
Abstract
Purpose
To establish a rat model of acute ischemic kidney injury by continually occluding the bilateral renal artery and renal veins, the functions of α-epithelial Na+ channel (α-ENaC) and aquaporin (AQP1) in lung injury induced by acute kidney injury (AKI) were examined and compared with lung injury induced by endotoxin.
Methods
Male Wistar rats were randomly divided into three groups: control group, AKI group, and sepsis group. The concentrations of AQP1 and α-ENaC in the lung tissue were detected. The concentrations of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) in the serum and bronchoalveolar lavage fluid were also detected.
Results
The arterial blood pH in AKI group and PaO2 in sepsis group decreased 2 h after the experiment. A significant pulmonary interstitial and alveolar space edema, which showed a typical pathological change in acute lung injury, was found in AKI and sepsis group 8 h after the experiment. Two hours after the experiment, the concentration of TNF-α and IL-6 in the serum and bronchoalveolar lavage fluid (BALF) in AKI and sepsis group increased, whereas the pulmonary expression of AQP1 and α-ENaC decreased. The pulmonary AQP1 and α-ENaC of the rats were negatively correlated with TNF-α and IL-6 in BALF. The relevance among AQP1, α-ENaC, TNF-α, and IL-6 in sepsis group was higher than that in AKI group.
Conclusion
The TNF-α and IL-6 levels increased significantly and the pulmonary expression of AQP1 and α-ENaC declined at the early stage of AKI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute kidney injury (AKI) is one of the common and complicated clinical problems. Approximately 30 % of critically ill patients are accompanied with AKI. Studies have proven that 5–6 % of intensive care unit patients are accompanied with AKI and require renal replacement therapy [1]. Despite the continuous improvements in dialysis and advanced treatments, the mortality of AKI remains as high as 40–60 %. Clinical studies have confirmed that the high mortality of AKI is closely related to acute lung injury (ALI) or acute respiratory distress syndrome (ARDS), which are complications of AKI [2, 3]. However, the mechanism by which AKI induces ALI remains obscure.
Tumor necrosis factor (TNF)-α is one of the first and the most important inflammatory mediators in inflammatory responses. Cytokines such as TNF-α and interleukin (IL)-6 constitute an inflammatory cell network and participate in the pathological process of trauma and many inflammatory diseases [4]. In ALI/ARDS induced by different animal models, the contents of TNF-α and IL-6 in serum, lung BAL fluid increased [5–8]. Studies have confirmed that inflammatory mediators vary in early-stage AKI patients [9–12], which may be the mechanism of AKI in inducing a distant organ injury. ALI is a diffuse lung injury characterized by noncardiogenic pulmonary edema, increased permeability of the endothelium and the alveolar epithelium, and decreased alveolar fluid clearance. Alveolar fluid clearance has two forms, namely active and passive transport, of which the former plays a major role. The active transport system for the Na+/H2O of the alveolar epithelium consists of epithelial Na+ channels (ENac), Na+–K+–ATPase, and aquaporin. In addition, recent studies have confirmed that aquaporin 1 (AQP1) and lung α-epithelial Na+ channel (α-ENaC) have important functions in maintaining the balance of liquid in the alveolar space. That is, the expressions of AQP1 and Na+ channel protein decline in ALI, which could induce ALI [13–16].
Current studies have found that AKI leads to changes in cytokines, inflammatory mediators, AQP1, and Na+ channel protein, thereby inducing ALI. Rabb et al. [17] reported that the expression of AQP and sodium channel protein decreased in acute renal failure and acute lung injury was induced. However, the mechanism behind the induction is not explicitly illustrated. In addition, the relevance among cytokines, AQP1, and Na+ channel protein, as well as the difference between ALI induced by AKI and ALI induced by common clinical etiology (e.g., ALI induced by sepsis), remains unknown. The present study observes the change rule in IL-6, TNF-α, AQP1, and Na+ channel protein, explores the pathological mechanism of AKI-induced ALI, and compares the differences in pathological mechanism between AKI-induced and endotoxin-induced ALI.
Previous studies of acute lung injury induced by acute kidney injury, the model of acute kidney injury, are mostly induced by unilateral or bilateral renal artery clipped a certain number of minutes and then removed to make a model of ischemia–reperfusion injury or bilateral nephrectomy [10–12]. Renal ischemia–reperfusion injury may cause acute kidney injury, but ischemia–reperfusion injury can also cause acute lung injury. Therefore, acute lung injury induced by renal ischemia–reperfusion injury does not mean that change is caused by acute kidney injury itself, is also caused due to ischemia–reperfusion injury. Bilateral nephrectomy can rule out the interference of a model of ischemia–reperfusion injury, but the experimental method would make the rat wounds larger, experimental manipulation is more difficult. In the present study, we used the method of clipping bilateral renal artery and vein to simulate bilateral nephrectomy, to create a model of acute kidney injury, to avoid an excessive surgical trauma, simple experiment operations. In addition, acute lung injury caused by ischemia reperfusion injury was reduced to the maximum extent.
Materials and methods
Animal models
Ninety male Wistar rats weighing 300–320 g were provided by the Experimental Animal Center of China Medical University. This study was performed strictly in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol was reviewed and approved by the Institutional Animal Care and Use Committee of the First Hospital of China Medical University.
The rats were anesthetized with 5 % chloral hydrate (300 mg/kg) and then subjected to tracheotomy. The arteria cervicalis and jugular vein of the rats were punctured, and a catheter was indwelled. Respiratory frequency, heart rate, systolic pressure, diastolic pressure, mean arterial blood pressure, and central venous pressure were monitored. As shown in Table 1, the rats were randomly divided into three groups 30 min after reaching a stable state.
Specimen collection
After the models were established, the rats in each group were sacrificed at 0, 2, 4, 6, and 8 h after the experiment started, with six rats for each time. Venous blood sample was collected to detect the cytokine. The chest was opened to expose the lungs, and bronchoalveolar lavage was given thrice to the left lung with 3 ml phosphate-buffered saline. The collected BALF was merged, and cytokines for detection were cryopreserved. The right lung lobes were preserved at −80 °C to detect AQP1 and α-ENaC.
IL-6 and TNF-α detection
The IL-6 and TNF-α levels in the blood serum and BALF were detected using double antibody sandwich ELISA. The detection was performed according to the manufacturer’s instructions (Shanghai Senxiong Biotech Industrial Corporation, Shanghai, China).
AQP1 detection
The chest was opened to remove the right lung lobe that was rapidly cryopreserved with liquid nitrogen. The homogenate was prepared, and then, the lung AQP1 concentration was detected using double antibody sandwich ELISA. The detection was performed according to the manufacturer’s instruction (Uscnlife Company).
Western blot
Protein was extracted from the right lung lobe and preserved at −80 °C after the protein concentration was detected. The protein sample (100 μg) was electrophoresed in 12 % SDS-PAGE gel, which was stopped when the interest protein approached the end of the gel. Subsequently, the protein was transferred to PVDF membranes under a constant voltage of 120 V at 4 °C. The membranes were sealed with 5 % skimmed milk powder at room temperature for 1 h. α-ENac first antibody at a dilution of 1:1,000 and GAPDH (an internal standard) first antibody at a dilution of 1:500 were added into the solution sequentially. The mixture was incubated overnight at 4 °C, and then, horseradish peroxidase-labeled second antibody at a dilution of 1:1,000 was added into the mixture, which had a color rendering after incubation at room temperature for 1 h. Images were taken using photographic photometry, and the experimental result was analyzed with Quantity One Software. Optical density value was scanned, which was shown at the ratio of interest protein-to-internal standard GAPDH.
Statistical analysis
The results were analyzed using SPSS 16.0, and data were presented as \( \overline{x} \) ± s. t Test was used to compare within groups, and the relevance between the two group values was examined with Pearson correlation. P < 0.05 was considered to indicate statistical significance.
Results
Rat arterial blood gas
Table 2 shows the variation tendency of the arterial blood gas of rats in the three groups. The arterial blood pH value in AKI group decreased 2 h after the experiment, and acidosis became obviously severe, which was significantly different with control group. The arterial blood CO2 exhibited a descending tendency, but no significant difference was found among the other two groups. The arterial partial pressure of oxygen in control and AKI group descended slightly after the experiment, but no statistical significance was found between the two groups. The pressure of oxygen in sepsis group descended evidently from the 2nd hour and was significantly different from that in the other two groups (P < 0.05).
Protein level in BALF and rats lung wet/dry weight (W/D) ratio
The protein level in BALF and the W/D ratio had no significant change in control group. By contrast, the protein level in BALF and the W/D ratio in AKI group increased 2 h after the experiment, which demonstrated a significant difference compared with those in control group. The protein level in BALF and the W/D ratio in sepsis group also increased 2 h after the experiment, which showed a significant difference compared with those in control and AKI group (Table 3).
Lung tissue structure
The alveoli of rats in control group were integrated 0 and 8 h after the experiment. No significant exudation in the alveoli and no pulmonary interstitial edema were found. The alveolar epithelium swelled in sepsis group 8 h after the experiment, the alveolar wall broadened, and blood capillary became dilated and congested. A significant pulmonary interstitial and alveolar space edema was observed. Inflammatory corpuscles, red blood cells, and protein exudation in pulmonary alveoli increased significantly. Minor respiratory injury and alveoli disturbance could be partially observed in the visual field, indicating a typical pathological change in ALI. Inflammatory corpuscles, red blood cells, and protein exudation in pulmonary alveoli were also found in AKI group 8 h after the experiment. Pulmonary interstitial and alveoli edema in AKI group was less severe compared with those in sepsis group but more severe compared with those in control group (Fig. 1).

HE dye (×400) of rats lung tissue in three groups. a Control group: 0 h, b control group: 8 h, c AKI group: 8 h, d sepsis group: 8 h
TNF-α and IL-6 levels
As shown in Table 4, the TNF-α and IL-6 levels in the serum and BALF in AKI group increased 2 h after the experiment, which demonstrated a significant difference compared with control group (P < 0.05). The TNF-α and IL-6 levels in the serum and BALF in sepsis group increased 2 h after the experiment, which exhibited a significant difference compared with control and AKI group (P < 0.05).
AQP1 concentration
As shown in Table 5, the concentration of AQP1 in the lung tissue in AKI group decreased gradually 2 h after the experiment, which showed a significant difference compared with control group (P < 0.05). The concentration of AQP1 in the lung tissue in sepsis group decreased gradually 2 h after the experiment, which demonstrated a significant difference compared with control and AKI group (P < 0.05).
α-ENaC expression
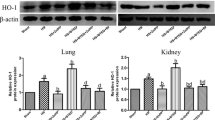
Figure 2 demonstrates the western blot results of α-ENaC in the lung tissues from the three groups. The α-ENaC level in the lung tissue from AKI group decreased gradually 2 h after the experiment, which had a significant difference compared with control group (P < 0.05). The α-ENaC level in the lung tissue from sepsis group decreased gradually 2 h after the experiment, which showed a significant difference compared with control and AKI group (P < 0.05).

The Western results of α-ENaC at different time in three groups
Relevance analysis between AQP1 and TNF-α and IL-6
Figure 3 illustrates the results from the relevance analyses of AQP1, TNF-α, and IL-6 in BALF. The AQP1 and TNF-α levels in the BALF of the rats were negatively correlated. The Pearson correlation coefficients of the AKI and sepsis groups were −0.490 and −0.509, respectively. IL-6 and AQP1 were also negatively correlated. The Pearson correlation coefficients of the AKI and sepsis groups were −0.546 and −0.574, respectively. The relevance between AQP1 and TNF-α and IL-6 in BALF in the sepsis group was higher than that in the AKI group.

The relevance analysis between the AQP1 and TNF-α and IL-6 in bronchoalveolar lavage fluid (BALF) in AKI group and sepsis group
Relevance analysis between α-ENaC and TNF-α and IL-6
Figure 4 demonstrates the results of the relevance analyses between the α-ENaC in the lung tissue and the TNF-α and IL-6 in the BALF. The α-ENaC levels in the rat lung tissue and the TNF-α and IL-6 in the BALF were negatively correlated. The Pearson correlation coefficients of the AKI and sepsis groups were −0.465 and −0.477, respectively. The IL-6 levels in the BALF and rat α-ENaC were also negatively correlated. The Pearson correlation coefficients of the AKI and sepsis groups were −0.412 and −0.445, respectively. The relevance between α-ENaC and TNF-α and IL-6 in the BALF sepsis group was higher than that in the AKI group.

The relevance analysis between α-ENaC and TNF-α and IL-6 in bronchoalveolar lavage fluid (BALF) in AKI group and sepsis group
Discussion
The protein level in the BALF increased significantly, and the lung W/D ratio decreased in the AKI group 2 h after the kidney injury. This finding is in agreement with the report of previous studies [18–21]. However, the protein level in the BALF in the AKI group was significantly lower than that in the endotoxin group. The lung W/D ratio in the AKI group was higher than that in the endotoxin group. This result suggests that although the oxygenation of the lungs was not affected at the early stage of AKI, the barrier function of the alveolar epithelium-endothelium was influenced. Moreover, the permeability of endothelial cells increased, and the lung epithelial cell liquid removal decreased. This result indicates that although ALI occurred, it was less severe than that caused by endotoxin. Previous studies reported that ALI occurs at the early stage of AKI, characterized by the infiltration of the inflammatory cells and hyperemia in alveolar space, and the changes in the pulmonary alveoli structure [18, 22].
During the experiment, the alveolar epithelium swelled, the alveolar wall broadened, and the blood capillary dilated and congested. A significant pulmonary interstitial and alveolar space edema was observed, and inflammatory corpuscles, red blood cells, and protein exudation in the pulmonary alveoli increased significantly. A number of inflammatory cells were infiltrated. Minor respiratory injury and alveoli disturbance could be partially observed in the visual field, showing a typical pathological change in the ALI. The pathological change in the AKI group was similar to that in the ALI caused by endotoxin. However, the infiltration of the inflammatory cells, red blood cells, and protein exudation, as well as alveoli disturbance, was less severe than that in endotoxin-induced ALI.
TNF-α is produced by mononuclear macrophage and functions through specific cell membrane-connecting receptors. TNF-α is the main cytokine that mediates ALI. TNF-α could induce the activation of lung endothelial cell, the migration of leukocyte, neutrophil degranulation, and the leakage of blood capillary. The accumulated edema fluid blocks the perfusion and oxygen exchange of the alveolar cell, which in turn causes ARDS. In addition, TNF-α could intercommunicate with many cytokines, which exhibit extensive action. The activated mononuclear macrophage in the blood was the main source of IL-6, which increased under the stimulation of LPS, IL-1β, and TNF-α. The TNF-α and IL-6 levels increase significantly in acute diseases, such as burns and operation traumas. Clinical studies [23] revealed that the TNF-α and IL-6 levels in the serum and BALF of ALI/ARDS patients increase, which is closely related to the pathogenesis of multiple organ failure. The present study found that the TNF-α and IL-6 levels in the rat serum and BALF increased significantly after AKI. This phenomenon may be because the accumulation of poisonous metabolic products in the body after AKI and operation trauma activates systemic inflammatory responses. The mediators of inflammation were decreased by the excretory function and inactivation of the kidney, significantly increasing the TNF-α and IL-6 levels in the rat blood circulation. Lungs have a huge blood-vascular system, in which the inflammatory mediators of blood circulation accumulate. As a result, TNF-α and IL-6 levels are increased and ALI is induced.
As proven by earlier studies, this experiment found that the systemic inflammatory response caused by intraperitoneal injection of endotoxin increased significantly the TNF-α and IL-6 levels in rat serum and BALF, which was more statistically significant than that of AKI. This phenomenon proved that both AKI and endotoxin activated inflammatory response and that the increase in cytokine level caused early-stage ALI. However, the initiating factors that induce systemic inflammatory response and the mechanism of the inflammatory mediators are different, resulting in different degrees of acute injury. AKI was caused by the accumulation of poisonous metabolic products in the body and operation trauma that activated systemic inflammatory response, such that the mediators of inflammation were decreased by the excretory function and inactivation of the kidney. This phenomenon increased the TNF-α and IL-6 levels, which in turn caused the occurrence of ALI, but the injury was less severe than that induced by endotoxin. The research on the pathophysiology of ARDS in two different conditions, sepsis and acute renal failure, has been carried out by Kim do et al. [24]. Part of their experimental results was similar to ours: acute kidney injury and sepsis caused inflammation factor increases. However, compared with sepsis, heat shock protein could play a more important role in acute kidney injury in their research. This study also found that the expression of AQP1 and α-ENaC in rat lungs after AKI decreased significantly, suggesting that the AQP1 and α-ENaC levels decreased at the early stage of AKI. This phenomenon may be one of the causes of AKI-induced ALI. Previous studies have suggested that one of the main pathological changes of lung injury is pulmonary edema. The transport of water has two main channels in the pulmonary alveoli of lung tissue. One is the passive transport by AQP, which is determined by the pressure difference between alveolar and pulmonary capillary pressure, as well as the osmotic gradient between the liquid in the pulmonary alveoli and plasma in addition to the quantity of AQP. The other channel is the active transport accompanied by Na. The Na-water active transport system in the alveolar epithelium was composed of ENaC, Na+–K+–ATPase, and AQP. Na+ was absorbed through the sodium channel of the lateral wall of alveolar epithelial cells and then was pumped into the pulmonary interstice by ENaC, Na+–K+–ATPase of the base lateral surface accompanied by the passive transport of water. In addition to accompanying the active the transport of Na, water is discharged from the aquaporin of the alveolar epithelial cells [13, 14]. Several studies have proven that the pulmonary expression of both AQP and ENaC decreases in ALI and ARDS [13–16, 22, 23].
The experiment also found that the TNF-α and IL-6 levels in rat BALF were negatively correlated with the expression of AQP1 and α-ENaC, suggesting that the expression of AQP1 and α-ENaC decreased during the process of acute injury, in which cytokines played an important role. The inflammatory reaction of rats became more severe after endotoxin injection, which induced a more significant increase in the TNF-α and IL-6 levels. Therefore, the expression of AQP1 and α-ENaC in the lungs of rats decreased more significantly in the endotoxin group. Dagenais et al. [25] also found experimentally that TNF-α could decrease the pulmonary expression of α-ENaC. In experiments, we found that the correlation coefficient of TNF-α or IL-6 and AQP1 or α-ENaC is only 0.4–0.5. It suggested that some interference factors regulated AQP1 and alpha ENaC has perhaps affected the experiments, such as the sample size, etc. The advanced research should been made in the future. A number of studies also proved that cytokines participate in the regulation of AQP [26]. In ARDS, the activated neutrophilic granulocyte and pulmonary alveolar macrophage migrate into the lungs and produce reactive oxygen such as TNF, TGF, and NO. The combination of these injurious factors and ferroheme increases the cGMP level in cells and then exerts effects through protein phosphorylation, in which cGMP depends. As a result, the pulmonary expression of AQP1 and α-ENaC decreases. In addition, stress reactions, high osmotic pressure, anoxia, pH, and Ca2+ could participate in regulating the expression of AQP1 and α-ENaC [27]. AKI disturbed the internal environment, such as the change in plasma osmotic pressure, oxidosis, and Ca2+ dislocation, which were more severe than that of the endotoxin group. This phenomenon is probably the reason why the relevance between TNF-α, IL-6 and AQP1 and α-ENaC was not as high as that of the endotoxin group. After AKI, aside from cytokines, the change in the plasma osmotic pressure, oxidosis, and Ca2+ dislocation, among others, participated in the regulation of AQP1 and α-ENaC.
In conclusion, the TNF-α and IL-6 levels increased significantly and the pulmonary expression of AQP1 and αENaC declined at the early stage of AKI, which may have caused the AKI-induced ALI.
References
Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C (2005) Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294:813–818. doi:10.1001/jama.294.7.813
Awad AS, Okusa MD (2007) Distant organ injury following acute kidney injury. Am J Physiol Renal Physiol 293:F28–F29. doi:10.1152/ajprenal.00159.2007
Paladino JD, Hotchkiss JR, Rabb H (2009) Acute kidney injury and lung dysfunction: a paradigm for remote organ effects of kidney disease. Microvasc Res 77:8–12. doi:10.1016/j.mvr.2008.09.001
Old LJ (1987) Tumour necrosis factor. Polypeptide mediator network. Nature 326:330–331. doi:10.1038/326330a0
Nick JA, Avdi NJ, Gerwins P, Johnson GL, Worthen GS (1996) Activation of a p38 mitogen activated protein kinase in human neutrophils by lipopolysaccharide. J Immunol 156:4867–4877
Rodrigo R, Trujillo S, Bosco C (2006) Biochemical and ultrastructural lung damage induced by rhabdomyolysis in the rat. Exp Biol Med (Maywood) 231:1430–1438
Rafi AQ, Zeytun A, Bradley MJ, Sponenberg DP, Grayson RL, Nagarkatti M, Nagarkatti PS (1998) Evidence for the involvement of Fas ligand and perforin in the induction of vascular leak syndrome. J Immunol 161:3077–3086
Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR (1999) Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 163:2217–2225
Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, Ruff SM, Zahedi K, Shao M, Bean J, Mori K, Barasch J, Devarajan P (2005) Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 365:1231–1238. doi:10.1016/S0140-6736(05)74811-X
Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H (2008) The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol 19:547–558. doi:10.1681/ASN.2007040469
Klein CL, Hoke TS, Fang WF, Altmann CJ, Douglas IS, Faubel S (2008) Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int 74:901–909. doi:10.1038/ki.2008.314
Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, Tao Y, Dursun B, Voelkel NF, Edelstein CL, Faubel S (2007) Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol 18:155–164. doi:10.1681/ASN.2006050494
Matthay MA, Fukuda N, Frank J, Kallet R, Daniel B, Sakuma T (2000) Alveolar epithelial barrier. Role in lung fluid balance in clinical lung injury. Clin Chest Med 21:477–490
Matthay MA, Clerici C, Saumon G (2002) Invited review: active fluid clearance from the distal air spaces of the lung. J Appl Physiol 93:1533–1541. doi:10.1152/japplphysiol.01210.2001
Matthay MA, Wiener JP (1990) Intact epithelial function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis 142:1250–1257. doi:10.1164/ajrccm/144.2.468a
Ware LB, Matthay MA (2001) Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163:1376–1383
Rabb H, Wang Z, Nemoto T, Hotchkiss J, Yokota N, Soleimani M (2003) Acute renal failure leads to dysregulation of lung salt and water channels. Kidney Int 63:600–606
Ishii T, Doi K, Okamoto K, Imamura M, Dohi M, Yamamoto K, Fujita T, Noiri E (2010) Neutrophil elastase contributes to acute lung injury induced by bilateral nephrectomy. Am J Pathol 177:1665–1673. doi:10.2353/ajpath.2010.090793
Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, Tao Y, Dursun B, Voelkel NF, Edelstein CL, Faubel S (2007) Acute renal failure after bilateral nephrectomy is associated with cytokine mediated pulmonary injury. J Am Soc Nephrol 18:155–164. doi:10.1681/ASN.2006050494
Hassoun HT, Grigoryev DN, Lie ML, Liu M, Cheadle C, Tuder RM, Rabb H (2007) Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol 293:F30–F40. doi:10.1152/ajprenal.00023.2007
Deng J, Hu X, Yuen PS, Star RA (2004) Alpha-melanocyte-stimulating hormone inhibits lung injury after renal ischemia/reperfusion. Am J Respir Crit Care Med 169:749–756. doi:10.1164/rccm.200303-372OC
Kramer AA, Postler G, Salhab KF, Mendez C, Carey LC, Rabb H (1999) Renal ischemia/reperfusion leads to macrophage-mediated increase in pulmonary vascular permeability. Kidney Int 55:2362–2367. doi:10.1046/j.1523-1755.1999.00460.x
Steinberg J, Halter J, Schiller H, Gatto L, Carney D, Lee HM, Golub L, Nieman G (2005) Chemically modified tetracycline prevents the development of septic shock and acute respiratory distress syndrome in a clinically applicable porcine model. Shock 24:348–356. doi:10.1097/01.shk.0000180619.06317.2c
Kim do J, Park SH, Sheen MR, Jeon US, Kim SW, Koh ES, Woo SK (2006) Comparison of experimental lung injury from acute renal failure with injury due to sepsis. Respiration 73:815–824
Dagenais A, Frechette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, Brochiero E, Masse C, Berthiaume Y (2004) Downregulation of ENaC activity and expression by TNF-alpha in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 286:L301–L311. doi:10.1152/ajplung.00326.2002
Towne JE, Krane CM, Bachurski CJ, Menon AG (2001) Tumor necrosis factor alpha inhibits aquaporin 5 expression in mouse lung epithelial cells. J Biol Chem 276:18657–18664. doi:10.1074/jbc.M100322200
Németh-Cahalan KL, Kalman K, Hall JE (2004) Molecular basis of pH and Ca2+ regulation of aquaporin water permeability. J General Physiol 123:573–580. doi:10.1085/jgp.200308990
Acknowledgments
Science and technology project plan of Shenyang city (F11-264-1-52). Fund for Scientific Research of The First Hospital of China Medical University, fsfh1103.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ma, T., Liu, Z. Functions of aquaporin 1 and α-epithelial Na+ channel in rat acute lung injury induced by acute ischemic kidney injury. Int Urol Nephrol 45, 1187–1196 (2013). https://doi.org/10.1007/s11255-012-0355-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-012-0355-1