
Abstract
Acute Kidney Injury (AKI) and Acute Lung Injury (ALI) are two everyday clinical conditions managed in hospitalization. With the concept that there is a continuous crosstalk between organs, any specific situation that injures one can in turn affect the other. In both scenarios, there is a systemic inflammatory response where chemokines produced act upon distant organs causing neutrophil infiltration, capillary leak, endothelial dysfunction, oxidative stress, among others. AKI leads to accumulation of toxic substances, fluid overload, electrolyte imbalance, and metabolic acidosis. In the lungs, this leads to edema, altered lung mechanics, and impaired gas exchange. The hypoxia and hypercapnia in ALI cause vasoconstriction of renal vessels and dilation of systemic vasculature which reduces the renal filtration rate. Mechanical Ventilation (MV) in this context is associated with an increase in mortality rates. Understanding the underlying pathological mechanisms of damage in each clinical situation that can lead to renal and/or respiratory failure can provide a solid foundation for recognizing the severity of any individual patient and what to expect from the clinical picture’s evolution.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
9.1 Introduction
The body’s anatomical and physiological characteristics offer the perfect organ communication pathway for coordination and homeostasis in order to provide a suitable environment. Broadly speaking, the circulatory and nervous system are the two main routes for hormones, chemical messengers, neurotransmitters, and countless mediators to reach distant organs in this “crosstalk” scenario and cause remote changes in either physiological conditions or pathological organ-specific diseases.
The kidney and lung have several characteristics and similarities that outline how both are bound to reach one another. They have an extensive capillary network since they receive all of the cardiac output with the purpose of filtrating or oxygenating the blood, respectively. They also have similar cell polarization and salt-water channels. This allows orchestration of crucial functions such as acid-base management, erythropoietin production, renin-angiotensin-aldosterone system (RAAS) activation for an adequate vascular tone, and fluid balance [1, 2]. The importance of acknowledging this relies upon the fact that when either of these two organs is injured, the other might suffer too, and hence needs special consideration when planning clinical care.
Understanding the underlying pathological mechanisms of damage in each clinical situation that can lead to renal and/or respiratory failure can provide a solid foundation for recognizing the severity of any individual patient and what to expect from the clinical picture’s evolution. Not only will it provide a greater awareness and anticipation for any possible mortal complication, but it will also enlighten the way for new ways of prevention.
9.2 Pathophysiology
A broad spectrum of diseases and pathological conditions can account for the cause of acute kidney injury (AKI) or acute lung injury (ALI) in any severely ill patient. Reinforcing the concept that there is a continuous crosstalk between organs, any specific situation that injures one can in turn affect the other. For example, pneumonia being a risk factor for developing AKI, or in an AKI scenario developing lung dysfunction is the most adverse outcome [3, 4]. There are some conditions not necessarily arousing from either organ that affects both, such as sepsis with multiorgan dysfunction (MOD) [5, 6]. In any case, patients who have acute respiratory distress syndrome (ARDS) have a greater chance of developing AKI (and vice versa), and also patients with both conditions can duplicate their mortality rates [2, 7]. The need for mechanical ventilation (MV) is higher in patients with AKI, and in this clinical scenario, the in-hospital mortality rate dramatically increases, as MV is an independent predictor of mortality [8].
Firstly, ALI is characterized by rapid alveolar damage and inflammation at neutrophils expenses. When the latest are activated, they trigger the formation of oxygen reactive species, cytokines, and proteases. This leads to epithelial damage and an increase in the alveolar-capillary barrier’s permeability [9]. ARDS includes bilateral radiographic opacities and reduced lung compliance with an increase in the physiological dead space that contributes to the characteristic hypoxemia of this setting. The Berlin definition also includes that the edema cannot be explained by cardiac failure or fluid overload. It also categorizes in mild, moderate, and severe, depending on the PaO2/FiO2 ratio of each patient [2, 10] (Table 9.1).
On the other hand, AKI develops due to an abrupt decline in kidney function that reflects the decrease of glomerular filtration rate (GFR) and retention of waste products such as creatinine and urea [11, 12]. It is diagnosed by a significant increase in serum creatinine and the reduction of urinary volume (Table 9.2).
There are little sensitive and quick biological markers that can be looked out for in these patients. The most commonly used markers to measure kidney function are serum creatinine and blood urea nitrogen (BUN). For ARDS, the diagnosis is based upon arterial blood gases. Recent experiments show that urine IL-6 and lung myeloperoxidase (MPO) activity are sensitive and rapid markers of kidney function and lung neutrophil activation, respectively. However they are still confined to studies in mice, enhancing the need for special attention and anticipation to the clinical deterioration symptoms or late markers [5].
9.3 Renal-Induced Lung Damage
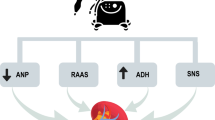
It is well known that AKI leads to accumulation of toxic substances such as urea, to fluid overload, electrolyte imbalance, and metabolic acidosis. In the lungs, this overall leads to altered lung mechanics and gas exchange, edema, and compensatory hyperventilation. Additionally, it is known that the kidney upregulates and secretes cytokines, hence kidney injury involves a systemic inflammation where chemokines produced act upon distant organs causing neutrophil infiltration, capillary leak, endothelial dysfunction, oxidative stress, among others. This proinflammatory state is being studied only recently and is opening many doors in understanding how and why the magnitude of the multi-organ damage in different scenarios [13, 14] (Fig. 9.1).

Kidney–lung crosstalk during acute kidney injury (AKI). DAMPs damage-associated molecular pattern
9.3.1 Cytokine Induced Damage
Cytokines are small peptides secreted by numerous cells that have autocrine, paracrine, and endocrine actions in order to modulate the immune system. Subfamilies include chemokines, lymphokines, interferon, interleukins, and all through cell signaling are responsible for events such as [15, 16]:
-
Neutrophil and lymphocyte migration and adhesion
-
Caspase activation and subsequent cell apoptosis
-
Oxidative stress
-
Changes in actin assembly and cytoskeleton (this leads to greater cellular gap and alteration of pulmonary endothelial-epithelial barrier)
-
Increased vascular permeability
-
Anti-inflammatory effects, which are usually unbalanced
Altogether are accountable for lung inflammation and ALI in the context of AKI. Blood testing and laboratory experiments help elucidate the specific effects of each cytokine. Taking into account the advancement of biological molecules and technology, counteracting or mimicking certain effects may eventually play a part in treatment and prevention [17].
IL-6 is a well-known and studied proinflammatory cytokine that rises in multiple acute and chronic conditions and serves as a reliable serum marker of inflammation. It stimulates the production of CXCL-1 and MIP-2, chemokines that through the CXCR2 receptor causes the infiltration of neutrophils which perpetuates cytokine production and the inflammatory response. It also causes lung endothelial cells to produce IL-8 which furthermore increases neutrophils in the alveolar space [1, 18].
It is proposed IL-8 plays a central role in the development of ALI in the context of AKI, as it was found to be increased in serum and bronchoalveolar fluid, and correlated with prolonged MV and mortality [19]. These two adverse outcomes were also shown in several studies in mice that measured and evidenced an increase in serum IL-6 after nephrectomy, ischemic AKI, or a sham operation [20]. Methodology in these studies also involved IL-6 deficient mice or neutralizing antibodies to block its action. In these cases, less lung inflammation, capillary leak, and reduced IL-8 levels were revealed, which were further restored when IL-6 was artificially injected, suggesting its crucial effect in inflammation and lung damage.
It is worth emphasizing that whenever the body carries out a certain action, its counteraction unleashes. IL-6, although being a proinflammatory cytokine, stimulates the production of IL-10, a quintessentially anti-inflammatory cytokine of the compensatory anti-inflammatory response syndrome (CARS). IL-10 has been shown to limit organ injury by downregulating inflammation in either infectious or noninfectious systemic inflammatory response syndrome (SIRS) [14]. Its primary source is CD4 T-lymphocytes of the spleen, also macrophages and B cells. Several studies with splenectomized or CD4 deficient mice have been conducted in order to mimic human AKI and the subsequent inflammatory response and have proven that IL-6 levels were higher, IL-10 lower, and lung inflammation was worse. In fact, it is known that patients with polymorphism that increases IL-10 production have reduced mortality in ALI and critically ill scenarios [4]. However, increased levels do not always foresee a better outcome. It was evidenced in splenectomized mice that IL-6, IL-8, and IL-10 were all higher in AKI with splenectomy in comparison to AKI alone and in this context lung injury was worse. In another cohort of patients, this increase in cytokines was associated with increased mortality and risk of sepsis [21]. The underlying approach may be that, both pro- and anti-inflammatory cytokines markedly elevated reflect a dysregulated and unbalanced inflammatory response, hence there is no coordinated reaction to the damage stimuli.
Tumor necrosis factor α (TNF-α) is another cytokine primarily involved in lung damage in AKI through a wide range of actions. It is produced by macrophages and, in our concern, accumulates in the lungs in order to cause neutrophil activation (and the subsequent release of more cytokines) and its degranulation, lymphocyte migration, and particularly activation of lung endothelial cells. In the latter, TNF-α signals the receptor TNRF1 and triggers the cell apoptotic pathway. This, together with the surrounding inflammation causes a disruption in the lung’s endothelium-epithelium barrier, which leads to edema that unable a proper gas exchange and clinically manifests as hypoxia in ARDS.
Damage-associated molecular patterns (DAMPs) are also released from kidney cells upon damage. They are not cytokines, but yet they can also activate immune cells and travel through the bloodstream to unleash distant injury. The consequences of mitochondrial DAMPs released in the context of kidney ischemia-reperfusion AKI convey the effects these molecules have on lung injury. It is known that the mitochondria plays an indispensable role in the cell’s function and survival, and it has been proven that these particular DAMPs lead to metabolic alteration of lung epithelial cells, particularly the fatty-acid oxidation pathway. Furthermore, they promote neutrophil infiltration and pulmonary endothelial cell’s apoptosis [22].
9.3.2 Immune Cells
The cytokines mentioned above are only part of a much bigger network. They are secreted by a number of specialized immune cells in the context of AKI as an inflammatory triggering event. Spreading distantly through the bloodstream, they act as chemokines attracting other immune cells to a specific organ, and activating them as well. In this reciprocal way, they perpetuate the inflammatory response and may potentially lead to organ injury.
Neutrophils are key mediators of lung injury in AKI. Infiltration of this type of cells have been studied in multiple animal models and were reported to be elevated in early phases of lung injury, mostly within the first 2 h after kidney injury [13, 19]. Chemoattraction leads to migration toward the vascular endothelium, and the accumulation likely leads to the formation of a plug, causing congestion and microvascular occlusion.
Once settled in the tissue, neutrophils release a series of enzymes, as well as oxygen reactive species, all which derive to cell and extracellular matrix damage, compromising tissue’s structure and functionality [23]. A specific protease that merits special mention is the neutrophil elastase (NE), which acts primarily on collagen, proteoglycans, and elastin found in the pulmonary cell’s membrane, and especially on the surrounding matrix. It has been studied that the NE activity increases substantially in AKI both systemically and in the lungs, demonstrating its implication in this crosstalk [18]. Sivelestat sodium hydrate is a specific NE inhibitor for clinical use in the context of ALI induced by systemic inflammation. It has been studied in mice models for specific AKI scenarios and evidenced reduced lung inflammation, furthermore enhancing its pathological role.
Methods that inhibit certain molecules can help elucidate its contribution to the pathology of a disease, as was seen with the neutralization of granulocyte colony-stimulating factor (G-CSF) that reduces lung damage in AKI. This is a cytokine involved in mature neutrophil activation and is also responsible for emergency granulopoiesis under stressful conditions, a situation that has been linked with lung injury [9].
Another emerging mechanism of damage proposed with neutrophil’s involvement is necroinflammation, where necrotic renal tubular cells lead to the release of histones as damage-associated molecular patterns. The citrullination of these histones potentiates the signaling for the formation of neutrophils extracellular traps, also known as NETs and NETosis [24]. Due to the anatomical distribution, through the vena cava and given the lung’s fine capillary network, they are likely to end up in the lungs. The NETs contain proinflammatory proteins, produce endothelial damage by itself, can activate the complement and coagulation cascade, and by auto-amplification induce other neutrophils to form more NETs. Studies revealed an increase in NET in ischemic and ischemia-reperfusion AKI models that can potentially be reduced by inhibitors such as anti-histone antibodies [19, 25].
Macrophages are another cellular lineage responsible for many of the pathological mechanisms of lung injury mentioned above. They come from monocytes that are recruited during inflammation and differentiate in order to secrete various cytokines that orchestrate the inflammatory response: IL-6, TNF-α, and G-CSF to name a few. This happens not only by the DAMPs exposed during AKI, but macrophages were evidenced to secrete G-CSF upon an increase of urea as well. They also up-regulate CXCL1 in the lungs, which as mentioned above causes neutrophil infiltration and activation [9, 26].
T-lymphocyte involvement reflects the bridge between innate and adaptive immunity. With regard to lung damage, T-cells are particularly implicated in lung cell apoptosis in AKI models. Cytotoxic CD8+ lymphocytes through the release of granzyme B, a serine protein present in its granules, and the Fas–Fas ligand interaction (two molecules involved in cell death regulation) cause caspase activation, which translates into cell death, vascular barrier dysfunction, and pulmonary edema [14, 27].
Cytokines are not the only products released by these immune cells. They are a major source of free radicals and oxygen reactive species that cause lipid peroxidation, mutations, and DNA damage which in turns leads to aberrant protein production for cell function and repair. It is furthermore aggravated in patients that require MV, as oxidative stress is related to high levels of oxygen administration. It is evidenced that antioxidant substances such as glutathione, beta-carotene, and vitamin C are reduced significantly in patients with ARDS [13, 23]. A constant environment of oxidative stress is also present in chronic kidney disease (CKD), and this can explain why there is evidence of DNA changes, fibrosis, cell apoptosis, and tissue damage in the lungs even when there is no objectifiable lung disease nor acute respiratory failure [28] (Table 9.3).
9.3.3 Ischemia Induced Damage
The role that an appropriate oxygen delivery has for suitable organ function and homeostasis cannot be put into question. In a kind of tendentious way, we usually attribute blood oxygenation merely to the lungs. Nevertheless, bearing the crosstalk mechanism in mind, it also depends on an adequate cardiac and kidney function. The kidneys are crucial for a proper vascular tone through renin secretion, maintaining a good preload by managing fluid balance, and for stimulating red blood cells formation via erythropoietin secretion [6]. An inadequate blood supply leads to organ stress, as it can happen in prerenal AKI, where hypovolemia causes ischemia; or after renal transplantation, as grafts undergo a period of ischemia and reperfusion.
The ischemia-reperfusion (IR) scenario causes cytokine production in the kidney, which as mentioned previously, evokes an inflammatory response and is accountable for the distant-organ damage. IR increases the mRNA expression of IL-6, TNF-α, and IL-1 not only in the kidney but in the lungs as well [29]. Another soluble mediator suggested to have a pathological role is osteopontin (OPN), a protein released from the immune cells activated in this context. It has a proinflammatory effect and can act synergistically with TNF-α in remote lung damage [30]. As inflammation itself is a solid explanation for distant organ dysfunction, studies are compelled to intervene in its progression. Anti-inflammatory effects of medications such as artesunate, or induction of heme oxygenase, which is normally up-regulated in stressful conditions because its byproducts have cytoprotective effects, have been proven to reduce lung damage in IR models. Even though the exact mechanisms are far from being established, they furthermore highlight the importance that inflammation has as a common factor for multiple adverse outcomes of AKI [11, 31].
9.3.4 Pulmonary Edema
We have already seen that the systemic inflammation, oxidative stress, and soluble mediators cause increased capillary permeability, leakage, and epithelium-endothelium barrier dysfunction, leading to the formation of a non-hydrostatic edema. Cytokines also mediate their effects by binding to cell receptors and changing the levels of phosphorylated proteins [32]. It is believed that this explains why it has been seen that in early stages of AKI, there is a down-regulation of sodium and aquaporin channels in the lungs. In physiological conditions, these energy dependent channels create an osmotic gradient which drives water from the alveoli inside the cells. Without a suitable amount, water will accumulate and contribute to pulmonary edema [13, 23].
It has also been known for decades that uremia causes serositis and fluid effusion. This can also happen in the pericardium, which in serious conditions may lead to cardiac tamponade and fluid overload in the lungs; or as a pleural effusion itself. The edema is also present in the alveoli as well, manifesting as a reduced carbon dioxide diffusion capacity (DLCO). Pathology autopsies even described lung affectation as “uremic pneumonitis,” characterized by hyaline membrane formation and inflammatory exudates [33]. It is worth noticing that uremic toxins also have remote effects that can perpetuate AKI and furthermore complicate the clinical scenario. Symptoms usually present are nausea and vomiting, and a common condition in uremic patients is platelet dysfunction that can lead to hemorrhage. If any of these ends up in a significant volume depletion, prerenal AKI can be seriously aggravated, feeding the damage cycle [34, 35].
Nonetheless, this is not the only cause of water accumulation in alveolar space and lung interstitium that impairs gas exchange and respiratory mechanics. In kidney failure, most of the patients present oliguria or an important reduction in the GFR. With the subsequent water retention, the heart becomes unable to manage this excess. Bearing that the concomitant electrolyte and acid/base imbalances deteriorate its contractile function, the fluid overload ends up in the lungs, as they can hold a greater volume due to their extensive capillary network and characteristic compliance. This condition has long been recognized as an important cause of respiratory failure and is associated with increased mortality [33].
9.4 Lung-Induced Renal Damage
Considering the bidirectional nature of the organ crosstalk concept, we can now fairly imagine that when the lung is under stress, remote repercussions are always a possibility. The association of ARDS and AKI has been studied and proved [36]. Having the concepts of distant lung damage during AKI, similar mechanisms of distant kidney damage in lung injury can be proposed, always adjusting to the biological plausibility and clinical conditions that may lead to them. Organ dysfunction in the context of ARDS has been investigated and specifically AKI has been pinpointed as a need for attention, simply because, as both organs receive all of the cardiac output they have an extensive capillary network which is prone to be exposed to circulating mediators [37] (Fig. 9.2).

Lung–kidney crosstalk during acute lung injury (ALI). GFR glomerular filtration rate, RAS renin-angiotensin system, VILI ventilator-induced lung injury, ARDS acute respiratory distress syndrome
9.4.1 Systemic Inflammation
IL-6, IL-1, IL-8, and TNF-α (mainly) together with the battery of immune cells mentioned in the renal-induced lung damage are not only limited to renal injury, but are the commanders of systemic inflammation arising from any injured organ. Proinflammatory mediators are also released during ALI and ARDS, affecting renal vascular tone by vasodilation of vessels and reducing perfusion pressure, as well as by activating apoptotic pathways in renal epithelial cells [6]. A particular mediator that promotes neutrophil infiltration and endothelial cell apoptosis, angiopoietin-2 (AGPT2) has been studied in ARDS pathogenesis. It acts as a marker of fluid overload [38] as it modulates cell permeability, it has been registered an increase in plasma levels in cases of shock and sepsis, and is even suggested as a biomarker for ARDS prediction and its severity [39]. Several studies evidenced the elevation of this growth factor in critically ill patients. Particularly, it has been independently associated with severe AKI only in patients who also had previously developed ARDS, suggesting a close interaction between these two organs in pathologic conditions [40].
The probability for a patient to develop AKI in this context depends on other variable factors that increase susceptibility to propagation of damage and inflammation. Age, moderate to severe ARDS (specially as it involves use of MV), hypertension, diabetes mellitus, heart failure, unconsciousness while hospitalization and elevation of D-dimer were some riskfactors associated with the likeliness of developing AKI during ARSD [41,42,43]. Although the exact mechanism of action has not yet been established, an elevated urinary IL-18 is a predictor of ARDS mortality, and it is a cytokine systemically elevated in obesity, suggesting that an increased body mass index can also account as a risk factor [44].
9.4.2 Ventilator-Induced Lung Injury
The origins of MV can be traced down to the late nineteenth century with subatmospheric body-enclosing boxes and when positive pressure ventilation gained its place after the polio epidemic. It is nowadays crucial in intensive care units (ICU), considered a life-saving therapy when used adequately [45]. However, as any action taken during clinical care, it can result in an adverse outcome. Ventilator-induced lung injury (VILI) is a well-established entity which arose from the study of the increased mortality in a number of people with ARDS being ventilated. There are four main mechanisms for VILI: barotrauma, volutrauma, atelectrauma, and biotrauma. Systematic reviews of invasive mechanical ventilation as a risk factor evidenced a distressing three-fold risk of developing AKI [46].
Reasonably if there is a notable trauma and/or atelectasis in the lungs that derives to a significant hypoxemia the kidneys will suffer in the context of a multiorgan dysfunction, specially being a highly metabolic organ susceptible to minimal changes in blood oxygen levels due to the amount of energy it requires to work. Anyhow, biotrauma deserves special attention when considering remote kidney injury during MV. It consists of a biological proinflammatory response of the lungs that can emerge from direct cell injury or indirectly by cell-signaling from the other three physical VILI mechanisms [47, 48]. Particular cytokines could be identified when scenarios of AKI during MV were replicated in animal models, as, for example, macrophage inflammatory protein (MIP-1α), NF-κB, VCAM-1 adhesion molecule, and vascular endothelial growth factor (VEGF) [7, 49]. The release of inflammatory mediators through the bloodstream reaches the kidneys, changing the expression of adhesion molecules, attracting immune cells, and even causing remote renal tubular cell apoptosis evidenced by increasing levels of circulating Fas–Fas ligand [50]. Using adequate levels of tidal volume (like the once adjusted by weight) has been studied as a protective ventilation strategy that reduces the AKI incidence during ARDS by creating a mild hypercapnic acidotic environment that acts as cytoprotective and anti-inflammatory [23].
Other mechanisms can be accounted for by the deterioration of ventilated patients, involving the decrease in the GFR. On the one hand, when high ventilatory pressures are used, this results in an increase of pressure that the right heart has to overcome. With a greater afterload, together with a reduced venous return, the cardiac output decreases, putting into danger an adequate renal perfusion. MV also activates the sympathetic nervous system and the RAAS, which drive to vasoconstriction, further decreasing the GFR [6].
9.4.3 Hypoxia and Hypercapnia
Hypercapnia has significant effects in the kidney by acting directly and indirectly in antagonistic ways with regard to changes of the vascular tone. Directly, high levels of CO2 cause constriction of intrarenal vessels. Indirectly, it causes dilation of the systemic vasculature. The net effect is a reduction in the renal blood flow which leads to cell hypoxia and hence loss of functional and metabolic processes, and a reduction in the GFR which will lead to fluid retention and accumulation of toxic substances. This has been evidenced in patients with chronic obstructive pulmonary disease (COPD), as they may usually present with fluid retention and reduced urinary sodium excretion. In extreme emphysematous cases, where intra-abdominal pressure is extremely elevated, veins are compressed and hence with less venous drainage kidney edema can develop which will furthermore aggravate the cell’s function [51].
Patients that present obstructive sleep apnea, another chronic condition, also show renal repercussions. Permanent nocturnal hypoxemia primarily induces a constant inflammatory state that is known to produce vascular disease such as atherosclerosis and is also related to hypertension [52]. The cytokines together with the vascular dysfunction, as already noted, can affect kidney function at various levels.
9.5 Conclusion
To only consider that organ communication involves a simple bidirectional cause and effect mechanism would be a reductionism. As it has been discussed, organ crosstalk is a vast network of simultaneous and consequent events, which are as complex in physiological conditions that aim to maintain homeostasis, as in a pathological state. AKI and ARDS are two everyday clinical conditions managed in hospitalization and intensive care units. When either of these major organs is injured first, we can think about the remote repercussions in two ways: functional and inflammatory. The first involves the direct effects of the loss of functionality of one organ; as it is in AKI, the accumulation of urea, fluid retention, and electrolyte imbalance will mainly lead to pulmonary edema; and in ARDS, the hypoxia will lead to renal ischemia and cell death. On the other hand, inflammation is elicited by most, if not all, of the damage mechanisms that lead to respiratory or kidney failure. Cytokines have the particularity of traveling through the bloodstream and being able to establish in distant organs, evoke inflammation, and immune cell activation. And even though here we focus specifically on the lungs and kidneys, the other organs also discussed in this book are as well-being altered, hence crosstalk comes from multiple directions.
The boundless communication pathways and mechanisms of remote organ damage in particular clinical conditions should not overwhelm when planning and delivering patient care. The objective of acknowledging this is being able to have a greater awareness of red flags and adapt to what to expect about each patient’s evolution, anticipating the possible aggravating conditions. Curiosity upon organ crosstalk and its components guides studies to unfold each time more the complexity of these interactions, enlightening the pathophysiological understanding but also giving rise to novel and beneficial therapeutic approaches. The findings of these studies are clinically relevant and show the importance of an integrative approach in the management of critical patients.
References
Domenech P, Perez T, Saldarini A, Uad P, Musso CG. Kidney-lung pathophysiological crosstalk: its characteristics and importance. Int Urol Nephrol. 2017;49(7):1211–5.
Seeley EJ. Updates in the management of acute lung injury: a focus on the overlap between AKI and ARDS. Adv Chronic Kidney Dis. 2013;20(1):14–20.
Du J, Abdel-Razek O, Shi Q, Hu F, Ding G, Cooney RN, Wang G. Surfactant protein D attenuates acute lung and kidney injuries in pneumonia-induced sepsis through modulating apoptosis, inflammation and NF-κB signaling. Sci Rep. 2018;8(1):15393. https://doi.org/10.1038/s41598-018-33828-7.
Kinsey GR. The spleen as a bidirectional signal transducer in acute kidney injury. Kidney Int. 2017;91(5):1001–3.
Bhargava R, et al. Acute lung injury and acute kidney injury are established by four hours in experimental sepsis and are improved with pre, but not post, sepsis administration of TNF-α antibodies. PLoS One. 2013;8(11):e79037.
Basu RK, Wheeler DS. Kidney–lung cross-talk and acute kidney injury. Pediatr Nephrol. 2013;28(12):2239–48.
Hepokoski M, et al. Ventilator-induced lung injury increases expression of endothelial inflammatory mediators in the kidney. Am J Physiol Renal Physiol. 2017;312(4):F654–60.
Faubel S, Edelstein CL. Mechanisms and mediators of lung injury after acute kidney injury. Nat Rev Nephrol. 2016;12(1):48–60.
Jing W, et al. G-CSF mediates lung injury in mice with adenine-induced acute kidney injury. Int Immunopharmacol. 2018;63:1–8.
Costa ELV, Amato MBP. The new definition for acute lung injury and acute respiratory distress syndrome: is there room for improvement? Curr Opin Crit Care. 2013;19:16. https://journals.lww.com/co-criticalcare/fulltext/2013/02000/the_new_definition_for_acute_lung_injury_and_acute.4.aspx.
Rossi M, et al. HO-1 mitigates acute kidney injury and subsequent kidney-lung cross-talk. Free Radic Res. 2019;53(9–10):1035–43.
Goyal A, Daneshpajouhnejad P, Hashmi MF, Bashir K, John BK. Acute kidney injury (nursing). Treasure Island, FL: StatPearls Publishing; 2021.
Lee SA, Cozzi M, Bush EL, Rabb H. Distant organ dysfunction in acute kidney injury: a review. Am J Kidney Dis. 2018;72(6):846–56.
Andres-Hernando A, et al. Circulating IL-6 upregulates IL-10 production in splenic CD4+ T cells and limits acute kidney injury-induced lung inflammation. Kidney Int. 2017;91(5):1057–69.
Zhu M, et al. Erythropoietin ameliorates lung injury by accelerating pulmonary endothelium cell proliferation via Janus kinase-signal transducer and activator of transcription 3 pathway after kidney ischemia and reperfusion injury. Transplant Proc. 2019;51(3):972–8.
Ma T, Liu XW, Liu Z. Function of the p38MAPK-HSP27 pathway in rat lung injury induced by acute ischemic kidney injury. Biomed Res Int. 2013;2013:981235. https://doi.org/10.1155/2013/981235.
de Abreu KLS, et al. Acute kidney injury in critically ill patients with lung disease: kidney-lung crosstalk. Rev Bras Ter Intensiva. 2013;25(2):130–6.
Ishii T, et al. Neutrophil elastase contributes to acute lung injury induced by bilateral nephrectomy. Am J Pathol. 2010;177(4):1665–73.
Teixeira JP, Ambruso S, Griffin BR, Faubel S. Pulmonary consequences of acute kidney injury. Semin Nephrol. 2019;39(1):3–16.
Ahuja N, et al. Circulating IL-6 mediates lung injury via CXCL1 production after acute kidney injury in mice. Am J Physiol Renal Physiol. 2012;303(6):F864–72.
Andrés-Hernando A, et al. Splenectomy exacerbates lung injury after ischemic acute kidney injury in mice. Am J Physiol Renal Physiol. 2011;301(4):F907–16.
Hepokoski M, et al. Altered lung metabolism and mitochondrial DAMPs in lung injury due to acute kidney injury. Am J Physiol Lung Cell Mol Physiol. 2021;320(5):L821–31.
Malek M, Hassanshahi J, Fartootzadeh R, Azizi F, Shahidani S. Nephrogenic acute respiratory distress syndrome: a narrative review on pathophysiology and treatment. Chin J Traumatol. 2018;21(1):4–10.
Tsourouktsoglou T-D, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of TLR4. Cell Rep. 2020;31(5):107602. https://doi.org/10.1016/j.celrep.2020.107602.
Hayase N, et al. Recombinant thrombomodulin prevents acute lung injury induced by renal ischemia-reperfusion injury. Sci Rep. 2020;10(1):289. https://doi.org/10.1038/s41598-019-57205-0.
Altmann C, et al. Macrophages mediate lung inflammation in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2012;302(4):F421–32. https://doi.org/10.1152/ajprenal.00559.2010.
Lie ML, White LE, Santora RJ, Park JM, Rabb H, Hassoun HT. Lung T lymphocyte trafficking and activation during ischemic acute kidney injury. J Immunol. 2012;189(6):2843–51. https://doi.org/10.4049/jimmunol.1103254.
Nemmar A, Karaca T, Beegam S, Yuvaraju P, Yasin J, Ali BH. Lung oxidative stress, DNA damage, apoptosis, and fibrosis in adenine-induced chronic kidney disease in mice. Front Physiol. 2017;8:896. https://doi.org/10.3389/fphys.2017.00896. eCollection 2017.
Tulafu M, et al. Atrial natriuretic peptide attenuates kidney-lung crosstalk in kidney injury. J Surg Res. 2014;186(1):217–25.
Zhao H, et al. Osteopontin mediates necroptosis in lung injury after transplantation of ischaemic renal allografts in rats. Br J Anaesth. 2019;123(4):519–30.
Liu Z, Qu M, Yu L, Song P, Chang Y. Artesunate inhibits renal ischemia-reperfusion-mediated remote lung inflammation through attenuating ROS-induced activation of NLRP3 inflammasome. Inflammation. 2018;41(4):1546–56. https://doi.org/10.1007/s10753-018-0801-z.
Ma T, Liu Z. Functions of aquaporin 1 and α-epithelial Na channel in rat acute lung injury induced by acute ischemic kidney injury. Int Urol Nephrol. 2013;45(4):1187–96. https://doi.org/10.1007/s11255-012-0355-1.
Doi K, Ishizu T, Fujita T, Noiri E. Lung injury following acute kidney injury: kidney–lung crosstalk. Clin Exp Nephrol. 2011;15(4):464–70.
Twardowski ZJ, Alpert MA, Gupta RC, Nolph KD, Madsen BT. Circulating immune complexes: possible toxins responsible for serositis (pericarditis, pleuritis, and peritonitis) in renal failure. Nephron. 1983;35(3):190–5.
Prezant DJ. Effect of uremia and its treatment on pulmonary function. Lung. 1990;168(1):1–14. https://doi.org/10.1007/bf02719668.
Darmon M, et al. Acute respiratory distress syndrome and risk of AKI among critically ill patients. Clin J Am Soc Nephrol. 2014;9(8):1347–53.
Kallet RH, Lipnick MS, Zhuo H, Pangilinan LP, Gomez A. Characteristics of nonpulmonary organ dysfunction at onset of ARDS based on the Berlin definition. Respir Care. 2019;64(5):493–501.
Fisher J, Douglas JJ, Linder A, Boyd JH, Walley KR, Russell JA. Elevated plasma angiopoietin-2 levels are associated with fluid overload, organ dysfunction, and mortality in human septic shock. Crit Care Med. 2016;44((11)):2018–27. https://doi.org/10.1097/ccm.0000000000001853.
van der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VWM, Groeneveld ABJ. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax. 2008;63(10):903–9.
Araújo CB, et al. Angiopoietin-2 as a predictor of acute kidney injury in critically ill patients and association with ARDS. Respirology. 2019;24(4):345–51.
Han H, Li J, Chen D, Zhang F, Wan X, Cao C. A clinical risk scoring system of acute respiratory distress syndrome-induced acute kidney injury. Med Sci Monit. 2019;25:5606–12.
Kaushik S, Villacres S, Eisenberg R, Medar SS. Acute kidney injury in pediatric acute respiratory distress syndrome. J Intensive Care Med. 2020;36:1084. https://doi.org/10.1177/0885066620944042.
Panitchote A, et al. Factors associated with acute kidney injury in acute respiratory distress syndrome. Ann Intensive Care. 2019;9(1):74. https://doi.org/10.1186/s13613-019-0552-5.
Soto GJ, Frank AJ, Christiani DC, Gong MN. Body mass index and acute kidney injury in the acute respiratory distress syndrome. Crit Care Med. 2012;40(9):2601–8.
Slutsky AS. History of mechanical ventilation. From Vesalius to ventilator-induced lung injury. Am J Respir Crit Care Med. 2015;191((10)):1106–15.
van den Akker JPC, Egal M, Groeneveld ABJ. Invasive mechanical ventilation as a risk factor for acute kidney injury in the critically ill: a systematic review and meta-analysis. Crit Care. 2013;17(3):R98.
Beitler JR, Malhotra A, Thompson BT. Ventilator-induced lung injury. Clin. Chest Med. 2016;37(4):633–46.
Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2013;369((22)):2126–36.
Wu Y, Wang L, Meng L, Cao G-K, Zhang Y. MIP-1α and NF-κB as indicators of acute kidney injury secondary to acute lung injury in mechanically ventilated patients. Eur Rev Med Pharmacol Sci. 2016;20(18):3830–4.
Imai Y, et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA. 2003;289(16):2104–12.
Husain-Syed F, Slutsky AS, Ronco C. Lung–kidney cross-talk in the critically ill patient. Am J Respir Crit Care Med. 2016;194(4):402–14.
Nicholl DDM, et al. Declining kidney function increases the prevalence of sleep apnea and nocturnal hypoxia. Chest. 2012;141(6):1422–30.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Capalbo, O.M., Simonovich, V. (2023). Kidney–Lung Crosstalk in Acute Kidney Injury. In: Musso, C.G., Covic, A. (eds) Organ Crosstalk in Acute Kidney Injury. Springer, Cham. https://doi.org/10.1007/978-3-031-36789-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-031-36789-2_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-36788-5
Online ISBN: 978-3-031-36789-2
eBook Packages: MedicineMedicine (R0)