
Abstract
von Willebrand disease (VWD) is the most common congenital bleeding disorder with a potential incidence of 1% of the general population. The disorder is classified into three main types: type 1 and type 3 as (respectively, partial and complete) quantitative deficiency of von Willebrand factor (VWF) and type 2 (with qualitative VWF defects). The bleeding tendency is highly variable, ranging from largely asymptomatic, mainly in mild type 1 VWD, to severe life-threatening hemorrhage, notably type 3 VWD. Diagnosis of VWD requires comprehensive laboratory testing aligned to appropriate clinical assessment. On-demand therapy is the mainstay of treatment in VWD, although long-term prophylaxis is emerging for those with recurrent and severe bleeding. Therapeutic choices in VWD depend to some extent on geographical location, but include VWF/FVIII concentrates, recombinant VWF, and desmopressin.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
von Willebrand disease (VWD) is reportedly the most common congenital bleeding disorder and may be inherited in an autosomal dominant or autosomal recessive manner [1]. The disorder is classified into six different types, with quantitative deficiency of von Willebrand factor (VWF) identified in type 1 (partial deficiency) and type 3 (complete deficiency), or qualitative defects in VWF identified in type 2 VWD, which comprises four separate (sub)types [2]. VWD may present as a heterogeneous bleeding disorder with variable bleeding tendency, depending on the VWD type, and the associated VWF deficiency/defect. In some patients, the bleeding symptoms may be so mild as to not easily distinguish these potential sufferers from unaffected individuals. However, in severely affected VWD individuals, life endangering bleeding (e.g., in central nervous system (CNS)) may occur. Mucocutaneous bleeding, including epistaxis and menorrhagia, are more typical presentations of the disorder, but other rare presentations also can be observed [1, 2]. As per the ISTH SSC (International Society on Thrombosis and Hemostasis Scientific Standardization Committee) classification of the disorder, and newer guidelines, affected individuals are characterized according to clinical manifestations and laboratory findings [3,4,5]. The severity of VWD is partially associated to the level and activity of the VWF. Although there are no standardized recommendations, as a practical guide, VWD can be considered “mild” when VWF activity levels are in the range 30–50 U/dL and/or factor VIII (FVIII) coagulant activity (FVIII:C): 40–60 U/dL, “moderate” when VWF activity levels are in the range 10–30 U/dL and/or FVIII:C 20–40 U dL–1, or “severe” when VWF activity levels are <10 U/dL and/or FVIII:C < 20 U/dL [5]. Based on such an approach, type 3 VWD, as well as some cases of type 1, type 2A, type 2M, and type 2N VWD, could be classified as severe forms of VWD [5].
Diagnosis of VWD is a global challenge, since a panel of accurate laboratory tests are required to align to a comprehensive and appropriate clinical assessment, and this approach is not available everywhere [5]. Significant progress has occurred in the laboratory diagnosis of VWD in recent years, but accurate diagnosis remains challenging for many health workers. The difficulty in diagnosis is due to several issues; these include (i) the lack of any definitive cut-offs to determine normal vs abnormal levels of VWF, thereby potentially identifying normal individuals vs VWD; (ii) the effect of different molecular changes in the VWF gene on VWF level and activity; (iii) other genetic modifiers and physiological factors that can reduce or increase plasma levels of VWF (e.g., blood group); (iv) the overlap of bleeding symptomology in normal individuals vs those with VWD; and (v) difficulties with performance and interpretation of laboratory tests [6, 7]. On-demand therapy is the current mainstay of treatment for people with VWD; however, long-term prophylaxis can significantly improve the quality of life in patients with severe bleeding. Desmopressin (DDAVP) is considered the main therapeutic option for patients with type 1 and a subset of patients with type 2 VWD with minor bleeding or undergoing minor surgical procedures. However, replacement therapy with VWF/FVIII concentrates represents the main therapeutic option in type 3 and most patients with type 2 VWD, as well as those responsive to desmopressin but with long-term treatment needs or undergoing major surgical procedures [7,8,9,10].
2 von Willebrand Factor Synthesis, Structure, and Function
These matters were comprehensively reviewed in the original chapter on VWD in the first edition of this book, and so will only be briefly summarized here [1].
2.1 von Willebrand Factor Biosynthesis
The biosynthesis of VWF comprises a series of sequential steps that ultimately permit incorporation of the protein in various cellular storage organelles. These steps include protein production, removal of a signal peptide, tail to tail dimerization, heat to head multimerization, N-linked glycosylation, maturation of N-glycan, O-linked glycosylation, formation of tubules, and incorporation to storage organelles [11].
2.2 von Willebrand Factor Structure
VWF is mapped to the tip of the short arm of chromosome 12. The VWF gene spans 178-kb and consists of 52 exons [12], reflecting a unique gene structure consisting of different repeated sequences [13]. VWF is a multimeric protein with unit molecular mass of 350-kDa, as coded by 9 kb mRNA and consisting of 2813 amino acids, including a 741 amino acid pro-peptide and a 2050 amino acid mature polypeptide [14]. VWF is synthesized as a precursor and processed in endoplasmic reticulum (ER) and Golgi apparatus in endothelial cells. After processing, the protein undergoes dimerization in the ER by disulfide bridging and cleavage into two components, the mature protein and a 97-kDa a pro-peptide. The pro-peptide is secreted and circulates independently of mature VWF [11].
VWF comprises several distinct domains, with some domains containing functional binding sites for FVIII, platelet receptors and for collagen. Defects in VWF may affect different domains, and thus lead to different qualitative forms of VWD. In brief, FVIII binds to the D-domain, and defects in this domain can lead to FVIII binding defects, or namely type 2N VWD. Binding to the main VWF receptor on platelets, glycoprotein Ib (GPIb), occurs in the A1 domain, and so defects in this domain can lead to GPIb binding defects, as captured within 2A, 2B, and 2M VWD. Binding to collagen, a protein housed within the subendothelial matrix, occurs within the A1 and A3 domains, and so defects in these domains can lead to collagen binding defects, as also captured within 2A, 2B, and 2M VWD [1].
2.3 Disulfide Bridging and Multimerization of von Willebrand Factor
About 8.3% of VWF is composed of the amino acid cysteine (234 of the 2813 residues), which is some fourfold higher than most other human proteins. In contrast to other domains, the triplicated domain A, where most of the adhesive function of VWF resides, has only six cysteine residues. The cysteine residues are otherwise mostly paired in disulfide bonds in the secreted protein; however, several unpaired cysteines remain and are essential for proper folding and secretion of VWF [15]. In ER, the subunits of pro-VWF undergo dimerization by disulfide bonds in C-terminal cysteine knot (CK) domains. This tail-to-tail dimerization needs only the sequence of the last 150 residues. Tail to tail pro-VWF is transported to Golgi and forms head-to-head dimerization by further disulfide bonding in the D3 domain. The VWF pro-peptide (domains D1 and D2) and also D’ are important in multimerization. After multimerization, the multimers are organized into a helical structure that permits the compaction of the protein, and its storage in the Weibel-Palade bodies of endothelial cells [11].
Regulation of the multimeric size of VWF is primarily mediated by the metalloproteinase ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), which cleaves VWF at a single site (Tyr1605-Met1606) in the A2 domain [16, 17]. It appears that domain D4, at residues from 1874 to 2813, has a role in binding to ADAMTS13 and this acts as the initial step for proteolysis of VWF by ADAMTS13 [18].
2.4 Post translational Modifications of von Willebrand Factor
During synthesis, VWF undergoes different post translational modifications including removal of the propeptide by the protease furin, completion of N and O-glycosylation, and sulfation of specific N-oligosaccharides [19]. The N-linked oligosaccharides of VWF differ from other proteins because they contain ABO blood group oligosaccharides [20].
2.5 Intracellular Storage and Secretion of VWF
Following synthesis, VWF is stored in α-granules of megakaryocytes/platelets or in Weibel-Palade bodies of endothelium. α-granules can still be formed in the absence of VWF, while the formation of Weibel-Palade bodies depends on the presence of VWF [21]. About 95% of the VWF formed is constitutively secreted as smaller multimers, while the remainder is stored as large multimers, with these having a greater number of active sites for interaction with platelets and vessel wall. Therefore, in an environment devoid of ADAMTS13 (i.e., in thrombotic thrombocytopenic purpura, TTP), the thrombogenic risk due to an accumulation of these larger molecules is high [22, 23]. After synthesis and packaging of VWF in Weibel-Palade bodies, a complex pathway is initiated that leads to secretion of VWF.
2.6 Biological Activities of von Willebrand Factor
Following injury to the endothelium, VWF binds to different components of sub-endothelium, as well as to platelets in order to sequester them to the injury site [24].
2.6.1 Interaction of von Willebrand Factor with Extracellular Protein
VWF is capable of binding to different types of collagen, including types I, II, III, IV, V, and VI. Domains A1 and A3 are responsible for interaction with the main components of extracellular matrix (especially collagen), with each domain capable of binding to different types of collagen [25, 26]. The A1 domain, which covers residues 497–716, binds to collagen type VI, while the A3 domain, comprising residues 910–1111, binds to collagen types I and III [27, 28]. Collagen types I and III support VWF-dependent platelet adhesion in high shear rate, whereas collagen type IV mostly supports platelet adhesion at lower shear rates.
2.7 Interaction of von Willebrand Factor with Glycoprotein Ibα
The A1 domain is responsible for binding to platelet glycoprotein (GP) Ib, a component of the platelet GPIb-V-IX receptor complex [29]. This interaction plays a significant role in platelet activation, platelet adhesion, and platelet aggregation, ultimately crucial for thrombus formation. In vivo, the initial platelet adhesion results from interaction between VWF and GpIbα under high fluid shear. In vitro, the bacterial glycopeptide antibiotic ristocetin can bind to VWF in laboratory tests, and thus facilitate platelet GPIbα binding under low shear. This property is exploited in several VWF tests, including ristocetin-induced platelet aggregation (RIPA) and the ristocetin cofactor (VWF:RCo) assay, both of which facilitate the laboratory estimation of VWF binding VWF to GpIb [30]. Botrocetin is a venom derived from the viper Bothrops jararaca, which can also activate platelets via GpIb binding, to facilitate VWF-dependent platelet aggregation in vitro [31]. The botrocetin-induced platelet aggregation (BIPA) is only selectively used in laboratory diagnostics [32]. There are several modern alternatives for the classical VWF:RCo assay, including the VWF GPIb binding assays (i) using recombinant GPIb and inert particles (latex or magnetic) to replace native platelets and GPIb otherwise used in the VWF:RCo, but still also using ristocetin (“VWF:GPIbR”), and (ii) using recombinant GPIb with gain of function variants and inert particles (latex or plastic wells) to replace native platelets and GPIb otherwise used in the VWF:RCo, but without need for use of ristocetin (“VWF:GPIbM”) [5, 6].
2.8 Stabilization and Transport of Coagulation Factor VIII
In patients affected by hemophilia A, caused by defects in the F8 gene, the FVIII level is decreased and the VWF level is normal. In contrast, in most of patients with VWD, concomitant reduction of both FVIII and VWF occurs, because the survival of FVIII depends on its interaction with VWF. This binding and protection of FVIII permits its delivery to sites of vascular injury, and after release, FVIII can participate in secondary hemostasis and facilitate the conversion of plasma fibrinogen to fibrin (i.e., clot formation) [33]. In this way, VWF contributes to secondary hemostasis. This FVIII binding activity can be assessed in vitro with a specific test called the VWF FVIII binding assay (VWF:FVIIIB) [5, 6].
2.9 Interaction of von Willebrand Factor with Integrin αIIbβ3 (GPIIb/IIIa)
Platelet GPIIb-IIIa, also known as the integrin αIIbβ3, is a surface receptor that, after platelet activation, can bind to different ligands, including fibrinogen, fibronectin, and VWF [34]. In domain C1 of VWF, the tetra peptide motif Arg-Gly-Asp-Ser (located in the C-terminal of the molecule) is a binding site for αIIbβ3 (GPIIb/IIIa). Although the major interaction for platelet adhesion is GbIb binding, inhibition of integrin αIIbβ3 (GPIIb/IIIa) also impairs platelet adhesion [31, 35, 36]. Thus, VWF–integrin αIIbβ3 (GPIIb/IIIa) interaction leads to further platelet adhesion and facilitation of platelet aggregation initiated through the binding of GpIb to VWF [36].
3 von Willebrand Disease
VWD, with an estimated incidence of ~1% according to epidemiological studies, is the most common congenital bleeding disorder, and is most often inherited in an autosomal dominant pattern. The disorder was first described by Erik von Willebrand in 1926, who named it “hereditary pseudohemophilia” [37]. VWD is caused by defects and/or deficiencies in VWF level, structure, or function. Patients with VWD present with variable bleeding symptoms, but mostly mucocutaneous. Epistaxis and bruising are the most common presentations in children, while menorrhagia and hematoma are common in adults, with severity and frequency of bleeding dependent on age, gender, and VWD type [2]. Plasma levels of VWF gradually increase with age, and this may ameliorate VWD diagnosis or effects in some cases. VWD is classified into three main types: types 1 and 3 represent partial and complete deficiency of VWF, respectively, while type 2 reflects qualitative defects further characterized into four subgroups. These comprise: (i) type 2A VWD, with loss of high and sometimes intermediate molecular weight VWF multimers; (ii) type 2B VWD, with increased affinity of VWF for GPIb; (iii) type 2M VWD, with defects in VWF function such as platelet adhesion but a relatively normal pattern of (i.e., no substantial decrease in high molecular weight) VWF multimers; and (iv) type 2N, with markedly decreased VWF binding affinity to FVIII (Table 3.1) [1, 3, 38].
Type 3 VWD, with an estimated incidence of 1 per one million in the general population in developed countries, is the rarest and the most severe form of disorder, while type 1 is the most commonly diagnosed form of VWD. However, in some developing countries, the reverse may be true, with type 3 VWD more often reported than type 1; this is due to consanguinity, where both parents may have heterozygous type 1 VWD, and also because of higher relative presentations to clinicians due to the severity of bleeding symptoms [39,40,41].
Types 1 and 2 VWD represent highly heterogeneous bleeding disorders, with bleeding tendency related to the circulating level of functional VWF and type of VWF defect. It is always important to determine whether the bleeding tendency is “life-long” or of recent onset, since the latter potentially indicates an acquired VWF abnormality rather than inherited VWD [42].
Diagnosis of VWD is a challenge worldwide, especially in mildest forms of the disease (where the boundary between normal and abnormal phenotypes is not clearly defined [4]), and in complex types, where misdiagnosis is common and can occur due to use of inferior methods or tests or ineffective test panels [5]. Correct diagnosis and classification of VWD is critical to correct patient management (Table 3.2).
3.1 Type 1 von Willebrand Disease
Type 1 is inherited in autosomal dominant manner with variable penetrance and a highly variable phenotype. The latest guidelines on the diagnosis of VWD [5, 43] identify type 1 VWD where VWF levels are <30 U/dL with concordance of functional VWF identified using a VWF GPIb binding assay (VWF:RCo, VWF:GPIbR, or VWF:GPIbM) (i.e., ratio of functional VWF/VWF:Ag >0.7). This is since most of such patients would be identified with a genetic variant in VWF. These guidelines [5, 43, 44] also recommend assigning a diagnosis of type 1 VWD where VWF levels are between 30 and 50 U/dL with concordance of functional VWF/VWF:Ag (i.e., >0.7) in those with a significant history of bleeding. The recent ASH ISTH NHF WFH guidelines categorize patients with levels between 30 and 50 U/dL as type 1 VWD only if they have a positive bleeding history, whereas previous guidelines may have classified them as having “low VWF as a risk factor for bleeding.” [5] A considerable number of factors are responsible for the highly variable clinical and laboratory phenotype in type 1 VWD. In some patients with type 1 VWD, a variant cannot be detected in the VWF gene. Genetic modifiers and physiological factors are major factors that can reduce plasma level of VWF, with one well-known genetic modifier outside the VWF gene being ABO blood group [45]. Plasma VWF level are ~25% lower in individuals with O blood group than non-O blood group. The severity of bleeding episodes in type 1 VWD largely depends on severity of the plasma VWF deficiency; thus, risk of severe bleeds is higher in patients with lower plasma levels of VWF. Plasma FVIII level is also reduced in parallel with VWF, and its deficiency can compound bleeding risk in VWD. Since type 1 VWD phenotypically defines an inherited bleeding disorder with partial quantitative VWF deficiency, both VWF:Ag and VWF activity fall in parallel; a functional abnormality of VWF (i.e., type 2 VWD) can essentially be excluded if all VWF activity/VWF:Ag ratios are around unity (i.e., > 0.7) [46]. With the use of more sensitive assays, a considerable number of patients with historical diagnosis of type 1 VWD based on older assays such as VWF:RCo may be shown to mild abnormalities of multimer structure or distribution [47]. Increased susceptibility of VWF to proteolytic cleavage may also contribute to bleeding severity in type 1 VWD. The Tyr1584Cys variant, for example, increases the susceptibility of VWF to cleavage by ADAMTS13. Desmopressin is generally suitable as the therapeutic choice in most patients with type 1 VWD, especially for short duration or minor treatments [47].
3.2 Type 2 von Willebrand Disease
Type 2 VWD is characterized by qualitative defects in structure and function of VWF and is further classified into one of four types: 2A, 2B, 2M, and 2N. Classically, type 2A has been considered the most common form, while 2N and 2B represent the rarest forms. However, the relative frequency of type 2 VWD depends in part on the geography in which the diagnosis is made, as well as the diagnostic test repertoire. For example, type 2N VWD is relatively more frequent in some parts of France and Italy, and type 2B diagnosis requires performance of RIPA, which if omitted may lead to diagnosis of such patients as 2A VWD [48, 49]. Type 2M VWD has classically been considered a rare type of VWD, but it is likely that many cases of historically defined 2A VWD are instead 2M VWD, not appropriately diagnosed due to limited testing.
Type 2 VWD is mostly transmitted in an autosomal dominant manner, except for type 2N, which has an autosomal recessive pattern of inheritance. Type 2 VWD is less common than type 1 VWD, and represents ~20% of all cases of VWD. Type 2 VWD is usually attributable to variants that either impair specific functional domains of VWF or which affect VWF multimer assembly or proteolysis. Type 2 VWD is diagnostically the most challenging form of VWD [50]. The hallmark of this type of disorder is low functional VWF/VWF:Ag ratio (<0.7). Functional VWF was mostly assessed historically as VWF:RCo; however, newer assays, including VWF:GPIbR, VWF:GPIbM, and VWF collagen binding (VWF:CB) are increasingly contributing to better more accurate diagnosis. Lack of large multimers is evident in types 2A and 2B VWD, with loss of intermediate multimers also often seen in type 2A VWD. Impaired RIPA in platelet-rich plasma (PRP) or in whole blood is evident in types 2A and 2M VWD, whereas an increased RIPA responsiveness is seen in 2B VWD [32].
3.2.1 Type 2A von Willebrand Disease
Type 2A is classically considered the most frequent type 2 VWD, comprising up to 5–10% of all cases with VWD and typically >40% of all type 2 VWD. Type 2A VWD represents a loss of high and intermediate multimers of VWF, due either to impaired VWF multimerization or increased susceptibility of multimers to degradation with ADAMTS13. The loss of high molecular weight multimers (HMWM) and often also intermediate molecular weight multimers (IMWM) leads to diminished activity of the binding domains for GPIb, collagen, and probably also GPIIb/IIIa [49].
3.2.2 Type 2B von Willebrand Disease
In type 2B VWD, gain-of function variants, usually in the A2 domain of VWF, cause an increased affinity of VWF for platelet GpIb. This is often identified in laboratory testing by elevated RIPA responsiveness. In vivo, the platelet-VWF complex is removed from plasma circulation, often leading to loss of HMWM and also (mild) thrombocytopenia. Additional features of 2B VWD include slightly decreased to normal levels of VWF:Ag and FVIII, and relatively decreased levels of VWF activity (i.e., ratios of VWF:RCo/Ag, VWF:GPIbR/Ag, VWF:GPIbM/Ag, and VWF:CB/Ag are all <0.7). However, not all cases present with this “classic” 2B VWD presentation, and occasionally, VWF multimeric pattern and functional VWF/VWF:Ag ratios may be normal than those with (“atypical”) type 2B. In 2B VWD patients in their second and third trimester of pregnancy, the pregnancy associated increased level of gain of function VWF may worsen the thrombocytopenia. A reduction in platelet counts may also be observed if desmopressin is administrated in patients with 2B VWD, and thus, desmopressin is often considered contraindicated in 2B VWD [51].
3.2.3 Type 2M von Willebrand Disease
Type 2M is mostly due to variants in the A1 domain of VWF, leading to conformational changes in VWF protein and decreased binding affinity to GpIb. VWF multimeric pattern is essentially normal, but platelet-dependent VWF activities are decreased. Rarely, 2M VWD variants can occur in the A3 domain, and thus also affecting collagen binding. Generally, type 2M VWD cases are identified by low VWF GPIb binding/Ag ratios (i.e., VWF:RCo/Ag, VWF:GPIbR/Ag or VWF:GPIbM/Ag <0.7), but without lack of HMWM or with normal VWF:CB/Ag ratio. Misdiagnosis is an important and largely under-recognized issue in type 2M VWD and a considerable number of clinicians or laboratories identify this type “incorrectly” as type 1 or type 2A VWD [4], often due to incomplete testing or an pre-assumption of 2A VWD. Patients with type 2M tent to present with mild to moderate bleeding tendency but severe bleeds also may occur [52].
3.2.4 Type 2N von Willebrand Disease
Type 2N VWD (VWD “Normandy”) was originally described in patients from the Normandy region of France, in 1989, as a variant of VWD caused by defects in the ability of VWF to bind FVIII [53]. This defect causes decreased plasma level of FVIII, thereby phenotypically resembling hemophilia A. However, the inheritance pattern of 2N VWD is autosomal recessive (whereas hemophilia A is sex-linked, being carried on the X-chromosome), and the defect is present on VWF (whereas in hemophilia A, the defect is in FVIII). Symptoms of type 2N VWD are similar to that of (mild) hemophilia A [54]. Phenotypically, the multimeric pattern is normal in type 2N VWD, and VWF:Ag and other functional assays (VWF:RCo, VFW:GPIbR, VWF:GPIbM, VWF:CB) are also normal, unless the 2N VWD is coinherited with a type 1 VWD. The diagnostic feature of 2N VWD is the level of FVIII, which is decreased due to an increased clearance (i.e., as the VWF does not bind FVIII, the FVIII is easily degraded in circulation). As noted, sometimes 2N VWD may arise as a duplex defect. For example, patients with a 2N VWD variant on one VWF gene, and another defect in the other VWF gene, may show more complex phenotypes and more severe/complex bleeding patterns. For example, if the second VWF gene carries a null variant, essentially mimicking a “heterozygous type 3 VWD,” then the resulting phenotype will express with lowered VWF levels [55].
3.2.5 Type 3 von Willebrand Disease
Type 3 VWD is the most severe form of VWD, albeit fortunately also the rarest type. By definition, type 3 VWD patients present with “undetectable” plasma and platelet level of VWF and also a low level of FVIII: C (<10 U dL−1). Clinical presentations and bleeding symptoms are similar to those of moderately severe hemophilia A, and a misdiagnosis of hemophilia A is possible if only FVIII is assessed [39].
4 Clinical Manifestations
Clinical manifestations and bleeding tendency among patients with VWD are highly variable and range from quite mild conditions to severe bleeding diathesis sufficient to require urgent medical intervention. Mucocutaneous bleeding such as epistaxis and menorrhagia represents the most typical presentation of VWD, with post-dental extraction bleeding the most common post-surgical bleeding event. Since VWF also binds to FVIII and facilitates platelet function, VWD may cause bleeding symptoms that are typical of platelet function disorders or mild to moderately severe hemophilia A or both [3, 56].
In some patients, especially in males, surgery may be the first hemostatic challenge that leads to “abnormal bleeding” and therefore facilitates the diagnosis in previously unrecognized cases.
A wide overlap can be observed between the bleeding diathesis of patients with mild VWD and the normal population; therefore, a proper bleeding history is a critical and crucial component in the diagnosis of VWD and should be done carefully. Women with VWD tend to be more symptomatic than men, because they are subject to increased hemostatic challenges (menstruation and childbirth). In children with mild VWD, the pattern of hemorrhagic symptoms is different from children with more severe congenital bleeding disorders, with life-threatening bleeds such as intracranial hemorrhage (ICH) and umbilical cord bleeding being more rare among young VWD patients. In children with VWD, the most common clinical presentations are bruising and epistaxis; however, as these presentations are also frequent in normal healthy children, VWD diagnosis is challenging [57]. Standard clinical presentations in adult patients with VWD, including menorrhagia and post-surgical bleeding, are not evaluable in the pediatric population. Standard bleeding assessment tools (BATs) and scoring systems may be useful for correct assessment of bleeding episodes among patients, because otherwise significant bleeding diathesis may be overlooked while minimal bleeding symptoms may be over emphasized [58].
In adults, hematoma, menorrhagia, and bleeding from minor wounds are the most frequent symptoms, depending on VWD type, disease severity and gender. Post-dental extraction and post-surgical bleeds are common and occur in about two-thirds of patients. Gastrointestinal (GI) bleeding is also reported in VWD, predominantly among adults, and this can sometimes be severe [59]. In some patients, especially those with severe VWD, epistaxis can be so severe as to require medical intervention with VWF concentrates or blood transfusion.
Postpartum hemorrhage (PPH) can also be observed among women with VWD, but with possibly lower frequency than “expected” because VWF levels increase markedly during pregnancy. Delayed PPH can occur due to gradual decrease of VWF level to baseline level post-delivery. Prolonged vaginal bleeding following normal vaginal delivery is a common presentation of women with VWD. Menorrhagia (>80 mL of blood loss per menstrual period) is a common and important bleeding symptom of women with VWD, and ~15% of women with menorrhagia have VWD. Therefore, menorrhagia is a sensitive but non-specific presentation of VWD in women, as sometimes accompanied by anemia and iron deficiency. Therefore, careful gynecological assessment of women with VWD is crucial [39, 58].
Bleeding symptoms tend to be milder in type 1 VWD and more severe in types 2 and type 3 VWD. In a recent international study, it was observed that the bleeding phenotype was significantly more severe in type 3 VWD than in type 1 VWD. The study found that epistaxis was the most common presentation in patients with type 3 VWD, followed by hemarthrosis in males and menorrhagia in females. Furthermore, the study estimated that the prevalence of CNS bleeding is more than 20 times higher in type 3 VWD than in type 1 VWD [60]. Since FVIII level is only slightly reduced in most types of VWD, spontaneous hemarthrosis or hematoma are rare in types 1, 2A, and 2B VWD, while in type 3 VWD, the severity of diathesis often resembles that of hemophilia. Severe life-threatening bleeding can occur in type 3 VWD and sometimes in patients with type 2 VWD, but is a rare presentation of type 1 VWD. ICH is a rare presentation of VWD that usually is only reported in type 3 VWD [39].
5 Diagnosis of von Willebrand Disease
VWD is diagnosed on clinical features, comprising personal and family history of bleeding or bruising, and confirmed by laboratory testing. As VWD is due to deficiencies or defects in the plasma protein VWF, a large adhesive protein with multiple activities, laboratory testing therefore needs to assess VWF level and activity using a panel of tests [8]. The more comprehensive this panel, the more likely the correct diagnosis or exclusion of VWD; the less comprehensive the assay panel, the more likely the misdiagnosis. The minimum recommended three-test panel comprises Factor VIII coagulant (FVIII:C), VWF:Ag, and VWF GPIb binding activity (i.e., VWF:RCo, VWF:GPIbR, or VWF:GPIbM); in our laboratory, we use a four-test panel comprising these plus VWF:CB. Additional assays including RIPA, VWF multimers, and VWF:FVIIIB are performed selectively, and where (i) low VWF activity/Ag ratios are evident (i.e., VWF:RCo/Ag, VWF:GPIbR/Ag, VWF:GPIbM/Ag, or VWF:CB/Ag <0.7; here reflexing to RIPA and VWF multimers) or (ii) where FVIII:C/VWF:Ag is low (i.e., <0.7; here potentially reflexing to VWF:FVIIIB). A complete diagnosis of VWD requires the classification of all six types of the disorder. Proper diagnosis and classification of VWD are crucial and have significant clinical consequences, as they can change the treatment strategy. Some therapeutic options, such as desmopressin or DDAVP, are effective for the management of type 2 VWD but are ineffective or potentially harmful for other types of the disorder [56]. The most often used assay for measuring VWF GPIb binding activity is the VWF:RCo, historically measuring the agglutination by VWF of fixed human platelets in the presence of ristocetin on an aggregometer, and more recently on automated analyzers [61]. This assay is now increasingly replaced using alternate assays based on binding of VWF to recombinant GPIb on inert particles (i.e., VWF:GPIgR or VWF:GPIbM) [61,62,63]. Although all these are VWF:GPIbB assays, VWF: RCo differs from both VWF:GPIbR and VWF:GPIbM [8]. Nevertheless, these assays comprise a single group reflecting assays of GPIb binding (i.e., classical VWF:RCo, as well as methodologies now defined as “VWF:GPIbR” and “VWF:GPIbM”), would be expected to broadly derive similar test results for VWD patients, and are thus essentially recognized to be “inter-changeable” in VWD diagnostics [8].
The anticipated test patterns in different types of VWD is summarized in Table 3.3. Type 1 VWD represents a partial quantitative deficiency of (functionally normal) VWF, so there is concordant decrease in VWF measured by any VWF assay (be it VWF:Ag, VWF:CB or GPIb binding), and the ratio of any one VWF assay to any other is close to unity (in practice, >0.7). Type 3 VWD represents a total loss of VWF, and all VWF test results will be close to 0 U/dL, albeit recognizing that lower limit VWF sensitivity issues means that some assays cannot detect to these low levels, and that such low levels of detected VWF precludes generation of assay ratios [6].
In contrast, type 2 VWD represents qualitative VWF defects/deficiencies, such that VWF activity is proportionally decreased below that of VWF:Ag; furthermore, the VWD type can be defined by the type of activity reduced. Type 2A VWD, defining a loss of HMW VWF multimers, patients express a relative reduction of all VWF activities sensitive to this loss (this includes both GPIb binding and VWF:CB assays). In practice, this is expressed as the ratio of VWF activity/VWF:Ag assays being lower than ~0.7. Type 2B VWD defines an increased affinity of VWF for GPIb, which often leads to loss of HMW VWF multimers and similar VWF test patterns to type 2A VWD. Type 2M VWD defines decreased VWF-dependent platelet adhesion without a selective deficiency of HMW VWF multimers. In type 2M VWD, there are specific changes in VWF function related to specific VWF variants. In practice, most type 2M variants affect GPIb binding, and less so collagen binding; thus, there is usually a low VWF:GPIb binding activity/VWF:Ag ratio, but the VWF:CB/Ag ratio may be normal. Type 2N VWD defines a decreased binding affinity for FVIII, as identified by the specific test VWF:FVIIIB. Phenotypically, however, these patients present similarly to those with hemophilia A, showing relatively lower FVIII:C to VWF:Ag ratios [8].
Including FVIII:C as part of a VWD diagnostic profile has many benefits. Since VWF protects FVIII, lower levels of VWF (i.e., VWD) will also generally mean lower levels of FVIII:C, and so represent additional bleeding risk. In type 3 VWD, for example, levels of FVIII:C are generally <10 U/dL. In type 1 VWD, the FVIII:C is generally proportional to VWF:Ag, and in type 2N VWD, FVIII:C is proportionally lower than VWF:Ag [8].
In summary, the recommended approach to diagnosis or exclusion of VWD can be aided by the use of algorithms, such as shown in Fig. 3.1 using a four-test panel comprising VWF:Ag, a VWF:GPIb binding assay (VWF:RCo, VWF:GPIbR, or VWF:GPIbM), VWF:CB, and FVIII:C. A four-test panel recommendation is made ahead of the more basic three-test panel based on decades of experience, and the high rate of diagnostic errors associated to smaller first line testing panels [6, 55,56,57,58,59,60,61,62,63,64,65,66,67,68]. Which tests within each category are employed by laboratories will depend on local availabilities (i.e., instrumentation and tests). VWF:Ag is mostly performed by latex immunoassay (LIA) procedure and less often by ELISA; however, chemiluminescence immuno-assay methodology is emerging and least problematic (CLIA). VWF:GPIb binding assays may be performed by platelet agglutination (VWF:RCo), latex agglutination (VWF:GPIbR, VWF:GPIbM), CLIA (VWF:GPIbR), or potentially by ELISA (VWF:GPIbM). VWF:CB testing is usually performed by ELISA, and increasingly by CLIA [64, 69]. Also, problems with reproducibility and low level VWF detection with classical VWF:RCo assays may drive usage of more modern VWF:GPIb binding assay alternatives such as VWF:GPIbR or VWF:GPIbM.

An algorithm for the diagnosis or exclusion of VWD using an initial four-test panel, as used at the Westmead laboratory
Once the methodology has been selected and validated, the recommended basic VWD four-test panel should be able to diagnose or exclude VWD, with additional investigations selected on a case by case basis, dependent on the results from the four-test panel (Fig. 3.1).
The basic caveat always remains that diagnosis or exclusion of VWD requires at the very least repeat testing using a fresh sample, due to preanalytical and also analytical limitations.
If all VWF tests are normal, as confirmed on repeat testing, then the patient either does not have VWD or has a form of VWD that is not able to be defined with current testing. Additional assays to define the bleeding disorder, inclusive of platelet function or additional factor assays, may be required. If all VWF tests are low, confirmed on repeat testing, but all VWF values are concordant (ratios of VWF:GPIb binding/Ag and VWF:CB/Ag both >0.7), then the patient has type 1 VWD if the level of VWF is <30 U/dL (or 30–50 U/dL with appropriate history as per the latest VWD diagnostic guidelines) [5]. In such cases, assessment of severity may be based on absolute VWF level; however, unlike the case for hemophilia [70], there is no available consensus for cut-off values defining severity of VWD.
If there is a low ratio of VWF:GPIb binding/Ag and/or VWF:CB/Ag, and this pattern is confirmed on repeat testing with a fresh sample, then the patient may have type 2A, 2B, or 2M VWD (or possibly platelet type [PT] VWD). Here, further tests should be undertaken (i.e., RIPA, VWF multimer assessment, depending on test findings and local test availability). In our experience, RIPA analysis is usually more important than multimer analysis; moreover, selection of the right test methodologies for VWF:Ag, VWF:CB and VWF:GPIb binding assays will often enable prediction of the VWF multimer pattern negating any need for its performance. Thus, a low ratio of VWF:GPIb binding/Ag plus a low ratio of VWF:CB/Ag usually points to a loss of HMW VWF, and thus likely 2A, 2B, or platelet type VWD. Instead, a low ratio of VWF:GPIb binding/Ag or VWF:CB/Ag (but not both), usually discounts a loss of HMW VWF and instead points to a type 2M VWD [8].
If all VWF test results are below the measuring range of the assays used, then this will create problems with clear diagnosis, but usually infers severe type 1 VWD or else type 3 VWD; therapy is similar in both cases, although clinical severity is often worse in type 3 VWD.
Finally, if the ratio of FVIII:C/VWF:Ag is low, this suggests either hemophilia A or 2N VWD. Hemophilia A is more common, and being sex-linked affects males more than females; however, misdiagnoses of both hemophilia A and 2N VWD, where the correct diagnosis was the other, do occur. Thus, testing by performance of a VWF:FVIIIB assay is recommended, and after repeat testing for confirmation, could include genetic analysis of F8 and/or VWF for final definitive verification.
Genetic analysis may also be useful where patients have been defined to be type 2A, 2B, 2M, or platelet type VWD, and is also typically successful when performed on such patients. Genetic analysis is useful in some type 3 VWD investigations, but generally not useful in type 1 VWD [71].
6 Molecular Basis of von Willebrand Disease
VWF is encoded by the VWF gene, which is located on the short arm of chromosome 12 (12p13.3), spans 180 kb, and consists of 52 exons, of which, exon 50 (40 kb) and exon 28 (1/3 kb) are considered the longest and smallest exons, respectively. The VWF pseudogene 1 (VWFP1), which is located on the long arm of chromosome 22 (22q11–13), spans ~21–29 kb and shows 97% homology with 23–34 exons of the VWF gene but encodes no functional transcript. The VWF gene is transcribed to an 8.8 kb mRNA, which translates to a 2813-amino acid pre-pro-VWF protein. Pre-pro-VWF comprises a signal peptide (pre) with 22 amino acids, a pro-peptide (pro) with 741 amino acids, and a 2050-amino acid mature protein. Any variant that leads to qualitative and/or quantitative abnormalities in VWF can be associated with VWD. Variants may affect different biosynthetic events, including gene expression (transcription, translation), post-translational processing, dimerization/multimerization mechanisms, proteolytic processing, storage, secretion processing, structure, clearance, and function of VWF. Different variant types are associated with VWD including: (1) those that involve transcription factor binding sites that lead to absent or reduced RNA transcription; (2) splice site mutations that disrupt the splice donor site and splice acceptor site of each intron, leading to exon skipping and production of shortened RNA and protein; (3) nonsense variants; (4) small deletions; (5) insertions; (6) duplications; (7) large deletions; and (8) missense variants. According to the ISTH-SSC VWF Online database (https://dbs.eahad.org/upcoming-vwf-db/) and many other studies, almost 600 separate variants have currently been reported in patients with VWD, about 80% of them are missense [37]. Patients with acquired von Willebrand syndrome (AVWS) VWD do not have hereditary VWF variants [72].
7 Treatment of von Willebrand Disease
Management of patients with VWD includes prevention or treatment of bleeding by correction of the hemostatic defects, which may be those of primary hemostasis (due to lack or decrease of VWF) or secondary hemostasis (due to FVIII deficiency). These can be achieved by increasing endogenous VWF (using desmopressin) or in unresponsive/contraindicated patients or long-term need, by infusion of exogenous VWF/FVIII (typically as plasma concentrates, although recombinant FVIII and VWF are available in some locations) [9, 73, 74]. In some developing countries, cryoprecipitate and fresh frozen plasma (FFP) might still be used; however, the risk of virus transmission and the need to transfuse a high volume of product reflect substantive obstacles for their use. Additional adjuvant therapies can also be employed for some situations, including antifibrinolytic therapy with tranexamic acid or epsilon aminocaproic acid, which can improve hemostasis in patients without altering their plasma VWF levels [9, 74].
On-demand therapy (meaning treatment of hemorrhage as soon as possible after onset of bleeding) remains the mainstay of treatment of patients with VWD. However long-term prophylaxis may be necessary for those with severe hemorrhages (e.g., type 3 VWD) to improve their quality of life by reducing annualized bleeding rates [9] and will be increasingly employed [75].
7.1 Desmopressin
Desmopressin (1-deamino-8-d-arginine vasopressin) (DDAVP) is synthetic analogue of vasopressin that causes an increase of endogenous plasma FVIII and VWF by facilitating their release from endothelial storage sites. Depending on location, desmopressin may be given intravenously, intranasally, or subcutaneously. Desmopressin is most likely to be useful in patients with baseline FVIII and functional VWF levels of 10–20 U/dL or more. Thus, desmopressin is most useful in management of patients with mild hemophilia or mild VWD (especially type 1 VWD). If required, patients can receive repeated doses of drug in 12 to 24 h intervals; however, repeated doses of desmopressin cause less effective responses, and eventual depletion of FVIII and VWF stores (called tachyphylaxis) [9, 73, 74]. A test dose of desmopressin is recommended before establishment of the magnitude and duration of the drug response in any given patient, since the genotype and phenotype of VWD can affect its effectiveness [76].
Although desmopressin is effective in some patients with type 2 VWD, mostly 2A and 2M, it is ineffective in the majority of these patients, and in patients with type 2B, it is considered contradicted because it may worsen thrombocytopenia and potentially increase the risk of bleeding. Type 3 VWD are unresponsive to desmopressin, due to lack of releasable stores of VWF in these patients [9, 76].
The use of desmopressin can be associated with certain side effects, including hypotension, cardiovascular complications, flushing, and hyponatremia. Hyponatremia can be prevented by limited fluid intake for 24 h after desmopressin administration, while other side effects are mostly due to vasodilating effects that can often be attenuated with intravenous infusion by slowing the rate [74, 76].
For intravenous purpose, 0.3 μg/kg in 50 mL is administrated over 30 min. FVIII:C, VWF:Ag and functional VWF (e.g., VWF:RCo) should be assessed pre-infusion and 1, 2, and/or 4 and 24 h post-infusion. Patients with a sufficient response to desmopressin have a two to five times increase from baseline levels and have FVIII and VWF levels above 50 U/dL at 1 h post-infusion. The levels remain above 30 U/dL at 4 h post-infusion but generally return to baseline levels by 24 h [4, 76, 77].
7.2 von Willebrand Factor Concentrates
Transfusion therapy using products containing virus inactivated concentrates of human VWF/FVIII represents the main therapeutic choice in patients unresponsive to desmopressin, or for long-term therapy use. These concentrates can be used for on-demand therapy (to stop bleeding when they occur), can prevent bleeding in surgery, or can be used for long-term secondary prophylaxis [78].
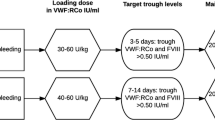
This treatment type reflects the current treatment of choice in patients with type 3 VWD, most patients with type 2 VWD, and a number of patients with type 1 VWD, (Table 3.4) [4, 10].
A number of different VWF/FVIII concentrates are available for use. The quantity of HMWM VWF and FVIII differs between products [1, 77]. It is important to avoid excessive concentrations of FVIII:C with injection of repeated doses in patients with VWD [4, 77]. Thus, the required dose of concentrate to use in patients may differ according to the product, and will also differ according to the situation (e.g., type and severity of bleeding episodes, type of planned surgery.
7.3 Recombinant von Willebrand Factor
Recombinant VWF (Vonicog alfa, Vonvendi (USA), Veyvondi (Europe)) is a relatively new product for management of VWD that is only available in some countries. Since the product is not exposed to ADAMTS13, it retains all sizes of VWF multimers including HMWM and ultra-large multimers [83]. Recombinant VWF has been approved by the US Food and Drug Administration (FDA) for on-demand therapy, surgical procedures, and prophylaxis [84]. It has also been approved by the European Medicines Agency (EMA), and multiple studies have demonstrated its safety and efficacy in managing VWD [85,86,87].
7.4 Prophylaxis
Prophylaxis should be considered for patients with type 3 VWD and recurrent hemarthrosis. Patients with recurrent GI bleeding and those with frequent epistaxis can also benefit from prophylaxis. Regular prophylaxis will decrease the number of bleeds and the severity of hemorrhages, prevent arthropathy, and improve quality of life in patients with VWD [77].
7.5 Surgery
Surgical procedures represent an important hemostatic challenge in patients with VWD; the majority of patients having major surgery will be managed by replacement VWF/FVIII therapy. Strategies for pre-operative management depends on type of VWD, type of surgery, baseline levels of VWF and FVIII, and therapeutic response to desmopressin [77].
7.6 Pregnancy and Delivery
VWF and FVIII levels increase two to three-fold during the second and third trimesters in patients with types 1 and 2 VWD and fall to baseline by 7–21 days post-partum; however, this increase is not observed in type 3 VWD. Also, in type 2 VWD, the increase in VWF level may not be associated with any substantive increase in functional VWF level, or in type 2B VWD, may lead to an increase in “abnormal VWF” that may worsen thrombocytopenia..Thus, management of pregnant women with VWF/FVIII may be required in types 2 and 3 VWD. A rapid decrease in plasma VWF and FVIII can also occur post-partum, and subsequently PPH, including delayed PPH, can also occur [88].
Generally, pregnant patients with VWD should be monitored for functional VWF (e.g., VWF:RCo, VWF:GPIbR, VWF:GPIbM, and VWF:CB) and FVIII:C once during the third trimester and within 10 days of expected date of delivery [89].
References
Dorgalaleh A, Tabibian S, Shiravand Y, Favaloro EJ. von Willebrand disease. In: Dorgalaleh A, editor. Congenital bleeding disorders: diagnosis and management. Cham, Switzerland: Springer Press; 2018. p. 57–102.
James PD, Goodeve AC. von Willebrand disease. Genet Med. 2011;13(5):365–76.
Sadler J, Budde U, Eikenboom J, Favaloro E, Hill F, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10):2103–14.
Laffan M, Brown S, Collins PW, Cumming A, Hill F, Keeling D, et al. The diagnosis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2004;10(3):199–217.
James PD, Connell NT, Ameer B, Di Paola J, Eikenboom J, Giraud N, Haberichter S, Jacobs-Pratt V, Konkle B, McLintock C, McRae S. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021;5(1):280–300.
Favaloro EJ, Pasalic L. Laboratory diagnosis of von Willebrand Disease (VWD): Geographical Perspectives. Semin Thromb Hemost. 2022;48(6):750–66.
Federici AB. Diagnosis of inherited von Willebrand disease: a clinical perspective. Semin Thromb Hemost. 2006;32(6):555–65.
Favaloro EJ, Pasalic L, Curnow J. Laboratory tests used to help diagnose von Willebrand disease: an update. Pathology. 2016;48(4):303–18.
Sadler J, Mannucci P, Berntorp E, Bochkov N, Boulyjenkov V, Ginsburg D, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84(2):160–74.
Mannucci PM, Chediak J, Hanna W, Byrnes J, Ledford M, Ewenstein BM, et al. Treatment of von Willebrand disease with a high-purity factor VIII/von Willebrand factor concentrate: a prospective, multicenter study. Blood. 2002;99(2):450–6.
Lenting PJ, Christophe OD, Denis CV. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015;125(13):2019–28.
Ginsburg D, Handin RI, Bonthron DT, Donlon TA, Bruns GA, Latt SA, et al. Human von Willebrand factor (vWF): isolation of complementary DNA (cDNA) clones and chromosomal localization. Science. 1985;228:1401–7.
Mancuso D, Tuley E, Westfield L, Worrall N, Shelton-Inloes B, Sorace J, et al. Structure of the gene for human von Willebrand factor. J Biol Chem. 1989;264(33):19514–27.
Federici AB, Lee CA, Berntorp EE, Lillicrap D, Montgomery RR. Von Willebrand disease: basic and clinical aspects. John Wiley & Sons; 2011.
Shapiro S, Nowak A, Wooding C, Birdsey G, Laffan M, McKinnon T. The von Willebrand factor predicted unpaired cysteines are essential for secretion. J Thromb Haemost. 2014;12(2):246–54.
Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood. 2008;112(1):11–8.
Zanardelli S, Crawley JT, Chion CKCK, Lam JK, Preston RJ, Lane DA. ADAMTS13 substrate recognition of von Willebrand factor A2 domain. J Biol Chem. 2006;281(3):1555–63.
Zanardelli S, Chion AC, Groot E, Lenting PJ, McKinnon TA, Laffan MA, et al. A novel binding site for ADAMTS13 constitutively exposed on the surface of globular VWF. Blood. 2009;114(13):2819–28.
Samor B, Michalski JC, Debray H, Mazurier C, Goudemand M, Halbeek H, et al. Primary structure of a new tetraantennary glycan of the N-acetyllactosaminic type isolated from human factor VIII/von Willebrand factor. FEBS J. 1986;158(2):295–8.
Canis K, McKinnon TA, Nowak A, Haslam SM, Panico M, Morris HR, et al. Mapping the N-glycome of human von Willebrand factor. Biochem J. 2012;447(2):217–28.
Haberichter SL, Merricks EP, Fahs SA, Christopherson PA, Nichols TC, Montgomery RR. Re-establishment of VWF-dependent Weibel-Palade bodies in VWD endothelial cells. Blood. 2005;105(1):145–52.
Sporn LA, Marder VJ, Wagner DD. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell. 1986;46(2):185–90.
George JN. Thrombotic Thrombocytopenic Purpura: From 1972 to 2022 and Beyond. Semin Thromb Hemost. 2022;48(8):926–36.
Ruggeri ZM, Mendolicchio G. Interaction of von Willebrand factor with platelets and the vessel wall. Hamostaseologie. 2015;35(3):211–24.
Roth GJ, Titani K, Hoyer LW, Hickey MJ. Localization of binding sites within human von Willebrand factor for monomeric type III collagen. Biochemistry. 1986;25(26):8357–61.
Cruz MA, Yuan H, Lee JR, Wise RJ, Handin RI. Interaction of the von Willebrand factor (vWF) with collagen Localization of the primary collagen-binding site by analysis of recombinant vWF a domain polypeptides. J Biol Chem. 1995;270(18):10822–7.
Pareti FI, Niiya K, McPherson J, Ruggeri Z. Isolation and characterization of two domains of human von Willebrand factor that interact with fibrillar collagen types I and III. J Biol Chem. 1987;262(28):13835–41.
Hoylaerts MF, Yamamoto H, Nuyts K, Vreys I, Deckmyn H, Vermylen J. von Willebrand factor binds to native collagen VI primarily via its A1 domain. Biochem J. 1997;324(1):185–91.
Fujimura Y, Titani K, Holland L, Russell S, Roberts J, Elder J, et al. von Willebrand factor. A reduced and alkylated 52/48-kDa fragment beginning at amino acid residue 449 contains the domain interacting with platelet glycoprotein Ib. J Biol Chem. 1986;261(1):381–5.
Scott J, Montgomery R, Retzinger G. Dimeric ristocetin flocculates proteins, binds to platelets, and mediates von Willebrand factor-dependent agglutination of platelets. J Biol Chem. 1991;266(13):8149–55.
Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67(1):395–424.
Wohner N, Legendre P, Casari C, Christophe O, Lenting P, Denis C. Shear stress-independent binding of von Willebrand factor-type 2B mutants p. R1306Q & p. V1316M to LRP1 explains their increased clearance. J Thromb Haemost. 2015;13(5):815–20.
Terraube V, O’donnell JS, Jenkins PV. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia. 2010;16(1):3–13.
Plow EF, Pierschbacher MD, Ruoslahti E, Marguerie GA, Ginsberg MH. The effect of Arg-Gly-Asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets. Proc Natl Acad Sci. 1985;82(23):8057–61.
Fressinaud E, Baruch D, Girma J-P, Sakariassen KS, Baumgartner HR, Meyer D. von Willebrand factor-mediated platelet adhesion to collagen involves platelet membrane glycoprotein IIb-IIIa as well as glycoprotein Ib. Transl Res. 1988;112(1):58–67.
Aoki T, Tomiyama Y, Honda S, Mihara K, Yamanaka T, Okubo M, et al. Association of the antagonism of von Willebrand factor but not fibrinogen by platelet αIIbβ3 antagonists with prolongation of bleeding time. J Thromb Haemost. 2005;3(10):2307–14.
Zolkova J, Sokol J, Simurda T, Vadelova L, Snahnicanova Z, Loderer D, Dobrotova M, Ivankova J, Skornova I, Lasabova Z, Kubisz P. Genetic Background of von Willebrand Disease: History, Current State, and Future Perspectives. Semin Thromb Hemost. 2020;46(4):484–500
Ginsburg D. von Willebrand disease. Williams hematology. 6th ed. Philadelphia: McGraw-Hill; 2001. p. 1813–28.
Eikenboom JC. Congenital von Willebrand disease type 3: clinical manifestations, pathophysiology and molecular biology. Best Pract Res Clin Haematol. 2001;14(2):365–79.
Favaloro EJ. Von Willebrand disease: local diagnosis and management of a globally distributed bleeding disorder. Semin Thromb Hemost. 2011;37(5):440–55.
Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD). Blood. 2007;109(1):112–21.
Colonne CK, Reardon B, Curnow J, Favaloro EJ. Why is misdiagnosis of von Willebrand disease still prevalent and how can we overcome it? A focus on clinical considerations and recommendations. J Blood Med. 2021;12:755–68.
Nichols WL, Rick ME, Ortel TL, Montgomery RR, Sadler JE, Yawn BP, James AH, Hultin MB, Manco-Johnson MJ, Weinstein M. Clinical and laboratory diagnosis of von Willebrand disease: a synopsis of the 2008 NHLBI/NIH guidelines. Am J Hematol. 2009;84(6):366–70.
Sadler JE, Rodeghiero F. Provisional criteria for the diagnosis of VWD type 1: on behalf of the ISTH SSC Subcommittee on von Willebrand factor. J Thromb Haemost. 2005;3(4):775–7.
Peake I, Goodeve A. Type 1 von Willebrand disease. J Thromb Haemost. 2007;5(s1):7–11.
Quiroga T, Goycoolea M, Belmont S, Panes O, Aranda E, Zuniga P, et al. Quantitative impact of using different criteria for the laboratory diagnosis of type 1 von Willebrand disease. J Thromb Haemost. 2014;12(8):1238–43.
Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101(6):2089–93.
Mazurier C, Goudemand J, Hilbert L, Caron C, Fressinaud E, Meyer D. Type 2N von Willebrand disease: clinical manifestations, pathophysiology, laboratory diagnosis and molecular biology. Best Pract Res Clin Haematol. 2001;14(2):337–47.
Sutherland JJ, O’Brien LA, Lillicrap D, Weaver DF. Molecular modeling of the von Willebrand factor A2 domain and the effects of associated type 2A von Willebrand disease mutations. J Mol Model. 2004;10(4):259–70.
Woods AI, Kempfer AC, Paiva J, Sanchez-Luceros A, Bermejo E, Chuit R, et al. Phenotypic Parameters in Genotypically Selected Type 2B von Willebrand Disease Patients: A Large, Single-Center Experience Including a New Novel Mutation. Semin Thromb Hemost. 2017;43(1):92–100.
Leebeek FW, Eikenboom JC. Von Willebrand’s disease. N Engl J Med. 2016;375(21):2067–80.
Castaman G, Federici A, Tosetto A, La Marca S, Stufano F, Mannucci P, et al. Different bleeding risk in type 2A and 2M von Willebrand disease: a 2-year prospective study in 107 patients. J Thromb Haemost. 2012;10(4):632–8.
Favaloro EJ, Mohammed S, Koutts J. Identification and prevalence of von Willebrand disease type 2N (Normandy) in Australia. Blood Coagul Fibrinolysis. 2009;20(8):706–14.
Perez Botero J, Pruthi RK, Nichols WL, Ashrani AA, Patnaik MM. von Willebrand disease type1/type 2N compound heterozygotes: diagnostic and management challenges. Br J Haematol. 2017;176(6):994–7.
Gadisseur A, Berneman Z, Schroyens W, Michiels JJ. Pseudohemophilia of Erik von Willebrand caused by homozygous one nucleotide deletion in exon 18 of the VW-factor gene. World J Hematol. 2013;2(4):99–108.
Federici A, Castaman G, Mannucci P. Guidelines for the diagnosis and management of von Willebrand disease in Italy. Haemophilia. 2002;8(5):607–21.
Federici AB, Bucciarelli P, Castaman G, Mazzucconi MG, Morfini M, Rocino A, et al. The bleeding score predicts clinical outcomes and replacement therapy in adults with von Willebrand disease. Blood. 2014;123(26):4037–44.
Rodeghiero F, Tosetto A, Abshire T, Arnold D, Coller B, James P, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–5.
Makris M. Gastrointestinal bleeding in von Willebrand disease. Thromb Res. 2006;118:S13–S7.
Tosetto A, Badiee Z, Baghaipour MR, Baronciani L, Battle J, Berntorp E, Bodó I, Budde U, Castaman G, Eikenboom JC, Eshghi P. Bleeding symptoms in patients diagnosed as type 3 von Willebrand disease: results from 3WINTERS-IPS, an international and collaborative cross-sectional study. J Thromb Haemost. 2020;18(9):2145–54.
Favaloro EJ. Diagnosing von Willebrand disease: a short history of laboratory milestones and innovations, plus current status, challenges, and solutions. Semin Thromb Hemost. 2014;40(5):551–70.
Just S. Laboratory Testing for von Willebrand Disease: The Past, Present, and Future State of Play for von Willebrand Factor Assays that Measure Platelet Binding Activity, with or without Ristocetin. Semin Thromb Hemost. 2017 Feb;43(1):75–91.
Bodo I, Eikenboom J, Montgomery R, Patzke J, Schneppenheim R, Di Paola J, von Willebrand P-d, factor activity. Nomenclature and methodology: communication from the SSC of the ISTH. J Thromb Haemost. 2015;13(7):1345–50.
Favaloro EJ, Bonar RA, Meiring M, Duncan E, Mohammed S, Sioufi J, et al. Evaluating errors in the laboratory identification of von Willebrand disease in the real world. Thromb Res. 2014;134(2):393–403.
Favaloro E, Bonar R, Mohammed S, Arbelaez A, Niemann J, Freney R, et al. Type 2M von Willebrand disease–more often misidentified than correctly identified. Haemophilia. 2016;22(3):e145–55.
Favaloro EJ, Bonar R, Kershaw G, Sioufi J, Baker R, Hertzberg M, et al. Reducing errors in identification of von Willebrand disease: the experience of the royal college of pathologists of australasia quality assurance program. Semin Thromb Hemost. 2006;32(5):505–13.
Favaloro EJ, Mohammed S. Towards improved diagnosis of von Willebrand disease: comparative evaluations of several automated von Willebrand factor antigen and activity assays. Thromb Res. 2014;134(6):1292–300.
Favaloro EJ, Mohammed S. Evaluation of a von Willebrand factor three test panel and chemiluminescent-based assay system for identification of, and therapy monitoring in, von Willebrand disease. Thromb Res. 2016;141:202–11.
Favaloro EJ, Dean E, Arunachalam S, Vong R, Mohammed S. Evaluating errors in the laboratory identification of von Willebrand disease using contemporary von Willebrand factor assays. Pathology. 2022;54(3):308–17.
Blanchette VS, Srivastava A. Definitions in Hemophilia: Resolved and Unresolved Issues. Semin Thromb Hemost. 2015;41(8):819–25.
Favaloro E. Genetic testing for von Willebrand disease: the case against. J Thromb Haemost. 2010;8(1):6–12.
Lillicrap D. The molecular genetics of von Willebrand disease. Hematology Meeting Reports (formerly Haematologica Reports); 2009.
Mannucci PM. How I treat patients with von Willebrand disease. Blood. 2001;97(7):1915–9.
Rodeghiero F, Castaman G, Tosetto A. How I treat von Willebrand disease. Blood. 2009;114(6):1158–65.
Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand Disease Prophylaxis Network (VWD PN). Haemophilia. 2013;19(1):76–81.
Castaman G, Goodeve A, Eikenboom J. Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica. 2013;98(5):667–74.
Curnow J, Pasalic L, Favaloro EJ. Treatment of von Willebrand Disease. Semin Thromb Hemost. 2016;42(2):133–46.
Thompson AR, Gill JC, Ewenstein B, Mueller-Velten G, Schwartz B. Successful treatment for patients with von Willebrand disease undergoing urgent surgery using factor VIII/VWF concentrate (Humate-P®). Haemophilia. 2004;10(1):42–51.
deWee EM, Leebeek FWG, EikenboomJCJ. Diagnosis and management of von Willebrand disease in The Netherlands. Semin Thromb Hemost. 2011;37(5):480–7.
Mannucci PM, Franchini M. The use of plasma-derived concentrates. In: Federici AB, Lee CA, Berntorp EE, Lillicrap D, Montgomery RR, editors. Von Willebrand disease. 2nd ed. West Sussex, UK: Blackwell Publishing Ltd; 2010.
Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14(2):171–232.
Dunkley S, Baker RI, Pidcock M, et al. Clinical efficacy and safety of the factor VIII/von Willebrand factor concentrate BIOSTATE in patients with von Willebrand’s disease: a prospective multicentre study. Haemophilia. 2010;16(4):615–24.
Gill JC, Castaman G, Windyga J, Kouides P, Ragni M, Leebeek FW, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015;126(17):2038–46.
Hancock JM, Escobar MA. An evaluation of von Willebrand factor (recombinant) therapy for adult patients living with severe type 3 von Willebrand disease. Expert Rev Hematol. 2023;16(3):157–61.
Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015;126(17):2038–46.
Leebeek F, Chapman M, Ploder B, Sytkowski A, Novack A, Ewenstein B. Treatment of gastrointestinal bleeding episodes with recombinant von Willebrand factor (rVWF) in patients with severe von Willebrand disease (VWD): sub-analysis from pivotal phase III on-demand study [abstract]. Res Pract Thromb Haemost. 2017;1(Suppl. 1):880.
Peyvandi F, Mamaev A, Wang JD, et al. Phase 3 study of recombinant von Willebrand factor in patients with severe von Willebrand disease who are undergoing elective surgery. J Thromb Haemost. 2019;17(1):52–62.
James A, Konkle B, Kouides P, Ragni M, Thames B, Gupta S, et al. Postpartum von Willebrand factor levels in women with and without von Willebrand disease and implications for prophylaxis. Haemophilia. 2015;21(1):81–7.
Simionescu AA, Buinoiu NF, Berbec N. Von Willebrand disease type 2 in pregnancy—a critical clinical association. Transfus Apher Sci. 2017;56(3):269–71.
Acknowledgments
The author acknowledges the contribution of Akbar Dorgalaleh, Yavar Shiravand, and Shadi Tabibian, who provided substantial input into the original chapter, upon which this revision is based. As the revision is built upon the shoulders of others, its incremental value would have been severely diminished without their original contribution.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Favaloro, E.J. (2023). von Willebrand Disease: An Update on Diagnosis and Treatment. In: Dorgalaleh, A. (eds) Congenital Bleeding Disorders . Springer, Cham. https://doi.org/10.1007/978-3-031-43156-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-43156-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-43155-5
Online ISBN: 978-3-031-43156-2
eBook Packages: MedicineMedicine (R0)