
Abstract
Effective utilization of carbon nanotubes (CNTs) as reinforcements in composites necessitates good interfacial bonding with the surround matrix material. The covalent functionalization of CNTs is an effective method to enhance this bonding. However, covalent bonds introduced by a functional group may alter the pristine structure of the CNT and lower its mechanical properties. Here we study the effect of hydrogen (–H), hydroxyl (–OH), carboxyl (–COOH), and amine (–NH2) functionalization on Young’s modulus of a single-wall CNT (SWCNT) using molecular mechanics (MM) simulations with the MM3 potential and the software TINKER. Both pristine and functionalized SWCNTs have been deformed in simple tension. From the strain energy of deformation vs. the axial strain curves, the value of Young’s modulus has been derived as a function of the functionalization group and the amount of functionalization. It is found that Young’s modulus decreases by about 30 % with 20 % of functionalization, the reduction is essentially proportional to the increase in the percentage of the functionalization material and is nearly the same for each of the four functional groups studied.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Researchers have employed experimental, analytical, density functional theory (DFT), molecular dynamics (MD) and molecular mechanics (MM) simulations to predict mechanical properties of carbon nanotubes (CNTs). Assuming that the wall thickness of a single-wall CNT (SWCNT) can be approximated as 0.34 nm, Treacy et al. (1996), Wong et al. (1997), and Krishnan et al. (1998) experimentally determined that Young’s modulus of a CNT is in terapascal (TPa) range. Xing et al. (2004) employed MD simulations to predict Young’s modulus of a SWCNT. Li and Chou (2003) computed elastic properties of CNTs using a combined structural mechanics and MM approach. Chang and Gao (2003) investigated size dependent elastic properties of SWCNTs through MM simulations. Sears and Batra (2004) determined the wall thickness, Young’s modulus, and Poisson’s ratio of CNTs using MM simulation with the MM3 potential and the software TINKER. They assumed that the response of a SWCNT in simple tension is energetically equivalent to that of a thin cylinder made of an isotropic and homogeneous material. Furthermore, the wall thickness of the cylinder equals that of the SWCNT. They found the wall thickness and Young’s modulus of a SWCNT to be 0.046 nm and 7.26 TPa, respectively. Shen and Li (2004) used MM potential and energy equivalent principle to determine values of five elastic constants of a CNT assuming the CNT as a transversely isotropic material with the centroidal axis of the tube as the axis of transverse isotropy. Batra and Sears (2007) proposed that the axis of transverse isotropy of a CNT is a radial line rather than the centroidal axis. By studying with the MM simulations radial expansion of a SWCNT, they showed that Young’s modulus in the radial direction is about 1/4th of that in the axial direction. Gupta and Batra (2008), Batra and Gupta (2008) predicted the wall thickness and the material moduli of a SWCNT by matching frequencies of bending, axial and torsional vibrations as well as that of radial breathing modes of a free–free SWCNT with those of the continuous cylinder of the same length and mean radius as the SWCNT. Wu et al. (2008) developed an atomistic based finite deformation shell theory for a SWCNT and found its stiffness in tension, bending, and torsion.
The SWCNTs due to their cylindrical shape, large length to diameter ratio, and high specific properties are potential candidates as reinforcements in composites. However, the effectiveness of SWCNTs as reinforcements depends on their uniform dispersion in and strong adhesion with the surrounding matrix. It has been very challenging to simultaneously meet these two requirements. The functionalization of SWCNTs appears to be an effective means of achieving good bonding between SWCNTs and the surrounding matrix. The surface properties of CNTs can be modified by either physical or chemical functionalization. The physical functionalization is achieved by attaching noncovalent groups, such as polymer, peptides or surfactants to the nanotubes. It is advantageous since it does not alter the pristine structure of nanotubes hence the mechanical properties of CNTs are not affected. However, these functional groups are attached to nanotubes with weak van der Waals interactions resulting in low load transfer efficiency between the nanotube and the matrix. In comparison, the chemical functionalization involving the covalent attachment of functional groups to atoms of CNTs provides relatively strong interfacial bonding between CNTs and the surrounding matrix thereby enhancing the load transfer efficiency. Haddon et al. used the nanotube-bound carboxylic acid groups for attaching long alkyl chains to SWCNTs via amide linkages (Chen et al. 1998; Hamon et al. 1999; Niyogi et al. 2001) and via carboxylate-ammonium salt ionic interactions (Chen et al. 2001). Khare et al. (2002) developed hydrogenated CNTs using electric discharge. Sun et al. showed that the esterification of the carboxylic acid groups could also be used to functionalize and solubilize nanotubes of any length (Riggs et al. 2000; Sun et al. 2001; Fu et al. 2001). Wilson et al. illustrated the use of anilines to functionalize nanotubes (Sun et al. 2001). Grujicic et al. (2006) investigated the effect of covalent functionalization of triple-walled CNTs on the efficiency of matrix-nanotube load transfer. Their results reveal that the covalent functionalization improves the load transfer efficiency especially when loads are applied in a direction orthogonal to the axis of the CNT. Experimental works (Cooper et al. 2002; Barber et al. 2003; Shofner et al. 2006; Buffa et al. 2007; Sun et al. 2008) have demonstrated the effectiveness of covalent functionalization in increasing the interfacial bonding strength between CNTs and polymer chains.
Although chemical functionalization is effective in enhancing the binding of CNTs with the surrounding matrix, it may damage the pristine structure of a CNT due to the introduction of covalent bonds, and reduce the mechanical stiffness of the CNT. Much of the research work dealing with the determination of Young’s modulus and other elastic properties of CNTs has considered pristine CNTs. Since the covalently functionalized CNTs would be better candidates for reinforcements in a composite than the pristine CNTs, it is important to determine the effect of covalent functionalization on Young’s modulus of a CNT. Zhang et al. (2008) used atomistic simulations to analyze mechanical properties of hydrogenated SWCNTs and found a decrease in Young’s modulus, strength, and ductility of the CNTs with an increase in the percentage of C–H bonds. Kuang and He (2009) computed Young’s moduli of vinyl functionalized SWCNTs using MM simulations with the condense-phase optimized molecular potential for atomistic simulation studies (COMPASS) force field and found that Young’s modulus depends on the density of the sp3 hybridized carbon atoms and chirality of the CNTs. Up to 33 % reduction in Young’s modulus was observed in their study with 38 % content of the functionalizing material. Ling et al. (2012) predicted Young’s modulus of functionalized CNTs using MM and MD simulations with the COMPASS force field and observed that Young’s modulus depends on the type of functionalizing material and the amount of functionalization. Recently, Milowska and Majewski (2013) studied effect of different functional groups on elastic properties of functionalized CNTs using DFT calculations. Their results showed that an increase in amount of covalently bound material to the wall of a CNT decreases Young’s, shear and bulk moduli. Here we have considered four functional groups namely, hydrogen (–H), hydroxyl (–OH), carboxyl (–COOH), and amine (–NH2) and compared their effects on the modulus of elasticity of a SWCNT with varying percentage of functionalization. The study has been carried out using MM simulations with the freely available software TINKER (Ponder 2000).
2 Molecular Mechanics Simulations
2.1 Force-Field
The MM3 potential (Allinger et al. 1989) with higher order expansions and cross-terms has been used to model interatomic interactions. This potential is suitable for studying deformations of CNTs because of similarities between sp2 bonds in the hexagonal structure of CNTs and the hexagonal structure of aromatic proteins for which the potential was originally developed. This potential describes the energy of the system as summation of the energies due to bonded and non-bonded interactions. The contributions for bonded interactions come from bond stretching \( (U_{s} ), \) in-plane angle bending \( (U_{\theta } ), \) out of plane bending \( (U_{\gamma } ), \) torsion \( (U_{\phi } ), \) and cross-interactions including stretch-bend \( (U_{sb} ), \) angle–angle \( (U_{{\theta \theta^{\prime}}} ) \) and stretch-torsion \( (U_{s\phi } ) \). The non-bonded interactions are van der Waals \( (U_{vdW} ) \) and dipole–dipole electrostatic \( (U_{{\mu \mu^{\prime}}} ) \). Equation (1) gives expressions for these energies.
Parameters r, θ and ϕ in Eq. (1) are shown in Fig. 1. Variable \( \gamma \) is the angle between the plane and one of the bonds. A subscript, 0, on a variable represents its value in the configuration of minimum potential energy. Variables \( \mu \) and \( \mu^{\prime} \)are bond centered dipole moments, \( \chi \) is the angle between two dipoles, \( \alpha \) and \( \alpha^{\prime} \) are angles made by two dipoles with the line connecting their centers, and R is the distance between their centers. The values of constants \( K{}_{s} \), \( K_{\theta } \), \( K_{\gamma } , \) \( K_{\theta \theta} ',{ V_{1}}, V_{2}, V_{3}, {K_{s \theta}}, K_{s \phi}, \varepsilon, r_v \)and D given by Ponder (2000) are used in this work.

Definitions of three molecular mechanics variables (Sears and Batra 2004)
2.2 Modeling of Functionalized SWCNTs
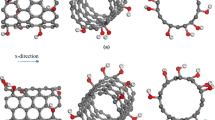
Functionalization of a zigzag (10, 0) finite length SWCNT of aspect ratio 12 has been considered as a model problem. The molecular structure of a functionalized SWCNT can be obtained from topologies of the pristine SWCNT and the functional group. In order to obtain the relaxed configuration of the SWCNT we minimize its potential energy by using the steepest decent algorithm with the root mean square (rms) gradient of 0.001. The diameter and the length of the relaxed tube are found to be 7.44 Å and 87.75 Å, respectively. The functional group is then positioned adjacent to a carbon atom of the relaxed CNT and the potential energy of the structure is minimized to obtain the relaxed configuration of the functionalized CNT. Note that the introduction of covalent bonds at the functional sites alters the hybridization of the affected carbon atoms from sp2 to sp3. The percentage of functionalization is defined as the ratio of the number of carbon atoms to which atoms of a functional group are attached to the total number of carbon atoms in the CNT. Four different functionalized CNTs have been studied with hydrogen, hydroxyl, carboxyl, and amine as the functionalizing groups. The carbon atoms to which a functional group is attached are randomly selected but the same set of atoms is used in each of the functionalized CNT for maintaining consistency in the numerical experiments. Figure 2 depicts schematics of the functional groups attached to a carbon atom of the CNT. The atoms in white, red, and blue color represent hydrogen, oxygen, and nitrogen atoms, respectively. It should be clear from sketches displayed in the figure that the covalent bond between the carbon atom and a functional group pulls out the carbon atom radially resulting in the distortion of the CNT at the functional site. The SWCNTs functionalized with these groups are schematically shown in Fig. 3.

Schematics of a carbon atom of the CNT attached with a hydrogen (–H) b hydroxyl (–OH), c carboxyl (–COOH), and d amine (–NH2) groups

Schematics of a (10, 0) SWCNT functionalized with 20 % a hydrogen (–H), b hydroxyl (–OH), c carboxyl (–COOH), and d amine (–NH2) groups
2.3 Virtual Simple Tension Experiments
While conducting virtual tension tests on the pristine and the functionalized SWCNT, its ends are left open and not saturated with hydrogen atoms which may change the bonding structure at the ends and lead to end effects. Furthermore, carbon atoms one diameter away from each end are fixed during simulations. The nanotubes are gradually deformed in tension and compression by a total of 3 % axial strain. After each displacement increment applied to atoms near the end faces, the tubes are allowed to equilibrate by minimizing their potential energies. The energy required to deform the tube equals its strain energy. The strain energy density is computed by dividing the strain energy by the volume of the CNT which is taken equal to that of a continuum cylinder of length and diameter equal to those of the relaxed pristine SWCNT and thickness equal to 0.34 nm. A polynomial through the data points is fitted by the least squares method. The first and the second derivatives of the strain energy density with respect to the axial strain yield the corresponding axial stress and Young’s modulus, respectively. This procedure is the same as that used by Sears and Batra (2004) to find Young’s modulus of a SWCNT.
3 Numerical Results
3.1 Validation of the Functionalization
In the relaxed configuration of the functionalized CNTs, the equilibrium bond length between the carbon atoms of the CNT and the adjoining atoms of the functional group, if present, has been computed from the equilibrated configurations. The C–H bond length in the hydrogen functionalized CNT is found to be 1.11, which agrees well with that (1.11) in the modified orthogonal tight binding model (Volpe and Cleri 2003) as well as the 1.12 given by the DFT calculations (Letardi et al. 2002). Methanol (CH3OH), acetic acid (CH3COOH), and amino methane (CH3NH2) are simple structures representing covalent bonds between a sp3 hybridized carbon and hydroxyl, carboxyl, and amine functional group, respectively. These structures can be generated using simplified molecular-input line-entry system (SMILES) specification (Weininger 1988; Weininger et al. 1989) as shown in Fig. 4. In order to validate the functionalization of the CNTs with hydroxyl, carboxyl, and amine groups, the bond angles and bond lengths of the functional groups in the relaxed configuration of the functionalized CNTs have been measured and compared with those in methanol, acetic acid and amino methane. In Tables 1, 2 and 3, we have listed values of these parameters for hydroxyl, carboxyl, and amine groups, respectively. It is evident that there is good agreement between the two sets of values.

Structures of a methanol, b acetic acid, and c amino methane generated using SMILES
3.2 Analysis of Relaxed Configurations
The number of iterations required in the steepest descent method to minimize potential energies of –H, –OH, –COOH and –NH2 functionalized CNTs is plotted in Figs. 5a, b and 6a, b respectively. Note that 0 % functionalization represents the pristine CNT. It can be observed from results depicted in Fig. 5a that when the CNT is functionalized with hydrogen, the potential energy of the relaxed functionalized structure is less than that of the relaxed pristine CNT, and this difference increases with an increase in the percentage of functionalization.

For four different percentages of functionalization, minimization of the potential energies of a SWCNT functionalized with a –H, b –OH

For four different percentages of functionalization, minimization of the potential energies of a SWCNT the functionalized with a –COOH, and b –NH2 groups
It is because the functionalization breaks the pi-bond of the sp3 hybridized carbon atoms and changes the atom type from alkene to the more stable alkane. Similar characteristics are observed with the hydroxyl and the amine functionalization while the potential energy curves for the carboxyl functionalized CNTs do not exhibit the same behavior due to the presence of double bonds in the –COOH group.
In Fig. 7a we have presented the contribution from each energy term of the MM3 potential to the total potential energies of the pristine CNT for the hydrogen functionalized CNTs in their relaxed configurations. These have been normalized with respect to corresponding energies of the relaxed pristine CNT.

Break down of total potential energy, U, in the individual energy components for the relaxed configurations of a hydrogen, b hydroxyl, functionalized CNTs with percentage of functionalization varying from 0 to 20 %
It can be observed from results included in the figure that the sum of the energies associated with the torsional and Van der Waals interactions are about 90 % of the total potential energy of the pristine CNT. The functionalization reduces these components of energies, thereby reducing the total potential energy and the reduction increases with an increase in the percentage of functionalization. Similar results for hydroxyl, carboxyl and amine functionalized CNTs are presented in Figs. 7a, b and 8a, b. Here, small effect of the electrostatic interactions has been observed on the total potential energies of the CNTs because of the presence of dipole moments in these functional groups. For the carboxyl functionalized SWCNT, the potential energy is not a monotonically increasing function of the amount of functionalization.

Break down of total potential energy, U, in the individual energy components for the relaxed configurations of a carboxyl, and b amine functionalized CNTs with percentage of functionalization varying from 0 to 20 %
3.3 Results from Virtual Experiments
In the calculation of Young’s modulus, as stated above, the thickness of a SWCNT is assumed to be 0.34 nm. The variation of the computed strain energy density with the axial strain for the pristine CNT is shown in Fig. 9. The third order polynomial fit to the strain energy density of the pristine CNT vs. the axial strain data with the regression coefficient of 1.0 is

Strain energy density of the pristine SWCNT vs. the axial strain computed using the MM3 potential
where W v is the strain energy density in J/m3 and ε is the nominal axial strain. Thus, expressions for the axial stress and the modulus of elasticity E in Pa are
The modulus of elasticity at zero axial strain equals 1.01 TPa. It compares well with that reported in the literature (Treacy et al. 1996; Wong et al. 1997; Krishnan et al. 1998). For the hydrogen functionalized CNT, the strain energy vs. the axial strain variation and the axial stress-axial strain curves for different percentages of functionalization are shown in Fig. 10a, b, respectively.

a Variation of the strain energy per atom with axial strain, and b axial stress-axial strain curves of –H functionalized SWCNT with the percentage of functionalization varied from 5 to 20 %
In computing the strain energy density, the volume of the functionalized CNT is taken equal to that of the pristine CNT. These plots reveal that the functionalization reduces the strain energy of deformation, and the slope (hence the modulus of elasticity) of the stress–strain curve for the functionalized tube is less than that of the pristine tube. Moreover, the modulus of elasticity decreases with an increase in the percentage of functionalization. This reduction in the modulus of elasticity could be due to the non-uniformities in the nanotube structure introduced by the functionalization that lead to localized deformation at the functional sites. Similar results for hydroxyl, carboxyl, and amine groups are exhibited in Figs. 11, 12 and 13, respectively.

a Variation of the strain energy per atom with axial strain, and b axial stress-axial strain curves of –OH functionalized SWCNT with the percentage of functionalization varied from 5 to 20 %

a Variation of the strain energy per atom with axial strain, and b axial stress-axial strain curves of –COOH functionalized SWCNT with the percentage of functionalization varied from 5 to 20 %

a Variation of the strain energy per atom with axial strain, and b axial stress-axial strain curves of –NH2 functionalized SWCNT with the percentage of functionalization varied from 5 to 20 %
Values of Young’s modulus of the functionalized CNTs for different percentages of functionalization listed in Table 4 suggest that they do not depend much upon the functionalization agent.
In Table 5, we have summarized similar results reported in the literature obtained using various computational methods and force fields along with the major results of the present study. It should be clear from the summary provided in Table 5 that the present results agree with those of other researchers. To better understand the effect of functionalization on Young’s modulus, the strain energies of the deformation in the tension tests of the pristine and the functionalized CNTs have been obtained in terms of their bonded and non-bonded energy components. In Figs. 14a, b and 15a, b we have depicted the contribution of individual energy terms to the total reduction in the strain energy of deformation due to hydrogen, hydroxyl, carboxyl, and amine functionalization, respectively. These values have been normalized with respect to those of the pristine CNT. These results indicate that the functionalization reduces the strain energies associated with bond-stretching and angle bending and increases the strain energies due to torsional, stretch-bend, and van der Waals interactions. The resulting effect is net reduction in strain energy and the reduction is proportional to the amount of functionalizing material attached to the wall of the CNT. Various components of the strain energy for 20 % of functionalization are plotted in Fig. 16. It can be concluded from these results that the type of functional group is found to have significant effect on strain energies associated with bond stretching and bond angle bending compared to other energy components but the same net effect is obtained for all functional groups considered here.

Break down of the total strain energy of deformation into the individual energy components for a hydrogen, b hydroxyl, functionalized CNTs with percentage of functionalization varying from 0 to 20 %

Break down of the total strain energy of deformation into the individual energy components for a carboxyl, and b amine functionalized CNTs with percentage of functionalization varying from 0 to 20 %

Break down of the total strain energy of deformation into the individual energy components for the CNTs functionalized (20 %) with different functional groups
4 Conclusions
We have studied the effect of covalent functionalization on Young’s modulus of a SWCNT. The functionalization localizes deformations of the CNT at the functionalized sites, which decreases the modulus of elasticity and this reduction increases with an increase in the percentage of functionalization. The type of functional group is found to have negligible effect on the value of Young’s modulus of the functionalized SWCNT. Approximately 30 % reduction in the value of Young’s modulus of the pristine CNT occurs for 20 % of functionalization.
References
Treacy MMJ, Ebesen TW, Gibson JM (1996) Exceptionally high Young’s modulus observed for individual carbon nanotubes. Nature (London) 381:678–680
Wong EW, Sheehan PE, Lieber CM (1997) Nanobeam mechanics: elasticity, strength and toughness of nanorods and nanotubes. Science 277:1971–1975
Krishnan A, Dujardin E, Ebbesen TW, Yianilos PN, Treacy MMJ (1998) Young’s modulus of single-walled nanotubes. Phys Rev B 58:14013–14019
Xing BW, Chun ZC, Zhao CW (2004) Simulation of Young’s modulus of single-walled carbon nanotubes by molecular dynamics. Physica B 352:156–163
Li C, Chou TW (2003) A structural mechanics approach for the analysis of carbon nanotubes. Int J Solids Struct 40:2487–2499
Chang T, Gao H (2003) Size-dependent elastic properties of a single-walled carbon nanotube via a molecular mechanics model. J Mech Phys Solids 51:1059–1074
Sears A, Batra RC (2004) Macroscopic properties of carbon nanotubes from molecular mechanics simulations. Phys Rev B 69:Art. No. 235406
Shen L, Li J (2004) Transversely isotropic elastic properties of single-walled carbon nanotubes. Phys Rev B 69:045414
Batra RC, Sears A (2007) Uniform radial expansion/contraction of carbon nanotubes and their transverse elastic moduli. Modell Simul Mater Sci Eng 15:835–844
Gupta SS, Batra RC (2008) Continuum structures equivalent in normal mode vibrations to single-walled carbon nanotubes. Comput Mater Sci 43:715–723
Batra RC, Gupta SS (2008) Wall thickness and radial breathing modes of single-walled carbon nanotubes. ASME J Appl Mech 75:Art. No. 0610101
Wu J, Hwang KC, Huang YJ (2008) An atomistic-based finite-deformation shell theory for single-wall carbon nanotubes. J Mech Phys Solids 56:279–292
Chen J, Hamon MA, Hu H, Chen Y, Rao AM, Eklund PC, Haddon RC (1998) Solution properties of single-walled carbon nanotubes. Science 282:95–98
Hamon MA, Chen J, Hu H, Chen Y, Itkis ME, Rao AM, Eklund PC, Haddon RC (1999) Dissolution of single-walled carbon nanotubes. Adv Mater 11:834–840
Niyogi S, Hu H, Hamon MA, Bhowmik P, Zhao B, Rozenzhak SM, Chen J, Itkis ME, Meier MS, Haddon RC (2001) Chromatographic purification of soluble single walled carbon nanotubes (s-SWNTs). J Am Chem Soc 123:733–734
Chen J, Rao AM, Lyuksyutov S, Itkis ME, Hamon MA, Hu H, Cohn RW, Eklund PC, Colbert DT, Smalley RE, Haddon RC (2001) Dissolution of full-length single-walled carbon nanotubes. J Phys Chem B 105:2525–2528
Khare B, Meyyappan M, Cassell AM, Nguyen CV, Han J (2002) Functionalization of carbon nanotubes with atomic hydrogen from a glow discharge. Nano Lett 2:73–77
Riggs JE, Guo Z, Carroll DL, Sun YP (2000) Strong luminescence of solubilized carbon nanotubes. J Am Chem Soc 122:5879–5880
Sun YP, Huang W, Lin Y, Fu K, Kitaygorodskiy A, Riddle LA, Yu YJ, Carroll DL (2001a) Soluble dendron-functionalized carbon nanotubes: preparation, characterization, and properties. Chem Mater 13:2864–2869
Fu K, Huang W, Lin Y, Riddle LA, Carroll DL, Sun YP (2001) Defunctionalization of functionalized carbon nanotubes. Nano Lett 8:439–441
Sun Y, Wilson SR, Schuster DI (2001b) High dissolution and strong light emission of carbon nanotubes in aromatic amine solvents. J Am Chem Soc 123:5348–5349
Grujicic M, Sun YP, Koudela KL (2006) The effect of covalent functionalization of carbon nanotube reinforcements on the atomic-level mechanical properties of poly-vinyl-ester-epoxy. Appl Surf Sci 253:3009–3021
Cooper CA, Cohen SR, Barber AH, Daniel WH (2002) Detachment of nanotubes from a polymer matrix. Appl Phys Lett 81:3873–3875
Barber AH, Cohen SR, Daniel WH (2003) Measurement of carbon nanotube-polymer interfacial strength. Appl Phys Lett 82:4140–4142
Shofner ML, Khabashesku VN, Barrera EV (2006) Processing and mechanical properties of fluorinated single-wall carbon nanotube–polyethylene composites. Chem Mater 18:906–9013
Buffa F, Abraham GA, Grady BP, Resasco D (2007) Effect of nanotube functionalization on the properties of single-walled carbon nanotube/polyurethane composites. J Polym Sci Part B Polym Phys 45:490–501
Sun L, Warren GL, O'Reilly JY, Everett WN, Lee SM, Davis D, Lagoudas D, Sue HJ et al (2008) Mechanical properties of surface-functionalized SWCNT/epoxy composites. Carbon 46:320–328
Zhang ZQ, Liu B, Chen YL, Jiang H, Hwang KC, Huang Y (2008) Mechanical properties of functionalized carbon nanotubes. Nanotechnology 19:395702
Kuang YD, He XQ (2009) Young’s moduli of functionalized single-wall carbon nanotubes under tensile loading. Compos Sci Technol 69:169–175
Ling CC, Xue QZ, Zhou XY (2012) Mechanical properties of functionalized carbon nanotube as reinforcements. Adv Mat Res 583:22–26
Milowska KZ, Majewski JA (2013) Elastic properties of functionalized carbon nanotubes. Phys Chem Ch Ph 15:14303–14309
Ponder JW (2000) Washington University in St. Loius, St. Louis
Allinger NL, Yuh YH, Lii JH (1989) Molecular mechanics. The MM3 force field for hydrocarbons. J Am Chem Soc 111:8551–8566 (and subsequent papers)
Volpe M, Cleri F (2003) Chemisorption of atomic hydrogen in graphite and carbon nanotubes. Surf Sci 544:24–34
Letardi S, Celino M, Cleri F, Rosato V (2002) Atomic hydrogen adsorption on a Stone-Wales defect in graphite. Surf Sci 496:33–38
Weininger D (1988) SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 28:31–36
Weininger D, Weininger A, Weininger JL (1989) SMILES. 2. Algorithm for generation of unique SMILES notation. J Am Chem Soc 29:97–101
Acknowledgments
This research was sponsored by the Army Research Laboratory and was accomplished under Cooperative Agreement Number W911NF-06-2-0014. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Army Research Laboratory or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation hereon.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Shah, P.H., Batra, R.C. (2014). Effect of Covalent Functionalization on Young’s Modulus of a Single-Wall Carbon Nanotube. In: Tserpes, K., Silvestre, N. (eds) Modeling of Carbon Nanotubes, Graphene and their Composites. Springer Series in Materials Science, vol 188. Springer, Cham. https://doi.org/10.1007/978-3-319-01201-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-01201-8_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-01200-1
Online ISBN: 978-3-319-01201-8
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)