
Abstract
Von Willebrand disease (VWD) is a defect of the primary hemostatic pathway that results from quantitative or qualitative deficiencies of the von Willebrand factor (VWF). Type 1 VWD is the commonest form and occurs due to reduced level of VWF protein. It typically presents with mucocutaneous bleeding, but symptoms can vary with severity of the disease.
Detailed bleeding history using bleeding assessment tools, family history, and screening laboratory investigation including platelet function analysis can aid diagnosis. Specific testing involves quantifying von Willebrand antigen and activity levels. VWD is managed by factor replacement and adjunctive therapies (e.g., anti-fibrinolytics and desmopressin analogue, DDAVP) while educating patients toward minimizing bleeding.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Case PresentationYou are seeing a 6-year-old girl in clinic today, referred to hematology for history of easy bruising and a workup initiated prior to tonsillectomy and adenoidectomy. She has a normal prothrombin time, mildly prolonged partial thromboplastin time at 36 seconds (per the lab reference), a ristocetin cofactor level of 25%, and a von Willebrand antigen level of 27%. Her factor VIII level is within the normal range (>50%) and so are her multimers. All other bleeding workup was also normal, including her platelet count.
Multiple-Choice Management Question
The next steps in planning for her upcoming surgery include:
-
(a)
Repeat von Willebrand profile to confirm diagnosis
-
(b)
Set up a trial for assessing response to DDAVP
-
(c)
Both A and B
-
(d)
Neither of A and B and plan for surgery with Humate-P™ considering the invasiveness of the surgery
Being a pre-surgical evaluation, we have enough time to complete the workup. This patient’s labs are consistent with a type 1 VWD, with moderate levels. Next steps will be to confirm the diagnosis and set up a desmopressin/DDAVP trial as DDAVP can be a viable therapeutic option (option c). The upcoming procedure should be postponed until such evaluation is carried out and a complete perioperative care plan can be communicated with the surgical and anesthesia teams, so as to avoid bleeding complications.
Introduction
Von Willebrand disease (VWD) was first described in 1926 by a Finnish physician, Erik Adolf von Willebrand, after encountering a 5-year-old girl with history of bleeding [1]. VWD is identified as a quantitative or functional defect of the von Willebrand factor (VWF). The von Willebrand protein, commonly referred to as von Willebrand antigen (VWF:Ag), plays essential roles in platelet activation and adhesion in primary hemostasis. It is also a chaperone protein for factor VIII (FVIII), stabilizing the FVIII and prolonging its circulating half-life four–sixfold [2]. The incidence of VWD varies between 0.8% and 1.3% [3].
Clinical Presentation
VWD is typically an abnormality of the primary hemostatic pathway. Patients often present with skin- and mucosal lining-related bleeding: epistaxis, gum and oral bleeding (prolonged bleeding after tooth extractions and from biting the cheek or lips, and mouth bleeding), easy bruising, gastrointestinal tract bleeding, and/or menorrhagia. The presentation can vary based on the severity and type of disease.
There are many clinical variants of VWD, and the broad categorization is into three types: 1, 2, and 3. Type 1 is the commonest of the types of VWD and is a quantitative insufficiency of VWF:Ag. This chapter will focus on type 1 disease. Type 2 is a qualitative defect of the VWF:Ag, whereas type 3 indicates negligible to no circulating VWF.
Type 1 VWD Pathophysiology
Type 1 VWD accounts for about 75% of all VWD cases [4]. It is primarily inherited in an autosomal dominant pattern and is a partial deficiency of otherwise normal and functional VWF. Severity of symptoms in type 1 VWD typically correlates with degree of deficiency of the factor. Type 1 VWD, hence, can further be subclassified based on the degree of quantitative deficiency of VWF:Ag. VWD is an inheritable bleeding disorder with its genetic basis being much better understood now, due to advances over the last four decades.
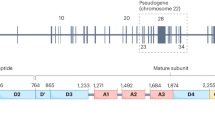
The von Willebrand (VW) protein is a large glycoprotein produced in endothelial cells and bone marrow megakaryocytes. Its production includes sophisticated processes involving initial synthesis, dimerization, glycosylation, and then multimerization, packaging for storage and subsequently cleaving, and release into circulation for its function (Fig. 9.1). The VW protein has excess of 2800 amino acids. The gene controlling this execution is the VWF gene, on short arm of chromosome 12, which is 178 kb and made up of 52 codons [5]. The mature VW protein is stored in the alpha granules of platelets and the Weibel-Palade (WP) bodies of the endothelial cells in vessel wall lining. The defects leading to VWD occur at many steps during this processing of the peptide. The majority of type 1 VWD is due to missense mutations in the coding regions, promoter, or splice sites in the VWF gene [6].

Structure of the von Willebrand protein with binding sites and sites of qualitative defects
The VWF protein, at baseline, circulates in a globular form and as a non-covalent complex with FVIII. Its activity is through two functions: first, it promotes hemostasis by mediating platelet adhesion to damaged vascular subendothelial matrix (e.g., collagen) and further platelet aggregation; second, it is the carrier protein for FVIII, protecting it from rapid proteolysis. The adhesive activity of VWF depends on the size of its multimers, with larger multimers being the most hemostatic. When there is exposure of sub-endothelium and modified flow dynamics with a vascular injury, a conformal change occurs in the VWF protein. As a result, the higher-molecular-weight multimers interact with collagen and platelet receptors with eventual regulatory participation from FVIII as well as in thrombus generation [7].
Besides vascular injury, there are a number of factors that can affect the circulating levels of VWF. Physiologically, the production of VWF in the body increases with age [8]. Also, pregnancy and high estrogen states can lead to increased VWF. Another hormonal pathway that can have an effect on VWF levels is thyroid, as hypothyroidism is associated with VWD. Stressors, such as a recent procedure or agitation due to blood draw, can increase the amount of VWF in the blood. A recent infection, inflammatory state, or anemia can also elevate VWF levels. Rigorous physical activity and exercise are known to raise the VWF level in the blood as well [9]. ABO blood groups can also exert a biological effect on VWF. People with type O blood have about 25% lower levels of VWF than people in blood groups A, B, and AB [10].
Type 1 Subclassification
Type 1 C – or “clearance type” – is a variant of type 1 VWD in which the survival of VWF is decreased due to increased clearance of the protein. It is also inherited in an autosomal dominant fashion and has another subtype known as the VWD Vicenza variant. The Vicenza-type VWD was originally described in patients from the Vicenza area in Italy and is notable for ultra-large multimers of VWF which are hypothesized to be subsequently cleared out of circulation, leaving behind very low level of plasma VWF despite the normal level of platelet VWF content [11].
Severe type 1 VWD is the quantitative deficiency of VWF, wherein significant clinical manifestations of bleeding may be present and can be seen at VWF:Ag and activity levels lower than 5 or 10 IU/dL. These patients usually have a correspondingly low FVIII, typically <20 IU/dL.
Differential Diagnosis
For patients presenting with mucocutaneous bleeding, consistent with a primary hemostatic-like defect, one must consider VWD in the differential. A detailed history and review of symptoms can help guide clinicians toward the diagnosis. To aid such history taking, multiple bleeding assessment tools have been designed, that help in screening patients for possible inherited bleeding diatheses. Several of these have been validated for VWD, namely, Vicenza bleeding score, the Pediatric Bleeding Questionnaire (PBQ), and ISTH/SSC Joint Working Group’s Bleeding Assessment Tool (ISTH-BAT), including their self-administrable versions, the Self-BAT and Self-PBQ [12]. The exact cutoffs for bleeding score obtained from these tools vary based on the type of instrument and patient’s age and gender, and continue to be an area of research. One may extrapolate from all these data that a score ≥2 can be considered abnormal for children [13] and can support initiating further workup for VWD.
Most bleeding assessment tools do not review the family history information. Hence, with a negative personal bleeding symptomatology, especially in the evaluation of children with none to minimal history of hemostatic challenges, family history is indispensable. If there is a known history of VWD or significant mucocutaneous bleeding in a close family member, it may be reasonable to proceed with initial evaluation, even with a low to normal patient bleeding score.
The differential for VWD includes abnormalities in platelet number, function- or adhesion-related defects, or dysfunction of collagen-platelet interaction. A quantitative defect of platelets, like immune thrombocytopenia, would also present in a similar bleeding phenotype. Disorders of collagen that interfere with platelet adhesion to sub-endothelium, e.g., Ehlers-Danlos syndrome, could also be difficult to discern from VWD in presentation.
Laboratory Findings
The evaluation for a bleeding disorder concerning for possible VWD, should start with a complete blood count (CBC). CBC can identify thrombocytopenia and other cellular abnormalities. Prothrombin time (PT) and partial thromboplastin time (PTT) are typically normal in VWD and can be part of the screen. In certain situations, PTT maybe prolonged, if the FVIII levels are low [14].
Platelet function analysis (PFA) can also detect more severe forms of VWD. It utilizes blood flowing through two different cartridges (containing either collagen plus ADP or collagen plus epinephrine). This test mimics the in vivo shear forces and activation of platelets followed by interaction with VWF, leading to hemostasis via clot/occlusion formation. In VWD, the PFA will be prolonged in both cartridges. Other platelet function issues can also give the same result though. Hence, prolonged PFA results are consistent with, but not specific for, VWD.
More specific testing for VWD to differentiate different subtypes, includes measurement of the von Willebrand protein itself (Table 9.1). This is achieved by carrying out an ELISA-based assay of the VWF:Ag and measurement of its function. The quantification of the VWF function is most commonly achieved by stimulating the platelet binding through use of antibiotic ristocetin as a cofactor [15]. The ristocetin-based testing for the VW function (VWF:RCo) can have high variability – in between different laboratories and within the same lab or same patients’ samples as well. It also does not directly measure physiologic function and is an indirect stimulator of VWF activity. Newer methodologies may prove more consistent and reliable. One of these is the glycoprotein IbM (GPIbM) testing, which utilizes a gain of function mutation-based assay, allowing the VWF to bind to platelets spontaneously, without needing ristocetin stimulation [16]. This assay is now commercially available from at least one reference laboratory in the United States. Factor VIII quantitative level is an important supplement to VWF functional evaluation, because it assesses the VWF’s ability to bind to FVIII, preventing its proteolysis. FVIII level is important in differentiating the type 2 subtypes and for diagnosis of type 3 VWD.
Testing of the VWF multimers is another specific test for VWD, but the multimer distribution is typically normal in type 1 VWD (Fig. 9.2). When the VWF:Ag and VWF:RCo are both reduced in a proportionate fashion, multimer testing is not required, unless the clinician has concerns for type 2M disease. Platelet function and multimer evaluation are expanded upon further in the following chapter, as platelet testing is essential for variants of VWD type 2. More details on genetic testing for VWD are provided in Chapter 10.

Multimer distribution patterns in various von Willebrand disease forms. VWD von Willebrand disease, TTP thrombotic thrombocytopenic purpura. (Used with permission from source: Richard Torres, Yuri Fedoriw. Laboratory Testing for von Willebrand Disease: Toward a Mechanism-Based Classification, Clinics in Laboratory Medicine, 2009-06-01, Volume 29, Issue 2, Pages 193–228, Copyright © 2009 Elsevier Inc.)
With regard to the VWF:Ag and VWF:RCo testing, what levels should be considered diagnostic for VWD continues to be a matter of debate. The United Kingdom and National Heart, Lung, and Blood Institute (NHLBI) guidelines advise VWF:Ag and activity of less than 30 IU/dL, as being diagnostic of VWD. These define the range of 30–50 IU/dL to be “low von Willebrand levels”, as patients in these ranges may also be experiencing symptoms that are more than the general population, but not severe enough to be called as disease [9, 17]. Canadian centers approach the diagnostic levels slightly differently and define levels <40 IU/dL as VWD. Per the NHLBI guidelines, those with low VWF levels, but a significant bleeding phenotype, may still be candidates for similar management as for those with VWD. For type 1C, the quantification of VWF propeptide can be helpful in making the correct diagnosis. There will be a higher circulating propeptide level than the VWF:Ag in type 1C.
For our patient in the clinical case, with ristocetin cofactor level of 25% and a von Willebrand antigen level of 27%, a diagnosis of type 1 disease could be made. Multimer testing is not necessary for type 1 disease, and this can be determined by noting that the VWF activity and antigen levels are proportionately decreased (i.e., VW activity/antigen ratio is >0.6).
During the initial workup for VWD, one must remain cautious of all the factors and stressors that can transiently increase the levels of VWF as discussed earlier. Patients with blood type O are known to have lower VWF levels but may accordingly have bleeding phenotypes correlating well with their levels. Also, when approaching the decision on timing of testing, age and recent stressors should be taken into account. In young children and infants with strong family history of VWD, timing of testing can be challenging. Testing and any elective major hemostatic challenges should be delayed beyond 6 months to a year of age, when better quantification of innate VWF can be done.
Management Options
The management of VWD varies significantly with the type and severity of disease. The management options include (1) replacement therapy, (2) supportive and adjunctive therapies, and (3) anticipatory guidance to minimize bleeding complications in patients.
Replacement Factors
Replacement products are utilized with a goal of improving circulating levels of VWF and FVIII. Humate-P™, Alphanate™, and Wilate™ are the plasma-derived products approved by Food and Drug Administration (FDA) for use for VWD in the United States. These products directly increase the circulating VWF and FVIII levels. They should be dosed based on their labeled content of VWF:RCo units, because each product is different for the content ratio of VWF:FVIII. Vonvendi™, the first recombinant VWD concentrate available in the United States, does not contain FVIII. It is not yet FDA approved for use in children.
Adjunctive Therapies
Desmopressin/DDAVP
Desmopressin or 1-deamino-8-D-arginine vasopressin (DDAVP), is a synthetic vasopressin analogue that leads to release of VWF and subsequently increased plasma VWF and FVIII concentrations. It acts as an agonist at the V2 receptors in the WP bodies and has no effect on the platelet-stored VWF.
Its successful use requires an evaluation of the patient’s ability to respond to it. Responsiveness is determined by “DDAVP challenge” testing. The challenge testing can be done with intranasal (IN) or intravenous (IV) DDAVP. Stimate® is the only brand of IN DDAVP that has sufficient concentration for VWD use. Young children (<5 years of age), who are unable to sniff on command, may be candidates for responsiveness testing through IV dosing. A baseline level of VWF:Ag and activity (RCo or GPIbM) and FVIII is obtained, followed by DDAVP administration. Then 1-, 2-, and 4-hour post levels are assessed to determine response. Successful response criteria include at least a twofold rise in VWF:Ag and VWF:RCo, in addition to achieving levels >30 IU/ml. In patients who do respond, if used too frequently, the response may wane once stored VWF is depleted. The most commonly studied frequency of use is every 24 hours for 3–4 doses. DDAVP should not be used any more than every 12 hours [9]. When used more frequently, tachyphylaxis may occur, and there is increasing risk of electrolyte disturbances. DDAVP’s side-effect profile includes flushing, tachycardia, edema, and headache. DDAVP leads to water retention, which can result in hyponatremia and, subsequently, seizures. Careful anticipatory guidance about fluid intake is essential to limit side effects. A guide for the allowable fluid volumes for weight is provided in Table 9.2.
Patients with history of severe headaches/migraines, autonomic dysfunction, and seizures may do poorly due to the side-effect profile of DDAVP, and careful counseling and risk-benefit discussion should be carried out with the family before the test.
Additionally, DDAVP is contraindicated in some type 2 variants of VWD (discussed in further detail in Chapter 10). In type 1C, a transient increase in the VWF levels is seen at the 1-hour post DDAVP dose testing, but the 4-hour post level is again low.
Antifibrinolytics
Synthetic lysine analogues, epsilon aminocaproic acid (EACA) and tranexamic acid (TXA), are agents that can be useful in stabilizing the fibrin clot in patients with defective hemostasis. They prevent fibrinolysis by inhibiting plasminogen activation. In bleeding from sites with higher concentration of natural fibrinolytics (e.g., nose, oropharynx, gastrointestinal tract), these prove effective in reducing clot breakdown. They can be used alone or as adjunctive therapy, with DDAVP or VWD concentrates.
EACA is available in a liquid formulation and pill form, but its high cost may prove prohibitive. TXA, on the other hand, is relatively cheaper but is only available in tablet form, which limits use in children. Both of these agents are available in injectable forms, for use in the perioperative and inpatient setting.
Other management approaches for specific bleeding manifestations in VWD (e.g., menorrhagia) are discussed in Chapter 10.
Comprehensive Care
VWD is a chronic condition that can affect many facets of the lives of patients. For patients with significant bleeding symptoms, quality of life can be poor as their symptoms may pose several limitations on their activities. Patients with VWD should be cared for in hemophilia treatment centers (HTCs). HTCs offer comprehensive care with multidisciplinary expertise. These centers have specialized experience in guiding the management of complicated scenarios like surgeries, trauma-related bleeding, access to research protocols, as well as newer developments in the management of bleeding disorders. Annual to biannual comprehensive visits, based on the frequency of bleeding symptoms, can help improve the general health maintenance for children with VWD.
Clinical Pearls and Pitfalls
-
Mucosal bleeding is the most common manifestation of von Willebrand disease.
-
Glycoprotein IbM testing is an alternative to ristocetin cofactor testing to measure von Willebrand factor activity.
-
Desmopressin affects fluid balance; thus, careful anticipatory guidance on fluid intake is essential to avoid side effects.
-
Stimate is the only brand of intranasal DDAVP appropriate for use in VWD.
-
Antifibrinolytic agents are a useful adjunctive therapy.
-
Von Willebrand levels can be affected by stress, exercise, and inflammation, so testing results should be interpreted with caution in these settings.
-
Patients who are pregnant or on estrogen-containing medications may have higher than baseline VW levels due to the estrogen effect.
References
Von Willebrand E. Hereditary pseudohemofilia. Fin Lakaresallsk Handl. 1926;68:87–112.
Lenting PJ, VAN Schooten CJ, Denis CV. Clearance mechanisms of von Willebrand factor and factor VIII. J Thromb Haemost. 2007;5(7):1353–60.
Werner EJ, Broxson EH, Tucker EL, Giroux DS, Shults J, Abshire TC. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr. 1993;123(6):893–8.
Haberichter SL, Castaman G, Budde U, Peake I, Goodeve A, Rodeghiero F, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). Blood. 2008;111(10):4979–85.
Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424.
Lillicrap D. Genotype/phenotype association in von Willebrand disease: is the glass half full or empty? J Thromb Haemost. 2009;7(Suppl 1):65–70.
Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–46.
Sanders YV, Giezenaar MA, Laros-van Gorkom BA, Meijer K, van der Bom JG, Cnossen MH, et al. von Willebrand disease and aging: an evolving phenotype. J Thromb Haemost. 2014;12(7):1066–75.
Nichols WL, Hultin MB, James AH. Von Willebrand disease (VWD): evidence- based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia. 2008;14:171–232.
Jenkins PV, O’Donnell JS. ABO blood group determines plasma von Willebrand factor levels: a biologic function after all? Transfusion. 2006;46(10):1836–44.
Mannucci PM, Lombardi R, Castaman G, Dent JA, Lattuada A, Rodeghiero F, et al. von Willebrand disease “Vicenza” with larger-than-normal (supranormal) von Willebrand factor multimers. Blood. 1988;71(1):65–70.
Bowman ML, James PD. Bleeding scores for the diagnosis of von Willebrand disease. Semin Thromb Hemost. 2017;43(5):530–9.
O’Brien SH. Bleeding scores: are they really useful? Hematology Am Soc Hematol Educ Program. 2012;2012:152–6.
Castaman G, Rodeghiero F. Advances in the diagnosis and management of type 1 von Willebrand disease. Expert Rev Hematol. 2011;4(1):95–106.
Vanhoorelbeke K, Cauwenberghs N, Vauterin S, Schlammadinger A, Mazurier C, Deckmyn H. A reliable and reproducible ELISA method to measure ristocetin cofactor activity of von Willebrand factor. Thromb Haemost. 2000;83(1):107–13.
Sharma R, Flood VH. Advances in the diagnosis and treatment of Von Willebrand disease. Blood. 2017;130(22):2386–91.
Laffan MA, Lester W, O’Donnell JS, Will A, Tait RC, Goodeve A, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014;167(4):453–65.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kaur, D., O’Brien, S.H. (2020). Classification and Management of Type 1 von Willebrand Disease. In: Dunn, A., Kerlin, B., O'Brien, S., Rose, M., Kumar, R. (eds) Pediatric Bleeding Disorders. Springer, Cham. https://doi.org/10.1007/978-3-030-31661-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-31661-7_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-31660-0
Online ISBN: 978-3-030-31661-7
eBook Packages: MedicineMedicine (R0)