
Abstract
Peptide receptor radionuclide therapy (PRRT) is a special type of radiopharmaceutical therapy (RPT) that employs peptide-based radiopharmaceuticals such as 177Lu-labeled agonists of somatostatin receptor subtype 2 (SST2) (e.g., [177Lu]Lu-DOTA-TOC and [177Lu]Lu-DOTA-TATE (Lutathera®)). SST2 are highly overexpressed in neuroendocrine tumors (NETs), which are most commonly found in the stomach, intestines, pancreas, and lungs. Until recently, it was thought that the internalization of the radiopharmaceutical was needed for effective SST-targeted RPT. However, radiolabeled SST antagonists have been shown to recognize more binding sites on SST-expressing tumor cells, produce higher tumor doses, and cause a greater number of DNA double strand breaks than agonists despite their poor internalization rates. As a result, several SST2 antagonists were synthesized and evaluated in a preclinical setting. Of these, [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-LM3 have produced the most promising results and were thus selected for clinical evaluation. Indeed, [177Lu]Lu-DOTA-JR11 produced tumor radiation doses in NET patients that were several times higher than either [177Lu]Lu-DOTA-TATE or [177Lu]Lu-DOTA-TOC, yielding an objective response rate between 30% and 45% as well as 1 year disease control rate of up to 90%. At the same time, however, [177Lu]Lu-DOTA-JR11 produced higher bone marrow toxicity than either [177Lu]Lu-DOTA-TATE or [177Lu]Lu-DOTA-TOC. Future developments in this area will include the use of SST2 antagonists together with α-emitting and combined β−/Auger electron-emitting radionuclides as well as the expansion of the radiolabeled antagonist approach to other receptor systems and other indications, such as small cell lung cancer, breast cancer, renal cell carcinoma, non-Hodgkin lymphoma, paraganglioma, pheochromocytoma, medullary thyroid cancer, meningioma, prostate cancer, and ovarian cancers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Neuroendocrine tumor (NET)
- Peptide receptor radionuclide therapy (PRRT)
- Theranostics
- Somatostatin receptor subtype 2 (SST2)
- SST2 antagonists
- Radiolabelled SST2 antagonists
- [177Lu]Lu-DOTA-JR11
- [177Lu]Lu-DOTA-LM3
1 The Fundamentals
Neuroendocrine tumors (NETs) are a group of tumors that arise from neuroendocrine cells and are most commonly found in the stomach, intestines, and pancreas (gastroenteropancreatic neuroendocrine tumors; GEP-NETs) as well as in the lungs (lung NETs) [1, 2]. As the majority of NETs are slowly growing tumors with almost no symptoms, up to 50% of cases are metastatic at diagnosis [3]. Somatostatin receptors (SST) have played a key role as molecular targets for both the diagnosis and treatment of NETs for almost 30 years. To date, five somatostatin receptor subtypes have been identified: SST1–5. Somatostatin receptor subtype 2 (SST2) is the predominant subtype. It is highly expressed in GEP-NETs and is expressed at lower levels in several other tumor types, including small cell lung cancer, lung NETs, breast cancer, renal cell carcinoma, non-Hodgkin lymphoma, paraganglioma, pheochromocytoma, medullary thyroid cancer, and meningioma [4].
Peptide receptor radionuclide therapy (PRRT) is a special type of radiopharmaceutical therapy (RPT) predicated on the use of radiolabeled peptides such as the 177Lu-labeled SST2 agonists [177Lu]Lu-DOTA-TOC and [177Lu]Lu-DOTA-TATE (Lutathera™). Although PRRT is one of the most efficient treatments for the management of NETs, it predominantly stabilizes—rather than cures—the disease [5]. There is thus an unmet need to improve PRRT with more effective radiopharmaceuticals. Until recently, it was thought that the internalization of the radiolabeled agonists was required for SST-targeted RPT. Yet in 2006, Ginj et al. proposed the paradigm shifting idea that radiolabeled SST antagonists may perform better than agonists despite their lack of internalization [6]. Indeed, there is compelling evidence that 177Lu-labeled SST2 antagonists—e.g., [177Lu]Lu-DOTA-JR11 = [177Lu]Lu-OPS201 = [177Lu]Lu-satoreotide tetraxetan—bind to many more SST2 sites on the cell surface [7], resulting in much higher tumor doses and thus greater treatment potential than 177Lu-labeled SST2 agonists [8,9,10].
2 The Details
2.1 A Short History of Peptide Receptor Radionuclide Therapy (PRRT)
After its introduction in the early 1990s, PRRT was gradually improved through a series of steps to enhance the treatment outcomes of patients with GEP-NETs:
-
1.
The introduction of PRRT with radiolabeled SST agonists such as [111In]In-DTPA-octreotide and, subsequently, the advent of improved SST agonists labeled with β-emitting radionuclides, primarily [90Y]Y- and [177Lu]Lu-DOTA-TOC as well as [177Lu]Lu-DOTA-TATE (Lutathera™) [11, 12].
-
2.
The invention of SST-targeted scintigraphy and, later, SST-targeted single photon emission computed tomography/computed tomography (SPECT/CT) and positron emission tomography/computed tomography (PET/CT) with radiolabeled SST agonists such as [111In]In-DTPA-octreotide (for scintigraphy and SPECT/CT) and [68Ga]Ga-DOTA-TOC and [68Ga]Ga-DOTA-TATE (for PET/CT). SST imaging allows for the sensitive detection of NETs as well as the identification of patients who will benefit from PRRT [13]. Along these lines, PRRT became one of the best examples of clinical theranostics, the use of one radiopharmaceutical (e.g., [68Ga]Ga-DOTA-TATE) to identify tumors with high SST2 expression and a second based on the same vector (e.g., [177Lu]Lu-DOTA-TATE) to deliver a therapeutic payload.
-
3.
The evaluation of PRRT in the NETTER-1 study: a randomized, controlled phase III trial with both an intervention arm—[177Lu]Lu-DOTA-TATE (Lutathera™) plus high-dose somatostatin analog octreotide LAR—and a control arm (only high-dose somatostatin analog octreotide LAR). The NETTER-1 study demonstrated the superiority of PRRT relative to treatment with octreotide LAR [14]. Based on the NETTER-1 study, PRRT with Lutathera™ was approved by the FDA (U.S. Food and Drug Administration) and EMA (European Medicines Agency) for the treatment of patients with GEP-NETs.
-
4.
The introduction of radiolabeled SST antagonists that are able to recognize more bindings sites on SST-expressing tumor cells show favorable pharmacokinetics and produce higher radiation doses to tumor tissue than agonists despite their very poor internalization rates [6, 8].
2.2 Preclinical Development of Radiolabeled SST Antagonists
SST antagonists were initially developed both for studying the pharmacology and mechanism of the natural hormone somatostatin and for enhancing the secretion of hormones such as growth hormone and insulin. Their design was based on modifications of the cyclic octapeptide octreotide. Octreotide is a truncated and stabilized version of the natural peptide somatostatin-14 (SS-14, Fig. 16.1) that activates SST receptors upon binding and internalizes inside cells as part of a peptide-receptor complex. Critically, the majority of the known radiolabeled SST peptide agonists are based on octreotide. The main structural features of octreotide—d-Phe2-c(Cys3-Phe7-d-Trp8-Lys9-Thr10-Cys14)-Thr(ol)15 (the amino acid numbers correspond to those for SS-14)—are as follows:
-
1.
The tetrapeptide Phe7-d-Trp8-Lys9-Thr10 is essential for the biological activity of SS-14, but l-Trp has been replaced by d-Trp to stabilize the peptide vis a vis enzymatic degradation (Fig. 16.1).
-
2.
As in SS14, the disulfide bridge protects the conformation of the active tetrapeptide.
-
3.
The d-Phe further protects the enzymatically vulnerable N-terminus of the peptide while a hydroxyl functionality lies at the C-terminus.

The evolution of [177Lu]Lu-DOTA-JR11. In the somatostatin-14 sequence, the red amino acids indicate the essential amino acids for receptor recognition. The color code also indicates chirality: red for l-amino acids and green for d-amino acids. The blue structure shows the DOTA chelator
Certain modifications to this structure have been identified as critical for turning a given peptide from an agonist into an antagonist, thereby inhibiting (or entirely preventing) internalization. Specifically, the following characteristics have been determined to favor antagonism:
-
1.
The inversion of chirality of amino acids 2 and 3 (i.e., from d-Phe2 to l-Phe2 and from l-Cys3 to d-Cys3) (Fig. 16.1) [15].
-
2.
The introduction of a substituted phenylalanine—e.g., p-NO2-Phe2 or p-Cl-Phe2—in the first position.
-
3.
The introduction of large hydrophobic aromatic amino acids—e.g., 2Nal15 (3-(2-naphthyl)alanine) or Tyr15 (both l- or d-configuration)—at the C-terminus [16, 17].
Taken together, these combinations contribute to antagonistic properties by weakening biological efficacy while maintaining high SST2 affinity. The first SST2 antagonist, namely BASS—(AcNH-p-NO2-Phe2-cyclo(D-Cys3-Tyr7-D-Trp8-Lys9-Thr10-Cys14)-D-Tyr15-NH2—came out of such a combination.
A number of different modifications have been made to BASS to tune its affinity, SST selectivity, and stability. Two particularly enticing modifications were the inclusion of [1] a carbamoyl functionality (the literature suggested that amide bond-rich moieties are favorably recognized by G-protein coupled receptors) and [2] a urea functionality (which provides structural stabilization via an increase in intra- and intermolecular hydrogen bonds) [18]. A dipeptide bearing both of these modifications—Aph(Hor)-d-Aph(Cbm), in which H-Aph(Hor)-NH2 = 4-amino-phenylalanine(l-hydroorotic acid) and H-d-Aph(Cbm)-NH2 = d-4-amino-phenylalanine(carbamoyl)—was used in the development of gonadotropin releasing hormone (GnRH) antagonists [18]. The question, of course, was whether these functionalities could be implemented in SST antagonists? In BASS, the amino acids 2, 3 14, and 15 were already “tailored” to antagonism. This left only the tetrapeptide (i.e., Tyr7-d-Trp8-Lys9-Thr10) in which these new inserts could be tried. Along these lines, amino acid 7 (position 3 in octreotide) has shown tolerability in terms of substitution, with a number of high affinity SST agonists arising out of its substitution [19]. In the case of the antagonists, the substitution of Tyr7 with carbamoyl-residues did not alter binding affinity and selectivity for SST2 but did improve hydrophilicity. Furthermore, the substitution of D-Trp8 by d-Aph(Cbm) clearly improved affinity as well as selectivity for SST2 [20]. A series of antagonists were developed with various combinations of the aforementioned characteristics [20]. The analog featuring the substitution of d-Trp8 with d-Aph(Cbm) as well as p-Cl-Phe2 in the first position—i.e., p-Cl-Phe2-c(d-Cys3-Tyr7-d-Aph(Cbm)8-Lys9-Thr10-Cys14)d-Tyr15-NH2—is known as LM3 [21]. In contrast, the analog with amino acids 7 and 8 replaced with the dipeptide Aph(Hor)-d-Aph(Cbm) as well as p-Cl-Phe2 in the first position—p-Cl-Phe2-cyclo[d-Cys3-Aph(Hor)7-d-Aph(Cbm)8-Lys9-Thr10-Cys14]-d-Tyr15-NH2—is known as JR11 [20]. JR11 was conjugated to DOTA via its N-terminus and labeled with lutetium-177 (Fig. 16.1) [22].
2.3 The Preclinical Evaluation of [177Lu]Lu-DOTA-JR11
DOTA-modified JR11 was initially complexed with various (radio)metals, including as indium, yttrium, lutetium, gallium, and copper [21, 22]. These early studies clearly demonstrated the high affinity of the JR11 conjugates for SST2 (Table 16.1). The affinities of Lu- and Y-DOTA-JR11 (IC50 = 0.73 ± 0.15 and 0.47 ± 0.05 nM, respectively) were comparable to that of DOTA-JR11 alone (IC50 = 0.72 ± 0.12 nM). However, both In- and Cu-DOTA-JR11 exhibited reduced affinities for the receptor (IC50 = 3.8 ± 0.7 and 29 ± 2.7 nM, respectively). Ga-DOTA-JR11 also displayed a reduced affinity, but this value could be improved by employing the NODAGA chelator to create Ga-NODAGA-JR11 (IC50 = 1.2 ± 0.2 nM) [22]. Biodistribution experiments in mice bearing SST2-expressing xenografts—i.e., a human embryonic kidney cell line transfected with human SST2 HEK-SST2)—further underscored the importance of the radiometal [23]. To wit, [177Lu]Lu-DOTA-JR11 (β−-and γ-emitter; mean energy = 149 keV, Table 16.2) was assessed head-to-head with [90Y]Y-DOTA-JR11 (β—emitter; mean energy = 934 keV, Table 16.2) and [111In]In-DOTA-JR11 (γ emitter, a frequent imaging surrogate for 90Y. The two therapeutic variants—[177Lu]Lu-DOTA-JR11 and [90Y]Y-DOTA-JR11—showed very similar biodistributions and pharmacokinetic profiles. As expected, [90Y]Y-DOTA-JR11 delivered a higher tumor dose due to the higher energy of its β− particles. However, the long tumor retention of DOTA-JR11 is better suited for the longer half-life of 177Lu (t1/2 = 162 h). Interestingly, significant differences were observed between the biodistributions of [90Y]Y-DOTA-JR11 and [111In]In-DOTA-JR11, both with respect to their accumulation in tumor tissue and healthy organs such as stomach, pancreas, and adrenals. Taken together, these data suggest that 111In-DOTA-JR11 may not be a suitable companion imaging agent for 90Y-DOTA-JR11. The most notable result of these studies, however, stemmed from the head-to-head comparison between [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-TATE (Fig. 16.2a–c) [23]. [177Lu]Lu-DOTA-JR11 showed significantly higher tumor uptake than [177Lu]Lu-DOTA-TATE at all time points (from 1 h to 7 d post-injection). Yet even more importantly, the former exhibited a longer residence time in the tumor than the latter. Together, these phenomena resulted in 2.5 times higher tumor radiation dose for [177Lu]Lu-DOTA-JR11 compared to [177Lu]Lu-DOTA-TATE (Fig. 16.2a).

In vivo comparison of [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-TATE in nude mice bearing HEK-SST2 xenografts (Human Embryonic Kidney cells transfected with the human somatostatin receptor subtype 2). (a) AUC (area under the curve) of [177Lu]Lu-DOTA-JR11 (red) and [177Lu]Lu-DOTA-TATE (blue). The AUC represents the tumor uptake integrated over time, which is directly proportional to the radiation dose to the xenograft. The tumor uptake is given as % injected activity per gram tumor tissue (%IA/g). (b) tumor-to-kidney ratios integrated over time for [177Lu]Lu-DOTA-JR11 (red) and [177Lu]Lu-DOTA-TATE (blue). Pharmacokinetic data for A and B were generated from parallel independent biodistribution data collected 1, 4, 24, 72 and 168 h after the injection of [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-TATE. (c) The impact of the amount of injected peptide of [177Lu]Lu-DOTA-JR11 (red) and [177Lu]Lu-DOTA-TATE (blue) on tumor uptake. *p ≤ 0.05. (d) [177Lu]Lu-DOTA-JR11 nanoSPECT/CT images 4 h after the injection of different amounts of peptide (20, 200 and 2000 pmol)
Two critical aspects of the therapeutic use of radiopharmaceuticals are their renal and hematological toxicity. The higher radiation dose to the tumor of [177Lu]Lu-DOTA-JR11 in the aforementioned study was accompanied by a 1.8-fold higher radiation dose to the kidneys and a 1.5-fold higher radiation dose to the bone marrow [23]. To wit, the tumor-to-kidney radiation dose ratio remained higher (by a factor of 1.3) for [177Lu]Lu-DOTA-JR11 compared to [177Lu]Lu-DOTA-TATE (Fig. 16.2b), while the tumor-to-bone marrow dose ratio was also in favor of [177Lu]Lu-DOTA-JR11 by a factor of 1.7. Importantly, the escalation of the mass of injected peptide from 10 pmol to 200 pmol to 2000 pmol significantly suppressed the background uptake of both [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-TATE, especially in SST2-expressing tissues such as the stomach, pancreas, and bone marrow primarily due to the saturation of the receptors in these tissues. Surprisingly, this mass dose escalation did not affect the tumoral uptake of [177Lu]Lu-DOTA-JR11 but significantly reduced that of [177Lu]Lu-DOTA-TATE (Fig. 16.2c). Ultimately, increasing the amount of peptide injected produced excellent tumor-to-background activity concentration ratios for [177Lu]Lu-DOTA-JR11 (Fig. 16.2d). Consequently, increasing the amount of peptide administered with [177Lu]Lu-DOTA-JR11 may improve its safety profile in the clinic by reducing its accumulation (and thus radiation dose) in the bone marrow and other organ healthy tissues.
2.4 Preclinical Therapy Studies with [177Lu]Lu-DOTA-JR11
The therapeutic efficacy of [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-TATE was compared in mice bearing H69 human small cell lung cancer xenografts [24]. The mice were given only a single 30 MBq dose (300 pmol) of each radiotherapeutic. The higher tumor uptake of [177Lu]Lu-DOTA-JR11 compared to [177Lu]Lu-DOTA-TATE in this tumor model as well as the former’s longer tumoral residence time combined to produce a tumor radiation dose for [177Lu]Lu-DOTA-JR11 (1.8 ± 0.7 Gy/MBq) 4.4 times higher than that of [177Lu]Lu-DOTA-TATE (0.36 ± 0.07 Gy/MBq). Treatment with [177Lu]Lu-DOTA-JR11 also resulted in a higher median survival rate (71 days vs 61 days) and a 1.4 times greater delay in tumor growth than [177Lu]Lu-DOTA-TATE, thought the latter was not statistically significant (Fig. 16.3a, b).

Outcome of nude mice bearing H69 xenografts after treatment with 30 MBq [177Lu]Lu-DOTA-JR11 (a) or 30 MBq [177Lu]Lu-DOTA-TATE (b). Data are from Dalm et al. [24]. (c) and (d) Outcome of nude mice bearing BON-SST2 xenografts after treatment with 2 × 20 MBq [177Lu]Lu-DOTA-JR11 (c) or 2 × 30 MBq [177Lu]Lu-DOTA-TOC (d). Data are from Albrecht et al. [25]. (a) and (c) show tumor growth. (b) and (d) show the corresponding Kaplan–Meier survival curves
In another therapy study, [177Lu]Lu-DOTA-JR11 was compared to [177Lu]Lu-DOTA-TOC in an orthotopic xenograft model using human pancreatic BON cells transfected with the human SST2 (BON-SST2) [25]. The study showed that treatment with [177Lu]Lu-DOTA-JR11 produces a significant tumor growth delay and longer survival compared to [177Lu]Lu-DOTA-TOC (Fig. 16.3c, d). The median survival rate was 1.7 times longer for the mice treated with [177Lu]Lu-DOTA-JR11 compared to those treated with [177Lu]Lu-DOTA-TOC (207 days vs 126 days). Furthermore, the improved therapeutic outcome of [177Lu]Lu-DOTA-JR11 was achieved despite using a 30% reduced therapeutic activity compared to [177Lu]Lu-DOTA-TOC (20 MBq vs 30 MBq per cycle, 2 cycles in an interval of 3 weeks). This reduction in activity was necessary due to the higher toxicity of [177Lu]Lu-DOTA-JR11 compared to [177Lu]Lu-DOTA-TOC. Finally, [177Lu]Lu-DOTA-JR11 showed superior targeting properties compared to [177Lu]Lu-DOTA-TATE in an estrogen receptor-positive patient-derived breast cancer mouse model with endogenous SST2 expression [26]. This study confirmed that the antagonist produces significantly higher tumor uptake than the agonist and suggests breast cancer may be an additional indication for [177Lu]Lu-DOTA-JR11.
Overall, the higher tumoral uptake and longer residence time of [177Lu]Lu-DOTA-JR11 compared to SST2 agonists (i.e., [177Lu]Lu-DOTA-TATE or [177Lu]Lu-DOTA-TOC) produces higher tumor doses, more favorable tumor-to-kidney activity concentration ratios, and an enhanced therapeutic effect [23,24,25]. Mansi et al. evaluated the characteristics that lead to the observed differences between SST2 antagonists and agonists on a cellular level [7]. While both [177Lu]Lu-DOTA-JR11 ([177Lu]Lu-OPS201) and [177Lu]Lu-DOTA-TATE exhibited comparable dissociation constant (KD) values of 0.15 ± 0.003 and 0.08 ± 0.02 nM, respectively, [177Lu]Lu-DOTA-JR11 recognized four times more binding sites than [177Lu]Lu-DOTA-TATE [maximum binding sites (Bmax) of 0.37 ± 0.02 vs. 0.09 ± 0.001 nM, respectively]. This could explain, at least partially, its higher accumulation in the SST2-expressing tumors. In addition, [177Lu]Lu-DOTA-JR11 showed faster association, slower dissociation, and longer cellular retention than [177Lu]Lu-DOTA-TATE in vitro. These characteristics could further explain the higher tumor uptake and retention that lead to the enhanced therapeutic efficacy of [177Lu]Lu-DOTA-JR11 compared to [177Lu]Lu-DOTA-TATE, regardless of their localization at the sub-cellular level (cell surface vs internalized, respectively). Interestingly, when [177Lu]Lu-DOTA-TATE bound to SST2 was challenged with an excess of either [natLu]Lu-DOTA-TATE or [natLu]Lu-DOTA-JR11, both non-labelled compounds were able to completely displace the [177Lu]Lu-DOTA-TATE from the receptor and prevent its rebinding. On the contrary, when [177Lu]Lu-DOTA-JR11 bound on SST2 was challenged with an excess of [natLu]Lu-DOTA-TATE, the latter was not able to displace it entirely or prevent its rebinding. This could only be prevented by the antagonist itself. These findings indicate that the antagonist binds not only to more SST2 binding sites but also to sites that are not recognized by the agonist. This hypothesis might have a clinical impact, as NETs are often treated with long-acting somatostatin agonists such as octreotide or lanreotide that are commonly interrupted before the administration of radiolabeled somatostatin agonists such as [177Lu]Lu-DOTA-TATE in order to avoid SST2 saturation. This practice is based on the assumption that the two agonists compete for the same somatostatin receptor sites. These observations on displacement/rebinding suggest that the interruption of somatostatin agonists before PRRT (which can worsen patient symptoms) may not be necessary when the radiolabeled somatostatin analog is an antagonist.
There are still other microscopic characteristics that may explain the gain in therapeutic efficacy associated with using antagonists. The therapeutic efficacy of radiopharmaceuticals is linked to radiation-induced DNA damage. The timing and degree of DNA double strand break (DSB) induction were quantified for [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-TATE using the number of p53-binding protein 1 (53BP1) foci per nucleus over time in SST2-transfected U2OS cells treated with both radiopharmaceuticals [24]. In line with the differences in their cellular uptake, [177Lu]Lu-DOTA-JR11 produced at least 60% more DSBs than [177Lu]Lu-DOTA-TATE, and this increased level remained over time despite the fact that [177Lu]Lu-DOTA-JR11 accumulates primarily on the cell membrane while [177Lu]Lu-DOTA-TATE accumulates mainly in the cytoplasm (i.e., closer to the nucleus and DNA). The radiation effects of [177Lu]Lu-DOTA-JR11 were also assessed by analyzing the cell-cycle distribution of the BON-SST2 cells after incubation with [177Lu]Lu-DOTA-JR11 or [177Lu]Lu-DOTA-TOC [25]. [177Lu]Lu-DOTA-JR11 caused an activity-dependent increase in the number of cells in the G2/M phase as well as a corresponding decrease in the number of cells in the G0/G1 phase. In contrast, same dose of [177Lu]Lu-DOTA-TOC did not affect the cell cycle. This is in line with the increased number of DNA double-strand breaks caused by [177Lu]Lu-DOTA-JR11 compared to [177Lu]Lu-DOTA-TATE [24].
2.5 Potential of Radiolabeled SST Antagonists for Novel Indications of PRRT
The improved tissue binding of radiolabeled SST2 antagonists compared to agonists was demonstrated using human tumor specimens. Human tissue samples from nine different tumors were analyzed via in vitro autoradiography to compare the binding of [125I]I-JR11 vs. [125I]I-Tyr3-octreotide [27] and [177Lu]Lu-DOTA-BASS vs. [177Lu]Lu-DOTA-TATE [28]. In all cases, the radiolabeled SST2 antagonist bound to more SST2 sites, with an antagonist:agonist binding ratio between 3.8 and 21.8 (Fig. 16.4). Such significantly increased binding is likely to increase the therapeutic efficacy of radiolabeled SST2 antagonists. Indeed, this increased binding capacity could make tumors other than GEP-NETs targets for SST2 antagonist RPT despite their relatively low SST2 expression. These tumors—none of which are currently routinely treated with PRRT—include small cell lung cancer, lung NETs, breast cancer, renal cell carcinoma, non-Hodgkin lymphoma, paraganglioma, pheochromocytoma, medullary thyroid cancer, and meningioma.

2.6 Clinical Translation of Radiolabeled SST Antagonists
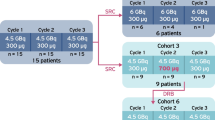
As we have noted, there is preclinical evidence that radiolabeled SST2 antagonists generate higher tumor doses and larger numbers of DNA double strand breaks than agonists, resulting in better treatment efficacy [23, 24]. Yet the question remains: will this difference translate to the clinic? Indeed, the SST2 antagonist [177Lu]Lu-DOTA-JR11 (a.k.a. [177Lu]Lu-OPS201, [177Lu]Lu-satoreotide tetraxetan) was superior to the agonist [177Lu]Lu-DOTA-TATE in a single-center, prospective first-in-human study (phase 0 study) with 4 patients who had advanced, metastatic NET [8]. The most relevant findings of this study were a 3.5-fold higher median tumor dose for [177Lu]Lu-DOTA-JR11 compared to [177Lu]Lu-DOTA-TATE as well as >twofold higher tumor-to-kidney dose ratios with the former. Furthermore, [177Lu]Lu-DOTA-JR11 produced tumor doses of up to 487 Gy and moderate adverse events, with one grade 3 thrombocytopenia after treatment with 3× ~5 GBq (total 15.2 GBq). The other three patients received two to three cycles with a total administrated radioactivity between 5.9 and 13.7 GBq [8]. In another trial, however, Reidy-Lagunes et al. described grade 4 hematotoxicity (leukopenia, neutropenia, and thrombocytopenia) in 4 of 7 patients with NETs treated with 2 × ~7.4 of [177Lu]Lu-DOTA-JR11 (total radioactivity between 10.5 and 15.0 GBq) [9]. As a result, their single-center phase I study (NCT02609737) was suspended, and the protocol was modified to limit the cumulative absorbed bone marrow dose resulting in less bone marrow toxicity. The most important results of this study are summarized in Table 16.3.
[177Lu]Lu-DOTA-JR11 (177Lu-satoreotide tetraxetan) is currently being evaluated in a phase I/II multicenter study (NCT02592707 and NTC05017662) in patients with rapidly progressive NETs [29] and in a retrospective single center study comparing the tumor and organ dosimetry of [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-TOC in the same patients with advanced NETs (Fig. 16.5). Based on preclinical findings by Nicolas et al., those studies were performed with 2–4 times higher amounts of peptide than previous studies in order to reduce the radiation dose to SST2-positive normal tissues [23]. [177Lu]Lu-DOTA-JR11’s “sister” compound, [177Lu]Lu-DOTA-LM3, was also evaluated in a single-center compassionate use study [30]. Table 16.3 displays the most important published clinical findings on [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-LM3. In summary, [177Lu]Lu-DOTA-JR11 yields several times higher tumor radiation doses than [177Lu]Lu-DOTA-TATE and [177Lu]Lu-DOTA-TOC in the same patients [8], resulting in objective response rates (ORR) between 30% and 45% and 1 year disease control rates (DCR) between ~75% and 90% (Table 16.3). Yet at the same time [177Lu]Lu-DOTA-JR11 produces higher bone marrow toxicity [9]. Overall, [177Lu]Lu-DOTA-JR11 is a valuable alternative to [177Lu]Lu-DOTA-TATE and [177Lu]Lu-DOTA-TOC. However, it remains to be evaluated if [177Lu]Lu-DOTA-JR11 improves upon the treatment efficacy and therapeutic indices of its agonist cousins.

Patient with advanced metastatic lung NETs who received [177Lu]Lu-DOTA-TOC and [177Lu]Lu-DOTA-JR11 treatment at an interval of 10 weeks: (a) post-treatment MIP images of quantitative SPECT at 48 and 168 h post-injection as well as (b, c) quantitative SPECT/CT images acquired 48 h after the injection of 7.4 GBq [177Lu]Lu-DOTA-TOC. (d) Post-treatment MIP images of quantitative SPECT at 48 and 168 h post-injection as well as (E and F) quantitative SPECT/CT images acquired 48 h after the injection of 3.7 GBq [177Lu]Lu-DOTA-JR11. The SUV window threshold was 10 for all images. Large arrows show one liver metastasis in segment VIII (a, b, d, e), and small arrows show one bone metastasis in the left acetabulum (a, c, d, f). The radiation dose to the liver segment VIII metastasis was 3.4 Gy/GBq with [177Lu]Lu-DOTA-TOC and 12.6 Gy/GBq with [177Lu]Lu-DOTA-JR11. The radiation dose to the left acetabulum metastasis was 1.5 Gy/GBq with [177Lu]Lu-DOTA-TOC and 9.9 Gy/GBq with [177Lu]Lu-DOTA-JR11. The mean radiation dose to the kidneys was 0.3 Gy/GBq with [177Lu]Lu-DOTA-TOC and 0.8 Gy/GBq with [177Lu]Lu-DOTA-JR11. Asterisks indicate kidneys (a, d). Half the dose of [177Lu]Lu-DOTA-JR11 was injected relative to the dose of [177Lu]Lu-DOTA-TOC due to the former’s higher dose to the kidneys and other organs. Abbreviations: MIP maximum intensity projection, SPECT single photon emission computed tomography, SPECT/CT combined SPECT with computed tomography, SUV standardized uptake value
3 Something Extra
3.1 Controversial Issues
Disease control rate and toxicity profile are the main criteria for evaluating the therapeutic performance of a radiopharmaceutical. The main dose-limiting organs of PRRT with radiolabeled somatostatin agonists are the kidneys and the bone marrow, with an accepted upper threshold radiation dose of 23 Gy for kidneys and 2 Gy for the bone marrow. It is worth mentioning, however, that these values originate from external beam radiotherapy. Therefore, the translation of these radiation dose values to radiopharmaceuticals leaves much to be desired, as radiopharmaceuticals irradiate the kidneys and bone marrow for a much longer period of time but with less energy.
So far, [177Lu]Lu-DOTA-JR11 has shown much higher tumor radiation doses compared to [177Lu]Lu-DOTA-TATE and [177Lu]Lu-DOTA-TOC. However, its therapeutic potential seems to be limited by its higher radiation doses to the bone marrow and kidneys. That said, even though [177Lu]Lu-DOTA-JR11 has produced higher radiation doses to the kidneys compared to [177Lu]Lu-DOTA-TATE and [177Lu]Lu-DOTA-TOC, the former’s tumor-to-kidney radiation dose ratio remains higher [8] (Fig. 16.5). Furthermore, in most PRRT protocols, amino acid infusions are used in order to reduce renal injury. Taken together, the kidney toxicity profile of [177Lu]Lu-DOTA-JR11 does not seem to raise additional concerns compared to [177Lu]Lu-DOTA-TATE and [177Lu]Lu-DOTA-TOC.
Bone marrow toxicity is a slightly different story, as there is no “bone marrow protection” strategy akin to the infusion of amino acids for the kidneys. In the NETTER-1 study, 3% of the [177Lu]Lu-DOTA-TATE group population showed treatment-related serious adverse events of grade 3 or worse, and 2% developed myelodysplastic syndrome after long-term follow-up [31]. According to the current clinical data, SST2 antagonists such as [177Lu]Lu-DOTA-JR11 [9] and [177Lu]Lu-DOTA-LM3 (summarized in Table 16.3) produced more hematological toxicity than agonists such as [177Lu]Lu-DOTA-TATE. It has also been shown that human hematopoietic cells express SST2, especially primitive CD34+ cells [32]. This might be the reason for the more pronounced cytotoxicity of [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-LM3, as both compounds show a higher capacity for SST2 binding than [177Lu]Lu-DOTA-TATE. However, the pathology of higher bone marrow toxicity with radiolabeled SST2 antagonists is not yet understood.
Finally, to wrap up our consideration of toxicity, it is important to note that the high accumulation of [177Lu]Lu-DOTA-JR11 in tumor cells allows for the administration of lower amounts of radioactivity without reductions in treatment efficacy (Fig. 16.5). This has multiple advantages, including lowering radiation doses to the kidney and bone marrow, reducing radiation exposure to the patient and hospital personnel, reducing the cost of per dose, and limiting the amount of radioactive waste produced.
4 The Future
The use of SST antagonists has the potential to offer patients a new and improved theranostic option. Below we have listed four possible future developments for the field:
-
1.
Several tumors other than GEP-NETs are candidates for theranostic studies with SST antagonists, including small cell lung cancer, lung NETs, breast cancer, renal cell carcinoma, non-Hodgkin lymphoma, paraganglioma, pheochromocytoma, medullary thyroid cancer, and meningioma. Along these lines, the evaluation of [177Lu]Lu-DOTA-JR11 in patients with advanced meningiomas is already planned (NCT04997317).
-
2.
Radiolabeled SST2 antagonists cause more bone marrow toxicity than agonists, as they likely exhibit more pronounced SST2 specific binding to hematopoietic cells. But the pathological mechanism of this phenomenon is not fully understood yet. A better understanding of this mechanism would likely aid in the design of radiolabeled SST antagonists that are less toxic to the bone marrow.
-
3.
To date, only lutetium-177 has been used as a radionuclide in conjunction with SST antagonists. The use of alternative radionuclides may decrease bone marrow toxicity and increase tumor toxicity. For example, α-emitters deliver a mean energy of >6000 keV within a maximal range of only 0.06–0.1 mm, resulting in a high linear energy transfer (LET) of ~100 keV/μm (~500 times greater than β−-emitters) (Table 16.2). Due to their high LET, α-emitters principally cause double-strand breaks (DSB) to DNA, the most toxic damage to the cell. Therefore, α-emitters such as actinium-225 and lead-212 are good candidates for use with SST antagonists (Table 16.2).
Auger electron-emitting radionuclides pose yet another option. These radionuclides have high LET, but it is difficult for them to produce DSB unless they are in very close proximity to the cell nucleus. Unfortunately, the specific nuclear accumulation of SST antagonists remains a challenge given their low rate of internalization [33]. However, recent research suggests that the cell membrane is more sensitive to the emission of Auger electrons than the cytoplasm [34]. Therefore, terbium-161—a combined β−- and Auger electron-emitter (Table 16.2)—is also a very promising candidate for the labeling of SST antagonists, as antagonists accumulate mainly on the cell membrane. Indeed, Borgna et al. use clonogenic in vitro assays to demonstrate that [161Tb]Tb-DOTA-LM3 induces a ~ 100 times higher tumor cell death rate than [177Lu]Lu-DOTA-LM3 [33]. The evaluation of [161Tb]Tb-DOTA-LM3 in a phase 0 study is ongoing in patients with GEP-NETs (NCT05359146).
-
4.
Several other receptor systems are also likely suitable for the antagonist approach, for example, targeting the gastrin-releasing peptide receptor (GRP) in patients with prostate cancer, breast cancer, small cell lung cancer, and ovarian cancer [35]. In a compassionate use program, a radiolabeled GRP antagonist—[177Lu]Lu-RM2—was successfully evaluated in 4 patients with metastatic castration-resistant prostate cancer [36] and a prospective open-label phase I/II is ongoing using another radiolabeled GRP antagonist: [177Lu]Lu-NeoB (NCT03872778).
-
5.
Last but not least, larger-scale randomized phase II/III studies evaluating radiolabeled DOTA-JR11/DOTA-LM3 and other promising radiolabeled SST antagonists are needed in order to prove their superiority over agonists in patients with GEP-NETs or other tumors with SST2 expression.
5 The Bottom Line
-
1.
Radiolabeled SST antagonists recognize more binding sites on SST-expressing tumor cells than agonists.
-
2.
Several SST2 antagonists were synthesized for preclinical evaluation. [177Lu]Lu-DOTA-JR11 and [177Lu]Lu-DOTA-LM3 showed the most promising results and were selected for further clinical studies.
-
3.
The SST2 antagonist [177Lu]Lu-DOTA-JR11 showed several times higher tumor radiation doses in patients than [177Lu]Lu-DOTA-TATE or [177Lu]Lu-DOTA-TOC and produced a high objective response rate between 30% and 45% as well as a 1-year disease control rate of up to 90%.
-
4.
In clinical studies, [177Lu]Lu-DOTA-JR11 produced higher bone marrow toxicity than [177Lu]Lu-DOTA-TATE or [177Lu]Lu-DOTA-TOC.
-
5.
Future developments in this field will include the use of SST2 antagonists together with α- and β−/Auger electron-emitting radionuclides, the use of radiolabeled SST2 antagonists for RPT in tumors beyond GEP-NETS, and the expansion of the use of radiolabeled antagonists to other receptor systems.
References
Kloppel G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2011;18(Suppl 1):S1–16.
Caplin ME, Baudin E, Ferolla P, Filosso P, Garcia-Yuste M, Lim E, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604–20.
Zandee WT, Kamp K, van Adrichem RC, Feelders RA, de Herder WW. Effect of hormone secretory syndromes on neuroendocrine tumor prognosis. Endocr Relat Cancer. 2017;24(7):R261–R74.
Fani M, Mansi R, Nicolas GP, Wild D. Radiolabeled somatostatin analogs-a continuously evolving class of radiopharmaceuticals. Cancers (Basel). 2022;14(5):1172.
Ambrosini V, Kunikowska J, Baudin E, Bodei L, Bouvier C, Capdevila J, et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur J Cancer. 2021;146:56–73.
Ginj M, Zhang H, Waser B, Cescato R, Wild D, Wang X, et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci U S A. 2006;103(44):16436–41.
Mansi R, Plas P, Vauquelin G, Fani M. Distinct in vitro binding profile of the somatostatin receptor subtype 2 antagonist [(177)Lu]Lu-OPS201 compared to the agonist [(177)Lu]Lu-DOTA-TATE. Pharmaceuticals (Basel). 2021;14(12):1265.
Wild D, Fani M, Fischer R, Del Pozzo L, Kaul F, Krebs S, et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: a pilot study. J Nucl Med. 2014;55(8):1248–52.
Reidy-Lagunes D, Pandit-Taskar N, O’Donoghue JA, Krebs S, Staton KD, Lyashchenko SK, et al. Phase I Trial of Well-Differentiated Neuroendocrine Tumors (NETs) with Radiolabeled Somatostatin Antagonist (177)Lu-Satoreotide Tetraxetan. Clin Cancer Res. 2019;25(23):6939–47.
Fani M, Nicolas GP, Wild D. Somatostatin Receptor Antagonists for Imaging and Therapy. J Nucl Med. 2017;58(Suppl 2):61S–6S.
Fjalling M, Andersson P, Forssell-Aronsson E, Gretarsdottir J, Johansson V, Tisell LE, et al. Systemic radionuclide therapy using indium-111-DTPA-D-Phe1-octreotide in midgut carcinoid syndrome. J Nucl Med. 1996;37(9):1519–21.
Ambrosini V, Fani M, Fanti S, Forrer F, Maecke HR. Radiopeptide imaging and therapy in Europe. J Nucl Med. 2011;52(Suppl 2):42S–55S.
Baumann T, Rottenburger C, Nicolas G, Wild D. Gastroenteropancreatic neuroendocrine tumours (GEP-NET) – imaging and staging. Best Pract Res Clin Endocrinol Metab. 2016;30(1):45–57.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of (177)Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–35.
Bass RT, Buckwalter BL, Patel BP, Pausch MH, Price LA, Strnad J, et al. Identification and characterization of novel somatostatin antagonists. Mol Pharmacol. 1996;50(4):709–15.
Raynor K, Murphy WA, Coy DH, Taylor JE, Moreau JP, Yasuda K, et al. Cloned somatostatin receptors: identification of subtype-selective peptides and demonstration of high affinity binding of linear peptides. Mol Pharmacol. 1993;43(6):838–44.
Hocart SJ, Jain R, Murphy WA, Taylor JE, Coy DH. Highly potent cyclic disulfide antagonists of somatostatin. J Med Chem. 1999;42(11):1863–71.
Jiang G, Stalewski J, Galyean R, Dykert J, Schteingart C, Broqua P, et al. GnRH antagonists: a new generation of long acting analogues incorporating p-ureido-phenylalanines at positions 5 and 6. J Med Chem. 2001;44(3):453–67.
Ginj M, Schmitt JS, Chen J, Waser B, Reubi JC, de Jong M, et al. Design, synthesis, and biological evaluation of somatostatin-based radiopeptides. Chem Biol. 2006;13(10):1081–90.
Cescato R, Erchegyi J, Waser B, Piccand V, Maecke HR, Rivier JE, et al. Design and in vitro characterization of highly sst2-selective somatostatin antagonists suitable for radiotargeting. J Med Chem. 2008;51(13):4030–7.
Fani M, Del Pozzo L, Abiraj K, Mansi R, Tamma ML, Cescato R, et al. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: the chelate makes the difference. J Nucl Med. 2011;52(7):1110–8.
Fani M, Braun F, Waser B, Beetschen K, Cescato R, Erchegyi J, et al. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J Nucl Med. 2012;53(9):1481–9.
Nicolas GP, Mansi R, McDougall L, Kaufmann J, Bouterfa H, Wild D, et al. Biodistribution, pharmacokinetics, and dosimetry of (177)Lu-, (90)Y-, and (111)In-labeled somatostatin receptor antagonist OPS201 in comparison to the agonist (177)Lu-DOTATATE: the mass effect. J Nucl Med. 2017;58(9):1435–41.
Dalm SU, Nonnekens J, Doeswijk GN, de Blois E, van Gent DC, Konijnenberg MW, et al. Comparison of the therapeutic response to treatment with a 177Lu-Labeled somatostatin receptor agonist and antagonist in preclinical models. J Nucl Med. 2016;57(2):260–5.
Albrecht J, Exner S, Grotzinger C, Prasad S, Konietschke F, Beindorff N, et al. Multimodal imaging of 2-Cycle PRRT with (177)Lu-DOTA-JR11 and (177)Lu-DOTATOC in an orthotopic neuroendocrine xenograft tumor mouse model. J Nucl Med. 2021;62(3):393–8.
Dalm SU, Haeck J, Doeswijk GN, de Blois E, de Jong M, van Deurzen CHM. SSTR-mediated imaging in breast cancer: is there a role for radiolabeled somatostatin receptor antagonists? J Nucl Med. 2017;58(10):1609–14.
Reubi JC, Waser B, Macke H, Rivier J. Highly increased 125I-JR11 antagonist binding in vitro reveals novel indications for sst2 targeting in human cancers. J Nucl Med. 2017;58(2):300–6.
Cescato R, Waser B, Fani M, Reubi JC. Evaluation of 177Lu-DOTA-sst2 antagonist versus 177Lu-DOTA-sst2 agonist binding in human cancers in vitro. J Nucl Med. 2011;52(12):1886–90.
Nicolas GP, Ansquer C, Lenzo NP, McEwan S, Wild D, Hicks RJ. An international open-label study on safety and efficacy of 177Lu-satoreotide tetraxetan in somatostatin receptor positive neuroendocrine tumours (NETs): an interim analysis. Ann Oncol. 2020;31(supplement 4):S771.
Baum RP, Zhang J, Schuchardt C, Muller D, Macke H. First-in-humans study of the SSTR antagonist (177)Lu-DOTA-LM3 for peptide receptor radionuclide therapy in patients with metastatic neuroendocrine neoplasms: dosimetry, safety, and efficacy. J Nucl Med. 2021;62(11):1571–81.
Strosberg JR, Caplin ME, Kunz PL, Ruszniewski PB, Bodei L, Hendifar A, et al. (177)Lu-Dotatate plus long-acting octreotide versus highdose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021;22(12):1752–63.
Oomen SP, van Hennik PB, Antonissen C, Lichtenauer-Kaligis EG, Hofland LJ, Lamberts SW, et al. Somatostatin is a selective chemoattractant for primitive (CD34(+)) hematopoietic progenitor cells. Exp Hematol. 2002;30(2):116–25.
Borgna F, Haller S, Rodriguez JMM, Ginj M, Grundler PV, Zeevaart JR, et al. Combination of terbium-161 with somatostatin receptor antagonists-a potential paradigm shift for the treatment of neuroendocrine neoplasms. Eur J Nucl Med Mol Imaging. 2022;49(4):1113–26.
Pouget JP, Santoro L, Raymond L, Chouin N, Bardies M, Bascoul-Mollevi C, et al. Cell membrane is a more sensitive target than cytoplasm to dense ionization produced by auger electrons. Radiat Res. 2008;170(2):192–200.
Cescato R, Maina T, Nock B, Nikolopoulou A, Charalambidis D, Piccand V, et al. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J Nucl Med. 2008;49(2):318–26.
Kurth J, Krause BJ, Schwarzenbock SM, Bergner C, Hakenberg OW, Heuschkel M. First-in-human dosimetry of gastrin-releasing peptide receptor antagonist [(177)Lu]Lu-RM2: a radiopharmaceutical for the treatment of metastatic castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. 2020;47(1):123–35.
Schottelius M, Simecek J, Hoffmann F, Willibald M, Schwaiger M, Wester HJ. Twins in spirit – episode I: comparative preclinical evaluation of [(68)Ga]DOTATATE and [(68)Ga]HA-DOTATATE. EJNMMI Res. 2015;5:22.
Reubi JC, Schar JC, Waser B, Wenger S, Heppeler A, Schmitt JS, et al. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med. 2000;27(3):273–82.
Kong G, Hicks RJ. Peptide receptor radiotherapy: current approaches and future directions. Curr Treat Options in Oncol. 2019;20(10):77.
Muller C, Domnanich KA, Umbricht CA, van der Meulen NP. Scandium and terbium radionuclides for radiotheranostics: current state of development towards clinical application. Br J Radiol. 2018;91(1091):20180074.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Wild, D., Fani, M. (2023). Case Study #6: [177Lu]Lu-DOTA-JR11: A Somatostatin Receptor Subtype 2 Antagonist for Radiopharmaceutical Therapy. In: Bodei, L., Lewis, J.S., Zeglis, B.M. (eds) Radiopharmaceutical Therapy. Springer, Cham. https://doi.org/10.1007/978-3-031-39005-0_16
Download citation
DOI: https://doi.org/10.1007/978-3-031-39005-0_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-39004-3
Online ISBN: 978-3-031-39005-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)