
Abstract
As cancer is the leading cause of death worldwide, there is an urgent necessity to discover novel diagnostic and prognostic biomarkers as well as therapeutic targets for this dreadful disease. The progression of various malignancies can be the result of abnormalities in multiple epigenetic regulations. Over the past decades, major epigenetics categories, including histone modification, DNA methylation, noncoding RNAs, and chromatin remodeling, have been reported to be involved in tumor genesis and development. Therefore, epigenetic changes can be used as clinical biomarkers for the diagnosis of cancer and to predict the prognosis. Moreover, epigenetic regulators have emerged as promising drug targets for cancer therapy. This review delineates the latest evidence in epigenetics alterations in cancer and discusses their potential contribution to the diagnosis, prognosis, and therapy of cancer. These diagnostic, prognostic, and therapeutic strategies based on epigenetics might bring hope to reducing the high fatality rate of malignancies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Epigenetics
- DNA methylation
- Histone modification
- Noncoding RNAs
- Chromatin remodeling
- Diagnosis and prognosis of cancer
- Therapeutic strategy
1 Epigenetics and Cancer
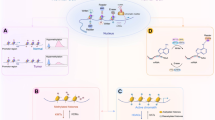
Classical genetics assumes that the molecular basis of heredity is nucleic acid, and the genetic information of life is stored in the base sequences of nucleic acids. Changes in the base sequences will cause changes in the phenotype of an organism, which can be transmitted from one generation to the next. However, with the development of genetics, it has been found that modifications at the DNA, histone, and chromosome levels can also cause changes in gene expression patterns that can be inherited. Modifications that alter the genome without affecting the DNA sequence can not only affect the development of the individual but also be passed on to future generations, and epigenetics refers to such changes in gene expression levels based on nongenetic sequence changes (Skvortsova et al. 2018). Epigenetics is currently divided into several categories, including histone modification, DNA methylation, noncoding RNAs (ncRNAs), and chromatin remodeling (Akone et al. 2020; Zhang et al. 2020a) (Fig. 4.1).

Applications of epigenetics in cancer include histone modifications, DNA methylation, noncoding RNAs, and chromatin remodeling. (a) Histone modifications. Histone modifications such as acetylation and methylation are among the most widely affected epigenetic pathways in tumors. (b) DNA methylation. Loss of DNA methylation results in abnormal transcription of target genes. (c) Noncoding RNA. Noncoding RNA regulation can alter the transcription and translation of oncogene targets. (d) Chromatin remodeling. Regulation of chromatin remodeling factor SWI/SNF
1.1 Histone Modification
Histone modifications mainly include acetylation, methylation, phosphorylation, and ubiquitination (Zhu et al. 2021). These modifications can recruit recognition proteins that recognize the modification sites (Zhang and Pradhan 2014), which in turn recruit other transcription factors or can form complexes with numerous physiological functions for transcriptional regulation (Lambert et al. 2018). Acetylation and methylation are among the most widely affected genetic pathways in tumors (Taby and Issa 2010), and many proteins that modify specific histones or bind specific histone modification sites have dysregulated activity in tumors (Rice et al. 2007). Histone acetyltransferase (HAT) and histone deacetylase (HDAC) are capable of acetylating or deacetylating a variety of nonhistone proteins, including p53, retinoblastoma (Rb), and myelocytomatosis oncogene (MYC) (Dang and Wei 2021; Lafon-Hughes et al. 2008; Wagner et al. 2014). HDAC is overexpressed in a variety of tumors, resulting in loss of histone acetylation and silencing of tumor suppressor gene expression (Yoon and Eom 2016). HDAC is divided into four classes, Among them, 11 subtypes, including class I, II, and IV, are all Zn2+-dependent proteins; seven subtypes of class III, Sir1–7, use nicotinamide adenine dinucleotide (NAD+) as the catalytic active site (Li and Seto 2016; Porter et al. 2017; Sixto-López et al. 2020). HDAC inhibitors are potential antitumor compounds, and many studies have shown that abnormal expression of HDACs is associated with a variety of tumors. By analyzing the expression of HDACs in 13 tumors (chronic lymphocytic leukemia, gastric cancer, breast cancer, colon cancer, liver cancer, medulloblastoma, non-small-cell lung cancer (NSCLC), lymphoma, neuroblastoma, ovarian cancer, pancreatic cancer, prostate cancer, and kidney cancer), the expression of class I HDACs was found in 11 types of tumors, indicating that class I HDACs might play a key role in tumorigenesis and invasion, and might be a promising antitumor target (Chun 2015). Histone methylation occurs at the N-terminal lysine or arginine residues of H3 and H4 histones (Yi et al. 2017). Mutations or altered expression of histone methyl modifications and methyl-binding proteins are associated with increased incidence of a variety of different cancers. For example, H3K27me3 methyltransferase is upregulated in some cancers, including prostate cancer, breast cancer, and lymphoma (Duan et al. 2020). Importantly, activating point mutations in Enhancer of zeste homolog 2 (EZH2) were recently found to be associated with B-cell lymphomas, which is consistent with the notion that EZH2 is oncogenic (Duan et al. 2020). Therefore, epigenetic drugs targeting acetyl and methyl groups may have clinical implications in cancer therapy (Fig. 4.1a).
1.2 DNA Methylation
In tumors, genome-wide and individual gene methylation patterns are often altered. DNA methylation can add methyl groups to DNA molecules without changing the DNA sequence, thereby regulating the effect of genetic expression. Many recent studies have shown that DNA aberrant methylation is closely related to the occurrence, development, and carcinogenesis of tumors. Aberrant DNA methylation in malignancies is mainly caused by DNA hypermethylation or hypomethylation (Nishiyama and Nakanishi 2021). Genome-wide hypomethylation is frequently detected in tumor genomes, which is often considered a hallmark of cancer cells (Dong et al. 2014). In contrast, DNA hypermethylation often leads to transcriptional repression and reduced gene expression, often occurring in specific CpG-enriched regions that result in the silencing of tumor suppressor genes (Pfeifer 2018). Therefore, changes in DNA methylation levels and changes in specific gene methylation levels can be used as tumor diagnostic indicators. DNA hydroxymethylation, another type of DNA modification, is produced in mammals mainly by sequential oxidative catalytic reactions of the ten-eleven translocation (TET) gene family, and the expression of TET family member TET2 is reduced in various hematopoietic malignancies, including acute myeloid leukemia and myeloproliferative disorders (Heiblig et al. 2015). Similar to DNA methylation, methylation modification packages also appear on RNA, including N6-methyladenosine (m6A), 5-methylcytosine (m5C), N1-methyladenosine (m1A), N3-methylcytosine (m3C), and N7-methylguanosine (m7G) (Yang et al. 2021). Among them, m6A is the most common RNA modification in mammals, which is related to a variety of malignancies, such as acute myeloid leukemia (AML) (Kumar et al. 2021), glioblastoma (Cui et al. 2017), breast cancer (Shi et al. 2020b), and hepatoblastoma (Liu et al. 2019; Ma et al. 2019; Sun et al. 2019b). In the process of m6A methylation, methyltransferase-like 3(METTL3) is the key methyltransferase, which can affect tumor formation by regulating the m6A modification in mRNA through key oncogenes or tumor suppressor genes (Wang et al. 2020b) (Fig. 4.1b).
1.3 Noncoding RNAs
With the development of genomics and bioinformatics, especially the massive application of high-throughput sequencing technologies, scientists have discovered an increasing number of nonprotein-coding transcription units like ncRNAs. Long noncoding RNAs (lncRNAs) play a vital role in diverse important biological processes, and lncRNAs can regulate gene expression in a variety of cells during early mammalian development (Fatica and Bozzoni 2014). Alterations of lncRNAs in cancer cells have also been found to be closely associated with tumor formation, progression, and metastasis. It has also been found that ncRNAs, especially microRNA (miRNA), are involved in the development of inflammatory responses and they are important for stabilizing and maintaining the genotypic characteristics of some cell types (Li et al. 2016). miRNA can affect oncogene expression, induce apoptosis, and participate in downstream regulation of oncogenes in tumor cells. In B-cell chronic lymphocytic leukemia (B-CLL), the cluster consisting of miR-15a and miR-16-1 is frequently absent or down-expressed, and this change is associated with the development of B-CLL (Bottoni et al. 2005). P53 is a well-known tumor suppressor gene, and its tumor suppressor effect partly comes from the transcriptional activation of tumor suppressor miRNA-miR-34a. In tumorigenesis, p53 often shows low expression, resulting in transcriptional repression of miR-34a (Shi et al. 2020a). Epigenetic mechanisms are also important causes of altered miRNA expression in cancer. During cancer development, lncRNAs are involved in the regulation of multiple epigenetic complexes that repress or activate gene expression. For example, lncRNA can bind to multiprotein complexes to regulate carcinogenesis. Polycomb repressive complex 1 (PRC1) and polycomb repressive complex 2 (PRC2) are known oncogenes that can cause many malignancies. The lncRNA, named Focally amplified lncRNA on chromosome 1 (FAL1), can bind to B-cell-specific Moloney murine leukemia virus insertion site1 (BMI1), a subunit of PRC1. In ovarian cancer, FAL1 has been reported to accelerate cancer progression and shorten patient survival time. The binding of FAL1 to BMI1 prevents BMI1 degradation to stabilize the PRC1 complex, which allows PRC1 to occupy and repress the promoters of target genes such as p21, leading to cell cycle dysregulation and increased chances of tumorigenesis (Hu et al. 2014) (Fig. 4.1c).
1.4 Chromatin Remodeling
During DNA transcription, chromatin changes from a tight superhelical structure to an open sparse structure, the structural change that does not alter the DNA base sequence is called chromatin remodeling (Goldberg et al. 2007). Chromatin remodeling is an important mechanism in epigenetic modification patterns, and chromatin remodeling regulates processes such as gene transcription, DNA repair, and programmed cell death. The chromatin remodeling enzyme amplification in liver cancer1 (ALC1), a potential oncogene, is activated in the presence of both poly (ADP-ribose) polymerase 1 (PARP1) and NAD+, driving nucleosomes to restructure chromatin (Ooi et al. 2021). Interestingly, chromatin remodeling complex ISWI complexes remodel in nucleosome arrays and nucleosome free zones, thereby regulating gene expression (Kwon et al. 2016), heterochromatin establishment and replication (Culver-Cochran and Chadwick 2013), DNA repair (Atsumi et al. 2015), as well as the coordination of rRNA gene expression (Erdel and Rippe 2011). Studies have shown that SWI/SNF subunits are highly mutated in a variety of cancers, including ovarian cancer, pancreatic cancer, kidney cancer, liver cancer, and bladder cancers (Kadoch et al. 2013) (Fig. 4.1d).
2 Epigenetics in Cancer Diagnosis
Notably, aberrant epigenetic modifications in organisms are usually closely associated with the occurrence and development of multiple cancers. For instance, DNA methylation, one of the first discovered epigenetic forms, is involved in a variety of cellular physiological functions and plays an essential role in the occurrence of diseases, especially cancer. Some studies have reported that the occurrence of specific types of cancer can be detected earlier by detecting changes in DNA methylation; thus, DNA methylation has a very high potential to be used as a biomarker for cancer diagnosis (Michalak et al. 2019). Importantly, a big data-based DNA methylation analysis showed that DNA methylation is tumor-specific and allows better detection of the primary tumor site, serving as a powerful diagnostic marker for primary cancers and leading to more precise and personalized therapies (Moran et al. 2016). Currently, lung cancer and colorectal cancer (CRC) are the top two causes of cancer death worldwide, which are often diagnosed at an advanced stage, missing the best time for therapy. DNA methylation, one of the most intensively studied epigenetic forms, is expected to significantly contribute to the early diagnosis of lung cancer and CRC. For example, a series of novel epigenetic regulatory molecules have been reported to help improve the diagnostic efficacy of standard clinical markers for diagnosis of lung cancer (Diaz-Lagares et al. 2016). Moreover, genome-wide hypomethylation is frequently captured in the early stages of CRC (Jung et al. 2020). In summary, in-depth studies of epigenetics hold the promise of greatly improving the early clinical diagnosis of multiple cancers and reducing the persistently high cancer mortality rate.
2.1 DNA Methylation as a Biomarker of Cancer Diagnosis
As mentioned above, DNA methylation has exciting potential to be a biomarker in cancer diagnosis. A genome-wide methylation analysis based on whole-genome bisulfite sequencing (WGBS) data and validated with methylation data from the Cancer Genome Atlas (TCGA) lung cancer cohort showed that some well-known methylation biomarkers for lung cancer, namely, SHOX2, POU4F2, BCAT1, HOXA9, and PTGDR, were all captured significantly. In addition, two novel hypermethylated genes, HIST1H4F and HIST1H4I, were significantly observed in both gene sets, with the area under the curve (AUC) of 0.89 and 0.90, respectively, reflecting the potential of both HIST1H4F and HIST1H4I as biomarkers for lung cancer diagnosis. Interestingly, the predictive potency of the combination was greater than that of the individual genes with an AUC of 0.95. Interestingly, TCGA pan-cancer methylation analysis showed that hypermethylation of HIST1H4F has the potential as a diagnostic biomarker for multiple cancers with AUCs of 0.9–1(Dong et al. 2019a). According to a new integrated epigenomic-transcriptomic analysis of lung cancer, eight novel hypermethylated driver genes, namely, PCDH17, IRX1, ITGA5, HSPB6, TBX5, ADCY8, GALNT13, and TCTEX1D1, were identified and validated with for predicting an AUC of 0.965 in lung cancer patients, demonstrating reliable clinical diagnostic value (Sun et al. 2021). Moreover, ITPKA gene body methylation could also be regarded as a novel diagnostic biomarker for lung cancer with an AUC of 0.93 in the TCGA-lung cancer cohort (Wang et al. 2016). Hitherto, there are two FDA-approved methylation-related diagnostic biomarkers in CRC, of which SEPT9 is a single-gene methylation biomarker and NDRG4 and BMP3 are multigene methylation biomarkers, both of which currently demonstrate compelling clinical value. In addition, SDC2, VIM, APC, MGMT, SFRP1, SFRP2, and NDRG4 are the most frequently reported methylation biomarkers with promising applications in the early clinical diagnosis of CRC (Müller and Győrffy 2022). In breast cancer, hypomethylation of SEPTIN7, TRIM27, LIMD2, and LDHA is often associated with malignant phenotype, while APC, RARB, GSTP1, DAPK, and SFN are frequently methylated in patients, and all these dysregulated methylation genes are of great diagnostic value (Sher et al. 2022). In a recent genome-wide methylation analysis of 91 esophageal squamous cell carcinoma (ESCC) cases in China, aberrant methylation of six genes was found to be associated with ESCC progression, namely, PAX9, THSD4, TWIST1, EPB41L3, GPX3, and COL14A1, and was similarly validated in TCGA data (Xi et al. 2022). In a meta-analysis of malignant mesothelioma, APC, miR-34b/c, and WIF1 were found to be promising diagnostic biomarkers, and further exploration of their diagnostic capabilities is necessary (Vandenhoeck et al. 2021). In cervical cancer, a modeling analysis based on TCGA methylation data yielded four potential diagnostic markers, namely, RAB3C, GABRA2, ZNF257, and SLC5A8, with AUCs of 94.2, 100, 100, and 100% in the Gene Expression Omnibus (GEO) dataset to distinguish between cancer and paracancerous tissue, which showed exciting diagnostic results (Xu et al. 2019) (Fig. 4.2).

The application of epigenetics in early cancer diagnosis. Currently, the potential of many epigenetic forms as early diagnostic markers has been widely reported in multiple cancers. Various cancer diagnostic markers have emerged in the four major epigenetic forms, DNA methylation, histone modification, non-encoding RNA, and imprinted genes
2.2 Histone Modification as a Biomarker of Cancer Diagnosis
Recently, abnormalities in histone modifications have been frequently captured in malignancies, reflecting the powerful potential of histone modifications in cancer diagnosis (Riedel et al. 2015). The results of a recent study suggest that upregulation of Histone-lysine N-methyltransferase SETD7 implies the occurrence of CRC and could serve as a potential biomarker for CRC (Duan et al. 2018). In addition, by detecting abnormalities in histone H3K27me3 and histone H4K20me, melanoma can be definitively diagnosed in a population with benign nevi (Davis et al. 2020) (Fig. 4.2).
2.3 Non-encoding RNAs as Biomarkers of Cancer Diagnosis
Notably, as an early cancer diagnosis strategy, the detection of emerging molecular ncRNAs has gradually made good progress. In NSCLC patients, a set of diagnostic lncRNAs HAGLR, ADAMTS9-AS2, LINC00261, MCM3AP-AS1, TP53TG1, C14orf132, LINC00968, LINC00312, TP73-AS1, LOC344887, LINC00673, SOX2-OT, AFAP1-AS1, and LOC730101 were reported to distinguish cancer and paraneoplastic tissues with a high AUC as 0.98 ± 0.01, reflecting a strong clinical application value (Sulewska et al. 2022). In gastric cancer, lncRNAs H19, HOTAIR, UCA1, PVT1, and LINC00152 were identified as potential diagnostic biomarkers (Fattahi et al. 2020). In addition, abnormalities in miRNAs are often captured in carcinogenesis. In CRC, miR-21, miR-9, miR-155, miR-17, miR-19, let-7, miR-24, miR-181b, miR-21, miR-183, let-7 g, miR-17, and miR-126 are all promising biomarkers for early diagnosis of CRC (Moridikia et al. 2018). Moreover, in breast cancer, upregulation of PIWI-interacting RNAs (piRNAs) piR-20,485, piR-20,582, and piR-20,365 could be used as early diagnostic biomarkers (Maleki Dana et al. 2020) (Fig. 4.2).
2.4 Imprinted Genes as Biomarkers of Cancer Diagnosis
Importantly, imprinted genes are also contributors to cancer diagnosis. Overexpression of imprinted SLC22A18 and SLC22A18AS gene has been reported to promote the occurrence and progression of NSCLC, and both genes can be used for early diagnosis of NSCLC (Noguera-Uclés et al. 2020). Another study showed that the imprinted genes GNAS, GRB10, SNRPN, and HM13 are also diagnostic markers for early-stage lung cancer, which are expected to translate to the clinic (Zhou et al. 2021). Similarly, the imprinted genes GNAS, GRB10, and SNRPZ also showed convincing diagnostic performance in another study (Shen et al. 2020) (Fig. 4.2).
3 Epigenetic in Cancer Prognosis
The epigenetic variation of cancer patients may enable the tumor to obtain the ability to adapt to the treatment, which will lead to a poor prognosis. In recent years, the research on epigenetics in cancer prognosis mainly focuses on the prognostic grading of cancer patients and the development of specific prognostic markers (Wong et al. 2019). Using epigenetic differences such as DNA methylation to classify patients and provide different targeted treatment methods is a research hotspot at present. The change in DNA methylation level exists in many cancers. Therefore, the identification and development of prognostic markers can be used to indicate tumor metastasis, recurrence, and 5-year survival rate. Now, the latest genome-wide epigenomics method makes it feasible to construct a comprehensive map of cancer methylation groups and may bring a standardized method for epigenetic prediction of cancer prognosis (Grady et al. 2021). The research content of epigenetics in cancer prognosis can be roughly divided into four aspects: DNA methylation, histone modification, chromatin remodeling, and functional noncoding RNA. This chapter will discuss the above four parts.
3.1 DNA Methylation in Cancer Prognosis
DNA methylation is an important part of epigenetic research, and differences in DNA methylation enable altered transcription of gene expression. Studies have shown that most of cancer patients, mainly gastric cancer, CRC, and liver cancer, tend to have some degree of changes in DNA methylation status (Mehdipour et al. 2020). Therefore, identifying abnormally methylated genes may provide new ideas for developing prognostic markers for cancer. Through studies of different liver cancer patients, it has been found that DNA methylation changes might contribute to a poorer prognosis. For instance, treatment with α-Interferon for liver cancer patients usually cannot achieve better curative effect when the patient’s miR-26a (an epigenetic marker) expression is high. Markers of tissue DNA methylation are equally important in the prediction of liver cancer prognosis and can serve as potential prognostic biomarkers for the staging of hepatocellular carcinoma mainly: keratin 19,5-hydroxymethylcytosine. In hepatocellular carcinoma, an analysis targeting the promoter methylation status of 105 possible tumor suppressor genes found that low methylation frequency might increase the risk of the poor prognosis (Tricarico et al. 2020). Meanwhile, the detection of DNA methylation abnormalities has also achieved certain results in the diagnosis and prognosis of gastrointestinal tumor. For example, the CpG island methylator phenotype (CIMP), first discovered in CRC, might be a potential prognostic marker for CRC as well as gastric cancer. One study, which analyzed more than 600 CRC patients, suggested that CIMP was potentially associated with poor prognosis in microsatellite stable CRC patients (Liu et al. 2020). It may become a new basis for patient prognostic stratification through methylation profiling of cancer patient samples as well as existing therapeutic targets. Evidence has been presented that there is epigenetic differential regulation of Lag3 by DNA methylation in tumor cells and normal immune cells, and Niklas et al. found that methylation of the promoter resulted in lower amounts of Lag3 expression, which negatively correlated with poor prognosis (Klumper et al. 2020). O6 methylguanine DNA methyltransferase (MGMT) gene promoter methylation levels and can be used as a basis for histologic stratification of patients with glioblastoma (GBM), as well as for prediction of posttreatment survival (Mansouri et al. 2019). An important reason that can lead to a poor prognosis of cancer is that tumors may acquire drug resistance in treatment, and an increasing number of studies have shown that epigenetic modification of mRNA has a certain link with tumor drug resistance. mRNA modifications include m6A, and Fukumoto T. et al. found that m6A modification in fzd10 mRNA is positively correlated with its stability and may be associated with resistance to PARP inhibitors in BRCA mutated epithelial ovarian cancer (EOC) (Fukumoto et al. 2019).
3.2 Histone Modifications in Cancer Prognosis
Histone modification refers to the process by which histones undergo methylation, acetylation, phosphorylation, and other modifications under the action of related enzymes. An increasing number of studies have shown that the modification of histones is inseparable from the occurrence and development of cancer. Similar to methylation of DNA, in which methylation of histones is more widespread in histone modification studies. Currently, some scholars focus their eyes on the synergistic roles of DNA and histone modifications in cancer progression, and G9a histone methyltransferase and DNA methyltransferase I were found to be significantly overexpressed in hepatocellular carcinoma. Both are synergistically associated with poor prognosis in hepatocellular carcinoma, and thus, intervention in hepatocellular carcinoma at the epigenetic level may be achieved through inhibition of G9a and DNMT1 (Barcena-Varela et al. 2019). Studies targeting epigenetic alterations have found that histone methylation and modifying enzymes may play a role in prognosis prediction in various cancers. Methylation of histone H3K27me3 has been associated with breast cancer migration and may serve as a prognostic marker (Hsieh et al. 2020). Studies have shown that inhibition of euchromatin histone-lysine-n-methyltransferases I and II (EHMT1/2) may reverse partial ovarian cancer resistance to PARP inhibitors (Watson et al. 2019). Histone modifications are closely associated with epigenetic alterations in gene expression and have significant research potential in both the identification of cancer subtypes and the development of predictive markers for patient survival. CDX2 is a prognostic biomarker for colorectal, and the histone deacetylases HDAC4 and HDAC5 can repress CDX2 expression (Graule et al. 2018). Nowadays, new concepts reveal that the tumor microenvironment may be associated with histone modifications, and a persistent hypoxic microenvironment has been found in pancreatic cancer capable of altering histone methylation (Li et al. 2021). The hypoxic tumor microenvironment serves as a potential judgment for cancer malignancy, which also provides a new direction for predicting the prognosis of tumors through histone modification status. The role of epigenetics in multiple myeloma has similarly received attention from investigators, and some modifying enzymes that alter the acetylation status of histones may be involved in the progression of multiple myeloma. The development of inhibitors targeting histone deacetylases may offer new therapeutic possibilities for multiple myeloma (Ohguchi et al. 2018).
3.3 Histone Variants and Chromatin Remodeling in Cancer Prognosis
Chromatin remodeling is a process in which chromatin unfolds during gene-initiated expression and depends on three dynamic properties of nucleosomes, including remodeling, enzyme induced covalent modification, and repositioning. In which histone variants can be incorporated into nucleosomes and displace existing nucleosomal subunits, a process that is tightly regulated by chromatin remodeling factors, such as the SWR1 complex. At the same time, these variants can also affect chromatin remodeling and thus transcriptional regulation. In addition to mutations in genes involved in chromatin remodeling that are frequently observed in many types of cancer, for example, studies of chromatin remodeling genes suggest that SMARCA4 may be involved in neuroblastoma tumorigenesis, numerous studies have also shown that histone variants can predict prognosis in various cancers. John Blenis and Ana P. Gomes et al. collaborate to discover that the histone H3 variant H3.3, under the action of histone chaperones that regulate metastasis, deposits at metastasis to induce transcription factor promoters and promote tumor metastasis. Standard histone intercalation is reduced in chromatin, leading to the deposition of poor prognosis genes in tumors (Gomes et al. 2019).
The family of chromatin regulators plays an important role in chromatin remodeling. Among these, cBAF is the most abundant of the SWI/SNF complexes. ARID1A is the largest subunit homologous to cBAF, and its gene is mutated at a frequency of up to 50–60% during carcinogenesis. There are studies showing partial or complete inactivation of arid2 expressed protein in liver cancer. Researchers have found that the C2H2 domain of ARID2 can recruit DNMT1 to the promoter of the transcription factor Snail, which elevates DNA methylation and inhibits the transcription of snail, thereby inhibiting epithelial mesenchymal transition in liver cancer cells. Therefore, mutations in the C2H2 domain of ARID2 promote liver cancer metastasis and reduce the 5-year survival rate of patients with liver cancer (Jiang et al. 2020).
Additional studies identified the chromatin structure regulators SND1 and RHOA as independent predictors of poor prognosis in glioma patients. SND1 can remodel chromatin conformation, allowing transcriptional upregulation of RHOA. Thus, it activates the Cyclin/CDK signaling pathway, which enables the G1/S phase transition of the glioma cell cycle and promotes glioma cell proliferation, migration, and invasion (Yu et al. 2019). In the development of CRC, the chromatin remodeling genes PRMT1 and SMARCA4 have higher expression; thus, inhibition of PRMT1/SMARCA 4 may be used as an intervention strategy to prolong the overall survival of patients with intestinal cancer (Yao et al. 2021). Whereas in hepatic cell carcinoma (HCC) cells, overexpression of the chromatin remodeler Hells epigenetically silences multiple tumor suppressor genes, thereby promoting HCC cell proliferation and migration, thus manifesting as more aggressive and worse patient outcomes in clinicopathological features (Law et al. 2019).
3.4 Noncoding RNAs in Cancer Prognosis
ncRNAs refer to RNAs that do not encode proteins. In the development of tumors, ncRNAs are involved in the process of their proliferation, differentiation, and metastasis, which play an extremely important role in the prognosis of tumors. Currently, a variety of ncRNAs are found to serve as tumor markers, becoming a hotspot of tumor research in recent years. miRNAs contain only 22–24 nucleotides, but by imperfect base complementarity, miRNAs can match and silence multiple mRNAs. In terms of tumor therapy, mir-302a affects tumor migration with the acquisition of drug resistance. Studies have suggested its role as a candidate prognostic predictor in CRC by targeting NFIB and CD44 (Sun et al. 2019a). Circular RNAs (circRNAs) do not have covalently closed loops at the 5 ‘and 3’ ends, but studies have found their ability to participate in transcription, regulation of translation, and localization of proteins. In the occurrence of multiple cancers, circRNAs can act on miRNAs, for example, CIRS-7 promotes CRC progression by blocking the tumor suppressive effect of miR-7. While in bladder cancer, circACVR2A acted as a miRNA sponge to regulate miR-626 to inhibit cancer cell proliferation and metastasis. This provides a new idea for the prognosis and treatment of bladder cancer (Dong et al. 2019b). In addition, it was demonstrated through autophagy-related experiments that the level of circCDYL increased in the tissues of breast cancer, thus promoting the level of autophagy in breast cancer cells and reducing the survival of breast cancer patients with curative effect, and this process was associated with miR-1275-ATG7/ULK1-AUTOP (Liang et al. 2020b). lncRNAs are a class of RNAs >200 bp in length that lack an effective open reading frame, sequestering little or no protein coding sequence. Analysis through multiple data means in recent years has shown that the expression of lncRNAs is associated with prognosis in multiple tumors (Zhang et al. 2020b). Acting with YBX1 in cells by specifically expressing lncRNA DSCAM-AS1 in tumors, adversely affecting the prognosis of tumors. In vitro and in vivo experiments have demonstrated the important regulatory roles of lncRNAs in tumorigenesis and development. In recent years, lncRNA research has become a current research hotspot. Multiple understudied lncRNAs were found to potentially play a role in tumor prognosis. LINC02273 contributed to the metastasis of breast cancer and increased the metastasis associated protein hnRNPL, which in turn activated the AGR2 axis, providing new protein markers for the cure and prognosis of breast cancer (Xiu et al. 2019). Whereas in CRC, lncRNA LINRIS may serve as an independent biomarker for its prognosis, which plays an important role in CRC by inhibiting aerobic glycolysis (Wang et al. 2019). It is believed that with the development of biotechnology, more novel ncRNAs will be found to function in cancer treatment and play more important roles in the prognosis of patients (Fig. 4.3).

Impact of epigenetic alterations on cancer prognosis. In the absence of alterations in the nuclear DNA sequence, tumor tissues exhibit diverse biological features through epigenetic modification pathways such as DNA methylation, histone modification, chromatin remodeling, and functional ncRNAs. Different degrees of epigenetic modification can alter cancer cell proliferation speed, migration ability and even lead to drug resistance. Therefore, DNA methylation, histone modification status, and so on can be used to predict the prognostic performance of cancer patients
4 Epigenetics in Cancer Therapy
As previously mentioned, epigenetic dysregulation has a far-reaching impact on gene expression, DNA replication, and DNA repair, which is closely associated with tumorigenesis and tumor progression. Unlike genetic mutations, epigenetic changes in cancer epigenome are mainly enzyme-catalyzed and probably reversible, which provides ideal targets for cancer treatment (Bianco and Gevry 2012; Lopez-Camarillo et al. 2019). So far, most epigenetic drugs are designed to modulate DNA methylation- and histone acetylation-related enzymes, including DNA methyltransferases, histone acetyltransferases, histone deacetylases, and histone methyltransferases, which are known as of importance targets of cancer treatment (Miranda Furtado et al. 2019).
4.1 Targeting DNA Methylation-Related Enzymes in Cancer Treatment
DNA methylation is the main epigenetic mechanism and a well-publicized epigenetic marker, in which the cytosine bases in CpG island are covalently modified by methyl groups. The DNMT family of enzymes, including DNMT1, DNMT3a, and DNMT3b, plays a pivotal role in the methylation process by catalyzing the transfer of a methyl group of s-adenosyl-l-methionine to DNA (Bestor 2000; Li et al. 1992; Okano et al. 1999; Schapira and Arrowsmith 2016). Hereunto, many DNMT inhibitors were discovered. For example, azacytidine and decitabine have been originally used to treat myelodysplastic syndrome and subsequently used in the treatment of chronic myelomonocytic (Derissen et al. 2013). Both can inactivate DNMT by forming an irreversible covalent complex with it (Stresemann and Lyko 2008). In addition, many other types of inhibitors, such as non-nucleoside chemicals, were developed in recent years. Procaine has been found to keep DNMT from interacting with DNA, whereby the promoter regions of CDKN2A and RAR β are less methylated (Li et al. 2018). In gastric cancer cells, procaine was detected to promote apoptosis and inhibit proliferation, suggesting the therapeutic potential of procaine on cancer as a DNMT inhibitor. MC3343, however, was found to exhibit proliferative activity on osteosarcoma cells by causing cell cycle arrest at the G0-G1 or G2-M phases, which can be attributed to its inhibitory activity against DNMT1, DNMT3a, and DNMT3b expression and biological activity (Manara et al. 2018). As an analog of MC3343, MC3353 acts as a DNMT inhibitor, displaying strong demethylation ability and providing reactivation of the silenced gene (Zwergel et al. 2019). By modulating the genes involved in osteoblast differentiation, MC3353 showed its activity in primary osteosarcoma cell models. In addition, compounds 3b and 4a can inhibit DNMT and therefore impair acute myelogenous leukemia cells KG1 and CRC cells HCT116 proliferation (Pechalrieu et al. 2020).
4.2 Targeting Histone Acetylation-Related Enzymes in Cancer Treatment
Being a crucial epigenetic regulation, the acetylation of histone lysine affects cell differentiation and proliferation by intervening the interaction between transcription factors and the regulatory sequence of oncogenes (Kulka et al. 2020). This process is generally controlled by related enzymes including KATs and KDACs. Among them, potential epigenetic targets HATs and HDAC are responsible for adding and deleting acetyl group to lysine residues (Tapadar et al. 2020). They are also known as “Writers” and “Erasers” of epigenetic modifications. Besides, the bromodomain-containing proteins (BRDs) functions as “Readers” to decode those acetylated lysine and consequently recruit chromatin regulators to control gene expression (Hillyar et al. 2020; Wang et al. 2021).
HATs include several enzymes. Epigenetic therapies targeting HATs mainly focus on the GCN5-related n-acetyltransferase (GNAT) family, including GCN5 and p300/CBP-related factor (PCAF) (Trisciuoglio et al. 2018; Wang et al. 2021). As a selective and effective catalytic inhibitor of p300/CBP, A-485 competed with acetyl-CoA and thus selectively inhibited proliferation across lineage-specific tumor types, including androgen receptor-positive prostate cancer and several hematologic malignancies (Lasko et al. 2017). P300 and CBP highly express in five gastric cancer cell lines, in which compound C646 affects cell cycle and promotes cell apoptosis to exert antitumor effects by selectively inhibiting P300 and CBP (Wang et al. 2017). Garcinol was discovered to exhibit inhibitory activity against human esophageal cancer cell lines KYSE150 and KYSE450 for migration and invasion in a dose-dependent manner via blocking p300 and TGF-β1 signaling pathway, which influences the cell cycle and induces apoptosis (Wang et al. 2020a).
HDACs take charge of removing acetyl groups on lysine residues of histone proteins. Considering that HDACs are high expression in cancer cells, accumulating explorations have focused on HADCs. Up to now, 18 subtypes of HDACs have been discovered, and their inhibitors have been identified and applied in clinical trials, including vorinostat, romidepsin, belinostat, and panobinostat (Fan et al. 2021). Besides, several compounds are being studied. VS13, a quinoline derivative, can potently inhibit HDAC6 in nanomolar concentration, showing its anti-proliferation activity against uveal melanoma cell line (Nencetti et al. 2021). Unlike VS13, compound 12a selectively inhibited subtype HDAC2, and consequently restrained A549 cells from migration and colony formation, and induced apoptosis and G2/M cell cycle arrest (Wang et al. 2021; Xie et al. 2017). AES-135 possesses potent pancreatic cancer cells cytotoxicity in vitro and prolonging the survival time of the pancreatic cancer mouse model, which can be attributed to its inhibitory effect on HDAC3, HDAC6, and HDAC11 (Shouksmith et al. 2019). Generally, HDACs inhibitors can restrain tumor proliferation by affecting apoptosis, differentiation, cell migration, and cell cycle arrest (Wang et al. 2021). As the class III HDAC family, the sirtuins (SIRTs) family is also considered as a therapeutic target of cancer treatment, whose pharmacological inhibition remodels the chromatin state and results in a blurring of the boundaries between transcriptional activity and static chromatin (Manzotti et al. 2019; Wang et al. 2021). SIRTs are a series of nicotinamide adenine dinucleotide dependent enzymes, including intranuclear SIRT1, SIRT6, and SIRT7, intramitochondrial SIRT3–5, and cytoplasmic SIRT2 (Houtkooper et al. 2012). Recently, the high level of SIRT1 has been found to be associated with recurrence and poor prognosis in patients with lung adenocarcinoma, which is highly sensitive to the treatment of gefitinib, a tyrosine kinase inhibitor (TKI). It has been reported that SIRT1 inhibitor TV-6 can enhance TKI therapeutic effect. The combined administration of TV-6 and TKI results in tumor regression in xenograft mouse models and improved sensitivity of tumor cells to TKI (Sun et al. 2020). Besides, gastric carcinoma cell lines are also sensitive to TV-6 with activating p53 or inhibiting autophagic flux (Ke et al. 2020). TM, a thiomyristoyl lysine compound, has been discovered as antiproliferative agent against many human cancer cells and breast cancer mouse models. TM inhibits SIRT2, promoting the ubiquitination and degradation of c-Myc and therefore exhibiting anticancer activity (Jing et al. 2016). Selectively targeting SIRT2, γ-mangostin, a natural product, was found to inhibit MDA-MD-231 and MCF-7 breast cancer cells proliferation by improving the acetylation level of α-tubulin (Yeong et al. 2020). As a SIRT1 activator, compound F0911–7667 induces autophagic cell death via the AMPK-mTOR-ULK complex and induces mitochondrial phagocytosis through the SIRT1-PINK1-Parkin pathway in glioblastoma cells (Yao et al. 2018). SIRT6 agonists UBCS039 and MDL-800 can induce autophagic cell death in several human tumors by activating the deacetylation of SIRT6 (Huang et al. 2018; Iachettini et al. 2018).
BRDs is a highly conserved protein superfamily containing eight families, in which the bromodomain and extra-terminal (BET) families are the most widely studied, containing BRD2, BDR3, BRD4, and BRDT (Wang et al. 2021). BRDs serve on readers of histone acetylation, causing epigenetic regulation of gene expression (Liang et al. 2020a). Many small molecules targeting BRDs have been discovered to develop cancer therapies. In phase I clinical trials, small-molecule pan-BET inhibitor ABBV-075 was discovered to be promising in tolerance and therapeutic activity in highly pretreated patients with refractory solid tumors (Piha-Paul et al. 2019). Further chemical modifications of ABBV-075 obtained compound 38, which exhibited higher inhibitory efficiency than clinical candidate OTX-015 (Li et al. 2020). In addition to pan-inhibitors, selective BRD inhibitors have also been identified. For instance, small-molecule FL-411 has been reported to be involved in autophagy-related cell death via targeting BRD4, which blocked BRD4-AMPK interaction and thereby activating the autophagic pathway in breast cancer (Ouyang et al. 2017). Bioavailable chemicals GSK452, GSK737, and GSK217 were screened to have ideal solubility, cell efficacy, as well as pharmacokinetics (Aylott et al. 2021). It has been reported that dBET6, a BET protein degrading agent, possesses antitumor activity against hematology and solid cancer and presents higher efficiency than first generation drugs, such as dBET1 and JQ1 (Bauer et al. 2021). Moreover, bBET6 also lowered the resistance of cancer to immuno- and chemotherapy.
4.3 Targeting Other Epigenetic Biomarkers in Cancer Treatment
Other important elements of epigenetic regulation are histone methylation enzymes, including histone H3K79 methyltransferase DOT1L, H3K27 methyltransferase enhancer of zeste homolog 2 EZH2, and histone lysine demethylases (KDMs) (Wang et al. 2021). It has been reported that DOT1L promoted triple-negative breast cancer (TNBC) progression via the binding between c-Myc and p300 acetyltransferase (Cho et al. 2015; Wang et al. 2021). DOT1L inhibitor PsA-309 was identified to have an inhibitory effect on the proliferation of human breast cancer cell lines MDA-MB-231 and Hs578T via selectively suppressing H3K79 methylation (Byun et al. 2019). PROTAC E7 was discovered to inhibit EZH2. E7-mediated degradation of EZH2 resulted in decreased proliferation rates in diffuse large B-cell lymphoma cell WSU-DLCL-2, human lung cancer cell A549, and NSCLC cell NCI-H1299 (Wang et al. 2021). Besides, inhibiting histone demethylation-related KDMs can also be a therapeutic strategy in cancer treatment. By inhibiting KDM1A, ORY-1001 can compromise leukemic stem cell activity in acute leukemia (Maes et al. 2018). ORY-1001 impaired cancer cell proliferation and increased survival in rodent xenograft models of acute leukemia. In addition, KDM5A inhibitor compound 1 has been reported to suppress the demethylation of H3K4me3, leading to p16 and p27 accumulation, which consequently resulted in cell cycle arrest and inhibited proliferation of various KDM5A-overexpressing breast cancer cells (Yang et al. 2019). Additionally, it has been proposed that miRNAs associated with cancer are therapeutic targets of cancer. Several miRNA inhibition therapies have been developed, for example, the combination treatment of anti-miRNA oligonucleotides with a low dose of sunitinib exhibited a significant synergistic antitumor effect in pancreatic ductal adenocarcinoma cells (Song et al. 2017) (Fig. 4.4).

Epigenetic interventions in cancer therapy. Currently, most epigenetic drugs are designed to inhibit DNA methylation and histone acetylation-related enzymes, including DNMTs, HATs, HDACs, KDMs, as well as histone acetylation readers BRDs, histone methylases DOT1L, and EZH2, suppressing tumorigenesis and malignant progression by inhibiting epigenetic dysregulation
5 Conclusions and Perspectives
To date, cancer incidence and mortality rates remain high worldwide; therefore, the discovery of novel cancer diagnostic and prognostic biomarkers and the search for emerging and effective targeted therapies are major attention worldwide. Epigenetics is closely related to the occurrence and development of many types of cancers, and abnormalities in multiple epigenetic regulations often lead to the progression of malignancies; therefore, an in-depth analysis of the key mechanisms of epigenetics in cancer development will help in the early diagnosis of cancer and greatly improve cancer treatment. At present, several epigenetic biomarkers have made great progress in the research of cancer diagnosis, prognosis, and treatment, but how to translate them into clinical practice is still a challenge for us.
Abbreviations
- ALC1:
-
Amplification in liver cancer 1
- AML:
-
Acute myeloid leukemia
- ARID2:
-
AT-rich interactive domain 2
- AUC:
-
Area under the curve
- B-CLL:
-
B-cell chronic lymphocytic leukemia
- BET:
-
Bromodomain and extra-terminal
- BMI1:
-
B-cell-specific Moloney murine leukemia virus insertion site1
- BRDs :
-
Bromodomain-containing proteins
- CIMP:
-
CpG island methylator phenotype
- circRNAs:
-
Circular RNA
- CRC:
-
Colorectal cancer
- DNMT1:
-
DNA methyltransferase 1
- EHMT1/2:
-
Euchromatin histone-lysine-n-methyltransferases I and II
- EOC:
-
Epithelial ovarian cancer
- ESCC:
-
Esophageal squamous cell carcinoma
- EZH2:
-
Enhancer of zeste homolog 2
- FAL1:
-
Focally amplified lncRNA on chromosome 1
- GBM:
-
Glioblastoma
- GEO:
-
Gene Expression Omnibus
- GNAT:
-
GCN5-related n-acetyltransferase
- HAT:
-
Histone acetyltransferase
- HCC:
-
Hepatocellular carcinoma
- HDAC:
-
histone deacetylase
- hnRNPL:
-
Heterogeneous nuclear ribonucleoprotein L
- KDMs:
-
Histone lysine demethylases
- lncRNAs:
-
Long noncoding RNAs
- m1A:
-
N1-methyladenosine
- m3C:
-
N3-methylcytosine
- m5C:
-
5-methylcytosine
- m6A:
-
N6-methyl-adenosine
- m7G:
-
N7-methylguanosine
- METTL3:
-
Methyltransferase-like 3
- MGMT:
-
O6 methylguanine DNA methyltransferase
- miRNA:
-
MicroRNA
- MYC:
-
Myelocytomatosis oncogene
- NAD+:
-
Nicotinamide adenine dinucleotide
- ncRNAs:
-
Noncoding RNAs
- NSCLC:
-
Non-small-cell lung cancer
- PARP1:
-
Poly (ADP-ribose) polymerase 1
- PCAF:
-
p300/CBP-related factor
- piRNAs:
-
PIWI-interacting RNAs
- PR C1:
-
Polycomb repressive complex 1
- PR C2:
-
Polycomb repressive complex 2
- SIRTs:
-
Sirtuins
- SND1:
-
Staphylococcal nuclease domain-containing protein 1
- TCGA:
-
The Cancer Genome Atlas
- TET:
-
Ten-eleven translocation
- TKI:
-
Tyrosine-kinase inhibitor
- TNBC:
-
Triple-negative breast cancer
- WGBS:
-
Whole-genome bisulfite sequencing
- YBX1:
-
Y-box binding protein 1
References
Akone SH, Ntie-Kang F, Stuhldreier F, Ewonkem MB, Noah AM, Mouelle SEM, Müller R (2020) Natural products impacting DNA methyltransferases and histone deacetylases. Front Pharmacol 11:992
Atsumi Y, Minakawa Y, Ono M, Dobashi S, Shinohe K, Shinohara A, Takeda S, Takagi M, Takamatsu N, Nakagama H et al (2015) ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient γH2AX foci formation. Cell Rep 13:2728–2740
Aylott HE, Atkinson SJ, Bamborough P, Bassil A, Chung CW, Gordon L, Grandi P, Gray JRJ, Harrison LA, Hayhow TG et al (2021) Template-hopping approach leads to potent, selective, and highly soluble Bromo and Extraterminal domain (BET) second Bromodomain (BD2) inhibitors. J Med Chem 64:3249–3281
Barcena-Varela M, Caruso S, Llerena S, Alvarez-Sola G, Uriarte I, Latasa MU, Urtasun R, Rebouissou S, Alvarez L, Jimenez M et al (2019) Dual targeting of histone methyltransferase G9a and DNA-methyltransferase 1 for the treatment of experimental hepatocellular carcinoma. Hepatology 69:587–603
Bauer K, Berghoff AS, Preusser M, Heller G, Zielinski CC, Valent P, Grunt TW (2021) Degradation of BRD4–a promising treatment approach not only for hematologic but also for solid cancer. Am J Cancer Res 11:530–545
Bestor TH (2000) The DNA methyltransferases of mammals. Hum Mol Genet 9:2395–2402
Bianco S, Gevry N (2012) Endocrine resistance in breast cancer: from cellular signaling pathways to epigenetic mechanisms. Transcription 3:165–170
Bottoni A, Piccin D, Tagliati F, Luchin A, Zatelli MC, degli Uberti EC (2005) miR-15a and miR-16-1 down-regulation in pituitary adenomas. J Cell Physiol 204:280–285
Byun WS, Kim WK, Han HJ, Chung HJ, Jang K, Kim HS, Kim S, Kim D, Bae ES, Park S et al (2019) Targeting histone methyltransferase DOT1L by a novel Psammaplin a analog inhibits growth and metastasis of triple-negative breast cancer. Mol Ther Oncolytics 15:140–152
Cho MH, Park JH, Choi HJ, Park MK, Won HY, Park YJ, Lee CH, Oh SH, Song YS, Kim HS et al (2015) DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat Commun 6:7821
Chun P (2015) Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res 38:933–949
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG et al (2017) M(6)a RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep 18:2622–2634
Culver-Cochran AE, Chadwick BP (2013) Loss of WSTF results in spontaneous fluctuations of heterochromatin formation and resolution, combined with substantial changes to gene expression. BMC Genomics 14:740
Dang F, Wei W (2021) Targeting the acetylation signaling pathway in cancer therapy. Paper presented at: seminars in cancer biology (Elsevier)
Davis LE, Shalin SC, Tackett AJ (2020) Utility of histone H3K27me3 and H4K20me as diagnostic indicators of melanoma. Melanoma Res 30:159–165
Derissen EJ, Beijnen JH, Schellens JH (2013) Concise drug review: azacitidine and decitabine. Oncologist 18:619–624
Diaz-Lagares A, Mendez-Gonzalez J, Hervas D, Saigi M, Pajares MJ, Garcia D, Crujerias AB, Pio R, Montuenga LM, Zulueta J et al (2016) A novel epigenetic signature for early diagnosis in lung cancer. Clin Cancer Res 22:3361–3371
Dong S, Li W, Wang L, Hu J, Song Y, Zhang B, Ren X, Ji S, Li J, Xu P et al (2019a) Histone-related genes are Hypermethylated in lung cancer and Hypermethylated HIST1H4F could serve as a Pan-cancer biomarker. Cancer Res 79:6101–6112
Dong W, Bi J, Liu H, Yan D, He Q, Zhou Q, Wang Q, Xie R, Su Y, Yang M et al (2019b) Circular RNA ACVR2A suppresses bladder cancer cells proliferation and metastasis through miR-626/EYA4 axis. Mol Cancer 18:95
Dong Y, Zhao H, Li H, Li X, Yang S (2014) DNA methylation as an early diagnostic marker of cancer. Biomed Rep 2:326–330
Duan B, Bai J, Qiu J, Wang J, Tong C, Wang X, Miao J, Li Z, Li W, Yang J, Huang C (2018) Histone-lysine N-methyltransferase SETD7 is a potential serum biomarker for colorectal cancer patients. EBioMedicine 37:134–143
Duan R, Du W, Guo W (2020) EZH2: a novel target for cancer treatment. J Hematol Oncol 13:104
Erdel F, Rippe K (2011) Chromatin remodelling in mammalian cells by ISWI-type complexes–where, when and why? FEBS J 278:3608–3618
Fan W, Zhang L, Wang X, Jia H, Zhang L (2021) Discovery of potent histone deacetylase inhibitors with modified phenanthridine caps. J Enzyme Inhib Med Chem 36:707–718
Fatica A, Bozzoni I (2014) Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15:7–21
Fattahi S, Kosari-Monfared M, Golpour M, Emami Z, Ghasemiyan M, Nouri M, Akhavan-Niaki H (2020) LncRNAs as potential diagnostic and prognostic biomarkers in gastric cancer: a novel approach to personalized medicine. J Cell Physiol 235:3189–3206
Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, Liu P, Kossenkov AV, Showe LC, Jean S et al (2019) N(6)-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res 79:2812–2820
Goldberg AD, Allis CD, Bernstein E (2007) Epigenetics: a landscape takes shape. Cell 128:635–638
Gomes AP, Ilter D, Low V, Rosenzweig A, Shen ZJ, Schild T, Rivas MA, Er EE, McNally DR, Mutvei AP et al (2019) Dynamic incorporation of histone H3 variants into chromatin is essential for acquisition of aggressive traits and metastatic colonization. Cancer Cell 36:402–417 e413
Grady WM, Yu M, Markowitz SD (2021) Epigenetic alterations in the gastrointestinal tract: current and emerging use for biomarkers of cancer. Gastroenterology 160:690–709
Graule J, Uth K, Fischer E, Centeno I, Galvan JA, Eichmann M, Rau TT, Langer R, Dawson H, Nitsche U et al (2018) CDX2 in colorectal cancer is an independent prognostic factor and regulated by promoter methylation and histone deacetylation in tumors of the serrated pathway. Clin Epigenetics 10:120
Heiblig M, El Hamri M, Salles G, Thomas X (2015) Epigenetics and adult acute myeloid leukemia. In: Current Pharmacogenomics and Personalized Medicine (Formerly Current Pharmacogenomics), vol 13, pp 117–133
Hillyar C, Rallis KS, Varghese J (2020) Advances in epigenetic cancer therapeutics. Cureus 12:e11725
Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Bio 13:225–238
Hsieh IY, He J, Wang L, Lin B, Liang Z, Lu B, Chen W, Lu G, Li F, Lv W et al (2020) H3K27me3 loss plays a vital role in CEMIP mediated carcinogenesis and progression of breast cancer with poor prognosis. Biomed Pharmacother 123:109728
Hu X, Feng Y, Zhang D, Zhao SD, Hu Z, Greshock J, Zhang Y, Yang L, Zhong X, Wang LP et al (2014) A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell 26:344–357
Huang ZM, Zhao JX, Deng W, Chen YY, Shang JL, Song K, Zhang L, Wang CX, Lu SY, Yang XY et al (2018) Identification of a cellularly active SIRT6 allosteric activator. Nat Chem Biol 14:1118-+
Iachettini S, Trisciuoglio D, Rotili D, Lucidi A, Salvati E, Zizza P, Di Leo L, Del Bufalo D, Ciriolo MR, Leonetti C et al (2018) Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis 9:996
Jiang H, Cao HJ, Ma N, Bao WD, Wang JJ, Chen TW, Zhang EB, Yuan YM, Ni QZ, Zhang FK et al (2020) Chromatin remodeling factor ARID2 suppresses hepatocellular carcinoma metastasis via DNMT1-snail axis. Proc Natl Acad Sci U S A 117:4770–4780
Jing H, Hu J, He B, Abril YLN, Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T, Giannakakou P et al (2016) A SIRT2-selective inhibitor promotes c-Myc Oncoprotein degradation and exhibits broad anticancer activity. Cancer Cell 29:297–310
Jung G, Hernández-Illán E, Moreira L, Balaguer F, Goel A (2020) Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol 17:111–130
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45:592–601
Ke XY, Qin QS, Deng TY, Liao YY, Gao SJ (2020) Heterogeneous responses of gastric cancer cell lines to Tenovin-6 and synergistic effect with chloroquine. Cancers 12
Klumper N, Ralser DJ, Bawden EG, Landsberg J, Zarbl R, Kristiansen G, Toma M, Ritter M, Holzel M, Ellinger J, Dietrich D (2020) LAG3 (LAG-3, CD223) DNA methylation correlates with LAG3 expression by tumor and immune cells, immune cell infiltration, and overall survival in clear cell renal cell carcinoma. J Immunother Cancer 8:e000552
Kulka LAM, Fangmann PV, Panfilova D, Olzscha H (2020) Impact of HDAC inhibitors on protein quality control systems: consequences for precision medicine in malignant disease. Front Cell Dev Biol 8:425
Kumar S, Nagpal R, Kumar A, Ashraf MU, Bae YS (2021) Immunotherapeutic potential of m6A-modifiers and MicroRNAs in controlling acute myeloid Leukaemia. Biomedicine 9
Kwon SY, Grisan V, Jang B, Herbert J, Badenhorst P (2016) Genome-wide mapping targets of the metazoan chromatin remodeling factor NURF reveals nucleosome remodeling at enhancers, Core promoters and gene insulators. PLoS Genet 12:e1005969
Lafon-Hughes L, Di Tomaso MV, Méndez-Acuña L, Martínez-López W (2008) Chromatin-remodelling mechanisms in cancer. Mutation Res/Rev Mutation Res 658:191–214
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT (2018) The human transcription factors. Cell 172:650–665
Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V et al (2017) Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550:128–132
Law CT, Wei L, Tsang FH, Chan CY, Xu IM, Lai RK, Ho DW, Lee JM, Wong CC, Ng IO, Wong CM (2019) HELLS regulates chromatin remodeling and epigenetic silencing of multiple tumor suppressor genes in human hepatocellular carcinoma. Hepatology 69:2013–2030
Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926
Li H, Peng C, Zhu C, Nie S, Qian X, Shi Z, Shi M, Liang Y, Ding X, Zhang S et al (2021) Hypoxia promotes the metastasis of pancreatic cancer through regulating NOX4/KDM5A-mediated histone methylation modification changes in a HIF1A-independent manner. Clin Epigenetics 13:18
Li J, Meng H, Bai Y, Wang K (2016) Regulation of lncRNA and its role in cancer metastasis. Oncol Res 23:205–217
Li Y, Seto E (2016) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6
Li YC, Wang Y, Li DD, Zhang Y, Zhao TC, Li CF (2018) Procaine is a specific DNA methylation inhibitor with anti-tumor effect for human gastric cancer. J Cell Biochem 119:2440–2449
Li Z, Xiao S, Yang Y, Chen C, Lu T, Chen Z, Jiang H, Chen S, Luo C, Zhou B (2020) Discovery of 8-methyl-pyrrolo[1,2-a]pyrazin-1(2H)-one derivatives as highly potent and selective Bromodomain and extra-terminal (BET) Bromodomain inhibitors. J Med Chem 63:3956–3975
Liang DL, Yu YF, Ma ZH (2020a) Novel strategies targeting bromodomain-containing protein 4 (BRD4) for cancer drug discovery. Eur J Med Chem 200:112426
Liang G, Ling Y, Mehrpour M, Saw PE, Liu Z, Tan W, Tian Z, Zhong W, Lin W, Luo Q et al (2020b) Autophagy-associated circRNA circCDYL augments autophagy and promotes breast cancer progression. Mol Cancer 19:65
Liu A, Wu Q, Peng D, Ares I, Anadon A, Lopez-Torres B, Martinez-Larranaga MR, Wang X, Martinez MA (2020) A novel strategy for the diagnosis, prognosis, treatment, and chemoresistance of hepatocellular carcinoma: DNA methylation. Med Res Rev 40:1973–2018
Liu L, Wang J, Sun G, Wu Q, Ma J, Zhang X, Huang N, Bian Z, Gu S, Xu M et al (2019) M(6)a mRNA methylation regulates CTNNB1 to promote the proliferation of hepatoblastoma. Mol Cancer 18:188
Lopez-Camarillo C, Gallardo-Rincon D, Alvarez-Sanchez ME, Marchat LA (2019) Pharmaco-epigenomics: on the road of translation medicine. Adv Exp Med Biol 1168:31–42
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, Yuan W, Kan Q, Sun Z (2019) The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol 12:121
Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, Guibourt N, Perdones A, Lufino MMP, Somervaille TCP et al (2018) ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 33:495-+
Maleki Dana P, Mansournia MA, Mirhashemi SM (2020) PIWI-interacting RNAs: new biomarkers for diagnosis and treatment of breast cancer. Cell Biosci 10:44
Manara MC, Valente S, Cristalli C, Nicoletti G, Landuzzi L, Zwergel C, Mazzone R, Stazi G, Arimondo PB, Pasello M et al (2018) A Quinoline-based DNA methyltransferase inhibitor as a possible adjuvant in osteosarcoma therapy. Mol Cancer Ther 17:1881–1892
Mansouri A, Hachem LD, Mansouri S, Nassiri F, Laperriere NJ, Xia D, Lindeman NI, Wen PY, Chakravarti A, Mehta MP et al (2019) MGMT promoter methylation status testing to guide therapy for glioblastoma: refining the approach based on emerging evidence and current challenges. Neuro-Oncology 21:167–178
Manzotti G, Ciarrocchi A, Sancisi V (2019) Inhibition of BET proteins and histone deacetylase (HDACs): crossing roads in cancer therapy. Cancers 11
Mehdipour P, Murphy T, De Carvalho DD (2020) The role of DNA-demethylating agents in cancer therapy. Pharmacol Ther 205:107416
Michalak EM, Burr ML, Bannister AJ, Dawson MA (2019) The roles of DNA, RNA and histone methylation in ageing and cancer. Nat Rev Mol Cell Biol 20:573–589
Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C (2019) Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics 14:1164–1176
Moran S, Martínez-Cardús A, Sayols S, Musulén E, Balañá C, Estival-Gonzalez A, Moutinho C, Heyn H, Diaz-Lagares A, de Moura MC et al (2016) Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 17:1386–1395
Moridikia A, Mirzaei H, Sahebkar A, Salimian J (2018) MicroRNAs: potential candidates for diagnosis and treatment of colorectal cancer. J Cell Physiol 233:901–913
Müller D, Győrffy B (2022) DNA methylation-based diagnostic, prognostic, and predictive biomarkers in colorectal cancer. Biochim Biophys Acta Rev Cancer 1877:188722
Nencetti S, Cuffaro D, Nuti E, Ciccone L, Rossello A, Fabbi M, Ballante F, Ortore G, Carbotti G, Campelli F et al (2021) Identification of histone deacetylase inhibitors with (arylidene)aminoxy scaffold active in uveal melanoma cell lines. J Enzyme Inhib Med Chem 36:34–47
Nishiyama A, Nakanishi M (2021) Navigating the DNA methylation landscape of cancer. Trends Genet 37:1012–1027
Noguera-Uclés JF, Boyero L, Salinas A, Cordero Varela JA, Benedetti JC, Bernabé-Caro R, Sánchez-Gastaldo A, Alonso M, Paz-Ares L, Molina-Pinelo S (2020) The roles of imprinted SLC22A18 and SLC22A18AS gene overexpression caused by promoter CpG Island Hypomethylation AS diagnostic and prognostic biomarkers for non-small cell lung cancer patients. Cancers 12
Ohguchi H, Hideshima T, Anderson KC (2018) The biological significance of histone modifiers in multiple myeloma: clinical applications. Blood Cancer J 8:83
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
Ooi SK, Sato S, Tomomori-Sato C, Zhang Y, Wen Z, Banks CAS, Washburn MP, Unruh JR, Florens L, Conaway RC, Conaway JW (2021) Multiple roles for PARP1 in ALC1-dependent nucleosome remodeling. Proc Natl Acad Sci U S A 118
Ouyang L, Zhang L, Liu J, Fu LL, Yao DH, Zhao YQ, Zhang SY, Wang G, He G, Liu B (2017) Discovery of a small-molecule Bromodomain-containing protein 4 (BRD4) inhibitor that induces AMP-activated protein kinase-modulated autophagy-associated cell death in breast cancer. J Med Chem 60:9990–10012
Pechalrieu D, Dauzonne D, Arimondo PB, Lopez M (2020) Synthesis of novel 3-halo-3-nitroflavanones and their activities as DNA methyltransferase inhibitors in cancer cells. Eur J Med Chem 186:111829
Pfeifer GP (2018) Defining driver DNA methylation changes in human cancer. Int J Mol Sci 19:1166
Piha-Paul SA, Sachdev JC, Barve M, LoRusso P, Szmulewitz R, Patel SP, Lara PN, Chen XT, Hu BB, Freise KJ et al (2019) First-in-human study of Mivebresib (ABBV-075), an Oral Pan-inhibitor of Bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin Cancer Res 25:6309–6319
Porter NJ, Mahendran A, Breslow R, Christianson DW (2017) Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc Natl Acad Sci 114:13459–13464
Rice K, Hormaeche I, Licht J (2007) Epigenetic regulation of normal and malignant hematopoiesis. Oncogene 26:6697–6714
Riedel SS, Neff T, Bernt KM (2015) Histone profiles in cancer. Pharmacol Ther 154:87–109
Schapira M, Arrowsmith CH (2016) Methyltransferase inhibitors for modulation of the epigenome and beyond. Curr Opin Chem Biol 33:81–87
Shen R, Cheng T, Xu C, Yung RC, Bao J, Li X, Yu H, Lu S, Xu H, Wu H et al (2020) Novel visualized quantitative epigenetic imprinted gene biomarkers diagnose the malignancy of ten cancer types. Clin Epigenetics 12:71
Sher G, Salman NA, Khan AQ, Prabhu KS, Raza A, Kulinski M, Dermime S, Haris M, Junejo K, Uddin S (2022) Epigenetic and breast cancer therapy: promising diagnostic and therapeutic applications. Semin Cancer Biol 83:152–165
Shi X, Kaller M, Rokavec M, Kirchner T, Horst D, Hermeking H (2020a) Characterization of a p53/miR-34a/CSF1R/STAT3 feedback loop in colorectal cancer. Cell Mol Gastroenterol Hepatol 10:391–418
Shi Y, Zheng C, Jin Y, Bao B, Wang D, Hou K, Feng J, Tang S, Qu X, Liu Y et al (2020b) Reduced expression of METTL3 promotes metastasis of triple-negative breast cancer by m6A methylation-mediated COL3A1 up-regulation. Front Oncol 10:1126
Shouksmith AE, Shah F, Grimard ML, Gawel JM, Raouf YS, Geletu M, Berger-Becvar A, de Araujo ED, Luchman HA, Heaton WL et al (2019) Identification and characterization of AES-135, a Hydroxamic acid-based HDAC inhibitor that prolongs survival in an Orthotopic mouse model of pancreatic cancer. J Med Chem 62:2651–2665
Sixto-López Y, Bello M, Correa-Basurto J (2020) Exploring the inhibitory activity of valproic acid against the HDAC family using an MMGBSA approach. J Comput Aided Mol Des 34:857–878
Skvortsova K, Iovino N, Bogdanović O (2018) Functions and mechanisms of epigenetic inheritance in animals. Nat Rev Mol Cell Biol 19:774–790
Song J, Ouyang Y, Che J, Li X, Zhao Y, Yang K, Zhao X, Chen Y, Fan C, Yuan W (2017) Potential value of miR-221/222 as diagnostic, prognostic, and therapeutic biomarkers for diseases. Front Immunol 8:56
Stresemann C, Lyko F (2008) Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer 123:8–13
Sulewska A, Niklinski J, Charkiewicz R, Karabowicz P, Biecek P, Baniecki H, Kowalczuk O, Kozlowski M, Modzelewska P, Majewski P et al (2022) A signature of 14 long non-coding RNAs (lncRNAs) as a step towards precision diagnosis for NSCLC. Cancers 14
Sun JT, Li GF, Liu YW, Ma MY, Song KF, Li HX, Zhu DX, Tang XJ, Kong JY, Yuan X (2020) Targeting histone deacetylase SIRT1 selectively eradicates EGFR TKI-resistant cancer stem cells via regulation of mitochondrial oxidative phosphorylation in lung adenocarcinoma. Neoplasia 22:33–46
Sun L, Fang Y, Wang X, Han Y, Du F, Li C, Hu H, Liu H, Liu Q, Wang J et al (2019a) miR-302a inhibits metastasis and Cetuximab resistance in colorectal cancer by targeting NFIB and CD44. Theranostics 9:8409–8425
Sun T, Wu R, Ming L (2019b) The role of m6A RNA methylation in cancer. Biomed Pharmacother 112:108613
Sun X, Yi J, Yang J, Han Y, Qian X, Liu Y, Li J, Lu B, Zhang J, Pan X et al (2021) An integrated epigenomic-transcriptomic landscape of lung cancer reveals novel methylation driver genes of diagnostic and therapeutic relevance. Theranostics 11:5346–5364
Taby R, Issa JPJ (2010) Cancer epigenetics. CA Cancer J Clin 60:376–392
Tapadar S, Fathi S, Wu B, Sun CQ, Raji I, Moore SG, Arnold RS, Gaul DA, Petros JA, Oyelere AK (2020) Liver-targeting class I selective histone deacetylase inhibitors potently suppress hepatocellular tumor growth as standalone agents. Cancers (Basel) 12
Tricarico R, Nicolas E, Hall MJ, Golemis EA (2020) X- and Y-linked chromatin-modifying genes as regulators of sex-specific cancer incidence and prognosis. Clin Cancer Res 26:5567–5578
Trisciuoglio D, Di Martile M, Del Bufalo D (2018) Emerging role of histone acetyltransferase in stem cells and cancer. Stem Cells Int 2018:8908751
Vandenhoeck J, van Meerbeeck JP, Fransen E, Raskin J, Van Camp G, Op de Beeck K, Lamote K (2021) DNA methylation as a diagnostic biomarker for malignant mesothelioma: a systematic review and meta-analysis. J Thorac Oncol 16:1461–1478
Wagner T, Brand P, Heinzel T, Krämer OH (2014) Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim Biophys Acta 1846:524–538
Wang J, Wu M, Zheng D, Zhang H, Lv Y, Zhang L, Tan HS, Zhou H, Lao YZ, Xu HX (2020a) Garcinol inhibits esophageal cancer metastasis by suppressing the p300 and TGF-beta1 signaling pathways. Acta Pharmacol Sin 41:82–92
Wang T, Kong S, Tao M, Ju S (2020b) The potential role of RNA N6-methyladenosine in cancer progression. Mol Cancer 19:88
Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX, Liu J, Luo XJ, Meng Q, Pu HY et al (2019) LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer 18:174
Wang Y, Xie Q, Tan H, Liao M, Zhu S, Zheng LL, Huang H, Liu B (2021) Targeting cancer epigenetic pathways with small-molecule compounds: therapeutic efficacy and combination therapies. Pharmacol Res 173:105702
Wang YM, Gu ML, Meng FS, Jiao WR, Zhou XX, Yao HP, Ji F (2017) Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int J Oncol 51:1860–1868
Wang YW, Ma X, Zhang YA, Wang MJ, Yatabe Y, Lam S, Girard L, Chen JY, Gazdar AF (2016) ITPKA gene body methylation regulates gene expression and serves as an early diagnostic marker in lung and other cancers. J Thorac Oncol 11:1469–1481
Watson ZL, Yamamoto TM, McMellen A, Kim H, Hughes CJ, Wheeler LJ, Post MD, Behbakht K, Bitler BG (2019) Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin Epigenetics 11:165
Wong CC, Li W, Chan B, Yu J (2019) Epigenomic biomarkers for prognostication and diagnosis of gastrointestinal cancers. Semin Cancer Biol 55:90–105
Xi Y, Lin Y, Guo W, Wang X, Zhao H, Miao C, Liu W, Liu Y, Liu T, Luo Y et al (2022) Multi-omic characterization of genome-wide abnormal DNA methylation reveals diagnostic and prognostic markers for esophageal squamous-cell carcinoma. Signal Transduct Target Ther 7:53
Xie R, Yao Y, Tang P, Chen G, Liu X, Yun F, Cheng C, Wu X, Yuan Q (2017) Design, synthesis and biological evaluation of novel hydroxamates and 2-aminobenzamides as potent histone deacetylase inhibitors and antitumor agents. Eur J Med Chem 134:1–12
Xiu B, Chi Y, Liu L, Chi W, Zhang Q, Chen J, Guo R, Si J, Li L, Xue J et al (2019) LINC02273 drives breast cancer metastasis by epigenetically increasing AGR2 transcription. Mol Cancer 18:187
Xu W, Xu M, Wang L, Zhou W, Xiang R, Shi Y, Zhang Y, Piao Y (2019) Integrative analysis of DNA methylation and gene expression identified cervical cancer-specific diagnostic biomarkers. Signal Transduct Target Ther 4:55
Yang B, Wang JQ, Tan Y, Yuan R, Chen ZS, Zou C (2021) RNA methylation and cancer treatment. Pharmacol Res 174:105937
Yang GJ, Ko CN, Zhong HJ, Leung CH, Ma DL (2019) Structure-based discovery of a selective KDM5A inhibitor that exhibits anti-cancer activity via inducing cell cycle arrest and senescence in breast cancer cell lines. Cancers 11
Yao B, Gui T, Zeng X, Deng Y, Wang Z, Wang Y, Yang D, Li Q, Xu P, Hu R et al (2021) PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med 13:58
Yao ZQ, Zhang X, Zhen YQ, He XY, Zhao SM, Li XF, Yang B, Gao F, Guo FY, Fu LL et al (2018) A novel small-molecule activator of Sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis 9:767
Yeong KY, Khaw KY, Takahashi Y, Itoh Y, Murugaiyah V, Suzuki T (2020) Discovery of gamma-mangostin from Garcinia mangostana as a potent and selective natural SIRT2 inhibitor. Bioorg Chem 94:103403
Yi X, Jiang X, Li X, Jiang DS (2017) Histone lysine methylation and congenital heart disease: from bench to bedside (review). Int J Mol Med 40:953–964
Yoon S, Eom GH (2016) HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J 52:1–11
Yu L, Xu J, Liu J, Zhang H, Sun C, Wang Q, Shi C, Zhou X, Hua D, Luo W et al (2019) The novel chromatin architectural regulator SND1 promotes glioma proliferation and invasion and predicts the prognosis of patients. Neuro-Oncology 21:742–754
Zhang G, Pradhan S (2014) Mammalian epigenetic mechanisms. IUBMB life 66:240–256
Zhang W, Qu J, Liu GH, Belmonte JCI (2020a) The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol 21:137–150
Zhang Y, Huang YX, Wang DL, Yang B, Yan HY, Lin LH, Li Y, Chen J, Xie LM, Huang YS et al (2020b) LncRNA DSCAM-AS1 interacts with YBX1 to promote cancer progression by forming a positive feedback loop that activates FOXA1 transcription network. Theranostics 10:10823–10837
Zhou J, Cheng T, Li X, Hu J, Li E, Ding M, Shen R, Pineda JP, Li C, Lu S et al (2021) Epigenetic imprinting alterations as effective diagnostic biomarkers for early-stage lung cancer and small pulmonary nodules. Clin Epigenetics 13:220
Zhu D, Zhang Y, Wang S (2021) Histone citrullination: a new target for tumors. Mol Cancer 20:90
Zwergel C, Schnekenburger M, Sarno F, Battistelli C, Manara MC, Stazi G, Mazzone R, Fioravanti R, Gros C, Ausseil F et al (2019) Identification of a novel quinoline-based DNA demethylating compound highly potent in cancer cells. Clin Epigenetics 11:68
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (Grant No. 82172649) and Fundamental Research Funds for the Central Universities (Grant No. 2682021CX088).
Compliance with Ethical Standards
Conflict of Interest: The authors declare that they have no conflict of interest.
Research involving human participants and/or animals: This article does not contain any studies with human participants performed by any of the authors.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Fu, L., Liu, B. (2023). Epigenetics in the Diagnosis, Prognosis, and Therapy of Cancer. In: Kalkan, R. (eds) Cancer Epigenetics. Epigenetics and Human Health, vol 11. Springer, Cham. https://doi.org/10.1007/978-3-031-42365-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-42365-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-42364-2
Online ISBN: 978-3-031-42365-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)