Metallegierungen. Ann. Phys. 333(7), p291–p336 (1910)
Ioffe, A.: Semiconductor Thermoelements and Thermoelectric Cooling. Infosearch, London (1957)
Vineis, C.J., Shakouri, A., Majumdar, A., Kanatzidis, M.G.: Nanostructured thermoelectrics: big efficiency gains from small features. Adv. Mater. 22, 3970–3980 (2010)
Sootsman, J.R., Chung, D.Y., Kanatzidis, M.G.: New and old concepts in thermoelectric materials. Angew. Chem. Int. Ed. 48, 8616–8639 (2009)
Ioffe, A.: The revival of thermoelectricity. Sci. Am. 199, 31–37 (1958)
Walker, C.T., Pohl, R.O.: Phonon scattering by point defects. Phys. Rev. 131, 1433–1442 (1963)
Slack, G.A.: New materials and performance limits for thermoelectric cooling. In: Rowe, D.M. (ed.) CRC Handbook of Thermoelectrics, p. 407. CRC, Boca Raton, FL (1995)
Mahan, G.D., Sofo, J.O.: The best thermoelectric. Proc. Natl. Acad. Sci. U. S. A. 93, 7436–7439 (1996)
Mahan, G.D.: Good Thermoelectrics, Solid State Physics, vol. 51, pp. 82–157. Academic, New York (1997)
Xie, W., Tang, X., Yan, Y., Zhang, Q., Tritt, T.M.: Unique nanostructures and enhanced thermoelectric performance of melt-spun BiSbTe alloys. Appl. Phys. Lett. 94, 102111 (2009). doi:10.1063/1.3097026
Poudel, B., Hao, Q., Ma, Y., Lan, Y., Yan, X., Wang, D., Vashaee, D., Dresselhaus, M., Chen, G., Ren, Z.F.: High-thermoelectric performance of nanostructured bismuth antimony telluride bulk alloys. Science 320, 634–638 (2008)
Minnich, A.J., Dresselhaus, M.S., Ren, Z.F., Chen, G.: Bulk nanostructured thermoelectric materials: current research and future prospects. Energy Environ. Sci. 2, 466–479 (2009)
Vashaee, D., Shakouri, A.: Improved thermoelectric power factor in metal-based superlattices. Phys. Rev. Lett. 92(10), 106103 (2004)
Shakouri, A., et al.: Demonstration of electron filtering to increase the Seebeck coefficient in In0.53Ga0.47As∕In0.53Ga0.28Al0.19As superlattices. Phys. Rev. B 74, 205335 (2006)
Snyder, G.J., Toberer, E.S.: Complex thermoelectric materials. Nat. Mater. 7(2), 105–114 (2008). doi:10.1038/nmat2090
Pichanusakorn, P., Bandaru, P.: Nanostructured thermoelectrics. Mater. Sci. Eng. R 67, 19–63 (2010)
Dresselhaus, M.S., Chen, G., Tang, M.Y., Yang, R.G., Lee, H., Wang, D.Z., Ren, Z.F., Fleurial, J.-P., Gogna, P.: New directions for low-dimensional thermoelectric materials. Adv. Mater. 19, 1043–1053 (2007). doi:10.1002/adma.200600527
Satyala, N., Vashaee, D.: The effect of crystallite size on thermoelectric properties of bulk nanostructured magnesium silicide (Mg2Si) compounds. Appl. Phys. Lett. 100, 073107 (2012)
Satyala, N., Vashaee, D.: Detrimental influence of nanostructuring on the thermoelectric properties of magnesium silicide. J. Appl. Phys. 112(9), 093716–093716-11 (2012)
Pshenai-Severin, D.A., Federov, M.I., Samunin, A.Y.: The influence of grain boundary scattering on thermoelectric properties of Mg2Si and Mg2Si0.8Sn0.2. J. Electron. Mater. (2013). doi:10.007/s11664-012-2403-0
Heremans, J.P., Thrush, C.M., Morelli, D.T.: Thermopower enhancement in lead telluride nanostructures. Phys. Rev. B 70, 115334 (2004)
Heremans, J.P., Jovovic, V., Toberer, E.S., Saramat, A., Kurosaki, K., Charoenpakdee, A., Yamanaka, S., Snyder, G.J.: Enhancement of thermoelectric efficiency in PbTe by distortion of the electronic density of states. Science 32, 554 (2008)
Harman, T.C., Taylor, P.J., Walsh, M.P., LaForge, B.E.: Quantum dot superlattice thermoelectric materials and devices. Science 297(5590), 2229 (2002)
Venkatasubramanian, R., Silvona, E., Colpitts, T., O’Quinn, B.: Thin-film thermoelectric devices with high room-temperature figures of merit. Nature 413, 597 (2001)
Nolas, G.S., Slack, G.A., Modeli, D.T., Tritt, T.M., Ehrlich, A.C.: The effect of rare‐earth filling on the lattice thermal conductivity of skutterudites. J. Appl. Phys. 79, 4002 (1996)
Fluerial, J.P., Caillat, T., Borshchevsky, A.: Skutterudites: a new class of promising thermoelectric materials. In: Proceedings of the 13th International Conference on Thermoelectrics, p. 40, Kansas City, Missouri (1994)
Sales, B.C., Mandrus, D., Williams, R.K.: Filled skutterudite antimonides: a new class of thermoelectric materials. Science 272, 1325 (1996)
Brown, S.R., Kauzlarich, S.M., Gascoin, F., Snyder, G.J.: Yb14MnSb11: new high efficiency thermoelectric material for power generation. Chem. Mater. 18, 1873–1877 (2006)
Toberer, E.S., Christensen, M., Iversen, B.B., Snyder, G.J.: High temperature thermoelectric efficiency in Ba8Ga16Ge30. Phys. Rev. B 77, 075203 (2008)
Caillat, T., Fleurial, J.P., Borshchevsky, A.: Preparation and thermoelectric properties of semiconducting Zn4Sb3. J. Phys. Chem. Solids 58, 1119–1125 (1997)
Kurosaki, K., Kosuga, A., Muta, H., Uno, M., Yamanaka, S.: Ag9TITe5: a high-performance thermoelectric bulk material with extremely low thermal conductivity. Appl. Phys. Lett. 87, 061919 (2005)
Pei, Y., Wang, H., Snyder, G.J.: Band engineering of thermoelectric materials. Adv. Mater. 24, 6125 (2012). doi:10.1002/adma.201202919
Chen, G.: Thermal conductivity and ballistic-phonon transport in the cross-plane direction of superlattices. Phys. Rev. B 57, 14958 (1998)
Hicks, L.D., Dresselhaus, M.S.: Effect of quantum-well structures on the thermoelectric figure of merit. Phys. Rev. B 47, 12727 (1993)
Vashaee, D., Zhang, Y., Shakouri, A., Zeng, G., Chiu, Y.: Cross-plane Seebeck coefficient in superlattice structures in the miniband conduction regime. Phys. Rev. B 74, 195315 (2006)
Vashaee, D., Shakouri, A.: Electronic and thermoelectric transport in semiconductor and metallic Superlattices. J. Appl. Phys. 95(3), 1233–1245 (2004)
Zide, J.T.M., Klenov, D.O., Stemmer, S., Gossard, A., Zeng, G.H., Bowers, J.E., Vashaee, D., Shakouri, A.: Thermoelectric power factor in semiconductors with buried epitaxial semimetallic nanoparticles. Appl. Phys. Lett. 87, 112102 (2005)
Poudel, B., Hao, Q., Ma, Y., Lan, Y., Yan, X., Wang, D., Vashaee, D., Dresselhaus, M., Chen, G., Ren, Z.F.: High thermoelectric performance of nanostructured bismuth antimony telluride bulk alloys. Science Express Research Articles (2008). doi:10.1126/science.1156446
Zamanipour, Z., Shi, X., Dehkordi, A.M., Krasinski, J.S., Vashaee, D.: The effect of synthesis parameters on transport properties of nanostructured bulk thermoelectric p-type silicon germanium alloy. Physica Status Solidi (a) 209(10), 2049–2058 (2012)
Minnich, A.J., Lee, H., Wang, X.W., Joshi, G., Dresselhaus, M.S., Ren, Z.F., Chen, G., Vashaee, D.: Modeling study of thermoelectric SiGe nanocomposites. Phys. Rev. B 80, 155327 (2009)
Biswas, K., He, J., Blum, I.D., Wu, C.-I., Hogan, T.P., Seidman, D.N., Dravid, V.P., Kanatzidis, M.G.: High-performance bulk thermoelectrics with all-scale hierarchical architectures. Nature 489, 414–418 (2012)
Thermion Company: Implemented conventional bulk technologies to fabricate thermoelectric devices of ~200μm thick. http://www.thermion-company.com (2005)
Gould, C., Shammas, N.: A review of thermoelectric MEMS devices for micro-power generation, heating and cooling applications. In: Takahata, K. (ed.) Micro Electronic and Mechanical Systems. Available from: http://www.intechopen.com/books/micro-electronicand-mechanical-systems/a-review-of-thermoelectric-mems-devices-for-micro-power-generation-heating-andcooling-applications
Joshi, G., Lee, H., Lan, Y., Wang, X., Zhu, G., Wang, D., Gould, R.W., Cuff, D.C., Tang, M.Y., Dresselhaus, M.S., Chen, G., Ren, Z.: Enhanced thermoelectric figure-of-merit in nanostructured p-type silicon germanium bulk alloys. Nano Lett. 8(12), 4670 (2008)
Bathula, S., Jayasimhadri, M., Singh, N., Srivastava, A.K., Pulikkotil, J., Dhar, A., Budhani, R.C.: Enhanced thermoelectric figure-of-merit in spark plasma sintered nanostructured n-type SiGe alloys. Appl. Phys. Lett. 101, 213902 (2012)
Mingo, N., Hauser, D., Kobayashi, N.P., Plissonnier, M., Shakouri, A.: Nanoparticle-in-alloy approach to efficient thermoelectrics: silicides in SiGe. Nano Lett. 9(2), 711–715 (2009)
Rowe, D.M. (ed.): CRC Handbook of Thermoelectrics. CRC, Boca Raton, FL (1995)
Zhu, Y.T., Langdon, T.G.: The fundamentals of nanostructured materials processed by severe plastic deformation. J. Miner. 56(10), 58–63 (2004)
Segal, V.M.: Engineering and commercialization of equal channel angular extrusion (ECAE). Mater. Sci. Eng. A386, 269 (2004)
Saravanan, M., Pillai, R.M., Pai, B.C.: Equal channel angular pressing of aluminum alloys and composites—an overview. Aluminum India 5(2), 3–14 (2005)
Slarnova, K.: Accumulative roll-bonding: first experience with a twin-roll cast AA8006 alloy. J. Alloys Compd. 378(1–2), 322–325 (2004)
Tsuji, N., Saito, Y., Lee, S.-H., Minamino, Y.: ARB (accumulative roll-bonding) and other new techniques to produce bulk ultrafine grained materials. Adv. Eng. Mater. 5(5), 338 (2003)
Androulakis, J., Lin, C.H., Kong, H.J., Uher, C., Wu, C.I., Hogan, T., Cook, B.A., Caillat, T., Paraskevopoulos, K.M., Kanatzidis, M.G.: Spinodal decomposition and nucleation and growth as a means to bulk nanostructured thermoelectrics: enhanced performance in Pb1-xSnxTe–PbS. J. Am. Chem. Soc. 129, 9780 (2007)
Sootsman, J.R., Pcionek, R.J., Kong, H., Uher, C., Kanatzidis, M.G.: Strong reduction of thermal conductivity in nanostructured PbTe prepared by matrix encapsulation. Chem. Mater. 18, 4993 (2006)
Hsu, K.F., Loo, S., Guo, F., Chen, W., Dyck, J.S., Uher, C., Hogan, T., Polychroniadis, E.K., Kanatzidis, M.G.: Cubic AgPb m SbTe2+m : bulk thermoelectric materials with high figure of merit. Science 303, 818 (2004)
Poudeu, P.F.P., D’Angelo, J., Downey, A.D., Short, J.L., Hogan, T.P., Kanatzidis, M.G.: High thermoelectric figure of merit and nanostructuring in bulk p-type Na1−x Pb m Sb y Te m+2. Angew. Chem. Int. Ed. 45, 3835 (2006)
Blum, I.D., Isheim, D., Seidman, D.N., He, J., Androulakis, J., Biswas, K., Dravid, V.P., Kanatzidis, M.G.: Dopant distributions in PbTe-based thermoelectric materials. J. Electron. Mater. 41(6), 1583–1588 (2012)
Chen, S., Carroll, D.L.: Synthesis and characterization of truncated triangular silver nanoplates. Nano Lett. 2, 1003 (2002)
Maillard, M., Giorgio, S., Pileni, M.P.: Silver nanodisks. Adv. Mater. 14, 1084 (2002)
Hao, E., Kelly, K.L., Hupp, J.T., Schatz, G.C.: Synthesis of silver nanodiscs using polystyrene meso-spheres as templates. J. Am. Chem. Soc. 124, 15182 (2002)
Ibano, D., Yokota, Y., Tominaga, T.: Preparation of gold nanoplates protected by an anionic phospholipid. Chem. Lett. 32, 574 (2003)
Simakin, A.V., Voronov, V.V., Shafeev, G.A., Brayner, R., Bozon-Verduraz, F.: Nanodisks of Au and Ag produced by laser ablation in liquid environment. Chem. Phys. Lett. 348, 182 (2001)
Lu, W., Ding, Y., Chen, Y., Wang, Z.L., Fang, J.: Formation of PbSe nanocrystals: a growth toward nanocubes. J. Am. Chem. Soc. 127, 10112 (2005)
Wang, W., Poudel, B., Yang, J., Wang, D.Z., Ren, Z.F.: High-yield synthesis of single-crystalline antimony telluride hexagonal nanoplates using a solvothermal approach. J. Am. Chem. Soc. 127, 13792 (2005)
Bertini, L., Stiewe, C., Toprak, M., Williams, S., Platzek, D., Mrotzek, A., Zhang, Y., Gatti, C., Muller, E., Muhammed, M., Rowe, M.: Nanostructured Co1−xNixSb3 skutterudites: synthesis, thermoelectric properties, and theoretical modeling. J. Appl. Phys. 93, 438 (2003)
Liu, C.J., Yamauchi, H.: Thermoelectric power and resistivity of La1.8Sr0.2CaCu2O6 and the effects of O2 hot-isostatic-press annealing. Phys. Rev. B 51, 11826 (1995)
Wang, X.W., Lee, H., Lan, Y.C., Zhu, G.H., Joshi, G., Wang, D.Z., Yang, J., Muto, A.J., Tang, M.Y., Klatsky, J., Song, S., Dresselhaus, M.S., Chen, G., Ren, Z.: Enhanced thermoelectric figure of merit in nanostructured n-type silicon germanium bulk alloy. Appl. Phys. Lett. 93, 193121 (2008)
Kishimoto, K., Koyanagi, T.: Temperature dependence of the Seebeck coefficient and the potential barrier scattering of n-type PbTe films prepared on heated glass substrates by rf sputtering. J. Appl. Phys. 92(9), 2544 (2002)
Wang, H., Li, J.F., Nan, C.W., Zhou, M., Liu, W., Zhang, B.P., Kita, T.: High-performance Ag0.8Pb18+xSbTe20 thermoelectric bulk materials fabricated by mechanical alloying and spark plasma sintering. Appl. Phys. Lett. 88, 092104 (2006)
Li, H., Tang, X.F., Su, X.L., Zhang, Q.J.: Preparation and thermoelectric properties of high-performance Sb additional Yb0.2Co4Sb12+y bulk materials with nanostructure. Appl. Phys. Lett. 92, 202114 (2008)
Chen, G.: Recent trends in thermoelectric materials research III. In: Willardson, R.K., Weber, E.R. (eds.) Semiconductors and Semimetals, vol. 71, pp. 203–259. Academic, New York (2001)
Liu, H., Zheng, Z., Yang, D., Ke, X., Jaatinen, E., Zhao, J.C., Zhu, H.Y.: Coherent interfaces between crystals in nanocrystal composites. ACS Nano 4(10), 6219–6227 (2010)
Huang, X., Wang, X., Cook, B.: Coherent nanointerfaces in thermoelectric materials. J. Phys. Chem. C 114(49), 21003–21012 (2010)
Bartkowiak, M., Mahan, G.D.: Heat and electricity transport through interfaces. In: Tritt, T.M. (ed.) Recent Trends in Thermoelectric Materials, vol. 2. Semiconductors and Semimetals, vol. 70, pp. 245–271. Academic, New York (2001)
Swartz, E.T., Pohl, R.O.: Thermal boundary resistance. Rev. Mod. Phys. 61(3), 605–658 (1989)
Singh, J.: Physics of Semiconductors and Their Heterostructures. McGraw-Hill, Singapore (1996)
Lundstrom, M.: Fundamentals of Carrier Transport, 2nd edn. Cambridge University Press, Cambridge (2000)
Heremans, J.P., Thrush, C.M., Morelli, D.T.: Thermopower enhancement in PbTe with Pb precipitates. J. Appl. Phys. 98, 063703 (2005)
Ramu, A.T., Cassels, L.E., Hackman, N.H., Lu, H., Zide, J.M.O., Bowers, J.E.: Thermoelectric transport in the coupled valence-band model. J. Appl. Phys. 109, 033704 (2011)
Jacob, U., Vancea, J., Hoffmann, H.: Surface-roughness contributions to the electrical resistivity of polycrystalline metal films. Phys. Rev. B 41, 11852–11857 (1990)
Taylor, W.E., Odell, N.H., Fan, H.Y.: Grain boundary barriers in germanium. Phys. Rev. 93, 666–667 (1954)
Tanaka, T., Hayashi, S., Shibayama, K.: Thermal-depolarization-current study of composites of epoxy resin containing mica flakes. J. Appl. Phys. 48, 3478–3483 (1977)
Shuai, Z., Wang, L., Song, C.: Theory of Charge Transport in Carbon Electronic Materials. SpringerBriefs in Molecular Science. Springer, Berlin (2012). doi:10.1007/978-3-642-25076-7_4
Bardeen, J., Shockley, W.: Deformation potentials and mobilities in non-polar crystals. Phys. Rev. 80(1), 72–80 (1950)
Ward, A., Broido, D.A.: Intrinsic phonon relaxation times from first-principles studies of the thermal conductivities of Si and Ge. Phys. Rev. B 81, 085205 (2010)
Tritt, T.M. (ed.): Thermal Conductivity: Theory, Properties, and Applications. Springer, New York (2005)
Toberer, E.S., Zevalkink, A., Snyder, G.J.: Phonon engineering through crystal chemistry. J. Mater. Chem. 21, 15843 (2011)
Pettersson, S.: Calculation of the thermal conductivity of alkali halide crystals. J. Phys. C Solid State Phys. 20(8), 1047–1061 (1987)
Slack, G.A.: Solid State Physics, vol. 34. Academic, New York (1979)
Roufosse, M., Klemens, P.G.: Thermal-conductivity of complex dielectric crystals. Phys. Rev. B 7(12), 5379–5386 (1973)
Pohl, R.O.: Thermal conductivity and phonon resonance scattering. Phys. Rev. Lett. 8(12), 481–483 (1962)
Wagner, M.: Influence of localized modes on thermal conductivity. Phys. Rev. 131(4), 1443–1455 (1963)
Morelli, D.T., Jovovic, V., Heremans, J.P.: Intrinsically minimal thermal conductivity in cubic I-V-VI2 semiconductors. Phys. Rev. Lett. 101(3), 035901 (2008)
Hopkins, P.E., Duda, J.C., Norris, P.M.: Anharmonic phonon interaction at interfaces and contributions to thermal boundary conductance. J. Heat Transfer 133(6), 062401 (2011)
Rath, S., Sanyal, S.P.: Effect of strain on vibrational modes in strained layer superlattices. Pramana 41(1), 21–29 (1993)
Herring, C., Vogt, E.: Transport and deformation-potential theory for many-valley semiconductors with anisotropic scattering. Phys. Rev. 101(3), 944–961 (1956)
Bir, G., Pikus, G.: Symmetry and Strain-Induced Effects in Semiconductors. Wiley, New York (1974)
Henry, A.S., Chen, G.: Spectral phonon transport properties of silicon based on molecular dynamics simulations and lattice dynamics. J. Comput. Theor. Nanosci. 5(2), 141–152(12) (2008)
Chou, F.C., Lukes, J.R., Liang, X.G., Takahashi, K., Tien, C.L.: Molecular dynamics in microscale thermophysical engineering. Annu. Rev. Heat Transfer 10, 141–176 (1999)
Chen, G., Tasciuc, B., Yang, R.: Nanoscale heat transfer. In: Nalwa, H.S. (ed.) Encyclopedia of Nanoscience and Nanotechnology, vol. 7, p. 429. American Scientific Publishers, Stevenson Ranch, CA (2004)
Maiti, A., Mahan, G.D., Pantelides, S.T.: Dynamical simulations of non-equilibrium processes—heat flow and the Kapitza resistance across grain boundaries. Solid State Commun. 102(7), 517 (1997)
Stevens, R.J., Zhigilei, L.V., Norris, P.M.: Effects of temperature and disorder on thermal boundary conductance at solid-solid interfaces: nonequilibrium molecular dynamics simulations. Int. J. Heat Mass Transfer 50(19–20), 3977 (2007)
Zhong, H.L., Lukes, J.R.: Interfacial thermal resistance between carbon nanotubes: molecular dynamics simulations and analytical thermal modeling. Phys. Rev. B 74, 125403 (2006)
Kaddouri, H., Benet, S., Charar, S., Makowaska-Janusik, M., Tedenac, J.C., Kityk, I.V.: Simulation of thermoelectric properties of bismuth telluride single crystalline films grown on Si and SiO2 surfaces. Phys. Rev. B 62(24), 17108 (2000)
Chonan, T., Katayama, S.: Molecular-dynamics simulation of lattice thermal conductivity in Pb1-x Sn x Te and Pb1-x Ge x Te at high temperature. J. Phys. Soc. Jpn. 75(6), 064601 (2006)
Huang, X., Huai, X., Liang, S., Wang, X.: Thermal transport in Si/Ge nanocomposites. J. Phys. D. Appl. Phys. 42, 095416 (2009)
Datta, S.: Electronic Transport in Mesoscopic Systems. Cambridge University Press, Cambridge (1995)
Meir, Y., Wingreen, N.S.: Landauer formula for the current through an interacting electron region. Phys. Rev. Lett. 68(16), 2512–2515 (1992)
Bulusu, A., Walker, D.G.: Modeling of thermoelectric properties of semi-conductor thin films with quantum and scattering effects. J. Heat Transfer 129(4), 492–499 (2006)
Savic, I., Stewart, D.A., Mingo, N.: Thermal conduction mechanisms in boron-nitride nanotubes: few-shell versus all-shell conduction. Phys. Rev. B 78, 235434 (2008)
Savic, I., Mingo, N., Stewart, D.A.: Phonon transport in isotope-disordered carbon and boron-nitride nanotubes: is localization observable? Phys. Rev. Lett. 101, 165502 (2008)
Stewart, D.A., Savic, I., Mingo, N.: First-principles calculation of the isotope effect on boron nitride nanotube thermal conductivity. Nano Lett. 9(1), 81–84 (2009)
Kim, W., Wang, R., Majumdar, A.: Nanostructuring expands thermal limits. Nano Today 2(1), 40–47 (2007)
Tani, J.I., Kido, H.: Thermoelectric properties of Al-doped Mg2Si1−x Sn x (x ≦ 0.1). J. Alloys Compd. 466, 335–340 (2008)
Choi, S.M., Kim, K.H., Kim, I.H., Kim, S.U., Seo, W.S.: Thermoelectric properties of the Bi-doped Mg2Si system. Curr. Appl. Phys. 11(3 Suppl), S388–S391 (2011)
Akasaka, M., Iida, T., Matsumoto, A., Yamanaka, K., Takanash, Y., Imai, T., Hamada, N.: The thermoelectric properties of bulk crystalline n- and p-type Mg2Si prepared by the vertical Bridgman method. J. Appl. Phys. 104, 013703 (2008)
Satyala, N., Vashaee, D.: Modeling of thermoelectric properties of magnesium silicide (Mg2 Si). J. Electron. Mater. 41(6), 1785–1791 (2012)
Zaitsev, V.K., Fedorov, M.I., Gurieva, E.A., Eremin, I.S., Konstantinov, P.P., Samunin, A.Y., Vedernikov, M.V.: Highly effective Mg2Si1−x Sn x thermoelectrics. Phys. Rev. B 74, 045207 (2006)
Mars, K., Ihou-mouko, H., Pont, G., Tobola, J., Scherrer, H.: Thermoelectric properties and electronic structure of Bi- and Ag-doped Mg2Si1−x Ge x compounds. J. Electron. Mater. 38(7), 1360 (2009)
Kim, I.-H., Jung, J.-Y., Choi, S.-M., Seo, W.-S., Kim, S.-U.: Synthesis of thermoelectric Mg2Si by mechanical alloying. J. Korean Phys. Soc. 57(4), 1005–1009 (2010)
Tani, J.-I., Kido, H.: Thermoelectric properties of Sb-doped Mg2Si semiconductors. Intermetallics 15, 1202–1207 (2007)
Sakamoto, T., Iida, T., Kurosaki, S., Yano, K., Taguchi, H., Nishio, K., Takanashi, Y.: Thermoelectric behavior of Sb- and Al-doped n-type Mg2Si device under large temperature differences. J. Electron. Mater. 40(5), 629–634 (2010)
Savary, E., Gascoin, F., Marinel, S.: Fast synthesis of nanocrystalline Mg2Si by microwave heating: a new route to nano-structured thermoelectric materials. Dalton Trans. 39, 11074–11080 (2010)
Dube, D.C., Fu, M., Agrawal, D., Roy, R., Santra, A.: Rapid alloying of silicon with germanium in microwave field using single mode cavity. Mater. Res. Innov. 12(3), 119 (2008)
Nikitin, E.N., Bazanov, V.G., Tarasov, V.I.: Thermoelectric properties of solid solutions Mg2Si-Mg2Sn. Sov. Phys. Solid State 3(12), 2648 (1962)
Yang, M., Zhang, L., Shen, Q.: Synthesis and sintering of Mg2Si thermoelectric generator by spark plasma sintering. J. Wuhan Univ. Technol. Mater. Sci Ed 23(6), 870–873 (2008)
Lu, L., Lai, M.O., Hoe, M.L.: Formation of nanocrystalline Mg2Si and Mg2Si dispersion strengthened Mg-Al alloy by mechanical alloying. Nanostruct. Mater. 10(4), 551–563 (1998)
Fiameni, S., Battiston, S., Boldrini, S., Famengo, A., Agresti, F., Barison, S., Fabrizio, M.: Synthesis and characterization of Bi-doped Mg2Si thermoelectric materials. J. Solid State Chem. 193, 142–146 (2012)
Wang, L., Qi, X.Y., Xiong, W., Zhu, X.G.: Fabrication and mechanical properties of bulk nanocrystalline intermetallic Mg2Si. Mater. Sci. Eng. A 459, 216–222 (2007)
Aymerich, F., Mula, G.: Pseudopotential band structures of Mg2Si, Mg2Ge, Mg2Sn, and of the solid solution Mg2(Ge, Sn). Physica Status Solidi (b) 42(697) (1970)
Acknowledgement
We acknowledge the support of Air Force Office of Scientific Research (AFOSR) High Temperature Materials program under grant no. FA9550-10-1-0010 and the National Science Foundation (NSF) under grant no. 0933763.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Satyala, N., Norouzzadeh, P., Vashaee, D. (2014). Nano Bulk Thermoelectrics: Concepts, Techniques, and Modeling. In: Wang, X., Wang, Z. (eds) Nanoscale Thermoelectrics. Lecture Notes in Nanoscale Science and Technology, vol 16. Springer, Cham. https://doi.org/10.1007/978-3-319-02012-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-02012-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-02011-2
Online ISBN: 978-3-319-02012-9
eBook Packages: EnergyEnergy (R0)