
Abstract
Cationic antimicrobial peptides (CAMPs) are essential compounds of the innate immunity system possessed by humans. CAMPs protect the host by exerting bactericidal activity, molecular signaling, modulating the immune response, and facilitating the communication between innate and acquired immunity. Over the millennia, bacteria have developed mechanisms to circumvent the antimicrobial activity of CAMPs, thereby promoting their survival during infection. In this chapter, we focus on the mechanisms used by various bacterial pathogens to resist the antibiotic-like action of CAMPs and the consequences of such resistance.
Maira Goytia and Justin L. Kandler contributed equally.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Efflux Pump
- Minimal Inhibitory Concentration
- Bacterial Surface
- Teichoic Acid
- Cationic Antimicrobial Peptide
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Regardless of the host, signs of infection caused by a bacterial pathogen are typically noticed after damage to the host has occurred and symptoms are manifested. These symptoms of infection can arise from toxins produced by the pathogen or host inflammatory processes triggered when the host recognizes pathogenic bacteria or their associated virulence factors. Early during infection, mediators of innate immunity are brought to the front line of defense to combat the invader and protect the host. The efficacy of this response can determine the duration, spread, and severity of disease. Cationic antimicrobial peptides (CAMPs), also appropriately called “host defense peptides” (Brown and Hancock 2006), are important in this response as they can directly or indirectly exert antibacterial activity. Any successful pathogen must find ways to evade the direct action of CAMPs or risk having their numbers severely reduced or even eliminated.
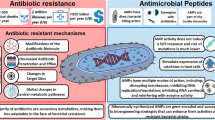
Although thousands of CAMPs exist in nature, humans confront bacteria with three main classes: the α- and β-defensins, the sole human cathelicidin termed LL-37, and peptides derived from protease digestion of proteins that perform important roles in other host response processes (e.g., cathepsin G). Unless otherwise stated, we focus this review on the mechanisms employed by bacterial pathogens to escape the action of gene-encoded CAMPs. It is important to note that CAMPs are, in essence, antibiotics. It is therefore not surprising that many of the general mechanisms developed by bacteria to resist classical antibiotics are in concept also used by bacteria to resist CAMPs. These mechanisms are summarized in Fig. 1.

Schematic representation of the mechanisms of resistance to cationic antimicrobial peptides (CAMP) used by Gram-negative and Gram-positive bacteria. Key: CAMP cationic antimicrobial peptide, CPS capsule polysaccharide, d -Ala d-alanine, l -Ara4N 4-amino-4-deoxy-l-arabinose, l -Lys l-lysine, LOS lipooligosaccharide, LPS lipopolysaccharide, PEA phosphoethanolamine. These examples are of specific mechanisms of CAMP resistance expressed by Gram-positive and Gram-negative bacteria and should not to be considered exhaustive
Studies on mechanisms of bacterial resistance to CAMPs have been facilitated by the ability to construct and use isogenic strains that differ by a single, defined mutation or the presence of a gene that impacts levels of bacterial susceptibility to CAMPs. To assess the significance of such differences, it is first important to have reproducible antibacterial assays, which are discussed below. It is difficult to use purified human CAMPs or synthetic versions, due to availability of materials and cost constraints, to select genetic variants, but these problems can be circumvented by the use of recombinantly produced CAMPs or commercially available compounds that mimic the bactericidal action of CAMPs; the cationic antimicrobial peptide polymyxin B has been a reliable CAMP substitute used by many investigators.
Understanding how bacteria can develop CAMP resistance requires the use of in vitro antimicrobial assays that are reproducible and relatively easy to use. Typically, these assays involve an assessment of direct colony forming unit reduction when bacteria are incubated in liquid media with purified CAMPs. Other standardized assays for determining minimal inhibitory concentrations (MIC) or minimal bactericidal concentrations (MBC) of CAMPs are also routinely employed (Institute Clinical and Laboratory Standard 2009). Typically, actively growing bacteria are diluted and incubated with CAMPs in broth or phosphate buffer-based solutions. These liquid media can be altered in pH and ionic strength to assess the impact of changes in these conditions on the killing efficacy of CAMPs. The radial diffusion agar overlay/underlay assay developed in the Lehrer laboratory (Qu et al. 1996) is also particularly useful and can provide quantitative results as well as allowing one to test how changes in ionic strength, presence of divalent cations, and pH impact CAMP activity. While these assays can collectively provide important information, laboratory growth media have little resemblance to the natural environments in which CAMPs must function in vivo. For instance, it is rare that the presence of other host compounds is taken into account, but these can either have positive or negative actions on the susceptibility to CAMPs. The presence of lysozyme or phospholipase A2 can enhance bacterial killing in CAMP assays. In our own studies with Neisseria gonorrhoeae, which typically (and often) infects the human genital tract, it was found that physiologically relevant levels of polyamines (e.g., spermine and spermidine) can decrease bacterial susceptibility to LL-37 (Goytia and Shafer 2010). Additionally, Dorschner and colleagues deduced that physiologically relevant levels of carbonate (CO 2−3 ) in laboratory media greatly increase the killing potential of numerous and varied CAMPs, but not of the anionic skin-derived antimicrobial peptide (AMP) termed dermcidin (Dorschner et al. 2006). They further found that Staphylococcus aureus responds to CO 2−3 through a dynamic transcriptional response, thinning its peptidoglycan via repression of the alternative sigma factor, sigB, which may explain the increased susceptibility of this pathogen to the tested peptides. Interestingly, others have shown that LL-37 adopts an α-helical (active) conformation in the presence of carbonate (Johansson et al. 1998), which may have played some part in the enhanced killing activity seen in the Dorschner study. Thus, CAMP behavior in the lab setting may not always reflect CAMP-bacteria interactions in vivo.
Environmental conditions (e.g., limitation of iron, anaerobiosis, local pH, ionic strength, and presence of divalent cations) can also significantly impact AMP activity against bacteria. The recent report (Schroeder et al. 2011) that anaerobic conditions can potentiate the antibacterial action of human β-defensin 1 (hBD-1) against commensal gut bacteria nicely illustrates this point. In this instance, reduction of the intramolecular disulfide bonds by the thioredoxin system significantly enhanced HBD-1 activity against anaerobic commensal bacteria, and this was proposed by the authors as a means used by the host to prevent their overgrowth. Interestingly, Nuding et al. found that similar anaerobic conditions can actually weaken the antibacterial action of the related peptide human β-defensin 3 (HBD-3) (Nuding et al. 2009). Such diversity of CAMP function may be a way for the body to fine-tune its gut microbiota population. Alternatively, conditions of hypoxia may induce, in the absence of a surrogate electron acceptor, a state of bacteriostasis for some bacteria. This has been observed in N. gonorrhoeae, resulting in gonococcal resistance to antimicrobial proteins and CAMPs (Casey et al. 1985). Bacteria also face iron-starvation conditions imposed by host iron-binding proteins and have a variety of response mechanisms to acquire iron (Ganz 2009). As an example, this environmental stress can influence the level of susceptibility of Streptococcus pyogenes to LL-37 (Froehlich et al. 2009).
Ex vivo and in vivo infection models have also been employed to gain insights regarding the significance of mechanisms of bacterial resistance to CAMPs. Perhaps the most widely used ex vivo model is that of polymorphonuclear leukocytes (PMNs). These professional phagocytic cells are used in monolayers or in suspension to evaluate changes in the intraleukocytic microbicidal activity that may be due to AMP resistance mechanisms. CAMPs (e.g., neutrophil defensins termed HNP 1–4 and LL-37) are, along with antimicrobial proteins such as the bactericidal/permeability-increasing (BPI) protein (Marra et al. 1990), lactoferrin, cathepsin G, CAP37, and lysozyme, important mediators of nonoxidative killing by PMNs (Sorrell et al. 1978; Spitznagel and Shafer 1985). These antimicrobial compounds are stored within the cytoplasmic specific and azurophilic granules and are delivered into the phagocytic vacuole after granule fusion and degranulation. Bacteria that inhibit phagosome-lysosome fusion can resist CAMPs and antimicrobial proteins (Scott et al. 2003). For instance, Salmonella presents a well-studied example where a protein (SipC) belonging to the Salmonella pathogenicity island-2 (SPI-2) is essential to prevent the fusion of Salmonella-containing vacuoles with host cell lysosomes (Uchiya et al. 1999). A recent study suggests that Salmonella recruits host proteins to the vacuole to inhibit the fusion (Madan et al. 2012). However, once CAMPs are delivered into the developing phagolysosome, they rapidly coat the bacterial surface and can achieve mg/mL concentrations, making it remarkable that any bacteria can survive inside the phagolysosome at all (Lehrer et al. 1988). With PMN models, it is possible to test inferences made regarding the significance of bacterial mutations that alter the susceptibility to isolated, PMN-derived CAMPs; specific examples are described below.
To test the idea that CAMP resistance mechanisms can enhance bacterial survival during infection, numerous studies have employed whole animal models. Mouse models of infection have been particularly useful in this respect, allowing investigators to readily test inferences they have drawn from in vitro and ex vivo tests. In this regard, mouse strains bearing knockout mutations in genes similar to human genes [e.g., CRAMP encoding the murine version of LL-37 (Nizet and Gallo 2002)] or knocked-in human CAMP genes [e.g., the HBD-5 used by N. Salzman and colleagues in their studies dealing with Salmonella typhimurium Salzman et al. (2003), Wehkamp et al. (2005)] have been used to test both the significance of CAMPs in host defense and if bacterial resistance mechanisms are important in promoting microbial survival during infection. Briefly, CRAMP knockout mice were more susceptible to invasive group A streptococcal infection than their CRAMP+/+ counterparts, while expression of HBD-5 provided mice with increased resistance to a lethal S. typhimurium infection.
To date, only a single report has appeared in the literature describing a human bacterial infection model for the purpose of studying the significance of a CAMP resistance mechanism. In this instance, Bauer et al. (Bauer et al. 2006; Bauer and Spinola 2000) utilized the forearm skin puncture model (Spinola et al. 2003) to study host responses to infection by Haemophilus ducreyi, the causative agent of the sexually transmitted infection chancroid. H. ducreyi is intrinsically resistant to CAMPs and uses two transport systems, the Mtr efflux and Sap importer systems, for this purpose; these systems are described in greater detail below. Loss of these systems significantly decreased the survival of H. ducreyi and lesion pathology in this model (Rinker et al. 2011; Mount et al. 2010), indicating that CAMP resistance is important for its ability to cause disease.
2 Mechanisms of Bacterial Resistance to CAMPs
How do we define CAMP resistance? This is not an easy question to answer, as “breakpoints” typically used to differentiate antibiotic-sensitive from antibiotic-resistant strains are seldom considered for CAMP studies undertaken by research laboratories. This matter is complicated by a number of issues: CAMPs can achieve very different concentrations depending on their location; local environmental conditions can be antagonistic or agonistic; inducible resistance can be displayed in the presence of sublethal levels of CAMPs, yet lost under normal conditions; and CAMPs can exert multiple mechanisms of killing which, for a given peptide, might differ depending on the target bacteria. The precise mechanism by which CAMPs kill bacteria is a matter of some controversy, and no unifying mechanism has been readily accepted by the CAMP research community. Certainly, CAMPs must first bind to the microbial surface and traverse the cell envelope. The events occurring post binding that result in bacterial death are where controversy exists, and it has not always been easy to separate direct killing from postmortem events. For instance, changes in membrane integrity and potential, inhibition of cell wall biosynthesis, and interaction of CAMPs with nucleic acids have been invoked as bactericidal events for certain CAMPs acting on a given bacterial target. Our purpose below is not to review how CAMPs kill bacteria, but rather describe how bacteria use constitutive and inducible mechanisms to circumvent their action.
In order for CAMPs to efficiently kill bacteria, they must reach their target in extracellular fluids or within intracellular compartments avoiding the action of peptidases/proteases, navigate past hydrophilic surface structures such as capsules and O-antigen chains of lipopolysaccharide (LPS), interact with negatively charged surface structures, insert into the cell envelope, reach the cytoplasmic membrane, and, in some instances, enter the cytosol. All of these steps provide opportunities for bacterial interference, which can decrease the susceptibility of the target microbe to CAMPs. Briefly, pathogens have evolved several strategies to circumvent the attack by CAMPs: (1) modulate CAMP gene expression, (2) degrade CAMPs by extracellular or intracellular peptidases/proteases, (3) trap CAMPs, (4) reduce binding of CAMPs to the cell surface, (5) export CAMPs by efflux pumps, and (6) alter intracellular targets. CAMP resistance mechanisms are typically expressed constitutively, but many are also under control of regulators of gene expression that respond to environmental cues. Below, we review examples of these strategies from several medically relevant pathogens. Table 1 summarizes specific examples from various pathogens, while Fig. 1 summarizes the different strategies described in this section. We review this subject by beginning with examples of downregulation of CAMP production and then follow CAMPs as they bind and enter target bacteria, providing descriptions of the various systems employed by different bacteria to avoid the killing action of these important peptides.
2.1 Bacterial Modulation of CAMP Gene Expression
Bacteria have developed novel and diverse strategies to modulate the availability of CAMPs in extracellular fluids and within phagolysosomes of phagocytes; the latter subject has been extensively reviewed elsewhere (Ray et al. 2009; Flannagan et al. 2009), and we will concentrate on studies dealing with bacterial modulation of CAMP production. It is important to note that modulation of CAMP production can have profound downstream effects on the overall host immune system, which can facilitate bacterial growth and dissemination during infection. Bacterial products can directly or indirectly modulate CAMP expression and activation of immune responses. For instance, Escherichia coli lipopolysaccharide (LPS) can increase mRNA production of human β-defensin 2 (HBD-2) via CD14-activation of neutrophils (Becker et al. 2000). Tada et al. showed that proteases from Porphyromonas gingivalis can cleave the macrophage CD14 outer-membrane receptor. CD14 recognizes pathogen-associated molecular patterns (PAMPs), and cleavage of this protein rendered macrophages unresponsive to the presence of this pathogen and prevented CAMP production (Tada et al. 2002). Additionally, uropathogenic E. coli (UPEC), collected from patients with urinary tract infections (UTI), express the so-called “curli” fimbriae that modulate the immune system of the host and provide resistance to LL-37. Curli, an amyloid-like fiber expressed in biofilms, promotes cell adherence, increases induction of IL-8 (a human proinflammatory cytokine), binds to LL-37 inhibiting its killing activity, and increases bacterial virulence in a mouse model (Kai-Larsen et al. 2010). Furthermore, Islam et al. showed that Shigella flexneri and S. dysenteriae downregulate and prevent expression of CAMPs such as LL-37 and HBD-1 by the host. Even though the molecular mechanism was not completely elucidated, the authors suggested a role for plasmid DNA from the bacteria (Islam et al. 2001). Zughaier et al. reported that Neisseria meningitidis capsular polysaccharide (CPS) can bind LL-37 and, as a consequence, dampen the host immune response against this strict human pathogen (Zughaier et al. 2010). Finally, N. gonorrhoeae can impair expression of LL-37 in cervical epithelial cell line ME180 (Bergman et al. 2005). This effect was observed with live bacteria, but not with dead gonococci nor with live commensal Neisseria species considered avirulent in a normal host. They concluded that a specific interaction took place between N. gonorrhoeae and the ME180 epithelial cell that suppressed expression of LL-37. The gonococcal structures responsible for this suppression of LL-37 production remain to be discovered.
2.2 Degradation of CAMPs
Bacteria can degrade CAMPs by proteolytic cleavage before they reach or pass the bacterial surface. CAMP degradation can occur extracellularly, by secreted and membrane-associated proteases, or intracellularly; the latter is facilitated by importers that deliver CAMPs to the bacterial cytosol, where they are degraded by peptidases and cytosolic proteases.
Studies with Salmonella enterica and Staphylococcus aureus have contributed significantly to our understanding of the role of bacterial peptidases/proteases in CAMP resistance. S. enterica expresses an outer-membrane protease, PgtE, which cleaves LL-37 and other linear CAMPs, and results in increased resistance to LL-37 in vitro and in vivo (Guina et al. 2000). PgtE is similar to the OmpT protein produced by E. coli, which cleaves protamine. E. coli ompT mutants are more susceptible to human protamine (Stumpe et al. 1998). S. aureus produces many proteases, and evidence has been presented that the action of aureolysin, a metalloprotease, and V8, a serine endopeptidase, can enhance its resistance to CAMPs. V8 cleaves and inactivates LL-37 (Sieprawska-Lupa et al. 2004) as well as complement proteins C3a and C4a, which have antimicrobial action (Zipfel and Reuter 2009). Aureolysin cleaves complement protein C3 at a nonphysiological site, rendering its cleavage products inactive (Laarman et al. 2011). Aureolysin-cleaved C3 protein is further degraded by host mechanisms, thus blocking the complement cascade, inactivating the antimicrobial activity of C3a, and preventing the targeting of S. aureus by the host immune response (Laarman et al. 2011). Schmidtchen et al. have published a series of elegant papers that collectively emphasize the role of proteases produced by medically important pathogens (and other relevant microorganisms), highlighting their importance for resistance to CAMPs and antimicrobial proteins (Schmidtchen et al. 2001, 2002). Briefly, these studies showed that elastase and alkaline protease from Pseudomonas aeruginosa, gelatinase from Enterococcus faecalis, a secreted cysteine proteinase (SpeB) from Streptococcus pyogenes, and a 50-kDa proteinase from Proteus mirabilis, possibly ZapA (Belas et al. 2004), can degrade LL-37 (Schmidtchen et al. 2001). As a secondary effect, these enzymes also degrade proteoglycans from the host’s extracellular matrix, releasing negatively charged dermatan and/or heparan sulfate, which significantly inhibits the antimicrobial activity of the α-defensin human neutrophil peptide 1 (HNP-1) (Schmidtchen et al. 2001), and of bactenecin-5 and bactenecin-7 (Park et al. 2001). An additional mechanism developed by S. pyogenes involves the bacterial membrane-associated protein GRAB, which binds α-2-macroglobulin (α2M), a host-derived protease inhibitor. α2M, in turn, binds the secreted bacterial protease SpeB, which maintains its proteolytic activity against CAMPs, preventing the action of CAMPs at their target site on the bacterial surface (Nyberg et al. 2004). Thus, S. pyogenes binding of host α2M appears to serve two purposes: (1) facilitate cleavage of CAMPs before they reach their target and (2) promote immunological mimicry by presenting self-antigens at the bacterial surface.
Bacteria can also degrade CAMPs intracellularly through the combined action of an importer, which delivers CAMPs to the cytosol, and intracellular peptidases normally used to cleave bacterial peptides and increase the available pool of amino acids. For example, non-typeable H. influenzae (NTHi), a commensal Gram-negative bacterium that can cause conjunctivitis, sinusitis, acute and chronic otitis media, and bronchitis (Erwin and Smith 2007), expresses the Sap (sensitivity to antimicrobial peptides) ABC transporter. This transporter, first identified and characterized in Salmonella (Groisman et al. 1992; Parra-Lopez et al. 1993), can increase bacterial resistance to CAMPs by 8-fold and is required for virulence of NTHi in a chinchilla model of otitis media (Mason et al. 2005). SapA binds chinchilla BD-1 as well as human CAMPs (e.g., LL-37, HBD-2 and HBD-3, and HNP-1) (Mason et al. 2011). The binding of CAMPs to SapA upregulates expression of the sap operon (sapABCDFZ) (Mason et al. 2005), promoting transfer of the CAMPs into the cytosol where they are degraded (Mason et al. 2006). Further research by this group proposed a mechanism where CAMPs are taken up by the periplasmic binding protein SapA then transferred to the cytoplasm through the SapBCDF transporter and SapZ accessory protein (Shelton et al. 2011). Interestingly, this mechanism might increase the intracellular levels of nutrients, since the amino acids from the degraded CAMPs could be recycled. SapA of H. ducreyi also increases bacterial resistance to LL-37, but not to α- and β-defensins. However, the most important effect of SapA in H. ducreyi was observed in the human forearm model of chancroid in that SapA production was found to increase virulence, probably by promoting resistance to the higher concentrations of LL-37 that are secreted at infection sites in the dermis (Mount et al. 2010).
2.3 Hindering CAMP Localization to the Bacterial Surface
When bacteria find themselves in environments rich in CAMPs, they have strategies other than proteolysis to neutralize or repulse CAMPs, thereby reducing their susceptibility to these antimicrobials. Non-proteolytic mechanisms of CAMP resistance include (1) the presence of CAMP-binding agents, (2) expression of hydrophilic bacterial biopolymers to retard the passage of amphipathic CAMPs in the electronegative bacterial surface, and (3) architectural constraints imposed by biofilms.
Binding or repulsion of CAMPs by extracellular compounds reduces their capacity to interact with negatively charged target sites on the bacterial surface. Several CAMP-binding compounds have been described. For example, staphylokinase (Sak) and the streptococcal inhibitor of complement (SIC) bind to and neutralize CAMPs such as HNP 1–3 and LL-37 in the extracellular milieu, effectively decreasing the local CAMP activity as much as 80 % (Jin et al. 2004; Pence et al. 2010). The production of extracellular polymers shields bacteria with an extra layer of protection against CAMPs. Polysaccharide intercellular adhesin (PIA) and poly-γ-glutamic acid (PGA) polymers, produced by staphylococci, inhibit HBD-3 and LL-37 activity (Kocianova et al. 2005; Vuong et al. 2004). It is thought that the charges present on these polymers are a triple threat as they are able to repulse similarly charged antimicrobials, neutralize and sequester oppositely charged antimicrobials, and behave as a mechanical barrier to their entry. Alginic acid produced by P. aeruginosa inhibits CAMPs in a similar fashion (Friedrich et al. 1999).
The production of capsule, or glycocalyx (literally, “sugar coat”), is a common defense mechanism also utilized by other human bacterial pathogens, including Neisseria meningitidis, Klebsiella pneumoniae, Legionella pneumophila, Streptococcus pneumoniae, S. aureus, and Bacillus anthracis. It is notable that many of these pathogens are the cause of mucosal and respiratory tract infections that may progress to the bloodstream, an environment where encapsulated organisms are at a distinct advantage over other bacteria (Yeaman and Yount 2003). However, not all prokaryotic glycocalyces are produced by the pathogen itself and can sometimes be stolen from the host (see pathogenic Neisseria below). In the environment, several species of both Eubacteria and Archaea produce S-layers, which is a glycoprotein shroud that completely surrounds the prokaryotic cell. Interestingly, some eubacterial S-layers can be glycosylated with up to 150 carbohydrate moieties per protein unit (Messner et al. 2008). S-layers may be another protective mechanism against CAMPs in the highly competitive soil and water microbiome.
As with proteolysis, biopolymer sequestration of CAMPs can still function even when physically separated from the cell. Certain Gram-negative bacteria can release membrane vesicles rich in CAMP-binding sites called “blebs,” and the act of blebbing could provide an extracellular sink for CAMPs. Some bacteria may also release negatively charged capsular polysaccharides (CPS) that titrate CAMPs by electrostatic interactions. Llobet et al. have described this mechanism of resistance in several clinically relevant pathogens, such as E. coli, K. pneumoniae, P. aeruginosa, and S. pneumoniae (Llobet et al. 2008). They show that CPS from different bacteria at concentrations as low as 1 μg/mL (E. coli, K. pneumoniae, P. aeruginosa) can increase the MIC of HNP-1 between 5- and 30-fold, regardless of the CPS source. However, this mechanism can be rendered inadequate by the presence of polycations that preferentially bind to CPS, freeing CAMPs to react with the bacterial cell wall and kill the targeted cell (Llobet et al. 2008). Additionally, Campos et al. showed that a K. pneumoniae mutant lacking CPS is more sensitive to CAMPs such as HNP-1, HBD-1, protamine sulfate, and polymyxin B, compared to the wild-type strain expressing CPS (Campos et al. 2004). They also showed that the CPS mutant binds more polymyxin B than the wild-type strain, suggesting that CPS protects the bacteria, either by mechanically shielding the bacteria or by titrating the CAMP. Moranta et al. injected wild-type or CPS mutant K. pneumoniae into mice and showed decreased levels of β-defensins produced in response to the wild-type strain, as compared to higher levels produced for the CPS mutant isogenic strain (Moranta et al. 2010). This suggested that CPS not only prevented the action of CAMPs at the surface of the bacteria, but also prevented signaling to the host immune system that would normally increase levels of β-defensins. In N. meningitidis, Jones et al. observed that expression of LOS and capsule is directly linked to increased resistance to LL-37 compared to the LOS-deficient and capsule-deficient mutants (Jones et al. 2009). Furthermore, they show that incubation of the wild-type bacteria with sublethal concentrations of LL-37 induced the expression of the capsule-associated genes siaC and siaD, which results in upregulation of capsule biosynthesis (Jones et al. 2009).
CAMPs are typically amphipathic molecules whose hydrophobic domain allows membrane insertion, an event required for killing the target microbe. Accordingly, the presence of bulky hydrophilic structures on the bacterial surface may hinder the migration of CAMPs from the extracellular milieu to the negatively charged bacterial surface structures that form the gateway to hydrophobic lipid bilayers. This may partly explain why the presence of the hydrophilic O-antigen in the LPS of Gram-negative enterics endows them with greater CAMP resistance than rough mutants lacking this hydrophilic glycopolymer. Indeed, early work with S. typhimurium (Rest et al. 1977) showed that when the O-antigen is lost and the inner core sugar chain is progressively truncated, bacterial susceptibility to granule extracts from human neutrophils increases proportionally. These granules are rich in defensins and cationic antimicrobial proteins (Spitznagel 1990). Although these “deep-rough” mutants have increased exposure of their negatively charged lipid A phosphate groups, which are important for CAMP binding, their surface is generally more hydrophobic. This characteristic is conducive for CAMP/membrane interactions and enhances the likelihood of membrane insertion.
The above mechanisms have been defined with planktonic bacteria. However, it is now recognized that many bacterial species form a specialized, highly organized community termed a biofilm. Due to their ultrastructural organization, bacteria within biofilms can exhibit increased resistance to antimicrobials, including CAMPs, compared to their planktonic counterparts (Anderl et al. 2000). Biofilms were correlated with persistent bacterial infections, such as chronic lung infection in cystic fibrosis (Singh et al. 2000). Antimicrobial resistance displayed by bacteria within biofilms appears to be due to mechanisms different from those typically observed in free-floating cells. Since biofilms are multicellular communities of bacteria encased in a hydrated matrix of polysaccharide, proteins, and/or nucleic acids, the capacity of antimicrobials to interact with all members of the community is reduced. Biofilms can also trap and inactivate CAMPs in the complex matrix imposed by its structure. Leid et al. demonstrated that biofilms from S. aureus can be penetrated by leukocytes that are active and secrete antimicrobial compounds (including CAMPs). However, though these leukocytes were able to phagocytose planktonic S. aureus, they could not engulf sessile cells (Leid et al. 2002). The authors suggested that the structure of the biofilm is more of a porous hydrogel than a fixed impenetrable structure. Three main hypotheses have been advanced to explain the increased antimicrobial resistance displayed by bacteria in biofilms (Stewart and Costerton 2001). One hypothesis invokes a less permeable and less diffusible environment created by the biofilm with negatively charged compounds (i.e., nucleic acid, polysaccharide) in the matrix that could retard CAMP diffusion. The second hypothesis emphasizes the ultrastructural architecture of the biofilm, where microenvironments might present unfavorable conditions of pH, salt, and anaerobiosis that render CAMPs inactive or inefficient. The third hypothesis speculates that bacteria go through a cell-differentiation process and reach a spore-like metabolic state while in the biofilm, which allows some of them to be resilient to higher concentrations of antibiotics. These hypotheses are not mutually exclusive, and other defense mechanisms described throughout this chapter could contribute to biofilm-mediated CAMP resistance. Another possible mechanism of resistance that needs to be considered is that biofilm formation may trigger an alternate gene expression profile that modulates resistance phenotypes.
Biofilm formation is a dynamic process and can be influenced by environmental conditions, including the presence of certain CAMPs, other host compounds, and bacterial gene products. In the first instance, LL-37 and lactoferrin can prevent biofilm formation by P. aeruginosa by promoting bacterial motility (Dean et al. 2011; Overhage et al. 2008). Due to its structural organization, the availability of molecular oxygen may differ at sites within the biofilm complex, and this could influence the antimicrobial action of CAMPs. Accordingly, Schroeder et al. found that hBD-1 was highly efficient against various human flora and select pathogens in its reduced form but lacked efficient antimicrobial activity in its oxidized form (Schroeder et al. 2011). This suggests that an oxidative environment (perhaps present at different degrees within biofilms) could preclude some CAMPs from their killing activity. Finally, a number of bacterial products directly enhance survival of CAMP attack in sessile cells. For example, inducible resistance can be controlled by two-component regulatory (TCR) systems (Mulcahy et al. 2008; Amer et al. 2010) and stand-alone regulators (Warner et al. 2007, 2008; Shafer et al. 2010); the hairlike surface appendage termed curli promotes formation of biofilm structures in UPEC strains of E. coli (Kai-Larsen et al. 2010); and periplasmic glucans may bind to CAMPs on their way to the cytoplasmic membrane and sequester them (Mah et al. 2003).
2.4 Envelope Modifications That Decrease CAMP Binding and Permeability
Nearly 50 years ago, Spitznagel and coworkers found that the antimicrobial, arginine-rich, cationic peptides present in neutrophil granules (now known as defensins) rapidly coat the surface of ingested bacteria (Spitznagel 1961; Zeya and Spitznagel 1966a, b). This electrostatic interaction between CAMPs and bacterial surface structures, and how bacteria modify these structures to inhibit said interaction, is perhaps the most studied mechanism of CAMP resistance (the reader is also directed to several excellent reviews especially those by Peschel 2002; Brogden 2005). In general, eukaryotic membranes are zwitterionic and have low affinity for CAMPs, which may provide them with some immunity to the lytic activity of these peptides. Prokaryotic cell surfaces, on the other hand, are typically negatively charged and so have a higher affinity for CAMPs. In order to prevent the deadly consequences of this attraction, many bacteria have evolved ways to decrease the net negative charge of their exteriors and modify the permeability of their membrane(s). Importantly, these mechanisms are not always specific to CAMPs and may provide protection against a broad spectrum of host and pharmacological cationic antimicrobials, including myeloperoxidase, phospholipase A2, lysozyme, vancomycin, moenomycin, and daptomycin (Peschel 2002).
Though both groups are negatively charged on their exteriors, Gram-negative bacteria are generally more resistant to CAMPs than Gram-positive bacteria, due to the presence of an outer membrane that can retard the passage of CAMPs to the inner membrane and cytoplasm. This is perhaps facilitated by LPS molecules that are held tightly together by (1) van der Waals interactions that exist between acyl chains and (2) salt bridges formed by divalent cations between neighboring carbohydrate chains and between lipid A phosphates. Early studies with deep-rough LPS mutants of S. typhimurium (Rest et al. 1977), and the pmrA mutants tested by Vaara and coworkers (Vaara et al. 1981; Helander et al. 1994) and Shafer et al. (1984), as well as Farley et al. (1987, 1988), support the concept that the availability of exposed, unsubstituted lipid A phosphate groups are critical to the ionic (and hydrophobic) interactions between CAMPs and the bacterial surface. More recent studies also support this model. For instance, a knockout insertion of a putative LPS synthesis gene galU in Campylobacter jejuni, a leading food-borne pathogen, decreased the length of LPS and reduced bacterial resistance to polymyxin B (Lin et al. 2009). Similarly, Bordetella bronchiseptica, an upper respiratory tract pathogen and close relative of B. pertussis (the etiologic agent of whooping cough), appears to require the addition of a negatively charged trisaccharide to LPS by the wlbA and wlbL genes for full resistance to several phylogenetically diverse CAMPs. It is thought that the uronic acid sugar moieties present in this trisaccharide shield the membrane from antimicrobial attack, perhaps by sequestering the peptides or providing a bulky barrier to entry (Banemann et al. 1998). Phosphorylcholine is produced by H. influenzae (Lysenko et al. 2000) and can increase the membrane fraction of zwitterionic phospholipids in the bacterial inner membrane. This would decrease the net negative charge normally present at the exoplasmic leaflet of the cytoplasmic membrane and slow the rate of CAMP self-promoted uptake (see below). Importantly, the investigators also observed modification of H. influenzae lipooligosaccharide (LOS) with phosphorylcholine. Such a modification is hypothesized to mimic host membranes (which contain phosphatidylcholine) and further reduce LL-37 binding. The viscosity of the periplasm may also contribute, since this space is densely packed with hydrophilic proteins that may nonspecifically hinder CAMPs on their way to the inner membrane and cytoplasm, similar to the nonspecific binding of drugs by plasma proteins in the human body (Silhavy et al. 2010).
Gram-negative bacteria can also decrease the net negative charge of their exterior by decorating LPS/LOS lipid A with positively charged small molecules. In N. meningitidis, N. gonorrhoeae, Helicobacter pylori, E. coli, and S. enterica serovar Typhimurium, the addition of phosphoethanolamine (PEA) not only removes the negative charge once provided by free lipid A phosphate, but also adds a positive charge, thereby decreasing the net negative charge of the outer membrane and perhaps membrane permeability as well (Lewis et al. 2009; Lee et al. 2004; Beceiro et al. 2011). In Campylobacter jejuni, PEA can be added to lipid A and to a flagellar protein serving two purposes: (1) increase CAMP resistance and (2) promote bacterial motility (Cullen et al. 2012). Alternatively, 4-amino-4-deoxy-l-arabinose (l-Ara4N) may be added to the same phosphates in some Gram-negative bacteria and provides resistance to CAMPs in a similar manner to phosphoethanolamine (Trent 2004). Bacteria may also use the LpxE lipid A phosphatase to simply remove phosphate from lipid A and reduce negative charge, a phenomenon seen in the plant symbiont Rhizobium leguminosarum, as well as the human pathogen H. pylori. LpxE orthologues are present in Francisella tularensis, Brucella melitensis, and Legionella pneumophila (Karbarz et al. 2003; Trent 2004). These modifications are not only important for bacterial survival, but also impact the immune response to LOS/LPS or “endotoxin,” one of the most potent inducers of septic shock (Peschel 2002). The regulation of these modifications has been very thoroughly studied in S. enterica and is controlled by combined efforts of the PhoP/PhoQ and PmrA/PmrB TCR systems (Lee et al. 2004; Gunn et al. 1998); these regulatory systems are discussed in more detail below.
In Gram-positive organisms, polyalditol, polyglycerol, or polyribitol phosphate polymers [teichoic acids (TA)] create a “continuum of negative charge” (Neuhaus and Baddiley 2003) with deprotonated phosphate residues present along each chain. The cationic antimicrobial lysosomal protein cathepsin G appears to use TA as a binding site on S. aureus (Shafer and Onunka 1989). Seminal work by Andreas Peschel and coworkers first characterized the CAMP repulsive effect caused by the d-alanylation of TA in S. aureus, an ability that endows this pathogen (and others) with decreased susceptibility to diverse CAMPs from different sources (Peschel et al. 1999). The enzymatic pathway encoded by the dltABCD operon processes the esterification of d-alanine to TA alditol residues and transforms TA into partly zwitterionic polymers, reducing the net negative charge at Gram-positive surfaces. The expression of the dlt operon in S. aureus is under the control of the Aps/GraRSX regulatory system (Li et al. 2007a, b). This appears to be a widespread defense mechanism present in other Firmicutes such as Bacillus, Enterococcus, and Streptococcus. It is also important to mention that S. aureus mutants lacking a functional dlt operon are more susceptible to the glycopeptide antibiotic vancomycin (Peschel et al. 2000), and in the future it may be possible to counter vancomycin-resistant S. aureus (VRSA) with drugs that block d-alanylation of TA.
Changes in membrane rigidity can also influence levels of CAMP resistance. PhoP/PhoQ control of pagP, which encodes a palmitoyltransferase that hepta-acylates S. enterica lipid A in response to stresses typically found in a phagosomal environment, can influence membrane rigidity and the capacity of CAMPs to productively insert into bacterial membranes. These stresses include low pH, varying Ca2+ and Mg2+ ionic strength, and high concentrations of CAMPs (Guo et al. 1998; Prost and Miller 2008). The MsbB protein in Vibrio cholerae plays an analogous role by adding an acyl chain to the same position as PagP. This modification was found to greatly enhance resistance to polymyxin B, LL-37 (and its mouse homolog CRAMP), and magainin 2 (Matson et al. 2010). Interestingly, msbB deletion mutants were unable to induce a TLR4 response in human embryonic kidney cells, which suggests that efficient recognition and binding of bacterial endotoxin is largely due to lipid A structure. This finding also supports the notion that CAMP resistance mechanisms may protect not only directly, but indirectly through modulating the host immune response. The actions of both PagP and MsbB ultimately increase the stability and hydrophobicity of the outer membrane and decrease its permeability, thus enhancing the fortitude of an already formidable barrier to CAMP entry (Peschel 2002). In S. aureus, pigment production through the crtOPQMN operon performs a similar function in CAMP resistance by increasing membrane rigidity (Mishra et al. 2011). The cold shock system of S. aureus may also be important in resistance as mutants lacking CspA (Katzif et al. 2003) and CspB (Duval et al. 2010) have reduced pigment levels and altered susceptibilities to certain antimicrobials, including cathepsin G-derived CAMPs.
Conversely, decreased membrane rigidity may also provide CAMP resistance. S. aureus tPMP (thrombin-induced platelet microbicidal protein)-resistant strains were consistently found to have greater membrane fluidity than their tPMP susceptible counterparts, caused by a preponderance of longer chain, unsaturated fatty acids (Bayer et al. 2000). It has also long been known that S. aureus membranes contain unsaturated sexa-, hepta-, and octa-isoprenoid menaquinones (Nahaie et al. 1984). Apart from their function as redox molecules in the electron transport chain, they also increase membrane fluidity. Thus, it has been hypothesized that large fluctuations in membrane fluidity to either extreme may distance membrane order from the “sweet spot” required for optimum CAMP bactericidal activity (Mishra et al. 2011; Yeaman and Yount 2003).
One of the most nonspecific and ubiquitous mechanisms of CAMP defense in bacteria is mediated by the MprF (multiple peptide resistance factor) protein (Peschel et al. 2001). Originally described by Peschel and colleagues (2001), it soon became clear that MprF provides significantly enhanced bacterial resistance to neutrophils and several evolutionarily distinct CAMPs. MprF is present in a wide variety of Gram-positive, Gram-negative, and acid-fast bacteria and is also present in the Archaea. This CAMP resistance mechanism is remarkable in that it only requires substrates that are abundant in the bacterial cell—charged tRNAs and membrane phospholipids—and is somewhat indiscriminant when recognizing tRNA donor and phospholipid acceptor molecules. This is thought to be why the MprF mechanism is so widespread (Roy and Ibba 2008). MprF is an integral cytoplasmic membrane protein and may add the positively charged l-lysine, l-alanine, and perhaps, in Mycobacterium tuberculosis, l-ornithine (Ernst and Peschel 2011; Khuller and Subrahmanyam 1970) amino acids to phosphatidylglycerol and cardiolipin (diphosphatidylglycerol). S. aureus MprF consists of (1) a transesterase domain which adds the amino acid residue to phosphatidylglycerol (PG) on the cytoplasmic leaflet of the membrane and (2) a “flippase” domain that flips the nascent lysyl-PG to the exoplasmic leaflet of the membrane where it can serve to repulse CAMPs by reducing the net negative charge of the membrane’s outer surface. It is noteworthy that MprF is the first flippase to be discovered in prokaryotes (Ernst and Peschel 2011). In M. tuberculosis, the lysX gene is actually a fusion of mprF and lysU, a lysyl-tRNA synthase. Here, lysyl-tRNA can be made at the cytoplasmic membrane level by the LysU domain and then shuttled into the MprF reactions described above (Maloney et al. 2009). Clostridium perfringens produces two MprF proteins, 1 and 2, which produce alanyl-PG and lysyl-PG, respectively (Roy and Ibba 2008). It is probable that, like other CAMP resistance mechanisms, the activities of MprF are under the control of two- or three-component regulatory systems; indeed, MprF appears to be under the control of the VirR protein of the VirRS TCR in Listeria monocytogenes (Mandin et al. 2005) and the Aps/GraRSX TCR in S. aureus (Li et al. 2007a, b; Otto 2009).
Intriguingly, it seems that CAMPs themselves are not the only stimuli that can induce CAMP resistance mechanisms in bacteria. A brief but elegant study by Dorrer and Teuber in the 1970s (Dorrer and Teuber 1977) demonstrated that phosphate starvation induced polymyxin B resistance in Pseudomonas fluorescens by increasing the membrane fraction of ornithylated lipids, which decreases the net negative charge of the bacterial envelope. Notably, it was later discovered that survival inside of macrophages induced the expression of phosphate importers 9.4-fold in Salmonella typhimurium (Valdivia and Falkow 1997). This might indicate that (1) low phosphate levels preclude the use of phosphate on membrane lipids and require that other groups (e.g., ornithine) provide the hydrophilic portion of the membrane lipid to maintain a stable bilayer structure and (2) the host environment may unwittingly hinder its own efforts to kill with CAMPs by inducing the production of these cationic lipid species.
Though the mechanisms described above allow the microorganism to change the envelope structure without dire consequences for growth, there are other CAMP resistance strategies that come at great fitness cost to the bacterium. In S. aureus, small colony variants (SCVs) are typically deficient in electron transport and have a diminished membrane potential (Δψ) (Yeaman and Yount 2003). They also arise much more readily (10,000-fold) in the host than in laboratory culture (Vesga et al. 1996). This suggests that slowing cell growth may represent a “niche-specific” defense mechanism, triggered by growth in a host, that allows bacteria to depolarize their membrane and decrease the rate of “self-promoted uptake” by host CAMPs (Peschel 2002) (see (Hancock 1997) for a description of this phenomenon). Interestingly, the S. aureus cspB mutant studied by Duval et al. described above, exhibited many of the characteristics of SCVs (Duval et al. 2010). In Gram-negative bacteria, reduced growth rates may also help stave off death by CAMP by reducing the occurrence of nascent septa that are a critical component during bacterial binary fission. Sochacki and colleagues used real-time fluorescence microvideography to show that rhodamine-labeled LL-37 consistently binds E. coli cells at their nascent septa first, then proceeds outward in a continuous “circumferential band” towards the distal poles of each developing daughter cell (Sochacki et al. 2011), though LL-37 was still able to bind to nonseptated cells.
2.5 Export of CAMPs
Even if CAMPs successfully traverse the formidable barriers described above, bacteria can still circumvent their action by the use of drug efflux pumps that capture and export structurally diverse antimicrobials after they breach the cell envelope. Drug efflux pumps are grouped into superfamilies based on their component stoichiometry, number of transmembrane regions in the transporter, energy source, and type of substrates recognized. Five superfamilies of efflux pumps are known: the resistance-nodulation-division (RND) superfamily, the major facilitator (MFS) superfamily, the multidrug and toxin extrusion (MATE) superfamily, the ATP-binding cassette (ABC) transporter superfamily, and the small multidrug resistance (SMR) superfamily (see (Piddock 2006) for an excellent review of bacterial efflux pumps). The MtrCDE efflux pump of N. gonorrhoeae is a member of the RND superfamily and was the first efflux pump shown to export CAMPs to the extracellular milieu (Shafer et al. 1998). This pump has been studied in detail and will be described later (see below); the analogous pump in N. meningitidis also can export CAMPs (Tzeng et al. 2005). Other Gram-negative pathogens have been found to use efflux pumps to resist CAMPs. Yersinia enterocolitica can protect itself from CAMP activity by expressing the RosAB efflux pump, which is induced upon growth at 37 °C. The RosA pump activity is powered by the potassium antiporter RosB and is thought to provide resistance through (1) efflux of CAMPs and (2) acidification of the cytoplasm (Bengoechea and Skurnik 2000). K. pneumoniae expresses the AcrAB-TolC efflux pump that mediates resistance to human antimicrobial peptides, as an AcrB mutant was more sensitive to HBD-1 and HBD-2 (Padilla et al. 2010). The homologous efflux pump in E. coli, AcrAB-TolC, is arguably the most structurally characterized efflux pump to date (Murakami et al. 2006; Husain and Nikaido 2010; Symmons et al. 2009) and will likely become an invaluable tool for the design and testing of an emerging class of antibiotics, the efflux pump inhibitors (EPIs) (Lomovskaya and Bostian 2006).
Efflux pumps also function in Gram-positive bacteria for CAMP resistance. For instance, the EpiFEG efflux pump of S. epidermidis exports and increases staphylococcal resistance to various CAMPs. EpiFEG is an ABC transporter that is known to export bacterial-derived CAMPs, such as gallidermin, nisin, and epidermin (Otto et al. 1998). The MefE/Mel efflux pump possessed by certain strains of S. pneumoniae is a mechanism used by this pathogen to develop resistance to macrolides. Expression of this pump was found (Zähner et al. 2010) to be inducible by 14- and 15-membered macrolides as well as LL-37/CRAMP and that such induction enhanced pneumococcal resistance to macrolides and LL-37. Maximal constitutive and inducible cathelicidin resistance expressed by pneumococci required a functional MefE/Mel pump system, although it is yet to be determined if these CAMPs are actual pump substrates. It also appears that some efflux pumps might actually enhance CAMP resistance independently of their efflux capacity. In S. aureus, QacA (a plasmid-encoded multidrug MFS efflux pump) mediates resistance to tPMP, but does not affect levels of resistance to HNP-1 or protegrin-1 (Kupferwasser et al. 1999). Curiously, later studies found that QacA-mediated resistance to tPMP was not due to efflux activity and that membrane fluidity seemed to diminish slightly in strains bearing the qacA gene (Bayer et al. 2006), but the exact mechanism of how QacA expression protects against tPMP remains to be determined.
Of note is a very intriguing pattern that has emerged among the Gram-positive bacteria that teams ABC-transporter efflux pumps and TCR systems into very close functional associations called “resistance modules.” Each component of the module is dependent on the other for resistance against antimicrobial peptides. Extensive phylogenetic analysis suggests a coevolution of efflux pumps and TCR systems in the phylum Firmicutes (over 250 resistance modules are estimated). In this partnership, sensor domain-deficient inner membrane histidine kinases (IMHKs) still relay signals through their cognate response regulators, but recognition of the environmental stimulus is carried out by a neighboring permease/transporter protein in the membrane (Dintner et al. 2011). Well-characterized examples of such TCR/efflux pump couplings include the BceRS TCR and BceAB pump in Bacillus subtilis and Streptococcus mutans (Bernard et al. 2007; Ouyang et al. 2010) and the BraRS TCR and BraED pump in S. aureus (Hiron et al. 2011). The VraED pump also appears to play a role in S. aureus resistance, but is required only for efflux and not for sensing.
2.6 Modification of Internal Targets
Since the antimicrobial action of most CAMPs is independent of stereochemistry, it has generally been thought that they do not recognize targets with a chiral center (Bessalle et al. 1990; Wade et al. 1990). Furthermore, their killing activity has been linked to processes such as loss of membrane integrity and depolarization. Due to these broad-spectrum killing mechanisms, CAMPs have been likened to “dirty bombs” in contrast to the “smart bomb”-like action of many antibiotic drugs (Peschel and Sahl 2006). This analogy may not be completely correct as it is now clear that CAMPs may also kill bacteria by interfering with internal cellular functions like DNA/RNA/protein synthesis, protein folding, peptidoglycan polymerization, and septum formation (Hale and Hancock 2007; Cudic and Otvos 2002). Just as the bacterial envelope can accumulate changes to hinder CAMP attack, antimicrobial stress selects for mutants containing modified and thus less CAMP-accessible cytoplasmic targets. One example of this is a mutation in the gyrB gene in E. coli. gyrB encodes DNA gyrase, a type II topoisomerase that maintains a level of DNA supercoiling necessary for replication, transcription, and recombination. GyrB is a target of the well-known class of antibiotics called quinolones, and also the bacteriocin microcin B17, which is produced by E. coli. del Castillo and colleagues (2001) found that mutation of residue W751 to hydrophilic amino acids like lysine or arginine imparts a great deal of resistance to microcin B17. The authors hypothesize that this residue may be located on the entry gate through which the intact DNA can be transported (T-segment) and that microcin B17 normally binds and inhibits GyrB activity, leading to cell death (del Castillo et al. 2001).
2.7 Inducible Mechanisms of CAMP Resistance
Inducible mechanisms of CAMP resistance allow bacteria to promptly respond to stressful changes in their environments. TCR systems sense potentially harmful changes, orchestrate a response to the imposed stress, and adapt gene expression to the new context. TCR systems consist of a sensor histidine kinase on the inner membrane and a cytoplasmic regulatory protein. Typically, the sensor kinase detects a signal in the environment, becomes autophosphorylated, and in turn phosphorylates the cognate intracellular regulator, activating it. The activated regulator binds to DNA and alters the expression of different genes. Numerous and functionally distinct TCR systems are found in bacteria. The PhoP/PhoQ TCR system was initially studied in S. typhimurium (Fields et al. 1989; Groisman et al. 1989; Miller et al. 1989), and homologs were subsequently found and studied in P. aeruginosa (Macfarlane et al. 1999), in various Enterobacteriaceae, and in Neisseria, where it is named MisR/MisS (Tzeng et al. 2004).
The PhoP/PhoQ and the PmrA/PmrB are well-studied examples of TCRs that have important roles in CAMP resistance. Under favorable conditions, PhoQ is bound by divalent cations such as Mg2+ and Ca2+ in the environment and is not active. At low concentrations of divalent cations, PhoQ phosphorylates PhoP, which in turn regulates many genes involved in AMP resistance, such as pagP, pgtE, slyA, and pmrD (Roland et al. 1994). PmrD is required for activation of the PmrA/PmrB TCR system (Otto 2009; Guina et al. 2000; Macfarlane et al. 2000, 1999; Navarre et al. 2005). When CAMPs are present in the media, it is thought that they displace divalent cations bound to an acidic patch of PhoQ and induce activation of the TCR system (Prost and Miller 2008). In S. enterica, PhoP/PhoQ is activated by CAMPs and upregulates genes related to CAMP resistance such as pagP, pagL, and lpxO (Bader et al. 2005; Hancock and McPhee 2005). Using a mutant strain of S. enterica showing increased resistance to polymyxin B, azurocidin, and CAP57 (Shafer et al. 1984) due to a pmrA mutation, Roland et al. identified the TCR system PmrA/PmrB (Roland et al. 1993). This TCR regulates expression of genes involved in CAMP resistance such as pmrHFIJKLM (or pbg operon), cld, and cptA, which are responsible for LPS modifications (Gunn 2008). In P. aeruginosa, PmrA/PmrB is induced by low concentrations of Mg2+ and by LL-37, which promotes expression of genes, such as pbgP, pbgE, and ugd, involved in LPS modification and resistance to polymyxin B (Gunn and Miller 1996; McPhee et al. 2003; Moskowitz et al. 2004). It was shown that mutating pmrAB in P. aeruginosa rendered the bacteria hypersusceptible to killing by LL-37 or by other CAMPs such as polymyxin B (Lewenza et al. 2005).
Li et al. uncovered a novel regulatory system in S. epidermidis that they named aps for antimicrobial peptide sensor (Li et al. 2007a, b), also observed in S. aureus and named gra for glycopeptide resistance associated genes (Kuroda et al. 2000; Cui et al. 2005). The system consists of a TCR system with a sensor kinase (apsS) and a regulator (apsR) and a third protein with unknown yet essential function (apsX) (Li et al. 2007a, b). Their research showed that deletion of any or all of these components led to downregulation of the dlt and mprF genes, which modify cell surface structures and enhance resistance to CAMPs (Li et al. 2007b). Furthermore, Lai et al. have described agr and sarA in S. aureus and S. epidermidis as major regulators that are induced in the presence of the anionic AMP dermcidin. These gene regulators increase expression and proteolytic activity of the SepA metalloprotease in presence of dermcidin (Lai et al. 2007).
3 CAMP Resistance in Clinically Relevant Pathogens
In the above sections, we reviewed major mechanisms that bacteria have evolved to resist CAMPs. In order to highlight how such CAMP resistance systems can influence the efficacy of host resistance to infection and bacterial pathogenesis, we discuss them in the context of three major clinically relevant pathogens of public health concern. In this respect, we focus on the obligate human Gram-negative pathogens N. gonorrhoeae and N. meningitidis and on the Gram-positive bacteria S. aureus, particularly the methicillin-resistant strains (MRSA).
3.1 N. gonorrhoeae and N. meningitidis
N. gonorrhoeae and N. meningitidis are Gram-negative diplococci and strict human pathogens (Shafer et al. 2010). Gonococci (GC) cause the sexually transmitted infection gonorrhea. In contrast, meningococci (MC) are present as commensals in 8–25 % of the human population, but can cause bacterial meningitis and fulminant septicemia (Stephens 2009). Gonorrhea is the second most reported infection in the United States, though many cases are asymptomatic, and can enhance HIV transmission (Klotman et al. 2008; McNabb et al. 2008). Furthermore, although there are vaccines available for many MC serogroups that cause disease, a protective vaccine for serogroup B MC is still under development; no vaccine has been developed that blocks GC infection. Both of these pathogens are also becoming increasingly resistant to antibiotics (Shafer et al. 2010). Worryingly, a recent report described a strain of gonorrhea that is resistant to the last remaining first-line antibiotic used in empirical treatment, ceftriaxone (Ohnishi et al. 2011). Thus, the pathogenic Neisseria represent a significant threat to global health.
As strictly human pathogens, GC and MC have evolved remarkable and redundant mechanisms to defend themselves against host CAMPs. These include capsule production by MC (Jones et al. 2009; Spinosa et al. 2007); host-molecule “cloaking” using the highly anionic polymers heparin/heparan sulfate and short, cationic polyamines (Goytia and Shafer 2010; Jones et al. 2009; Seib et al. 2009); MC sequestration of LL-37 in the bacterial cytosol (Frigimelica et al. 2008); downregulation of host LL-37 production (Bergman et al. 2005); export of CAMPs by the MtrCDE efflux pump (Shafer et al. 1998); decoration of lipid A with PEA (Cox et al. 2003; Tzeng et al. 2005; Lewis et al. 2009); and hexa-acylation of lipid A (Tzeng et al. 2005). Some of these mechanisms have been shown to be under the control of the MisR/MisS TCR system, named for its regulation of meningococcal LOS inner core structure, which itself is necessary for resistance to CAMPs (Johnson et al. 2001; Newcombe et al. 2004; Tzeng et al. 2004). Other mechanisms may be induced by different inputs, e.g., upregulation of mtrCDE expression in GC upon exposure to hydrophobic pump substrates typically present at infection sites (Rouquette et al. 1999).
Is there evidence that any of these resistance mechanisms influence bacterial survival during infection? Briefly, yes: in support of this idea, elegant experiments performed in the laboratory of A. Jerse have shown that loss of the MtrC–MtrD–MtrE efflux pump due to its genetic inactivation decreased the ability of GC to survive in an experimental model of lower genital tract infection in female mice (Jerse et al. 2003). Further work by her group (Warner et al. 2007, 2008) showed that overexpression of MtrC–MtrD–MtrE increases fitness of GC during infection by nearly three orders of magnitude. In contrast, loss of the ability to activate transcription of mtrCDE decreased fitness in vivo by 500-fold. Finally, lptA mutants of GC that are unable to decorate their lipid A with PEA are more susceptible to CAMPs and less fit in vivo than the parental wild-type strain (Jerse, personal communication, 2011).
3.2 Staphylococcus aureus
S. aureus is a Gram-positive bacterium that has evolved to survive in a commensal capacity on the human host. It can be found on the skin and in the nares in 20 % of the population, but when staphylococci breach host defenses, they can cause many different illnesses, including skin infections, abscesses, and life-threatening diseases such as endocarditis, pneumonia, meningitis, toxic shock syndrome, and sepsis. Importantly, S. aureus is one of the most frequent causes of hospital- and community-acquired infections. The incidence of multiple antibiotic-resistant strains of S. aureus continues to increase, restricting the options for treatment. S. aureus has become one of the most difficult bacterial infections to treat as multidrug-resistant strains have emerged; a typical example is methicillin-resistant S. aureus, or MRSA. MRSA colonizes 2 % of the population, many of whom are immunocompromised due to age (e.g., the elderly and young children) or medical condition (e.g., pregnant women, HIV-positive, and cancer patients) (Kowalski et al. 2005). MRSA can cause life-threatening infections such as pneumonia, septicemia, and infections following surgery. MRSA resist most β-lactam antibiotics (penicillins and cephalosporins) including penicillin, methicillin, and amoxicillin. Furthermore, it is quite common to see resistance develop when MRSA infection is treated with macrolides and/or fluoroquinolones. Importantly, as of 2007, MRSA infections caused more deaths (>17,000) in the United States than HIV/AIDS (Klevens et al. 2007).
S. aureus has found strategies to impair all of the events associated with CAMP killing activity and sometimes in more than one way. For instance, S. aureus secretes proteolytic enzymes, V8 and aureolysin, which are able to degrade and inactivate CAMPs such as LL-37 (Sieprawska-Lupa et al. 2004). It was suggested that loss of these enzymes by molecular modification could render S. aureus more susceptible to CAMPs in vitro, ex vivo, and in vivo (Sieprawska-Lupa et al. 2004). Staphylokinase (Sak) is a secreted protein that sequesters LL-37 and increases virulence in vivo (Braff et al. 2007). Burlak et al. demonstrated that S. aureus express Sak in vivo, since injection of S. aureus in mouse elicited the production of specific antibodies against Sak (Burlak et al. 2007). S. aureus produces positively charged polysaccharide intercellular adhesin (PIA) and negatively charged poly-γ-glutamic acid (PGA) at its surface, which increases the net positive charge of the cell surface and, as it was described for S. epidermidis, could impair binding of positively charged CAMPs by electrostatic repulsion; however, other mechanisms might be involved as well, since PIA also protected S. aureus from the negatively charged AMP dermcidin (Vuong et al. 2004; Kocianova et al. 2005). Moreover, PIA, which is produced by the intercellular adhesion ica locus, has been involved in biofilm formation in S. aureus (Cramton et al. 1999). S. aureus can also express d-Ala and l-Lys at its surface, modifying the net charge, through the dlt operon and mprF gene, respectively (Peschel et al. 1999, 2001; Staubitz et al. 2004; Collins et al. 2002; Nishi et al. 2004). These mechanisms are also efficient against other CAMPs, such as those derived from lactoferrin and phospholipase A2 (Koprivnjak et al. 2002).
S. aureus regulates these genes and many others involved in CAMP resistance with Aps/GraRSX, an inducible system that is activated in presence of CAMPs (Li et al. 2007a; Kraus et al. 2008). Li et al. showed that a mutant with a deletion of aspS was less virulent in an intraperitoneal mouse infection model than the wild-type strain (Li et al. 2007a). Other inducible mechanisms described in S. aureus and implicated in CAMP resistance involve Agr and SarA (Huang 2006). Modifications of the membrane involve carotenoid production by the crtOPQMN operon, which can suppress nonoxidative host defenses mediated by CAMPs (Mishra et al. 2011). S. aureus is also able to prevent CAMP activity by expressing efflux pumps such as the plasmid-encoded QacA and the ABC transporter EpiFEG (though QacA-mediated resistance is independent of efflux activity). Finally, S. aureus is able to form biofilms, which are ultrastructures that promote bacterial resistance to AMPs and other killing agents. Internal targets are probable in S. aureus, since CAMPs are able to kill S. aureus without significant depolarization or disruption of the membrane (Koo et al. 2001).
4 Conclusions and Perspectives
Bacteria have constantly evolved novel mechanisms to overcome attacks by CAMPs. It seems that for every way CAMPs kill, bacteria have developed a resistance mechanism(s) in response. As mentioned above, the mechanisms of CAMP resistance are for all purposes similar to those developed by bacteria to resist classical antibiotics. At first glance, mechanisms of bacterial resistance to CAMPs would seem to favor the microbe and not the host. However, this view may be overly simplistic; most of the bacteria we interact with on a daily basis are not normally pathogenic, and many are associated with good health. If such commensally carried, helpful bacteria were to be reduced or eliminated in the presence of CAMPs, how would this impact our health? Perhaps CAMP resistance mechanisms evolved not as a way for pathogens to avoid elimination, but rather as a way for the helpful commensals to survive.
CAMPs have been promoted as a new class of therapeutic antimicrobials for treating multi-antibiotic-resistant pathogens, some of which cause infections that are becoming untreatable. Studies exploring various characteristics of CAMPs (charge, amphipathicity, hydrophobicity, etc.) (see Shprung et al. 2012) will help gain insight in the design of ever more efficient synthetic CAMPs. Alternatively, further research focusing on specific bacterial metabolic states could prevent formation of structures such as biofilms that are extremely hard to destroy and that increase the risk of chronic infections and antibiotic resistance development. In this respect, work in the Hancock laboratory on the ability of CAMPs to prevent formation of biofilms is especially important and could be exploited by attaching these peptides to medical devices (de la Fuente-Núñez et al. 2012). The continued advancement of these peptides as therapeutics will require additional studies to further analyze their potential short- and long-term toxic effects, their specificity, their pharmacokinetics, the appearance of resistance patterns, and immunomodulatory/immunostimulatory secondary effects.
Continued studies on mechanisms of CAMP resistance are also warranted. As therapeutic antimicrobial peptides pass through clinical trials, we can use the knowledge gained from such experiments to predict how bacteria will respond to their presence during treatment (which will likely be at higher levels than what occurs naturally) and if resistance (especially broad spectrum) will develop. We must, however, be cognizant of the possibility that resistance to administered CAMPs may negatively impact innate host defenses mediated by the natural CAMPs that function at different sites in the human body. How this might influence decisions to move forward with the therapeutic application of CAMPs is a matter for future consideration.
References
Abachin E, Poyart C, Pellegrini E, Milohanic E, Fiedler F, Berche P, Trieu-Cuot P (2002) Formation of D-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. Mol Microbiol 43(1):1–14
Akesson P, Sjoholm AG, Bjorck L (1996) Protein SIC, a novel extracellular protein of Streptococcus pyogenes interfering with complement function. J Biol Chem 271(2):1081–1088
Amer LS, Bishop BM, van Hoek ML (2010) Antimicrobial and antibiofilm activity of cathelicidins and short, synthetic peptides against Francisella. Biochem Biophys Res Commun 396(2):246–251. doi:S0006-291X(10)00742-4[pii]10.1016/j.bbrc.2010.04.073
Anderl JN, Franklin MJ, Stewart PS (2000) Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob Agents Chemother 44(7):1818–1824
Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI (2005) Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 122(3):461–472. doi:S0092-8674(05)00553-2[pii]10.1016/j.cell.2005.05.030
Banemann A, Deppisch H, Gross R (1998) The lipopolysaccharide of Bordetella bronchiseptica acts as a protective shield against antimicrobial peptides. Infect Immun 66(12):5607–5612
Bauer ME, Spinola SM (2000) Localization of Haemophilus ducreyi at the pustular stage of disease in the human model of infection. Infect Immun 68(4):2309–2314
Bauer ME, Townsend CA, Ronald AR, Spinola SM (2006) Localization of Haemophilus ducreyi in naturally acquired chancroidal ulcers. Microbes Infect 8(9–10):2465–2468. doi:S1286-4579(06)00230-9[pii]10.1016/j.micinf.2006.06.001
Bayer AS, Prasad R, Chandra J, Koul A, Smriti M, Varma A, Skurray RA, Firth N, Brown MH, Koo SP, Yeaman MR (2000) In vitro resistance of Staphylococcus aureus to thrombin-induced platelet microbicidal protein is associated with alterations in cytoplasmic membrane fluidity. Infect Immun 68(6):3548–3553
Bayer AS, Kupferwasser LI, Brown MH, Skurray RA, Grkovic S, Jones T, Mukhopadhay K, Yeaman MR (2006) Low-level resistance of Staphylococcus aureus to thrombin-induced platelet microbicidal protein 1 in vitro associated with qacA gene carriage is independent of multidrug efflux pump activity. Antimicrob Agents Chemother 50(7):2448–2454. doi:50/7/2448[pii]10.1128/AAC.00028-06
Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, Dhanji H, Chart H, Bou G, Livermore DM, Woodford N (2011) Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob Agents Chemother 55(7):3370–3379. doi:AAC.00079-11[pii]10.1128/AAC.00079-11
Becker MN, Diamond G, Verghese MW, Randell SH (2000) CD14-dependent lipopolysaccharide-induced beta-defensin-2 expression in human tracheobronchial epithelium. J Biol Chem 275(38):29731–29736. doi:10.1074/jbc.M000184200M000184200[pii]
Belas R, Manos J, Suvanasuthi R (2004) Proteus mirabilis ZapA metalloprotease degrades a broad spectrum of substrates, including antimicrobial peptides. Infect Immun 72(9):5159–5167. doi:10.1128/IAI.72.9.5159-5167.2004
Bengoechea JA, Skurnik M (2000) Temperature-regulated efflux pump/potassium antiporter system mediates resistance to cationic antimicrobial peptides in Yersinia. Mol Microbiol 37(1):67–80. doi:mmi1956[pii]
Bergman P, Johansson L, Asp V, Plant L, Gudmundsson GH, Jonsson AB, Agerberth B (2005) Neisseria gonorrhoeae downregulates expression of the human antimicrobial peptide LL-37. Cell Microbiol 7(7):1009–1017. doi:CMI530[pii]10.1111/j.1462-5822.2005.00530.x
Bernard R, Guiseppi A, Chippaux M, Foglino M, Denizot F (2007) Resistance to bacitracin in Bacillus subtilis: unexpected requirement of the BceAB ABC transporter in the control of expression of its own structural genes. J Bacteriol 189(23):8636–8642. doi:JB.01132-07[pii]10.1128/JB.01132-07
Bessalle R, Kapitkovsky A, Gorea A, Shalit I, Fridkin M (1990) All-D-magainin: chirality, antimicrobial activity and proteolytic resistance. FEBS Lett 274(1–2):151–155
Braff MH, Jones AL, Skerrett SJ, Rubens CE (2007) Staphylococcus aureus exploits cathelicidin antimicrobial peptides produced during early pneumonia to promote staphylokinase-dependent fibrinolysis. J Infect Dis 195(9):1365–1372. doi:JID37442[pii]10.1086/513277
Brogden KA (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3(3):238–250. doi:10.1038/nrmicro1098[pii] nrmicro1098
Brown KL, Hancock RE (2006) Cationic host defense (antimicrobial) peptides. Curr Opin Immunol 18(1):24–30. doi:S0952-7915(05)00199-8[pii]10.1016/j.coi.2005.11.004
Burlak C, Hammer CH, Robinson MA, Whitney AR, McGavin MJ, Kreiswirth BN, Deleo FR (2007) Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell Microbiol 9(5):1172–1190. doi:10.1111/j.1462-5822.2006.00858.x
Campos MA, Vargas MA, Regueiro V, Llompart CM, Alberti S, Bengoechea JA (2004) Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect Immun 72(12):7107–7114. doi:72/12/7107[pii]10.1128/IAI.72.12.7107-7114.2004
Casey SG, Shafer WM, Spitznagel JK (1985) Anaerobiosis increases resistance of Neisseria gonorrhoeae to O2-independent antimicrobial proteins from human polymorphonuclear granulocytes. Infect Immun 47(2):401–407
Collins LV, Kristian SA, Weidenmaier C, Faigle M, Van Kessel KP, Van Strijp JA, Gotz F, Neumeister B, Peschel A (2002) Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J Infect Dis 186(2):214–219. doi:JID010926[pii]10.1086/341454
Cox AD, Wright JC, Li J, Hood DW, Moxon ER, Richards JC (2003) Phosphorylation of the lipid A region of meningococcal lipopolysaccharide: identification of a family of transferases that add phosphoethanolamine to lipopolysaccharide. J Bacteriol 185(11):3270–3277
Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F (1999) The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun 67(10):5427–5433
Cudic M, Otvos L Jr (2002) Intracellular targets of antibacterial peptides. Curr Drug Targets 3(2):101–106
Cui L, Lian JQ, Neoh HM, Reyes E, Hiramatsu K (2005) DNA microarray-based identification of genes associated with glycopeptide resistance in Staphylococcus aureus. Antimicrob Agents Chemother 49(8):3404–3413. doi:10.1128/AAC.49.8.3404-3413.2005
Cullen TW, Madsen JA, Ivanov PL, Brodbelt JS, Trent MS (2012) Characterization of unique modification of flagellar rod protein FlgG by Campylobacter jejuni lipid A phosphoethanolamine transferase, linking bacterial locomotion and antimicrobial peptide resistance. J Biol Chem 287(5):3326–3336. doi:10.1074/jbc.M111.321737
de la Fuente-Núñez C, Korolik V, Bains M, Nguyen U, Breidenstein EB, Horsman S, Lewenza S, Burrows L, Hancock RE (2012) Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob Agents Chemother. 56(5):2696–704. doi:10.1128/AAC.00064-12
Dean SN, Bishop BM, van Hoek ML (2011) Susceptibility of Pseudomonas aeruginosa biofilm to alpha-helical peptides: D-enantiomer of LL-37. Front Microbiol 2:128. doi:10.3389/fmicb.2011.00128
del Castillo FJ, del Castillo I, Moreno F (2001) Construction and characterization of mutations at codon 751 of the Escherichia coli gyrB gene that confer resistance to the antimicrobial peptide microcin B17 and alter the activity of DNA gyrase. J Bacteriol 183(6):2137–2140. doi:10.1128/JB.183.6.2137-2140.2001
Dintner S, Staron A, Berchtold E, Petri T, Mascher T, Gebhard S (2011) Coevolution of ABC transporters and two-component regulatory systems as resistance modules against antimicrobial peptides in Firmicutes bacteria. J Bacteriol 193(15):3851–3862. doi:JB.05175-11[pii]10.1128/JB.05175-11
Dorrer E, Teuber M (1977) Induction of polymyxin resistance in Pseudomonas fluorescens by phosphate limitation. Arch Microbiol 114(1):87–89
Dorschner RA, Lopez-Garcia B, Peschel A, Kraus D, Morikawa K, Nizet V, Gallo RL (2006) The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J 20(1):35–42. doi:20/1/35[pii]10.1096/fj.05-4406com
Duval BD, Mathew A, Satola SW, Shafer WM (2010) Altered growth, pigmentation, and antimicrobial susceptibility properties of Staphylococcus aureus due to loss of the major cold shock gene cspB. Antimicrob Agents Chemother 54(6):2283–2290. doi:10.1128/AAC.01786-09
Ernst CM, Peschel A (2011) Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol 80(2):290–299. doi:10.1111/j.1365-2958.2011.07576.x
Erwin AL, Smith AL (2007) Nontypeable Haemophilus influenzae: understanding virulence and commensal behavior. Trends Microbiol 15(8):355–362. doi:S0966-842X(07)00109-6[pii]10.1016/j.tim.2007.06.004
Fan X, Goldfine H, Lysenko E, Weiser JN (2001) The transfer of choline from the host to the bacterial cell surface requires glpQ in Haemophilus influenzae. Mol Microbiol 41(5):1029–1036. doi:mmi2571[pii]
Farley MM, Shafer WM, Spitznagel JK (1987) Antimicrobial binding of a radiolabeled cationic neutrophil granule protein. Infect Immun 55(6):1536–1539
Farley MM, Shafer WM, Spitznagel JK (1988) Lipopolysaccharide structure determines ionic and hydrophobic binding of a cationic antimicrobial neutrophil granule protein. Infect Immun 56(6):1589–1592
Fields PI, Groisman EA, Heffron F (1989) A Salmonella locus that controls resistance to microbicidal proteins from phagocytic cells. Science 243(4894 Pt 1):1059–1062
Flannagan RS, Cosío G, Grinstein S (2009) Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7(5):355–366. doi:10.1038/nrmicro2128
Friedrich C, Scott MG, Karunaratne N, Yan H, Hancock RE (1999) Salt-resistant alpha-helical cationic antimicrobial peptides. Antimicrob Agents Chemother 43(7):1542–1548
Frigimelica E, Bartolini E, Galli G, Grandi G, Grifantini R (2008) Identification of 2 hypothetical genes involved in Neisseria meningitidis cathelicidin resistance. J Infect Dis 197(8):1124–1132. doi:10.1086/533456
Froehlich BJ, Bates C, Scott JR (2009) Streptococcus pyogenes CovRS mediates growth in iron starvation and in the presence of the human cationic antimicrobial peptide LL-37. J Bacteriol 191(2):673–677. doi:JB.01256-08[pii]10.1128/JB.01256-08
Frosch M, Weisgerber C, Meyer TF (1989) Molecular characterization and expression in Escherichia coli of the gene complex encoding the polysaccharide capsule of Neisseria meningitidis group B. Proc Natl Acad Sci USA 86(5):1669–1673
Ganz T (2009) Iron in innate immunity: starve the invaders. Curr Opin Immunol 21(1):63–67. doi:S0952-7915(09)00012-0[pii]10.1016/j.coi.2009.01.011
Gao LY, Laval F, Lawson EH, Groger RK, Woodruff A, Morisaki JH, Cox JS, Daffe M, Brown EJ (2003) Requirement for kasB in Mycobacterium mycolic acid biosynthesis, cell wall impermeability and intracellular survival: implications for therapy. Mol Microbiol 49(6):1547–1563. doi:3667[pii]
Goytia M, Shafer WM (2010) Polyamines can increase resistance of Neisseria gonorrhoeae to mediators of the innate human host defense. Infect Immun 78(7):3187–3195. doi:IAI.01301-09[pii]10.1128/IAI.01301-09
Groisman EA (2001) The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol 183(6):1835–1842. doi:10.1128/JB.183.6.1835-1842.2001
Groisman EA, Chiao E, Lipps CJ, Heffron F (1989) Salmonella typhimurium phoP virulence gene is a transcriptional regulator. Proc Natl Acad Sci USA 86(18):7077–7081
Groisman EA, Parra-Lopez C, Salcedo M, Lipps CJ, Heffron F (1992) Resistance to host antimicrobial peptides is necessary for Salmonella virulence. Proc Natl Acad Sci USA 89(24):11939–11943
Guina T, Yi EC, Wang H, Hackett M, Miller SI (2000) A PhoP-regulated outer membrane protease of Salmonella enterica serovar typhimurium promotes resistance to alpha-helical antimicrobial peptides. J Bacteriol 182(14):4077–4086
Gunn JS (2008) The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol 16(6):284–290. doi:S0966-842X(08)00105-4[pii]10.1016/j.tim.2008.03.007
Gunn JS, Miller SI (1996) PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J Bacteriol 178(23):6857–6864
Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI (1998) PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27(6):1171–1182
Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI (1998) Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 95(2):189–198. doi:S0092-8674(00)81750-X[pii]
Hale JD, Hancock RE (2007) Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev Anti Infect Ther 5(6):951–959. doi:10.1586/14787210.5.6.951
Hancock RE (1997) Peptide antibiotics. Lancet 349(9049):418–422. doi:S0140-6736(97)80051-7[pii]10.1016/S0140-6736(97)80051-7
Hancock RE, McPhee JB (2005) Salmonella’s sensor for host defense molecules. Cell 122(3):320–322. doi:S0092-8674(05)00754-3[pii]10.1016/j.cell.2005.07.023
Helander IM, Kilpelainen I, Vaara M (1994) Increased substitution of phosphate groups in lipopolysaccharides and lipid A of the polymyxin-resistant pmrA mutants of Salmonella typhimurium: a 31P-NMR study. Mol Microbiol 11(3):481–487
Hiron A, Falord M, Valle J, Debarbouille M, Msadek T (2011) Bacitracin and nisin resistance in Staphylococcus aureus: a novel pathway involving the BraS/BraR two-component system (SA2417/SA2418) and both the BraD/BraE and VraD/VraE ABC transporters. Mol Microbiol. doi:10.1111/j.1365-2958.2011.07735.x
Huang HW (2006) Molecular mechanism of antimicrobial peptides: the origin of cooperativity. Biochim Biophys Acta 1758(9):1292–1302. doi:S0005-2736(06)00041-1[pii]10.1016/j.bbamem.2006.02.001
Husain F, Nikaido H (2010) Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol Microbiol 78(2):320–330. doi:10.1111/j.1365-2958.2010.07330.x
Institute Clinical and Laboratory Standard (2009) Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, vol M07-A8, 8th edn. Clinical and Laboratory Standard Institute, Wayne, PA
Islam D, Bandholtz L, Nilsson J, Wigzell H, Christensson B, Agerberth B, Gudmundsson G (2001) Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat Med 7(2):180–185. doi:10.1038/84627
Jerse AE, Sharma ND, Simms AN, Crow ET, Snyder LA, Shafer WM (2003) A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect Immun 71(10):5576–5582
Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A (2004) Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol 172(2):1169–1176
Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B (1998) Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J Biol Chem 273(6):3718–3724
Johansson L, Thulin P, Sendi P, Hertzen E, Linder A, Akesson P, Low DE, Agerberth B, Norrby-Teglund A (2008) Cathelicidin LL-37 in severe Streptococcus pyogenes soft tissue infections in humans. Infect Immun 76(8):3399–3404. doi:10.1128/IAI.01392-07
Johnson CR, Newcombe J, Thorne S, Borde HA, Eales-Reynolds LJ, Gorringe AR, Funnell SG, McFadden JJ (2001) Generation and characterization of a PhoP homologue mutant of Neisseria meningitidis. Mol Microbiol 39(5):1345–1355. doi:mmi2324[pii]
Jones A, Georg M, Maudsdotter L, Jonsson AB (2009) Endotoxin, capsule, and bacterial attachment contribute to Neisseria meningitidis resistance to the human antimicrobial peptide LL-37. J Bacteriol 191(12):3861–3868. doi:JB.01313-08[pii]10.1128/JB.01313-08
Kai-Larsen Y, Luthje P, Chromek M, Peters V, Wang X, Holm A, Kadas L, Hedlund KO, Johansson J, Chapman MR, Jacobson SH, Romling U, Agerberth B, Brauner A (2010) Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog 6(7):e1001010. doi:10.1371/journal.ppat.1001010
Karbarz MJ, Kalb SR, Cotter RJ, Raetz CR (2003) Expression cloning and biochemical characterization of a Rhizobium leguminosarum lipid A 1-phosphatase. J Biol Chem 278(41):39269–39279. doi:10.1074/jbc.M305830200M305830200[pii]
Katzif S, Danavall D, Bowers S, Balthazar JT, Shafer WM (2003) The major cold shock gene, cspA, is involved in the susceptibility of Staphylococcus aureus to an antimicrobial peptide of human cathepsin G. Infect Immun 71(8):4304–4312
Khuller GK, Subrahmanyam D (1970) On the ornithinyl ester of phosphatidylglycerol of Mycobacterium 607. J Bacteriol 101(2):654–656
Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK (2007) Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298(15):1763–1771. doi:10.1001/jama.298.15.1763
Klotman ME, Rapista A, Teleshova N, Micsenyi A, Jarvis GA, Lu W, Porter E, Chang TL (2008) Neisseria gonorrhoeae-induced human defensins 5 and 6 increase HIV infectivity: role in enhanced transmission. J Immunol 180(9):6176–6185. doi:180/9/6176[pii]
Kocianova S, Vuong C, Yao Y, Voyich JM, Fischer ER, DeLeo FR, Otto M (2005) Key role of poly-gamma-DL-glutamic acid in immune evasion and virulence of Staphylococcus epidermidis. J Clin Invest 115(3):688–694. doi:10.1172/JCI23523
Koo SP, Bayer AS, Yeaman MR (2001) Diversity in antistaphylococcal mechanisms among membrane-targeting antimicrobial peptides. Infect Immun 69(8):4916–4922. doi:10.1128/IAI.69.8.4916-4922.2001
Koprivnjak T, Peschel A, Gelb MH, Liang NS, Weiss JP (2002) Role of charge properties of bacterial envelope in bactericidal action of human group IIA phospholipase A2 against Staphylococcus aureus. J Biol Chem 277(49):47636–47644. doi:10.1074/jbc.M205104200M205104200[pii]
Kowalski TJ, Berbari EF, Osmon DR (2005) Epidemiology, treatment, and prevention of community-acquired methicillin-resistant Staphylococcus aureus infections. Mayo Clin Proc 80(9):1201–1207, quiz 1208
Kraus D, Herbert S, Kristian SA, Khosravi A, Nizet V, Gotz F, Peschel A (2008) The GraRS regulatory system controls Staphylococcus aureus susceptibility to antimicrobial host defenses. BMC Microbiol 8:85. doi:1471-2180-8-85[pii]10.1186/1471-2180-8-85
Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, Gallo RL, Nizet V (2005) D-alanylation of teichoic acids promotes group a Streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol 187(19):6719–6725. doi:187/19/6719[pii]10.1128/JB.187.19.6719-6725.2005
Kupferwasser LI, Skurray RA, Brown MH, Firth N, Yeaman MR, Bayer AS (1999) Plasmid-mediated resistance to thrombin-induced platelet microbicidal protein in staphylococci: role of the qacA locus. Antimicrob Agents Chemother 43(10):2395–2399
Kuroda M, Kuwahara-Arai K, Hiramatsu K (2000) Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem Biophys Res Commun 269(2):485–490. doi:10.1006/bbrc.2000.2277
Laarman AJ, Ruyken M, Malone CL, van Strijp JA, Horswill AR, Rooijakkers SH (2011) Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J Immunol 186(11):6445–6453. doi:jimmunol.1002948[pii]10.4049/jimmunol.1002948
Lai Y, Villaruz AE, Li M, Cha DJ, Sturdevant DE, Otto M (2007) The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Mol Microbiol 63(2):497–506. doi:MMI5540[pii]10.1111/j.1365-2958.2006.05540.x
Lauth X, von Kockritz-Blickwede M, McNamara CW, Myskowski S, Zinkernagel AS, Beall B, Ghosh P, Gallo RL, Nizet V (2009) M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J Innate Immun 1(3):202–214. doi:10.1159/000203645
Lee H, Hsu FF, Turk J, Groisman EA (2004) The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J Bacteriol 186(13):4124–4133. doi:10.1128/JB.186.13.4124-4133.2004186/13/4124[pii]
Lehrer RI, Ganz T, Selsted ME (1988) Oxygen-independent bactericidal systems—mechanisms and disorders. Hematol Oncol Clin North Am 2(1):159–169
Leid JG, Shirtliff ME, Costerton JW, Stoodley P (2002) Human leukocytes adhere to, penetrate, and respond to Staphylococcus aureus biofilms. Infect Immun 70(11):6339–6345
Lewenza S, Falsafi RK, Winsor G, Gooderham WJ, McPhee JB, Brinkman FS, Hancock RE (2005) Construction of a mini-Tn5-luxCDABE mutant library in Pseudomonas aeruginosa PAO1: a tool for identifying differentially regulated genes. Genome Res 15(4):583–589. doi:15/4/583[pii]10.1101/gr.3513905
Lewis LA, Choudhury B, Balthazar JT, Martin LE, Ram S, Rice PA, Stephens DS, Carlson R, Shafer WM (2009) Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect Immun 77(3):1112–1120. doi:IAI.01280-08[pii]10.1128/IAI.01280-08
Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M (2007a) The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol Microbiol 66(5):1136–1147. doi:MMI5986[pii]10.1111/j.1365-2958.2007.05986.x
Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M (2007b) Gram-positive three-component antimicrobial peptide-sensing system. Proc Natl Acad Sci USA 104(22):9469–9474. doi:0702159104[pii]10.1073/pnas.0702159104
Lin J, Wang Y, Hoang KV (2009) Systematic identification of genetic loci required for polymyxin resistance in Campylobacter jejuni using an efficient in vivo transposon mutagenesis system. Foodborne Pathog Dis 6(2):173–185. doi:10.1089/fpd.2008.0177
Llobet E, Tomas JM, Bengoechea JA (2008) Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 154(Pt 12):3877–3886. doi:154/12/3877[pii]10.1099/mic.0.2008/022301-0
Lomovskaya O, Bostian KA (2006) Practical applications and feasibility of efflux pump inhibitors in the clinic–a vision for applied use. Biochem Pharmacol 71(7):910–918. doi:S0006-2952(05)00815-4[pii]10.1016/j.bcp. 2005.12.008
Lysenko ES, Gould J, Bals R, Wilson JM, Weiser JN (2000) Bacterial phosphorylcholine decreases susceptibility to the antimicrobial peptide LL-37/hCAP18 expressed in the upper respiratory tract. Infect Immun 68(3):1664–1671
Macfarlane EL, Kwasnicka A, Ochs MM, Hancock RE (1999) PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol Microbiol 34(2):305–316. doi:mmi1600[pii]
Macfarlane EL, Kwasnicka A, Hancock RE (2000) Role of Pseudomonas aeruginosa PhoP-PhoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 146(Pt 10):2543–2554
Madan R, Rastogi R, Parashuraman S, Mukhopadhyay A (2012) Salmonella acquires lysosome-associated membrane protein 1 (LAMP1) on phagosomes from golgi via SipC protein-mediated recruitment of host Syntaxin6. J Biol Chem 287(8):5574–5587. doi:10.1074/jbc.M111.286120
Mah TF, Pitts B, Pellock B, Walker GC, Stewart PS, O’Toole GA (2003) A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426(6964):306–310. doi:10.1038/nature02122nature02122[pii]
Maloney E, Stankowska D, Zhang J, Fol M, Cheng QJ, Lun S, Bishai WR, Rajagopalan M, Chatterjee D, Madiraju MV (2009) The two-domain LysX protein of Mycobacterium tuberculosis is required for production of lysinylated phosphatidylglycerol and resistance to cationic antimicrobial peptides. PLoS Pathog 5(7):e1000534. doi:10.1371/journal.ppat.1000534
Mandin P, Fsihi H, Dussurget O, Vergassola M, Milohanic E, Toledo-Arana A, Lasa I, Johansson J, Cossart P (2005) VirR, a response regulator critical for Listeria monocytogenes virulence. Mol Microbiol 57(5):1367–1380. doi:MMI4776[pii]10.1111/j.1365-2958.2005.04776.x
Marra MN, Wilde CG, Griffith JE, Snable JL, Scott RW (1990) Bactericidal/permeability-increasing protein has endotoxin-neutralizing activity. J Immunol 144(2):662–666
Mason KM, Munson RS Jr, Bakaletz LO (2005) A mutation in the sap operon attenuates survival of nontypeable Haemophilus influenzae in a chinchilla model of otitis media. Infect Immun 73(1):599–608. doi:73/1/599[pii]10.1128/IAI.73.1.599-608.2005
Mason KM, Bruggeman ME, Munson RS, Bakaletz LO (2006) The non-typeable Haemophilus influenzae Sap transporter provides a mechanism of antimicrobial peptide resistance and SapD-dependent potassium acquisition. Mol Microbiol 62(5):1357–1372. doi:MMI5460[pii]10.1111/j.1365-2958.2006.05460.x
Mason KM, Raffel FK, Ray WC, Bakaletz LO (2011) Heme utilization by nontypeable Haemophilus influenzae is essential and dependent on Sap transporter function. J Bacteriol 193(10):2527–2535. doi:JB.01313-10[pii]10.1128/JB.01313-10
Matson JS, Yoo HJ, Hakansson K, Dirita VJ (2010) Polymyxin B resistance in El Tor Vibrio cholerae requires lipid acylation catalyzed by MsbB. J Bacteriol 192(8):2044–2052. doi:JB.00023-10[pii]10.1128/JB.00023-10
McCoy AJ, Liu H, Falla TJ, Gunn JS (2001) Identification of Proteus mirabilis mutants with increased sensitivity to antimicrobial peptides. Antimicrob Agents Chemother 45(7):2030–2037. doi:10.1128/AAC.45.7.2030-2037.2001
McNabb SJ, Jajosky RA, Hall-Baker PA, Adams DA, Sharp P, Worshams C, Anderson WJ, Javier AJ, Jones GJ, Nitschke DA, Rey A, Wodajo MS (2008) Summary of notifiable diseases–United States, 2006. MMWR Morb Mortal Wkly Rep 55(53):1–92. doi:mm5553a1[pii]
McPhee JB, Lewenza S, Hancock RE (2003) Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol Microbiol 50(1):205–217. doi:3673[pii]
Messner P, Steiner K, Zarschler K, Schaffer C (2008) S-layer nanoglycobiology of bacteria. Carbohydr Res 343(12):1934–1951. doi:S0008-6215(08)00004-9[pii]10.1016/j.carres.2007.12.025
Miller SI, Kukral AM, Mekalanos JJ (1989) A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc Natl Acad Sci USA 86(13):5054–5058
Miller SI, Pulkkinen WS, Selsted ME, Mekalanos JJ (1990) Characterization of defensin resistance phenotypes associated with mutations in the phoP virulence regulon of Salmonella typhimurium. Infect Immun 58(11):3706–3710
Mishra NN, Liu GY, Yeaman MR, Nast CC, Proctor RA, McKinnell J, Bayer AS (2011) Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob Agents Chemother 55(2):526–531. doi:AAC.00680-10[pii]10.1128/AAC.00680-10
Moranta D, Regueiro V, March C, Llobet E, Margareto J, Larrarte E, Garmendia J, Bengoechea JA (2010) Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells. Infect Immun 78(3):1135–1146. doi:IAI.00940-09[pii]10.1128/IAI.00940-09
Moskowitz SM, Ernst RK, Miller SI (2004) PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J Bacteriol 186(2):575–579
Mount KL, Townsend CA, Rinker SD, Gu X, Fortney KR, Zwickl BW, Janowicz DM, Spinola SM, Katz BP, Bauer ME (2010) Haemophilus ducreyi SapA contributes to cathelicidin resistance and virulence in humans. Infect Immun 78(3):1176–1184. doi:IAI.01014-09[pii]10.1128/IAI.01014-09
Mulcahy H, Charron-Mazenod L, Lewenza S (2008) Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog 4(11):e1000213. doi:10.1371/journal.ppat.1000213
Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A (2006) Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443(7108):173–179. doi:nature05076[pii]10.1038/nature05076
Nahaie MR, Goodfellow M, Minnikin DE, Hajek V (1984) Polar lipid and isoprenoid quinone composition in the classification of Staphylococcus. J Gen Microbiol 130(9):2427–2437
Navarre WW, Halsey TA, Walthers D, Frye J, McClelland M, Potter JL, Kenney LJ, Gunn JS, Fang FC, Libby SJ (2005) Co-regulation of Salmonella enterica genes required for virulence and resistance to antimicrobial peptides by SlyA and PhoP/PhoQ. Mol Microbiol 56(2):492–508. doi:MMI4553[pii]10.1111/j.1365-2958.2005.04553.x
Neuhaus FC, Baddiley J (2003) A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev 67(4):686–723
Newcombe J, Eales-Reynolds LJ, Wootton L, Gorringe AR, Funnell SG, Taylor SC, McFadden JJ (2004) Infection with an avirulent phoP mutant of Neisseria meningitidis confers broad cross-reactive immunity. Infect Immun 72(1):338–344
Nishi H, Komatsuzawa H, Fujiwara T, McCallum N, Sugai M (2004) Reduced content of lysyl-phosphatidylglycerol in the cytoplasmic membrane affects susceptibility to moenomycin, as well as vancomycin, gentamicin, and antimicrobial peptides, in Staphylococcus aureus. Antimicrob Agents Chemother 48(12):4800–4807. doi:48/12/4800[pii]10.1128/AAC.48.12.4800-4807.2004
Nizet V, Gallo RL (2002) Surviving innate immunity. Trends Microbiol 10(8):358–359
Nuding S, Zabel LT, Enders C, Porter E, Fellermann K, Wehkamp J, Mueller HA, Stange EF (2009) Antibacterial activity of human defensins on anaerobic intestinal bacterial species: a major role of HBD-3. Microbes Infect 11(3):384–393. doi:S1286-4579(09)00004-5[pii]10.1016/j.micinf.2009.01.001
Nyberg P, Rasmussen M, Bjorck L (2004) Alpha2-macroglobulin-proteinase complexes protect Streptococcus pyogenes from killing by the antimicrobial peptide LL-37. J Biol Chem 279(51):52820–52823. doi:C400485200[pii]10.1074/jbc.C400485200
Ohnishi M, Golparian D, Shimuta K, Saika T, Hoshina S, Iwasaku K, Nakayama S, Kitawaki J, Unemo M (2011) Is Neisseria gonorrhoeae Initiating a future era of untreatable gonorrhea?: detailed characterization of the first strain with high-level resistance to ceftriaxone. Antimicrob Agents Chemother 55(7):3538–3545. doi:AAC.00325-11[pii]10.1128/AAC.00325-11
Otto M (2009) Bacterial sensing of antimicrobial peptides. Contrib Microbiol 16:136–149. doi:000219377[pii]10.1159/000219377
Otto M, Peschel A, Gotz F (1998) Producer self-protection against the lantibiotic epidermin by the ABC transporter EpiFEG of Staphylococcus epidermidis Tu3298. FEMS Microbiol Lett 166(2):203–211. doi:S0378109798003334[pii]
Ouyang J, Tian XL, Versey J, Wishart A, Li YH (2010) The BceABRS four-component system regulates the bacitracin-induced cell envelope stress response in Streptococcus mutans. Antimicrob Agents Chemother 54(9):3895–3906. doi:AAC.01802-09[pii]10.1128/AAC.01802-09
Overhage J, Campisano A, Bains M, Torfs EC, Rehm BH, Hancock RE (2008) Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun 76(9):4176–4182. doi:10.1128/IAI.00318-08IAI.00318-08[pii]
Padilla E, Llobet E, Domenech-Sanchez A, Martinez-Martinez L, Bengoechea JA, Alberti S (2010) Klebsiella pneumoniae AcrAB efflux pump contributes to antimicrobial resistance and virulence. Antimicrob Agents Chemother 54(1):177–183. doi:AAC.00715-09[pii]10.1128/AAC.00715-09
Park PW, Pier GB, Hinkes MT, Bernfield M (2001) Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 411(6833):98–102. doi:10.1038/3507510035075100[pii]
Parra-Lopez C, Baer MT, Groisman EA (1993) Molecular genetic analysis of a locus required for resistance to antimicrobial peptides in Salmonella typhimurium. EMBO J 12(11):4053–4062
Pence MA, Rooijakkers SH, Cogen AL, Cole JN, Hollands A, Gallo RL, Nizet V (2010) Streptococcal inhibitor of complement promotes innate immune resistance phenotypes of invasive M1T1 group A Streptococcus. J Innate Immun 2(6):587–595. doi:000317672[pii]10.1159/000317672
Peschel A (2002) How do bacteria resist human antimicrobial peptides? Trends Microbiol 10(4):179–186. doi:S0966842X02023338[pii]
Peschel A, Sahl HG (2006) The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol 4(7):529–536. doi:nrmicro1441[pii]10.1038/nrmicro1441
Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274(13):8405–8410
Peschel A, Vuong C, Otto M, Gotz F (2000) The D-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob Agents Chemother 44(10):2845–2847
Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA (2001) Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with L-lysine. J Exp Med 193(9):1067–1076
Piddock LJ (2006) Multidrug-resistance efflux pumps—not just for resistance. Nat Rev Microbiol 4(8):629–636. doi:nrmicro1464[pii]10.1038/nrmicro1464
Prost LR, Miller SI (2008) The Salmonellae PhoQ sensor: mechanisms of detection of phagosome signals. Cell Microbiol 10(3):576–582. doi:CMI1111[pii]10.1111/j.1462-5822.2007.01111.x
Qu X-D, Harwig SSL, Oren A, Shafer WM, Lehrer RI (1996) Susceptibility of Neisseria gonorrhoeae to protegrins. Infect Immun 64(4):1240–1245
Ray K, Marteyn B, Sansonetti PJ, Tang CM (2009) Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7(5):333–340. doi:10.1038/nrmicro2112
Rest RF, Cooney MH, Spitznagel JK (1977) Susceptibility of lipopolysaccharide mutants to the bactericidal action of human neutrophil lysosomal fractions. Infect Immun 16(1):145–151
Rinker SD, Trombley MP, Gu X, Fortney KR, Bauer ME (2011) Deletion of mtrC in Haemophilus ducreyi increases sensitivity to human antimicrobial peptides and activates the CpxRA regulon. Infect Immun 79(6):2324–2334. doi:IAI.01316-10[pii]10.1128/IAI.01316-10
Roland KL, Martin LE, Esther CR, Spitznagel JK (1993) Spontaneous pmrA mutants of Salmonella typhimurium LT2 define a new two-component regulatory system with a possible role in virulence. J Bacteriol 175(13):4154–4164
Roland KL, Esther CR, Spitznagel JK (1994) Isolation and characterization of a gene, pmrD, from Salmonella typhimurium that confers resistance to polymyxin when expressed in multiple copies. J Bacteriol 176(12):3589–3597
Rouquette C, Harmon JB, Shafer WM (1999) Induction of the mtrCDE-encoded efflux pump system of Neisseria gonorrhoeae requires MtrA, an AraC-like protein. Mol Microbiol 33(3):651–658. doi:mole1517[pii]
Roy H, Ibba M (2008) RNA-dependent lipid remodeling by bacterial multiple peptide resistance factors. Proc Natl Acad Sci USA 105(12):4667–4672. doi:0800006105[pii]10.1073/pnas.0800006105
Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL (2003) Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 422(6931):522–526. doi:10.1038/nature01520nature01520[pii]
Schmidtchen A, Frick IM, Bjorck L (2001) Dermatan sulphate is released by proteinases of common pathogenic bacteria and inactivates antibacterial alpha-defensin. Mol Microbiol 39(3):708–713. doi:mmi2251[pii]
Schmidtchen A, Frick IM, Andersson E, Tapper H, Bjorck L (2002) Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol 46(1):157–168
Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, Buchner J, Schaller M, Stange EF, Wehkamp J (2011) Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 469(7330):419–423. doi:nature09674[pii]10.1038/nature09674
Scott CC, Botelho RJ, Grinstein S (2003) Phagosome maturation: a few bugs in the system. J Membr Biol 193(3):137–152. doi:10.1007/s00232-002-2008-2
Seib KL, Serruto D, Oriente F, Delany I, Adu-Bobie J, Veggi D, Arico B, Rappuoli R, Pizza M (2009) Factor H-binding protein is important for meningococcal survival in human whole blood and serum and in the presence of the antimicrobial peptide LL-37. Infect Immun 77(1):292–299. doi:IAI.01071-08[pii]10.1128/IAI.01071-08
Shafer WM, Onunka VC (1989) Mechanism of staphylococcal resistance to non-oxidative antimicrobial action of neutrophils: importance of pH and ionic strength in determining the bactericidal action of cathepsin G. J Gen Microbiol 135(4):825–830
Shafer WM, Casey SG, Spitznagel JK (1984) Lipid A and resistance of Salmonella typhimurium to antimicrobial granule proteins of human neutrophil granulocytes. Infect Immun 43(3):834–838
Shafer WM, Qu X, Waring AJ, Lehrer RI (1998) Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci USA 95(4):1829–1833
Shafer WM, Folster JP, Nicholas RA (2010) Molecular mechanisms of antibiotic resistance expressed by the pathogenic Neisseria. In: Genco CA, Wetzler L (eds) Neisseria. Molecular mechanisms of pathogenesis. Caister Academic, Norfolk, pp 245–268
Shelton CL, Raffel FK, Beatty WL, Johnson SM, Mason KM (2011) Sap transporter mediated import and subsequent degradation of antimicrobial peptides in Haemophilus. PLoS Pathog 7(11):e1002360. doi:10.1371/journal.ppat.1002360
Shprung T, Peleg A, Rosenfeld Y, Trieu-Cuot P, Shai Y (2012) Effect of PhoP-PhoQ activation by broad repertoire of antimicrobial peptides on bacterial resistance. J Biol Chem 287(7):4544–4551. doi:10.1074/jbc.M111.278523
Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J (2004) Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother 48(12):4673–4679. doi:48/12/4673[pii]10.1128/AAC.48.12.4673-4679.2004
Silhavy TJ, Kahne D, Walker S (2010) The bacterial cell envelope. Cold Spring Harb Perspect Biol 2(5):a000414. doi:cshperspect.a000414[pii]10.1101/cshperspect.a000414
Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP (2000) Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407(6805):762–764. doi:10.1038/35037627
Slayden RA, Barry CE 3rd (2002) The role of KasA and KasB in the biosynthesis of meromycolic acids and isoniazid resistance in Mycobacterium tuberculosis. Tuberculosis (Edinb) 82(4–5):149–160. doi:S1472979202903331[pii]
Sochacki KA, Barns KJ, Bucki R, Weisshaar JC (2011) Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc Natl Acad Sci USA 108(16):E77–E81. doi:10.1073/pnas.1101130108
Sorrell TC, Lehrer RI, Cline MJ (1978) Mechanism of nonspecific macrophage-mediated cytotoxicity: evidence for lack of dependence upon oxygen. J Immunol 120(2):347–352
Spinola SM, Bong CT, Faber AL, Fortney KR, Bennett SL, Townsend CA, Zwickl BE, Billings SD, Humphreys TL, Bauer ME, Katz BP (2003) Differences in host susceptibility to disease progression in the human challenge model of Haemophilus ducreyi infection. Infect Immun 71(11):6658–6663
Spinosa MR, Progida C, Tala A, Cogli L, Alifano P, Bucci C (2007) The Neisseria meningitidis capsule is important for intracellular survival in human cells. Infect Immun 75(7):3594–3603. doi:IAI.01945-06[pii]10.1128/IAI.01945-06
Spitznagel JK (1961) The effects of mammalian and other cationic polypeptides on the cytochemical character of bacterial cells. J Exp Med 114:1063–1078
Spitznagel JK (1990) Antibiotic proteins of human neutrophils. J Clin Invest 86(5):1381–1386. doi:10.1172/JCI114851
Spitznagel JK, Shafer WM (1985) Neutrophil killing of bacteria by oxygen-independent mechanisms: a historical summary. Rev Infect Dis 7(3):398–403
Starner TD, Swords WE, Apicella MA, McCray PB Jr (2002) Susceptibility of nontypeable Haemophilus influenzae to human beta-defensins is influenced by lipooligosaccharide acylation. Infect Immun 70(9):5287–5289
Staubitz P, Neumann H, Schneider T, Wiedemann I, Peschel A (2004) MprF-mediated biosynthesis of lysylphosphatidylglycerol, an important determinant in staphylococcal defensin resistance. FEMS Microbiol Lett 231(1):67–71. doi:10.1016/S0378-1097(03)00921-2S0378109703009212[pii]
Stephens DS (2009) Biology and pathogenesis of the evolutionarily successful, obligate human bacterium Neisseria meningitidis. Vaccine 27(Suppl 2):B71–B77. doi:S0264-410X(09)00635-5[pii]10.1016/j.vaccine.2009.04.070
Stewart PS, Costerton JW (2001) Antibiotic resistance of bacteria in biofilms. Lancet 358(9276):135–138. doi:S0140673601053211[pii]
Stumpe S, Schmid R, Stephens DL, Georgiou G, Bakker EP (1998) Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli. J Bacteriol 180(15):4002–4006
Symmons MF, Bokma E, Koronakis E, Hughes C, Koronakis V (2009) The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci USA 106(17):7173–7178. doi:0900693106[pii]10.1073/pnas.0900693106
Tada H, Sugawara S, Nemoto E, Takahashi N, Imamura T, Potempa J, Travis J, Shimauchi H, Takada H (2002) Proteolysis of CD14 on human gingival fibroblasts by arginine-specific cysteine proteinases from Porphyromonas gingivalis leading to down-regulation of lipopolysaccharide-induced interleukin-8 production. Infect Immun 70(6):3304–3307
Trent MS (2004) Biosynthesis, transport, and modification of lipid A. Biochem Cell Biol 82(1):71–86. doi:10.1139/o03-070o03-070[pii]
Tzeng YL, Datta A, Ambrose K, Lo M, Davies JK, Carlson RW, Stephens DS, Kahler CM (2004) The MisR/MisS two-component regulatory system influences inner core structure and immunotype of lipooligosaccharide in Neisseria meningitidis. J Biol Chem 279(33):35053–35062. doi:10.1074/jbc.M401433200M401433200[pii]
Tzeng YL, Ambrose KD, Zughaier S, Zhou X, Miller YK, Shafer WM, Stephens DS (2005) Cationic antimicrobial peptide resistance in Neisseria meningitidis. J Bacteriol 187(15):5387–5396. doi:187/15/5387-a[pii]10.1128/JB.187.15.5387-5396.2005
Uchiya K, Barbieri MA, Funato K, Shah AH, Stahl PD, Groisman EA (1999) A Salmonella virulence protein that inhibits cellular trafficking. EMBO J 18(14):3924–3933. doi:10.1093/emboj/18.14.3924
Ulvatne H, Haukland HH, Samuelsen O, Kramer M, Vorland LH (2002) Proteases in Escherichia coli and Staphylococcus aureus confer reduced susceptibility to lactoferricin B. J antimicrob chemother 50(4):461–467
Vaara M, Vaara T, Jensen M, Helander I, Nurminen M, Rietschel ET, Makela PH (1981) Characterization of the lipopolysaccharide from the polymyxin-resistant pmrA mutants of Salmonella typhimurium. FEBS Lett 129(1):145–149
Valdivia RH, Falkow S (1997) Fluorescence-based isolation of bacterial genes expressed within host cells. Science 277(5334):2007–2011
Vesga O, Groeschel MC, Otten MF, Brar DW, Vann JM, Proctor RA (1996) Staphylococcus aureus small colony variants are induced by the endothelial cell intracellular milieu. J Infect Dis 173(3):739–742
Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M (2004) A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem 279(52):54881–54886. doi:M411374200[pii]10.1074/jbc.M411374200
Wade D, Boman A, Wahlin B, Drain CM, Andreu D, Boman HG, Merrifield RB (1990) All-D amino acid-containing channel-forming antibiotic peptides. Proc Natl Acad Sci USA 87(12):4761–4765
Warner DM, Folster JP, Shafer WM, Jerse AE (2007) Regulation of the MtrC-MtrD-MtrE efflux-pump system modulates the in vivo fitness of Neisseria gonorrhoeae. J Infect Dis 196(12):1804–1812. doi:10.1086/522964
Warner DM, Shafer WM, Jerse AE (2008) Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE Efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol Microbiol 70(2):462–478. doi:MMI6424[pii]10.1111/j.1365-2958.2008.06424.x
Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H Jr, Fellermann K, Ganz T, Stange EF, Bevins CL (2005) Reduced paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA 102(50):18129–18134. doi:0505256102[pii]10.1073/pnas.0505256102
Yeaman MR, Yount NY (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55(1):27–55. doi:10.1124/pr.55.1.2
Zähner D, Zhou X, Chancey ST, Pohl J, Shafer WM, Stephens DS (2010) Human antimicrobial peptide LL-37 induces MefE/Mel-mediated macrolide resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother 54(8):3516–3519. doi:10.1128/AAC.01756-09
Zeya HI, Spitznagel JK (1966a) Cationic proteins of polymorphonuclear leukocyte lysosomes. I. Resolution of antibacterial and enzymatic activities. J Bacteriol 91(2):750–754
Zeya HI, Spitznagel JK (1966b) Cationic proteins of polymorphonuclear leukocyte lysosomes. II. Composition, properties, and mechanism of antibacterial action. J Bacteriol 91(2):755–762
Zipfel PF, Reuter M (2009) Complement activation products C3a and C4a as endogenous antimicrobial peptides. Int J Pept Res Th 15(2):87–95. doi:10.1007/s10989-009-9180-5
Zughaier SM, Svoboda P, Pohl J, Stephens DS, Shafer WM (2010) The human host defense peptide LL-37 interacts with Neisseria meningitidis capsular polysaccharides and inhibits inflammatory mediators release. PLoS One 5(10):e13627. doi:10.1371/journal.pone.0013627
Acknowledgments
We wish to thank Lane Pucko for help in manuscript preparation and P. Rather for critical review. The senior author (W. M. S.) is indebted to J.K. Spitznagel, M.D., for his mentorship and introducing him to CAMP research 30 years ago. This work was supported by the NIH grants R37 AI021150 and U19 AI031496 and a VA Merit Award from the Department of Veterans Affairs. W. M. S. was supported in part by a Senior Research Career Scientist Award from the Department of Veterans Affairs.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Basel AG
About this chapter
Cite this chapter
Goytia, M., Kandler, J.L., Shafer, W.M. (2013). Mechanisms and Significance of Bacterial Resistance to Human Cationic Antimicrobial Peptides. In: Hiemstra, P., Zaat, S. (eds) Antimicrobial Peptides and Innate Immunity. Progress in Inflammation Research. Springer, Basel. https://doi.org/10.1007/978-3-0348-0541-4_9
Download citation
DOI: https://doi.org/10.1007/978-3-0348-0541-4_9
Published:
Publisher Name: Springer, Basel
Print ISBN: 978-3-0348-0540-7
Online ISBN: 978-3-0348-0541-4
eBook Packages: MedicineMedicine (R0)