
Abstract
Neuropathies are a major branch of neuromuscular disorders, and they vary greatly with regards to their aetiology, pathophysiology, and clinical presentation. Therefore, different classifications and subclassifications are used to describe different aspects of these diseases. Motor and sensory disturbances are common symptoms and signs of peripheral neuropathies.
In this chapter, the questions of what kind of complaints, physical and laboratory examination findings are detected in neuropathies, and what are the inflammatory, toxic, idiopathic, and genetic causes of neuropathy are answered. Differential diagnosis methods are briefly explained. Examples of patients with their histopathological images, mainly nerve biopsy, and changes observed in muscle in neuropathies are presented. At the end of the chapter, under the title “all about the pathology of neuropathies”, case examples, surprising cases, short clinical histories, and histopathological pictures are presented.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Neuropathies are a major branch of neuromuscular disorders (NMDs), and they vary greatly with regard to their aetiology, pathophysiology and clinical presentation. Therefore, different classifications and subclassifications are used to describe different aspects of these diseases. Classifications are useful tools, helping clinicians to reach a diagnosis and decide on treatment. However, diagnosing and identifying the aetiology of neuropathies remains a challenge for clinicians. A multidisciplinary approach is essential at each step from diagnosis to treatment. Motor and sensory disturbances are common symptoms and signs. Of the multiple symptoms, neuropathic pain is the most disturbing, while autonomic involvement may threaten life. As expected, treatment includes aetiological and symptomatic measures. In this chapter we aim to cover the areas of epidemiology, aetiology, signs and symptoms, evaluation, diagnosis and treatment options.
Epidemiology
Peripheral neuropathy (PN) is a frequently seen disorder especially in neurology clinics. The prevalence of peripheral neuropathies is reported to be 2.4% in the population; however, it increases to 8% in the elderly [1]. The worldwide increase of obesity, diabetes and aging has also contributed to the increased occurrence of PN over the years [2, 3]. Diabetic neuropathy (DN) is the most common cause of distal symmetrical sensorimotor polyneuropathy (50%) since it affects almost half of diabetic patients [1, 2]. Idiopathic or cryptogenic PN is the second most common group that is associated with metabolic syndrome and prediabetes [2]. Concerning infections, leprosy is still the leading cause worldwide especially in Southeast Asia [1]. Charcot-Marie-Tooth disease type 1A is the most common genetic polyneuropathy (PNP). In adults carpal tunnel syndrome is the most frequently seen entrapment neuropathy (EN) [1, 5]. The risk factors for developing chronic neuropathies depend on age and socioeconomic status within the studied populations [3]. The terms polyneuropathy, peripheral neuropathy and neuropathy have different meanings. However, they are frequently used in place of each other. Peripheral neuropathy is an inclusive term which encompasses polyneuropathy and any other disorder of the peripheral nervous system such as mononeuropathy, radiculopathy and plexopathy. Plexopathy is a disorder of a network of nerves known as a plexus. It generally develops in the brachial or lumbosacral plexus which sends signals from the spinal cord to the upper or lower extremities. Symptoms of plexopathies include pain, muscle weakness and sensory deficits such as numbness. Radiculopathy can be described as damage to nerve roots in the area where they leave the spine. This condition generally results from disc degeneration, disc herniation or other trauma [1, 4, 5]. Neuropathy is described as the ‘damage, disease, or dysfunction of one or more nerves especially of the peripheral nervous system’ in the Merriam-Webster medical dictionary [6]. Polyneuropathy is a more specific term meaning generalised involvement of peripheral nerves due to the same pathophysiologic mechanism in a relatively symmetrical distribution, with distal nerves being affected more severely [7]. Deciding a polyneuropathy differential diagnosis should be completed carefully to exclude mononeuropathies, mononeuropathy multiplex and some central nervous system diseases. The term mononeuropathy refers a single nerve injury usually with a local cause such as trauma or a compressive lesion. Mononeuropathy multiplex indicates multiple single-nerve involvement occurring simultaneously or consecutively due to a vasculitic process [7].
Aetiology and Classification
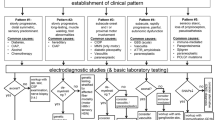
The aetiology of peripheral neuropathies is diverse and sometimes overlapping. Classification can be based on pathology, aetiology, function, distribution or according to electrophysiological parameters. Discriminating involvement patterns can be very useful in deciding the differential diagnosis of neuropathies (Fig. 10.1). A simple approach that may be used for classifying neuropathies is summarised in Table 10.1. The biopsy findings in some peripheral neuropathies are not familiar to pathologists because nerve biopsy examination is almost never utilised when determining a differential diagnosis of certain disorders such as radiculopathy, plexopathy and mononeuropathies.

Involvement patterns in different neurological disorders: (a) polyneuropathy, (b) transverse spinal cord lesion, (c) ipsilateral motor defects and contralateral sensory deficits with pain below the level of hemisection of the spinal cord (Brown-Sequard syndrome), (d) mononeuropathy multiplex, (e) saddle-shaped anaesthesia in compression of the cauda equina nerves, (f) cross-sensory defect in brain stem lesion and (g) sensory defect in lesions of the thalamus and its surroundings
Mononeuropathies
Mononeuropathies of the Upper Limbs
There a several mononeuropathies of the upper limbs. The most common are briefly discussed in this section.
Median Nerve
Carpal tunnel syndrome (CTS) is the most common neuropathy in adults, and it is more frequent in females than males. The median nerve is compressed while passing through the carpal tunnel, formed by the transverse carpal ligament in the wrist. Initially the symptoms include tingling and hypoaesthesia over the sensory areas of the median nerve with some radiation to the forearm, especially during sleep. In later stages, muscle weakness of the thenar muscles, which are innervated by the median nerve, occurs. CTS is more frequently seen in patients with diabetes and hypothyroidism. A bilateral presence of CTS should be a warning to search for these metabolic conditions [8]. Pronator teres syndrome is entrapment of the median nerve by the pronator muscle or fibrous structures in the forearm. Median neuropathy (Fig. 10.2) in the forearm may be the first sign of mononeuropathy multiplex [9, 10].

Median nerve compound muscle action potential amplitude is low (3.4 mV), and nerve conduction velocity (51.1 m/s) and distal latency (3.5 ms) are normal, compatible with axonal neuropathy
Ulnar Nerve
Entrapment of the ulnar nerve at the level of the elbow is the most frequent lesion of the ulnar nerve, and this level is frequently referred to as the cubital tunnel. However not all the lesions at the elbow are in the cubital tunnel. There are three sites: above the elbow, at the ulnar groove and the cubital tunnel. Electrophysiological examination shows the exact localisation where clinical findings are similar for all three sites. Guyon tunnel syndrome is entrapment of the ulnar nerve at the level of the wrist. The ulnar dorsal branch of the ulnar nerve leaves the main trunk before entering the tunnel so that it is saved from compression. This feature helps to identify the localization of the lesion [5].
Radial Nerve
Radial nerve entrapments are not as frequent as median and ulnar nerve entrapments. The most frequent lesion occurs due to compression at the spiral groove. Famous ‘Saturday night palsy’ is the result of compression of the radial nerve between the humerus and a solid object. The main clinical finding is wrist drop, meaning inability to extend the wrist and digits. The triceps muscle is saved but brachioradialis muscle involvement usually occurs. Sensory deficit is less prominent. It is usually a benign condition which can rarely progress to axonal degeneration. Fracturing the humerus is another potential cause of radial nerve injury at the proximal level which may require surgical exploration. The posterior interosseous nerve is a pure motor branch of the radial nerve. Lesions distal to the elbow can cause posterior interosseous neuropathy which causes wrist drop, but it spares the brachioradialis muscle [2].
Proximal Neuropathies
There are proximal neuropathies which are not encountered frequently such as suprascapular, axillary and long thoracic nerve neuropathies. The suprascapular nerve is a sensorimotor nerve which innervates the supraspinatus and infraspinatus muscles. Entrapment at the suprascapular notch may cause weakness of innervated muscles and shoulder pain. If impingement occurs at the spinoglenoid notch, there are only motor symptoms without pain. If it is due to trauma, the axillary nerve may also be involved. Long thoracic neuropathy is important to consider for the differential diagnosis with suspected brachial plexopathy because it arises from the fifth to seventh cervical roots, proximal to the brachial plexus. Damage of the long thoracic nerve results in winging of the scapula (Fig. 9.3). It is noteworthy that scapular wing occurs in other conditions such as accessory neuropathy, dorsal scapular neuropathy, cervical radiculopathy and in some myopathies. Direct trauma, stretching and inflammation may cause long thoracic neuropathy. The axillary nerve is prone to traumatic injuries due to shoulder dislocation or humerus fractures. Clinical features include sensory impairment of a sharply demarcated area over the lateral shoulder with weakness in arm abduction and external rotation. Spinal accessory nerve (a cranial nerve) dysfunction affects the upper extremity because it supplies motor fibres to the trapezius muscle as well as the sternocleidomastoid muscle. Paralysis of the trapezius muscle causes winging of the scapula and a deficit in shoulder elevation. Iatrogenic traumas are common which include radical neck dissection for malignant diseases and biopsy of lymph nodes. External blunt traumas may be a cause of injury too [11].
Mononeuropathies of the Lower Limbs
The lower limbs are innervated via lumbar and sacral roots, spinal nerves, the lumbosacral plexus and peripheral nerves. Major nerves that arise from the lumbar plexus include the femoral nerve, obturator nerve, saphenous nerve and lateral femoral cutaneous nerve. Lower lumbar and upper sacral fibres converge to form the sciatic nerve which has tibial and peroneal components. Inferior and superior gluteal nerves are also distal branches of the lumbosacral plexus which innervate posterolateral hip muscles. Pelvic floor muscles are innervated by the pudendal nerve which only has sacral fibres. Perinea sensation is also carried by the pudendal nerve.
Lower extremity neuropathies occur mostly due to chronic compressive lesions. Acute transection, inflammation, infection, radiation injury and ischaemia are other conditions that affect lower limb nerves less frequently. Detailed history taking and through neurologic examination are a great help when determining a diagnosis and differential diagnosis. Peripheral neurologic symptoms classically involve muscle power, sensation and reflexes. Specifically questioning the time of onset, evolution of symptoms, related events and associated disorders is very important. Pain is the most frequent symptom that brings patients to the doctor. Localisation and radiation of pain should be checked. Identifying sensory abnormalities is important as it helps to localise the lesion. Therefore, the dermatomal and radicular distribution of sensory impairment should be examined [12]. Electrophysiological investigations are usually, but not always, useful for reaching a diagnosis and differential diagnosis. Superimposed disorders, such as dropped foot and suspected radicular involvement in a diabetic patient with lumbar disc herniation, present considerable difficulty for electrophysiologists. Imaging methods are used for locating lesion sites and for defining the nature of the lesions [7, 13].
Peroneal Nerve
A peroneal nerve lesion at the fibular head is the most common entrapment in the lower limbs. The common peroneal nerve passes through a fibro-osseous tunnel at the fibular head. Lesions of adjacent structures may cause compression of the nerve. Prolonged squatting, lying in bed or crossing of the legs may cause both stretching and compression. Dropped foot is the most prominent finding which causes a ‘steppage’ gait [4]. Sensory impairment of the skin over the dorsolateral foot and lateral side of the shin is present, but pain is usually absent. The deep peroneal nerve may be compressed at the level of the ankle where it passes underneath the retinaculum fibres. Motor deficit is restricted to the extensor digitorum brevis muscle which dorsally flexes the toes. Sensory loss occurs over the skin web between the great and second toe [12, 13].
Tibial Nerve
Tibial nerve entrapment at the level of the ankle behind the medial malleolus is called tarsal tunnel syndrome. The tibial nerve is compressed between the bones and flexor retinaculum, while passing underneath tendons of the flexor muscles [13]. Pain over the anterior two thirds of the sole of the foot, which is worse especially during walking, is the most prominent symptom. Sensory disturbances at the same area are reported. Over the retinaculum, there is a positive Tinel’s sign which describes a tingling or prickling feeling brought on by the percussion of a damaged nerve. This sign also denotes the regeneration of nerves. Motor signs include weakness of flexion and abduction of the toes. The medial and lateral plantar nerves may be exposed to compression while they pass through the sole of the foot. With this, sensory complaints are more common than motor. Morton’s neuroma is also a painful neuropathy which affects interdigital nerves [12].
Femoral Nerve
A femoral nerve lesion at the level of the inguinal ligament may be caused by lymphadenopathy, haematoma or other space-occupying lesions as well as hip fractures or hip replacement. Pain radiating over the anteromedial side of the thigh, medial shin and arch of foot and weakness of the quadriceps muscle are the reported symptoms. Lateral femoral nerve lesions cause meralgia paresthetica syndrome. Tight belts, tight garments, sitting in the same position for a long time, abdominal obesity and diabetes either alone or in combination may cause sensory symptoms over the lateral aspect of the thigh. Since the lateral femoral cutaneous nerve is purely sensory, there is no motor deficit. If clinical diagnosis is definite with normal neurological examination, other than sensory deficit over the lateral thigh, electrophysiological examination is not necessary. It is difficult to obtain lateral femoral cutaneous nerve sensory nerve action potentials (SNAPs) in overweight people, even on the asymptomatic side.
Sciatic Nerve
The sciatic nerve is a large, deeply localised nerve, and so injury due do external trauma is not frequently seen. However, injuries can occur due to intramuscular injections or compression caused by a deep haematoma, abscess or pelvic mass. Hip dislocation fractures may also damage the sciatic nerve at a proximal site. Piriformis syndrome causes controversies from time to time. Hypertrophic piriformis muscle or a variation of nerve course, due to something like penetrating muscle bulk, may cause symptoms in certain positions. If present, electrophysiological findings are of the sciatic nerve, not the piriformis muscle. At the level of the mid-thigh, femur fractures and vascular lesions may harm the sciatic nerve. Electrophysiological examinations help to determine the site of injury.
Lower extremity nerves can be affected by polyneuropathy or mononeuropathy multiplex and entrapment neuropathies concurrently. Careful neurological and electrophysiological examination is important when deciding a differential diagnosis. When pathological findings are present in one location, homologous nerves should be examined. If necessary, examination should be extended.
Plexopathies
Upper Limb Plexopathies
The brachial plexus is formed by fibres arising from the fifth cervical to first thoracic spinal roots. Fibres are organised as trunks, cords and peripheral nerves which form a mesh. Due to its complex structure, brachial plexus lesions are difficult to localise and differentiate from other disorders. One peripheral nerve may contain fibres from different roots, trunks and cords. Postganglionic sympathetic fibres also join the motor and sensory fibres of the brachial plexus [1]. Different types of pathological processes can affect the brachial plexus such as compression, transaction, ischaemia, inflammation, metabolic abnormalities, neoplastic processes and radiation. Symptoms of brachial plexus lesions depend on the time course. Acute onset symptoms include severe pain over the shoulder that radiates to the upper arm. Chronic cases complain of numbness and increasing weakness of certain muscles of the upper limbs. Trauma, metabolic and inflammatory processes cause acute presentations, while neoplastic involvement or radiation therapy produces a more insidious onset. Electrophysiological examinations are useful for forming a diagnosis and partial differential diagnosis. Imaging studies are also important, especially if neoplastic processes or structural abnormalities are suspected [11, 13, 14].
Traumatic Brachial Plexus Lesions
Traumatic brachial plexus lesions are frequently due to accidents in adults, usually motorcycle accidents and falls, while difficult birth is the main cause in children [11]. Open traumas are associated with lesions of other structures like bone fractures, blood vessel lacerations and haematomas which complicate the diagnosis and management. Root avulsions are caused by forceful stretching of the nerve fibres which results in detachment from the spinal cord. Root avulsion and plexopathy can occur together. A diligent electrophysiological examination is needed in these conditions [13, 14].
Neurogenic Thoracic Outlet
Neurogenic thoracic outlet is less frequent than expected. It usually involves the medial cord and inferior trunk causing thenar atrophy and sensory deficit over the medial side of the hand and forearm. Electrophysiological parameters are compatible with these symptoms showing motor involvement of intrinsic hand muscles innervated by the median nerve with the absence of ulnar and medial antebrachial nerve SNAPs.
Backpack Palsy
Backpack palsy usually presents with unilateral weakness of an arm and/or shoulder. Carrying weight in a backpack or using baby carriers on the back may cause upper trunk injury [14].
Neuralgic Amyotrophy
Neuralgic amyotrophy, also known as parsonage-Turner syndrome and idiopathic brachial plexopathy, is regarded as an inflammatory illness which can be recurrent. First symptoms include pain over the shoulder and arm which is followed by weakness of the muscles, occurring within a day or 2 weeks. Muscle atrophy appears later in some muscles because brachial plexus involvement is patchy in neuralgic amyotrophy. The long thoracic, suprascapular, musculocutaneous, radial, anterior interosseous and axillary nerves are more frequently affected. In some cases, the homologous limb is also affected simultaneously or on a different occasion. Single-nerve involvement, for example, the anterior interosseous nerve, may be the sole finding which can simulate mononeuropathy multiplex. Phrenic nerve involvement occurs in approximately 8% of neuralgic amyotrophy patients [15]. Recovery is slow and can take between 1 and 3 years. However, it may be incomplete in 30% of patients. Biopsy is not performed routinely so verifying histopathological data is not possible.
Hereditary Brachial Plexopathy (Hereditary Neuralgic Amyotrophy)
This is an autosomal dominant disorder caused by a variant of the septin 9 gene on chromosome 17. Hereditary brachial plexopathy patients may have some dysmorphic features like close set eyes, short stature, small face, unusual skin folds and creases on the neck. Four patients with hereditary brachial plexopathy had nerve biopsy during attacks, and in two of them inflammatory changes were seen [14, 16]. Infections, surgical operations, trauma or giving birth may cause exacerbations. Treatment with corticosteroids is reported to improve symptoms, especially pain. During the acute phase, intravenous immunoglobulin (IVIG) treatment may be beneficial [14, 16].
Pancoast Tumours
These may be associated with plexopathy and Horner syndrome. Brachial plexopathy in a patient who had surgery and radiotherapy for malignancy raises questions of malignant invasion and radiation plexopathy. Prominent pain suggests neoplastic invasion, while fasciculations and myokymia in needle EMG favours radiation plexopathy.
Diabetic Brachial Plexus Neuropathy
This is rare and usually occurs with lumbosacral plexopathy which is more severe and draws more attention [14].
Lower Limb Plexopathies
Lumbosacral Plexopathy
The anterior rami of the L1-S4 roots form the lumbar and sacral plexus. As usual for plexus lesions, the symptoms appear asymmetrically. Weakness, pain and sensory disturbances are present in multiple adjacent dermatomes and myotomes. Lumbar plexus lesions affect flexion and adduction of the thigh and/or extension of the knee. Lumbosacral trunk and upper sacral plexus lesions usually involve abduction of the thigh, flexion of the knee and foot movements. Sensory involvement depends on the involved nerves distribution [12, 13].
Diabetic Amyotrophy/Idiopathic Lumbosacral Radiculoplexus Neuropathy
Diabetic amyotrophy, also called diabetic radiculoplexus neuropathy, differs from other diabetic neuropathies because the underlying mechanism is highly complex. The nerves and roots are involved in immune, inflammatory and vascular processes. Development of subacute and painful proximal muscle weakness with some degree of autonomic impairment constitute the clinical findings. Electrophysiological examination reveals sensory and motor conduction abnormalities and acute denervation activity in needle EMG. Corticosteroids, IVIG and plasma exchange are effective treatment choices [17, 18]. The only difference between idiopathic lumbosacral radiculoplexus neuropathy and diabetic amyotrophy is the absence of diabetes as the aetiological factor. Otherwise, the signs and symptoms are similar and so are the electrophysiological examination results [19].
Neoplastic Invasion
Neoplastic invasion of the lumbosacral plexus occurs due to expansion of primary or metastatic tumours from the organs close by. Colorectal, bone, testis, bladder, uterine and cervical cancers can cause lumbosacral plexus lesions. Pain is prominent. Iatrogenic lesions that occur during surgery are also possible. Compression due to abscess and haematoma masses overlying the psoas muscle can cause damage [19].
Radiculopathies
Upper Limb Radiculopathies
Upper limb nerves are derived from the fibres of the C5 through to T1 roots. The fibres of an individual root can innervate more than one muscle via same nerve or different nerves. Similarly, one individual muscle receives fibres from different roots through the same nerve. When a peripheral nerve is injured, the resulting deficiency in muscle strength is more prominent than that produced from a single-root lesion [20].
Cervical radiculopathy is a common cause of pain and weakness of the neck, shoulder and arm. Weakness of the muscle, dermatomal sensory deficits and reduced deep tendon reflexes are classical findings. Intervertebral disc herniation, spondylosis and degenerative changes of bony structures are common causes of cervical root compression. Traumas, tumours and infections may also cause radicular dysfunction. The presence of Lhermitte’s sign, increased deep tendon reflexes in the lower limbs, and an extensor plantar response suggest an associated myelopathy.
Electrophysiological findings in cervical radiculopathy include normal or near normal motor nerve conduction velocities (NCVs), normal sensory NCVs and sensory action potentials. Needle EMG findings may also be normal. If axonal degeneration is present, reduced recruitment with denervation potentials may be observed. Large motor unit potentials and reduced recruitment during maximal muscle contraction are compatible with a chronic course. Magnetic resonance imaging (MRI) of the cervical spine is usually the preferred choice for identifying structural abnormalities.
Lower Limb Radiculopathies
The symptoms of lumbosacral radiculopathy include pain, paraesthesia and muscle weakness. Pain and paraesthesia show a dermatomal distribution. When muscle weakness is not prominent, myotomal distribution is difficult to discern. Lumbosacral radiculopathy usually occurs due to intervertebral disc herniation and neural foraminal stenosis. Tethered cord, diastematomyelia, spina bifida and nonskeletal conditions such as infection, inflammation, neoplasm and vascular disease are defined aetiological factors [12]. MRI, computed tomography and electrophysiological methods are used for evaluating radiculopathy. Prompt radiological evaluation is necessary when neoplasia or an abscess is suspected or if acute progressing neurological deficits, urinary retention, saddle anaesthesia and bilateral neurologic symptoms are present [12, 21].
Mononeuritis Multiplex
Mononeuropathy multiplex is a lesion of two or more peripheral nerves that cannot be explained by other peripheral nerve disorders such as polyneuropathy, root or plexus injury. Mononeuritis multiplex is another term that is used to define same condition [2]. Asymmetric, non-length-dependent involvement of nerves and a subacute pattern constitute the characteristic features. Symptoms occur in different nerves simultaneously or consecutively. Patients may present with paraesthesia, deep, dull pain and weakness in a single peripheral nerve distribution followed by another nerve area. As the process continues, other nerves become involved so that clinical and electrophysiological findings resemble symmetric distal sensorimotor neuropathy [22]. Multifocal nerve infarctions cause mononeuritis multiplex. Systemic vasculitis and nonsystemic vasculitis are the most common aetiological factors. Vasculitis, which accompanies systemic diseases, has some additional symptoms such as weight loss, skin lesions and adult-onset asthma/sinusitis. A prospective study found that patients with systemic vasculitis who had vasculitic neuropathy at the onset of disease had a worse prognosis, even without poor prognostic factors [23].
Shin J. Oh et al. reported nine cases of peripheral neuropathy associated with vasculitis in patients who had malignant disease before or after a diagnosis of neuropathy [9]. They referred to a report of three cases by Torvik et al. dated 1968 as the first report concerning paraneoplastic vasculitis resulting in peripheral neuropathy with malignant diseases. In that article Torvik et al. stated that ‘The vasculitis of these cases may remain localised to muscles and peripheral nerves and leave the visceral organs intact’ [24]. At present paraneoplastic vasculitis is one of the accepted causes of asymmetric neuropathy or mononeuritis multiplex. Symptoms and signs are responsive to immune suppressant therapy. The most common treatment is pulse intravenous cyclophosphamide with corticosteroids, with transition to azathioprine [2, 25].
Besides vasculitis, diabetes, infection, toxicity and drug adverse effects must be investigated during evaluation [25, 26]. Multiple entrapment neuropathies, a family history of peripheral neuropathy and compression neuropathies are also important to distinguish from hereditary neuropathies accompanying autonomic signs which suggests amyloidosis. Possible aetiologic factors of mononeuritis multiplex are shown in Table 10.2. Electrophysiological tests reveal predominantly motor axonal involvement with denervation activity to some extent (Fig. 10.2). Even asymptomatic nerves should be examined to show involvement of other nerves. An extensive search for the aetiology is very important to identify treatable autoimmune and inflammatory causes. Patients with systemic vasculitis may have vasculitic neuropathy which presented as mononeuritis multiplex treated with corticosteroids and immunosuppressant drugs [26, 27].
Polyneuropathies
Hereditary Neuropathies
Hereditary neuropathies (HN) include a wide spectrum of motor, sensory and autonomic nerve involvement. Besides the peripheral nervous system, other organs may be affected. Different HNs share similar clinical and electrophysiological features, which renders the differential diagnosis difficult. It is possible that a patient, who has a HN, may also have a condition that causes acquired neuropathy. In this case, overlapping characteristics cause more complexity [16]. HNs are usually devoid of sensory symptoms and have an early age of onset. The onset is described subacutely by patients. However, symptoms such as pes cavus/pes planus, hammer toes and atrophy of distal limb muscles tell another story. Electrophysiological findings also support a chronic clinical course. The most common type is Charcot-Marie-Tooth (CMT) disease [2, 28].
Foot and toe deformities that can be seen in HNs include pes cavus, pes planus, claw toe, mallet toe, hammer toe and curly toe (Fig. 10.3). Pes cavus or claw foot means a foot with an abnormally high plantar longitudinal arch. In this condition, too much weight and stress are placed on the heel of the foot when walking. Different toe deformities can also be observed with pes cavus. Pes planus or flat foot is the loss of the medial longitudinal arch of the foot. In this condition, the medial arch of the foot comes closer to or in contact with the ground. The claw toe is a bending of the toe at the ball of the foot. At the middle joint, and sometimes the distal joint as well, the toe bends downward in a claw-like or curly appearance. Claw or curly toes can occur in any toe except the big toe. Hammer toe often presents along with hallux valgus which is also known as bunion deformity. The toe is bent in the middle joint causing a curling of the toe in this deformity. This is most common in the second toe but it can occur in any toe. A mallet toe is like a hammer toe except the joint involved is the distal joint instead of the middle joint, giving the toe a mallet-like appearance at the end [6, 18, 28].

Different feet and toe deformities: (a) claw foot, (b) normal foot, (c) flat foot, (d) claw toe, (e) mallet toe, (f) hammer toe and (g) curly toe
Hereditary polyneuropathies (HPNs) include a wide variety of motor, sensory, autonomic and other systems involvement with considerable overlap. The pathophysiological process eventually results in axonal degeneration and neurological dysfunction in all types of HPNs. The morbidity and mortality depend on the neural and systemic involvement. Overlapping genetic and acquired factors increase diagnostic complexity. Neurologic examination and neurophysiologic tests are important for diagnosis. Clinical features, electrophysiological characteristics, the mode of genetic transmission, metabolic deficiency and genetic loci are used for classifying HPNs [29]. The primary HPNs predominantly involve peripheral nerves and symptoms are due to dysfunction of those peripheral nerves. On the other hand, peripheral neuropathies, which are associated with other disorders, also affect the central nervous system and organs. Symptoms of other organ or system involvement may dominate, and peripheral nerve symptoms may go unnoticed (Table 10.3). Genetic diagnosis has improved parallel to next-generation sequencing. Symptoms which were classified as idiopathic or atypical previously may now have a definite diagnosis. Gene-specific therapies have also developed alongside genetic diagnosis. Antisense oligonucleotides, RNA interference, small molecule chaperones and viral gene delivery therapies appear as the new therapeutic options. Treatments for hereditary transthyretin amyloidosis and Fabry disease are already available. However symptomatic treatment and family counselling are the principal therapy options for most inherited neuropathy classes [29, 30].
Charcot-Marie-Tooth (CMT)
Advances in science and technology furthered our knowledge of inherited neuropathies leading to a change in the way they are classified. Peroneal muscular atrophy and hypertrophic interstitial neuritis became Charcot-Marie-Tooth and Déjérine-Sottas neuropathy. Dyck and Lambert described demyelinating, axonal and intermediate forms of neuropathies according to motor nerve conduction velocity. These include type 1 demyelinating (NCV below 38 m/s) and type 2 axonal (NCV over 38 m/s). An intermediate form (NCV between 38 and 45 m/s) was added as the need arose. However, due to confusion caused by overlapping phenotypes, the name hereditary motor sensory neuropathy (HMSN) came into use [28]. A new, more comprehensive classification, based mostly on associated genes, has been constructed upon CMT classification. With this evolution, the CMT eponym became more popular again. Subtypes of CMT are described and numbered 1 through 7, and there is also an X-linked category (CMTX). Each category has an assigned letter (CMT1A, CMT2B) which indicates the presence of an associated specific gene. Multiple genes have been found to cause CMT; however most cases have five pathogenic genes which are peripheral myelin protein gene 22 (PMP22), myelin protein zero (MPZ), gap junction protein beta 1 (GJB1/connexin 32) and Mitofusin-2 (MFN2) [29].
CMT1 is autosomal dominant demyelinating neuropathy which is the most common type of CMT. Typical findings are prominent distal muscle weakness and atrophy, reduced sensation, decreased deep tendon reflexes and different foot deformities such as pes cavus and hammer toes [18]. Electrophysiological examinations reveal symmetrical-reduced NCVs, between 15 and 38 m/s, without conduction block and temporal dispersion. Seven subtypes of CMT1 have been identified, with five causal genes [2, 4, 28]. CMT1A accounts for approximately 70% of CMT1 cases and more than 50% of all CMT cases. Electrophysiological findings are compatible with demyelinating neuropathy in the beginning; however by the time of diagnosis, signs and symptoms of secondary axonal degeneration appear. Neurological disability is caused by axonal degeneration rather than demyelination. Histopathological evidence of hypertrophic segmental degeneration and regeneration presents as ‘onion bulbs’. The severity of clinical findings is greater in patients with very low NCVs [29]. The causative mutations of the PMP22 gene are most often duplications. However, point mutations may also occur. While duplication causes overexpression of PMP22, point mutations cause a different distribution of PMP22 protein. There is no simple correlation between the expressed PMP22 protein levels and disease severity. Moreover, the severity of neurological findings differs highly within affected families or identical twins. This data suggests that other external factors like epigenetic and environmental changes may have effects on disease severity. An interesting finding is that besides the typical neuropathic symptoms, associated sleep apnoea may be observed in CMT1A patients with duplication of the PMP22 gene. Patients who have 1.5 Mb deletion of the PMP22 gene have hereditary neuropathy with pressure palsy [30].
CMT1B cannot be differentiated from CMT1A on clinical or pathological grounds. Genetic research showed that mutations of the myelin protein zero (MPZ) gene, which is also one of the principal genes of CMT, result in CMT1B. MPZ is a component of compact myelin which is important in maintaining myelin compaction and stability. On rare occasions MPZ mutations can be found in patients with late-onset axonal neuropathy (CMT2J). Adie tonic pupil may also be seen in some of the patients. Adie syndrome affects the pupils in either a unilateral or bilateral fashion. It was described as being almost synchronous with CMT1B by Adie, Morgan, Symonds and Holmes in 1931. This neurological disorder is characterised by a tonically dilated pupil that reacts slowly to light but shows a more definite response to accommodation (light-near dissociation). The affected pupil appears abnormally dilated at rest and shows sluggish pupillary constriction in bright light. Constriction is typically more notable with near reaction. It is caused by damage to the parasympathetic innervation of the eye due to different reasons including the HPNs [6, 28, 31].
CMT1C is an autosomal dominant demyelinating neuropathy which has minor symptoms and doesn’t cause significant disability. The causative mutation is in the lipopolysaccharide-induced tumour necrosis factor-alpha factor (LITAF) gene. CMT1D is very rare, accounting for less than 1% of CMT cases. Pathogenic variants of the EGR2 gene, which encodes for early growth response protein 2, cause this CMT form. Most patients have severe symptoms such as delayed motor development and breathing problems. CMT1E is caused by single nucleotide variants in the PMP22 gene. In addition to the classic CMT phenotype, sensorineural hearing loss is also observed in patients with CMT1E. CMT1F is very rare. The neurofilament light (NEFL) gene on chromosome 8p21 is defective in this type. Abnormalities in the same gene are also implicated in CMT2E. Roussy-Levy syndrome is a CMT1 phenotype with additional symptoms such as postural tremor, gait ataxia, distal muscle atrophy, pes cavus, areflexia and mild distal sensory loss. Genetic testing of different families found abnormalities that indicated CMT1A and CMT1B type diseases [28, 31].
CMT2 is an autosomal dominant axonal neuropathy. According to epidemiological studies, approximately 8–30% of the CMT cases are genetically confirmed as CMT2 type. The age of onset is usually in the second or third decade which is later compared to CMT1. However, an early onset form also exists with more severe symptoms. It is not possible to differentiate CMT1 and CMT2 on clinical grounds. Distal prominent loss of muscle strength, sensory deficits, reduced deep tendon reflexes, atrophy and deformities are classical findings. However, electrophysiological examination shows reduced CMAP and SNAP amplitudes with NCVs above 38 m/s which are compatible with axonal neuropathy. Needle EMG findings correlate a chronic course with chronic reinnervation potentials. Histopathological examination of the sural nerve reveals loss of large myelinated fibres, regeneration activity without demyelination and hypertrophic properties which are hallmarks of primary axonal injury. CMT2 has more than 30 subtypes and 33 genes have been reported to be involved. Ten more genes are associated with intermediate forms. Despite this, most of the patients with typical findings of axonal CMT do not have a genetic diagnosis. Due to abundance of subtypes, the more common ones are mentioned in this section. Mutations of the mitofusin 2 protein coding gene (MFN2) are responsible for subtype CMT2A. Besides typical characteristic findings of CMT, additional clinical features such as optic atrophy, hearing loss, vocal cord paralysis and diaphragmatic weakness are observed. Specific gene mutations are involved in various specific clinical findings such as CMT2A-MFN2 with optic atrophy, CMT2C-TRPV4 with vocal cord paralysis and CMT2B-RAB7A with prominent sensory loss plus foot ulceration plus mutilation due to the inability to feel pain. Weakness of distal upper limb muscles is more prominent than lower limb weakness in CMT2D-GARS1 cases. As genetic test results are expanding, the clinical spectrum of CMT2 enlarges with many overlaps between subtypes [28,29,30,31].
CMT3 consists of two disorders: Dejerine-Sottas syndrome and congenital hypomyelination neuropathy. Dejerine-Sottas syndrome is a severe demyelinating neuropathy which causes floppy infant syndrome. Characteristic findings are motor retardation with severe sensory impairment, distal and proximal weakness of limbs, absent deep tendon reflexes and ataxia. Electrophysiological examinations of peripheral nerves reveal extremely slow conduction velocities below 10 m/s. Scoliosis and contractures can also occur and progress during the disease, but walking ability is preserved through adult age. Clinically, Déjérine-Sottas syndrome is different from both classical CMT and congenital hypomyelination neuropathy. However, as new genetic forms of autosomal dominant or recessive patterns have been described, the genetic overlaps become harder to understand as these described genes are also involved in CMT1 and CMT4. There are pathogenic variants with mutations in the PMP22 gene, which is also involved in CMT1A, and the MPZ gene which is also involved in CMT1B. Congenital hypomyelination neuropathy is one of the causes of floppy infant syndrome. Infants are born with contractures [28, 32].
CMT4 is autosomal recessive. Manifestations of CMT4 include prominent distal muscle weakness and atrophy, sensory impairment and deformities like pes cavus. Electrophysiological examinations show slow NCVs below 40 m/s which is compatible with demyelinating neuropathy. Several subtypes of CMT4 have been reported in consanguineous and nonconsanguineous families [33].
X-linked CMT neuropathies make up around 10–15% of CMT cases. The CMTX1 subtype includes patient with mutations in the GJP1 gene. Almost 90% of CMTX patients have GJP1 mutations which result in connexin 32 (a gap junction protein) dysfunction. Since the mutation is X-linked, dominant females are also affected but less severely than males. Progressive weakness and atrophy of muscles, areflexia, sensory impairment and variable central nervous system symptoms are observed in affected patients. When questioned, patients report a history of frequent falls in their adolescence and early adulthood. Electrophysiological studies reveal intermediate NCVs with mildly prolonged latencies and low amplitudes of CMAPs. Mutations of the recently found PRPS1 gene result in Arts syndrome. This syndrome is part of a spectrum of PRPS-1-related disorders with reduced activity of the PRPP synthetase 1 enzyme which includes CMT5 and X-linked nonsyndromic sensorineural deafness [6, 28, 30]. The intermediate CMT category is reserved for cases which do not meet the complete criteria for either demyelinating or axonal neuropathy. This CMT variant is a rare form which causes controversy about its existence and classification. Due to this diversity of views, traditional categories are used as much as possible. Some X-linked types and autosomal recessive forms are in this category. Dominant intermediate CMT type A (DI-CMTA) and dominant intermediate CMT type C (DI-CMTC) have been described. Recessive forms of intermediate forms include RI-CMTB and RI-CMTC which are also defined [30].
Hereditary motor sensory neuropathy (HMSN) types 5, 6 and 7 were classically included in CMT and HMSN classifications, but currently these disorders are evaluated with the associated symptoms. Patients with autosomal dominant spastic paraparesis and sensory neuropathy are referred as HMSN 5. HMSN 6 refers to patients with dominant or recessive optic atrophy and motor sensory neuropathy. HMSN 7 refers to patients with retinitis pigmentosa and motor sensory neuropathy [30, 34].
Hereditary Sensory and Autonomic Neuropathies
Other groups of HNs are the hereditary sensory and autonomic neuropathies (HSAN). Sensory and autonomic features are predominantly seen in patients who have the disease [2]. Loss of large myelinated and unmyelinated fibres are prominent features of HSAN. Classification of HSAN is based on clinical characteristics and genetic grounds [35].
HSAN1 is autosomal dominant and is the most frequently seen form of HSAN. Clinical features include distal sensory loss followed by distal muscle weakness and atrophy. Facial sensation is preserved and foot ulcers are frequently seen. Onset is usually in early adulthood. Autonomic abnormalities vary in severity. The underlying pathology is progressive degeneration of motor neurons and dorsal root ganglia. Hearing loss and dementia have also been reported in some affected families. Genetically confirmed, four subtypes are reported. The genes that are implicated in HSAN1 are serine palmitoyltransferase, long-chain base subunit 1 (SPTLC1), SPTLC2, atlastin GTPase 1 (ATL1), ATL3 and DNA-metiltransferaz 1 (DNMT1). Electrophysiological examinations show axonal degeneration of motor and sensory fibres. Sensory neuron action potentials (SNAP) are of low amplitude or normal in the upper limbs, but usually they cannot be detected in the lower limbs. Variability of motor nerve conduction velocities from normal range to the demyelinating range with conduction blocks may cause diagnostic difficulty. This electrophysiological diversity may cause misdiagnosis in patients with prominent motor signs and scarce autonomic findings [36, 37].
HSAN2 is autosomal recessive and is characterised by loss of touch, pressure, pain and temperature sense. Involvement of large myelinated and small unmyelinated fibres cause clinical signs and symptoms. Recurrent infections, fractures of digits, osteomyelitis, spasticity and other autonomic disturbances are frequently encountered. The genes that are implicated in HSAN2 are WNK1/HSN2, FAM134B, KIF1A and SCN9A. Genetically defined, three subtypes are reported in the literature. Recently, a novel RETREG1 (FAM134B) founder allele has been linked to HSAN2B, and the resulting renal disease was identified in a Turkish family. Mutations causing loss of function result in insensitivity to pain and temperature, hearing loss and hyposmia, while gain of function mutations cause excess pain including erythromelalgia [38].
HSAN3 is also called familial dysautonomia and Riley-Day syndrome. HSAN3 is almost exclusively seen in children with Ashkenazi Jewish ancestry. Only a very limited number of cases are reported in other populations. It is an autosomal recessive disorder. The clinical course entails progressive sensorimotor neuropathy with autonomic features. Sympathetic autonomic involvement is responsible for most of the disturbances. Dysautonomic crises are sometimes difficult to manage. Myelin abnormalities are also found in the central nervous system causing dorsal column demyelination. Small stature, vertebral column abnormalities, lack of fungiform papillae causing a smooth tongue surface, dysarthria, mental disability and emotional lability are additional characteristics of the syndrome.
Congenital insensitivity to pain and anhidrosis describes the clinical characteristics of HSAN4 which is transmitted as an autosomal recessive trait. Severe insensitivity to temperature and pain causes injuries which lead to self-mutilation and osteomyelitis. Seizures and a mild/moderate mental impairment are also seen. HSAN5 is an autosomal recessive disorder. Loss of pain and temperature sensation is present, while other sensations are normal. HSAN6 is one of the causes of floppy infant syndrome. Autonomic abnormalities, failure to thrive and absent psychomotor development are characteristic findings. It is an autosomal recessive disorder. HSAN7 is an autosomal dominant disorder. Signs and symptoms appear at birth or during infancy. Congenital insensitivity to pain, excessive sweating, delayed motor development without cognitive impairment and gastrointestinal dysmotility are characteristic features. Due to the inability to feel pain, self-mutilation, joint dislocation, bone fractures and osteomyelitis are common disturbances. Muscle weakness is not a prominent finding. HSAN8 is inherited as an autosomal recessive trait. Pain insensitivity causes soft tissue injuries, self-mutilation and tooth loss. There are also some HSAN cases which cannot yet be classified [28, 35,36,37].
Hereditary Neuropathy with Pressure Palsy
Hereditary neuropathy with pressure palsy (HNPP) is a recurrent, episodic demyelinating neuropathy which is autosomal dominant. Patients typically present with single nerve dysfunction due to compression, usually at the usual sites for entrapment. The most frequently involved nerves are the axillary, median, radial, ulnar, peroneal and brachial plexus nerves. Sensorineural deafness and scoliosis are associated findings. The age of onset is usually in the second decade, but it can develop in early childhood or in the third decade. Isolated nerve palsies may occur successively with recovery taking days to months. Motor deficits can be persistent, so that in later stages, accumulated deficits may resemble symmetrical neuropathy with entrapment syndromes [28]. HNPP is also called tomaculous neuropathy due to the microscopic appearance of nerves in biopsy materials during tear fibre examination. PMP22 deletion and single nucleotide variants are found in HNPP which render HNPP allelic to CMT1A. The deletion in chromosome 17p11.2 results in decreased expression of the PMP22 gene [38].
Familial Amyloid Polyneuropathy (FAP)
Due to the emerging treatment options, diagnosis of familial amyloid polyneuropathy (FAP) has gained importance. Mutations of the transthyretin (TTR), apolipoprotein A1 (APOA1) and gelsolin (GSN) genes cause this disorder that can be potentially fatal [2]. Accumulation of fibril aggregates of amyloid precursor proteins in the peripheral nerves and other systemic organs results in early autonomic involvement, unexplained cardiomyopathy, bilateral carpal tunnel syndrome and a progressive course in patients with a family history [2, 28]. Length-dependent small fibre neuropathy that causes impaired temperature and pain sensation is the typical manifestation. Autonomic dysfunction may cause life-threatening cardiac arrhythmias with cardiac failure, especially in the elderly [28]. Nephrotic disease is another symptom of FAP. TTR mutations are the most common type. Val30Met was the first identified TTR mutation and is seen most frequently. However, there are more than 100 different amyloidogenic point mutations [2]. Autosomal dominant mutations cause phenotypic heterogeneity [39]. The patients who have the same mutations show different clinical signs and symptoms. They may also have a different age of onset. For example, in Portuguese patients, the age of onset is typically in the third decade, whereas in people from Northern European countries, onset occurs in the sixth decade [28]. Due to the heterogeneous aspects of the disease, TTR-FAP diagnosis is made usually long after the onset of the symptoms. If progressive sensorimotor neuropathy presents with any of the following findings such as a family history, autonomic dysfunction, cardiac failure, gastrointestinal symptoms, weight loss with unidentified cause, bilateral carpal tunnel syndrome, renal dysfunction or ocular involvement, TTR-FAP should be considered in the differential diagnosis [39]. Tafamidis meglumine is a drug approved in Europe and the United States which is administered orally and blocks mutated TTR misfolding and accumulation. The US Food and Drug Administration also approved two gene therapies, patisiran and inotersen. Liver transplantation, which was the only hope for TTR-amyloidosis patients before new molecular therapies were developed, has become a less preferred treatment [28].
Electrophysiological evaluation is very useful for differentiating hereditary and acquired neuropathies. Uniform nerve conduction velocity slowing is the usual finding in inherited neuropathies, whereas patchy slowing, partial conduction blocks and increased temporal dispersion are seen in acquired neuropathies. However, CMTX-GJB1 mutations constitute an exception. In patients with this mutation, nerve conduction abnormalities mimic abnormalities of acquired demyelinating neuropathies. The blink reflex (BR) is an electrical analogue of the clinical corneal reflex. When peripheral nerve conduction studies are ambiguous, blink reflex examination may provide a clue for diagnosing demyelinating HN. Irrespective of severity, a latency of R1 response which is more than 13 milliseconds supports the diagnosis [32]. Autonomic testing can be helpful when distinguishing CMT from other inherited neuropathies that have autonomic involvement such as TTR-FAD and HSAN. R-R interval changes during resting, deep breathing and the Valsalva manoeuvre, as well as a sympathetic skin response and orthostatic blood pressure changes, are accepted as useful tools [28].
Hereditary Brachial Plexus Neuropathy
Hereditary brachial plexopathy is a rare autosomal dominant disorder. Hereditary neuralgic amyotrophy (HNA) is another name for recurrent, painful brachial plexopathy. HNA and hereditary neuropathy with predisposition to pressure palsies (HNPP) are recurrent and episodical disorders [38]. The condition is caused by pathogenic variants in the septin 9 (SEPT9) gene. Childhood onset of hereditary brachial plexopathy is not unusual. Many patients exhibit a relapsing-remitting course characterised by attacks that resolve spontaneously, either completely or incompletely, leaving additive residual weakness. The disorder can also follow a progressive pattern. Physical exertion and pregnancy are reported triggering events. The attacks are heralded by pain and paraesthesia, followed by paresis of the shoulder and arm. While any nerve in the brachial plexus can be involved, injury to the upper part of the brachial plexus is the most frequent feature. The characteristic somatic features of hereditary brachial plexopathy include short stature, hypotelorism, a small face, unusual skin folds and creases on the neck [11, 38].
Giant Axonal Neuropathy
Giant axonal neuropathy (GAN) is a degenerative disorder which affects both the central and peripheral nervous systems. This is a severe autosomal recessive disorder. The genetic locus of the disease maps to 16q.24.1. Symptoms appear in early childhood with frequent falls and gait abnormalities. Weakness of the distal muscles and ataxia are also found. Central nervous system involvement signs include cerebellar dysfunction, spasticity and optic atrophy. A typical phenotype to develop the disorder consists of red and curly hair with a pale complexion and long eyelashes. Cognitive impairment is present in some cases. Electrophysiological findings are compatible with axonal neuropathy. The progressive course ends with death which is usually due to respiratory insufficiency [28, 38].
Acquired Neuropathies
Toxic Neuropathies
Toxic neuropathies (TNs) are an important category of acquired neuropathies, and they are caused by exogenous neurotoxic substances. Toxic substances enter the body via digestion and inhalation or via parenteral and transcutaneous routes. Acute, cumulative and delayed intoxications may occur [40]. Neurotoxic agents can be classified as environmental, occupational, recreational or iatrogenic [41]. Additionally, the increased survival rates of cancer patients has led to more chemotherapy-related problems. Toxic neuropathy is one of the frequent neurological disorders seen in cancer patients due to the toxic effects of chemotherapeutic agents. More than 30% of patients who are exposed to potentially neurotoxic agents have neuropathi disturbances. The highest prevalence is seen in patients who received platinum-based drugs, vinca alkaloids and taxanes [2, 40]. In developed countries most toxic neuropathy patients are exposed to these group agents. However, in developing countries, TNs are mostly caused by environmental and/or occupational toxic substances including arsenic, lead, mercury and organophosphorus. In addition, some manufacturing processes have moved to less-developed countries which have less control over the occupational hazards related to previously known toxic substances such as hexane, carbon disulphide and 1-bromopropane.
Axonal degeneration, demyelination, neuronopathy, ion channel dysfunction and other molecular pathways are associated with TN (Table 10.4). Most of the toxic substances predominantly cause axonal neuropathy which can be acute, subacute or chronic [42]. But there are no strict rules about this. N-hexane exposure can cause both axonal and demyelinating neuropathies. The effects caused by toxic substances may continue after the offending exposure is removed. This condition is called the ‘coasting effect’. Neuropathy due to isoniazid toxicity can occur in patients with tuberculosis. Isoniazid metabolites inactivate B6 and inhibit the enzymes which convert pyridoxine to active pyridoxal phosphate. Concurrent administration of vitamin B6 prevents the development of neuropathy. However, chronic overdose of B6 also causes axonal large and small fibre neuropathy in susceptible people [43, 44]. Chemotherapeutics, antimicrobials and antiretroviral drugs are also known to cause neuropathy. New treatments such as tumour necrosis factor inhibitors (infliximab) and immune check point inhibitors (ipilimumab) can cause TN.
Alcohol may cause toxic neuropathy by itself. Additionally heavy alcohol use is usually associated with nutritional deficiency which worsens neuropathic disorders. Distal symmetric axonal polyneuropathy is present in 25–66% of chronic alcoholics in the United States [2]. Organophosphate toxicity frequently encountered due to accidental or intentional exposure has severe systemic and neurological effects. One of the neurological effects is axonal neuropathy.
Nutritional Neuropathies
Neuropathies due to the deficiency of essential substances need to be quickly diagnosed and treated before irreversible changes occur. However, associated neurological and systemic disorders may overshadow the signs and symptoms of neuropathies. Nutritional deficiencies occur due to either insufficient intake or reduced absorption. Starvation, consuming foods poor in nutritional content and eating a restricted diet are the reasons for reduced intake. Reduced absorption can be caused by gastrointestinal diseases like inflammatory bowel disease or drugs which impair nutrition uptake. In some conditions, increased metabolic demand can also cause deficiency. Acute and subacute developments of malnutrition and weight loss require a prompt diagnosis and treatment to avoid persistent damage and dysfunction of the nervous system [40,41,42,43,44,45,46].
Thiamine (B1) is essential for the metabolism of carbohydrates and amino acids as it is a coenzyme for more than 24 enzymes. A deficiency of thiamine causes ‘wet’ beriberi with cardiovascular involvement, ‘dry’ beriberi involving peripheral nerves or Wernicke-Korsakoff syndrome which may result in dementia. Length-dependent, large fibre sensorimotor axonal neuropathy with reduced deep tendon reflexes is a characteristic finding. Autonomic dysfunction may also occur. Tropical ataxia is the result of a diet consisting of cassava which causes thiamine deficiency. Tropical ataxia symptoms consist of sensory ataxia, sensorineural loss of hearing and blindness due to bilateral optic atrophy. Chronic alcohol consumption may also cause neuropathy via thiamine deficiency and/or alcohol toxicity. An increased need for thiamine in acute metabolic stress should be kept in mind, especially for patients in intensive care units [45].
Pyridoxine (B6) is the generic name for pyridoxamine, pyridoxine, pyridoxal and their phosphorylated forms. These compounds function as co-enzymes in carbohydrate, lipid and amino acid metabolism as well as heme and neurotransmitter synthesis. Malabsorption, increased loss and use of certain medications are causative factors of B6 deficiency. B6 deficiency causes a length dependent, mostly sensory neuropathy with paraesthesia and sensory deficits. Occasional motor involvement is also reported. High levels of B6 also result in sensory neuronopathy which causes sensory ataxia [2, 40, 43, 46].
Folate (B9) deficiency is due to insufficient dietary intake, increased turn over, drug-associated deficient absorption/distribution and folate analogues. Folate acts as a coenzyme in the metabolism of 1-carbon units and DNA synthesis with cobalamin. The neuropathy of folate deficiency has been defined in only a few reports. It appears to be a length-dependent, symmetric, large fibre-predominant sensory neuropathy. Accompanying neurological findings resemble subacute combined degeneration which occurs in B2 deficiency. Megaloblastic changes of erythrocytes are also seen [43, 44].
Cobalamin’s (B12) main function is to serve as a cofactor in the methylation process which is very important for DNA synthesis, cell metabolism and erythrocyte function. It also takes part in myelination of the central and peripheral nervous systems. Therefore, deficiency results in demyelination of the posterior and lateral columns of the spinal cord and optic nerves. As expected, the deficiency of B12 causes both haematological and neurological disturbances. The most common deficiency is of vitamin B12 which may be due to insufficient intake or insufficient absorption. Starvation, hunger strikes, low socioeconomic status, inadequate nutrition and vegan diets are some of the causes of insufficient intake. Abnormal absorption is seen in pernicious anaemia, bariatric surgery, short bowel syndrome and gastric bypass surgery. Long-term metformin use can also cause low vitamin B12 levels with a high neuropathy prevalence [47]. Neuropathy is found in one fourth of the B12 deficient patients. Sensorimotor axonal neuropathy, sensory neuronopathy and small fibre neuropathy in a small group of patients are defined in patients with B12 deficiency. Subacute combined degeneration causes impaired proprioception and sensory ataxia with cognitive and psychiatric symptoms. Vitamin E and acquired copper deficiency are difficult to differentiate from B12 deficiency base on only clinical grounds [2].
Vitamin E includes different compounds; however, α-tocopherol is the most common form in human tissue. The functions of vitamin E are very important for survival. It is a cytoprotective antioxidant, it diminishes free radical concentrations in neural tissues, and it modulates glutamate toxicity. Vitamin E also takes part in the construction of cellular membranes, vesicular and cellular transport. Insufficient dietary intake and malabsorption are the main reasons of deficiency. Impairment of large sensory fibre function is reported. However, this is not expected to be a solitary manifestation of vitamin E deficiency because central nervous system dysfunctions such as spinocerebellar syndrome with ganglionopathy create more prominent symptoms. Anaemia and immune system dysfunction are also associated with vitamin E deficiency [44].
Copper is an important essential element which takes part in maintaining the structure and function of the haematopoietic and nervous systems. The most common cause of copper deficiency is surgical operations of the gastrointestinal system. Chronic haemodialysis and zinc toxicity are other frequent issues related with copper deficiency. Large fibre neuropathy occurs in deficient states, but copper deficiency-related vacuolar myelopathy is a more prominent disorder, mainly affecting elderly women [44].
Metabolic and Endocrine Neuropathies
Metabolic neuropathies consist of several peripheral nerve disorders due to the deficiency of organs or glands. The main underlying abnormality is the disorder of metabolic pathways. However, the pathogenetic mechanisms that result in neuropathy are not completely understood yet. There are various associated factors which can cause neuropathy by themselves or worsen present neuropathy. When there is an established metabolic disorder, the symptoms of neuropathy may easily be attributed to the known disease. There may be a causal relationship, but physicians must be cautious and should rule out other possibilities [48,49,50].
Hypothyroidism can cause multiple central and peripheral dysfunctions. Peripheral complications of hypothyroidism include entrapment neuropathies, polyneuropathy, neuromuscular junction disorders and myopathy. Carpal tunnel syndrome (CTS) incidence is higher in hypothyroid patients compared to the normal population. However, hypothyroidism is not the only factor which causes CTS in patients with hypothyroidism. Other factors such as high body mass index or the presence of other metabolic disorders appear to also contribute to CTS. Deposition of mucin in the perineurium and endoneurium of the median nerve and the deposition of mucopolysaccharides in synovial structures cause increased pressure in the carpal tunnel. CTS symptoms and findings do not differ clinically or electrophysiologically in patients with hypothyroidism. Replacement therapy for hypothyroidism may reverse the changes. However, some patients may be unresponsive. Screening of CTS patients for hypothyroidism is not performed in routine practice. However, it is easy, and thyroid-stimulating hormone and free T4 levels should be obtained, especially in patients with bilateral CTS. Sensorimotor polyneuropathy with stocking and glove distribution is another complication of hypothyroidism. Deep tendon reflexes have a longer relaxation time which is a characteristic finding of hypothyroidism. Painful neuropathy, which suggests small fibre involvement, is another presenting form of neuropathy. Symptoms and signs are reversible with replacement treatment. Besides this classical presentation of polyneuropathy, polyneuropathy with autoimmune features can be seen in hypothyroidism like CIDP, GBS and multifocal motor neuropathy. Hormone replacement therapy alone is not sufficient, and so that immunomodulatory therapies are applied [44].
Hyperthyroidism-associated sensory polyneuropathy, CTS and myopathy are also reported. Basedow’s paraparesis is used to define uniform subacute paraparesis that affects both distal and proximal muscles in patients with hyperthyroidism. Deep tendon reflexes are absent or reduced with severe hypotonia. However, sphincters and sensory examination are normal. Electrophysiological investigations identify reduced CMAPs and acute denervation which suggest axonal degeneration. However, histopathological examinations have not yet been reported [44].
Uremic polyneuropathy is commonly seen in patients with renal failure. In approximately 50% of the patients, neuropathy is asymptomatic. As kidney function decreases, neuropathy frequency increases, reaching up to 80% in end stage patients. The prevalence of uremic neuropathy is higher in female patients than male. Sensory symptoms that affect the distal limbs are the initial complaints. In later stages, motor findings appear. Electrophysiological findings are compatible with axonal and demyelinating features. Abnormalities of beat-to-beat variation (R-R interval) reflecting autonomic dysfunction generally correlate with axonal neuropathy. The cause of neuropathy is not precisely known; however, it may be related to a deficiency of thiamine, zinc and biotin and decreased transketolase activity. An increase of potentially toxic metabolites and hyperparathyroidism are suggested to contribute to neuropathy development because uremic neuropathy shares many features of toxic neuropathy [48].
Acromegaly is generally due to the overproduction of growth hormone from an eosinophilic pituitary adenoma. CTS and length-dependent sensory motor polyneuropathy are the most common peripheral nervous system complications.
Hepatic neuropathy incidence is variable in different reports. In most patients, neuropathy has a subclinical course with minimal symptoms. Length-dependent sensorimotor neuropathy and autonomic neuropathy are reported in patients with nonalcoholic hepatic failure. Autonomic dysfunction may be a life-threatening condition. Porphyrias are inherited metabolic disorders which can cause acute axonal neuropathy. Early diagnosis is important because severe complications such as quadriparesis, autonomic dysfunction and respiratory insufficiency may occur. The proximal upper limb is more frequently affected [49, 50].
Diabetic Neuropathies
Diabetes has become a public health issue in the twenty-first century [51,52,53]. The prevalence of diabetes in 2019 was estimated to be 9.3% which means 463 million people are living with diabetes worldwide. The prevalence is expected to rise 10.2% by 2030 and 10.9% by 2045 [51]. Around half of the patients with diabetes are affected by diabetic peripheral neuropathy (DPN). Therefore, increased DPN prevalence is also anticipated in the future [52]. DPN prevalence was found to be similar in type I diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM) patients in the Rochester Diabetic Neuropathy Study by using clinical and nerve conduction parameters. It is interesting that population-based studies using only clinical findings, without nerve conduction assessment, also showed very close prevalence rates for DPN. The European Diabetes Prospective Complications Study (EURODIAB Study) reported that major risk factors for DPN development were poor glycolic control, age and duration of diabetes. Modifiable cardiovascular risk factors such as hyperlipidaemia (especially triglyceridaemia), obesity and cigarette smoking are also involved in the development of DPN in some studies [52]. However, in a recent meta-analysis, body mass index, hyperlipidaemia and cigarette smoking were found to not be risk factors for DPN. The same study identified diabetic retinopathy as a risk factor for DPN [53]. The inflammatory immune response plays an important role in some types of DPN [17]. DPN is also known to increase mortality, independent of other causes of mortality. The relationship between mortality and diminished vibration sense in DM patients is a very interesting and important finding [51,52,53]. A widely accepted definition of DPN, described by Boulton, is ‘the presence of symptoms and/a sign of peripheral nerve dysfunction in people with diabetes after exclusion of other causes’ [54]. However, DPN has many subtypes and is classified based on different aspects. It should be noted that diabetes may cause any type of peripheral neuropathy. In addition, these different types of neuropathies, either with the same or diverse pathophysiology, can be found in the same patients [55]. DPN classification can be made according to the time course, involved nerve types, symmetry and site of involvement or pathophysiology [55, 56]. There is a simple classification list of DPN in Table 10.5 [57].
Distal symmetrical polyneuropathy (DSPN) with sensorimotor involvement is the most common presentation of DPN. DSNP can be separated into three categories according to the involved nerve fibres. These are predominantly large fibre neuropathy, predominantly small fibre and pure small fibre neuropathy. Treatment-induced neuropathy and hyperglycaemia-induced neuropathy of diabetes mellitus can cause an acute painful DSPN. Typical symptoms of DSPN are numbness, paraesthesia/dysesthesia, pain and a burning sensation which usually affects the distal lower limbs. Positive symptoms like pain appear unprovoked. Patients may feel lancinating pain in otherwise insensitive feet. DSPN is a length-dependent neuropathy which begins in the periphery, affects the feet bilaterally and progresses proximally [56, 58]. Sensory involvement is usually described in a ‘stocking glove’ fashion. The proximal parts of the limbs are less severely involved than the distal parts, and the upper limbs are rarely involved. If symptoms are present in the upper limbs, they are most probably due to an accompanying mononeuropathy [55]. Asymmetric involvement should be a warning sign to consider other diagnoses besides DSPN. Asymptomatic patients may comprise of 50% of diabetic patients, while 25% experience painful diabetic peripheral neuropathy (pDPN). The use of sensory examinations should not be underestimated, especially in asymptomatic patients. The presence of feet insensitivity creates a predisposition for injury and increased risk for ulceration. Motor involvement signs emerge later. Weakness of the lower limb muscles first affects the toes, then ankles and calves. Difficulty in daily living and self-care activities becomes prominent [56, 58]. Insidiously presenting autonomic neuropathy commonly accompanies DSPN. If there is no adequate intervention, a progressive and chronic course of autonomic neuropathy is inevitable. Quality of life is seriously affected by painful neuropathic pain and foot ulcerations [59]. Patients with pDPN are more likely to have amputations. Psychiatric morbidity such as depression and anxiety as well as sleep disorders appear as symptom progress. Therefore, psychosocial wellbeing is frequently affected [56]. The fast and maintained normalisation of blood glucose can cause treatment-induced diabetic neuropathy (TIND) which mainly affects sensory and autonomic nerves [17]. There is a risk of DSPN occurrence in prediabetic patients, and so some patients already have DSPN by the time of diagnosis [58]. Electrodiagnostic studies usually show mild-moderate slowing of sensory and motor NCVs. Lowered motor and sensory action potential amplitudes with denervation potentials, seen in needle EMG, are characteristic findings which are prominent in the lower limbs. Upper limb examinations may reveal normal results. Electrophysiological findings are especially important for the differential diagnosis if there is a possibility of other neuropathic symptoms.
For instance, diabetic patients are prone to developing entrapment neuropathies. Inadequate diagnosis and treatment are a major problem. Unfortunately, neither patients nor physicians are aware of the importance of the DSPN. Treatment of DSPN is a complex issue and so a holistic approach is preferred. In particular, for diabetic foot care, having a multidisciplinary team consisting of an endocrinologist, neurologist, psychiatrist, physiotherapist, psychologist, podiatrist, orthopaedic surgeon, orthotist, vascular surgeon, microbiologist, pain specialist and a specialist nurse is essential. First, the management of diabetes itself is a priority. Euglycemia may prevent the occurrence or progression of DSPN in type1 DM, but not in type 2 [59]. Reducing other cardiovascular risk factors is also included in the treatment regimen. Targeting the pathogenesis of DSPN is another aspect. Alpha lipoic acid, benfotiamine, epalrestat and actovegin are permitted for use in some countries. Symptomatic treatment constitutes most of the DSPN therapy. Neuropathic pain is one of the worst symptoms that affects quality of life. Analgesics, antidepressants, anticonvulsants and in resistant cases opioid drugs are the best choices for the treatment of neuropathic pain. Local capsaicin treatment is difficult to apply but is reported to be beneficial in some cases. Each treatment has its own limitations. Combination therapy may reduce the individual drug doses and side effects. Neuromodulation therapy like high-frequency (10 kHz) spinal cord stimulation may be beneficial for pharmacotherapy-resistant patients [57].
Diabetic autonomic neuropathy (DAN) involves small myelinated and unmyelinated fibres. Cardiac, gastrointestinal and urogenital system symptoms are frequent. Cardiac effects are evaluated using heart rate changes during deep breathing, the Valsalva manoeuvre and during R-R interval change recordings (Fig. 10.4) [17]. Besides cardiovagal impairment, adrenergic denervation of the heart also occurs. Sympathetic involvement is evaluated using single-photon emission tomography or positron emission tomography and sympathetic skin response recordings (Fig. 10.5). Cardiac abnormality prevalence is higher in type 1 diabetic patients. Cardiac autonomic neuropathy is associated with high mortality rates. Orthostatic hypotension is another manifestation of autonomic dysfunction. Baroreflex failure is due to both adrenergic and vagal dysfunction [60]. Postprandial hypotension occurs after a heavy meal with a high carbohydrate content. Due to sympathetic denervation, blood pools in the splanchnic mesenteric bed after such a meal which causes orthostatic symptoms. An exaggerated blood pressure increase in response to direct adrenergic agents is related to denervation hypersensitivity. Gastrointestinal motility disorders are also a major component of autonomic dysfunction. Delayed gastric emptying, constipation or diarrhoea is reported. In diabetic patients with chronic intestinal pseudo-obstruction, a majority were found to have an abdominal vagal neuropathy [61]. Bacterial overgrowth, ischaemia of intestinal mucosa and pancreatic exocrine insufficiency have also been implicated as causative factors [17]. The prevalence of neurogenic bladder in diabetic patients is expected to rise as the duration of diabetes prolongs. Decreased detrusor tone results in an increased bladder capacity. The reduced perception of bladder fullness with the presence of high post-voiding residual urine causes overflow incontinence. The symptoms and signs are due to parasympathetic involvement which is usually part of a generalised autonomic disorder. Insufficient emptying of the bladder can be demonstrated by ultrasonographic examination and urodynamic tests [17, 60]. Penile erection is a function of the parasympathetic system, while ejaculation requires sympathetic activation. Erectile dysfunction may be the earliest symptom of autonomic disorder in diabetic men. After initial parasympathetic dysfunction, sympathetic denervation develops which causes ejaculation failure or retrograde ejaculation. Besides autonomic failure, nitric oxide levels and vascular insufficiency, somatic sensation loss contributes to sexual dysfunction [17]. Sexual dysfunction in females has not been studied as well compared to males. Absent and decreased sympathetic skin response amplitudes were reported in diabetic women [62]. A decrease in libido and lubrication, painful intercourse and anorgasmia are reported sexual problems in diabetic women [17]. Sweating and thermoregulation may be impaired due to sympathetic degeneration. Quantitative sudomotor axon reflex and thermoregulatory sweat tests can be used to detect these abnormalities [17, 60]. Before diagnosing autonomic neuropathy, other causes in differential diagnosis must be excluded such as malignant or vascular disorders.

Normal R-R interval recording

Normal sympathetic skin responses recorded from the hand
Diabetic radiculoplexus neuropathy differs from other diabetic neuropathies because the underlying mechanism is highly complex. The nerves and roots are involved in immune, inflammatory and vascular processes. The development of subacute and painful proximal muscle weakness with some degree of autonomic impairment are common clinical findings. Electrophysiological examination reveals sensory and motor conduction abnormalities and acute denervation activity in needle EMG [17].
Infectious Neuropathies
There are many types of primary nerve infections, but infections can cause peripheral nerve dysfunction through different mechanisms.
Leprous neuropathy is a well-known disease, even in ancient times. Leprous neuropathy is one of the earliest symptoms of leprosy which appears as an impairment of sensation which may transform into painful neuropathy later in the course of the disease. In the tuberculoid form of leprosy, this is more likely to be a focal neuropathy. The affected nerves are usually located near the skin lesions. However, with the lepromatous form, a generalised neuropathy occurs. Claw hand, foot drop and lagophthalmos are frequent. The ulnar, median, common peroneal, tibial, facial, radial cutaneous and great auricular nerves are commonly affected nerves. Hypertrophic nerves can be palpated, especially the ulnar nerve at the elbow. Earliest findings include impairment of cold and warmth sensation and sensory nerve conduction abnormalities. Although there is a demyelinating pathology, the mechanism is not well understood [63].
Lyme disease can cause lymphocytic meningitis, facial nerve palsy and radiculoneuritis on top of other systemic involvements. Painful radiculoneuritis, also called Garin-Bujadoux-Bannwarth syndrome or Bannwarth syndrome, is more common in European countries. In some patients, facial nerve palsy and motor weakness with pleocytosis in cerebrospinal fluid can also be found [64].
Human immunodeficiency virus (HIV) can cause a distal symmetrical polyneuropathy (DSPN). This common neurologic complication creates considerable difficulty in daily living activities. The underlying pathology includes ganglionopathy, axonal degeneration and decreased intraepidermal fibre density. The effects of HIV infection on peripheral nerves occurs most likely due to the production of inflammatory cytokines and chemokines by infected monocytes and macrophages because HIV does not infect cells devoid of CD4 receptors such as neurons, dorsal root ganglia and Schwann cells. The envelope glycoprotein gp120 also has direct toxic effects on Schwann cells and dorsal root ganglia by initiating a chain reaction. Genetic susceptibility renders some patients prone to developing DSPN. Regeneration and axonal sprouting defects are also found. Symptoms like dizziness, fainting and bladder dysfunction are suggestive of autonomic involvement [65].
Cytomegalovirus (CMV) infection causes a progressive radiculopathy and mononeuritis multiplex in patients with HIV infection or in immunocompromised patients.
It was reported in a large meta-analysis investigating COVID-19-related neuropathies that SARS-CoV-2 does not cause viral neuropathy. However, peripheral neuropathy frequently occurs during COVID-19 infection. Neurotoxicity due to drugs used for treatment and immune mechanisms are suggested factors which can cause neuropathy. Compressive neuropathies are also reported to occur in patients with associated risk factors such as diabetes [66].
Paraneoplastic Neuropathies
Subacute sensory neuronopathy is mostly associated with small cell lung cancer (SCLC). It precedes the diagnosis in most cases [67]. Numbness and tingling sensations might be the first signs that progress to the impairment of proprioception, due to the involvement of large, myelinated axons. Impairment of sensory function begins in one limb and spreads to other limbs, the face and the trunk. Severe, disabling sensory ataxia and pseudoathetosis may prevent performing daily living activities. Patients rarely have mild ataxia but prominent painful neuropathy. Sensory nerve conduction velocities are normal or near normal, but SNAPs are low amplitude or absent. Motor nerves are usually found to be normal in electrophysiological examinations. However, in some cases, minor involvement is found, which presents a difficulty for differential diagnosis. Clinically, one absent or three low-amplitude SNAPs (<30% of normal), with less than two abnormal motor nerve conduction findings in the lower limbs, favour a diagnosis of sensory ganglionopathy. CSF findings are either normal or pleocytosis, oligoclonal bands and a high IgG index can be seen. SCLC-associated cases usually have anti-Hu antibodies. Anti-collapsin-response mediator protein 5 (CRMP5) antibodies are found in some cases [68, 69]. Co-occurrence of CRMP5 and anti-Hu antibodies is also possible. Superimposition of mixed axonal and demyelinating sensory motor neuropathy on sensory ganglionopathy can be found if the two antibodies are present together. Sensory ganglionopathy may also be associated with breast, prostate, colon cancers, lymphoma or uterine sarcomas. Symptoms improve when the primary malignant disease is treated. Immune modulatory therapies like IVIG, plasmapheresis and corticosteroids are of little help. Patients without a diagnosed malignancy should be followed up for 5 years [68].
Acute sensorimotor radiculoneuropathy occurs in patients with malignant diseases, especially in patients with lymphoma and solid tumours. Symptoms and signs of this disorder are identical to Guillain-Barré syndrome (GBS), and it is treated as GBS. The neuropathy can appear at any time during the course of the disease and may be a warning of relapse [67, 68].
Chronic sensorimotor neuropathy occurs in patients with a malignant disorder without any other identifiable cause. Patients are usually at a late stage of disease. Paraneoplastic antibodies are not present, and the onset is slow and not disabling. In the presence of paraneoplastic antibodies like CRMP5, neuropathy is progressive and disabling.
Neuropathies associated with lymphoproliferative disorders are associated with elevated paraprotein levels. These disorders are found in patients with multiple myeloma, POEMS and Waldenström macroglobulinaemia. The POEMS acronym stands for polyneuropathy, organomegaly, endocrinopathy, M protein, polyneuropathy and skin changes. Multiple myeloma-associated neuropathy is also associated with immunoglobulin light chain (AL) amyloidosis or POEMS and is axonal and demyelinating in nature. If neuropathy is the initial symptom, it may cause a misdiagnosis of CIDP. POEMS may be associated with multicentric Castleman disease. Waldenström macroglobulinaemia may be associated with neuropathy during the initial phase of disease, and antibodies against myelin-associated glycoprotein (MAG), GM1 gangliosid or asialo-GM1 ganglioside can be found in the sera of patients [70].
When it appears as a paraneoplastic syndrome, autonomic neuropathy is mostly associated with SCLC. Other malignancies associated with autonomic neuropathy are carcinomas of the pancreas, thyroid and rectum; Hodgkin’s lymphoma (HL) and carcinoid tumours of the lung. Anti-Hu, anti-CRMP5 and antibodies against ganglionic acetylcholine receptors (AChR) may be found in the sera of the patients.
Vasculitic neuropathy presents as mononeuropathy multiplex or in some cases proximal neuropathy. Nerve biopsy is useful for diagnosis and differential diagnosis. A diagnosis of vasculitis may predate a diagnosis of cancer. The associated malignant disorders include lymphomas and cancers of the lungs, prostate and endometrium [24, 67].
Checkpoint inhibitor-associated neuropathies are possible adverse events of these medications. Combined therapies (ipilimumab and nivolumab) increase the occurrence of adverse events. GBS like syndromes, cranial neuropathy, non-length-dependent neuropathy and autonomic involvement are commonly reported neuropathies. Meningitis may be accompanied with cranial neuropathy. Small fibre neuropathy with antineutrophil cytoplasmic antibody (ANCA)-related mononeuropathy multiplex, neuralgic amyotrophy and some other types may also occur. Mostly axonal neuropathy-compatible changes are revealed by electrophysiological examinations.
Paraneoplastic neuromyotonia, also called Isaacs syndrome, is due to peripheral nerve hyperexcitability. Muscle cramps, stiffness and increased sweating are the usual symptoms. EMGs reveal complex repetitive discharges and continuous muscle activity [67]. Findings of sensorimotor polyneuropathy are also observed. In patients with Isaacs syndrome, an underlying thymoma, SCLC and HL should be searched for. If central nervous system symptoms occur, it is called as Morvan syndrome.
Immune-Mediated Neuropathies
Multifocal motor neuropathy (MMN) is an immune-mediated and acquired disorder of peripheral nerves, which is defined by asymmetric motor involvement without sensory deficits [71]. Nerve conduction studies show conduction blocks in motor nerves, hence why it is also called multifocal motor neuropathy with conduction blocks. The worldwide prevalence is estimated to be approximately 0.6–2 per 100,000, and males are affected more than females (2.7:1). Age at diagnosis is usually between 30 and 50 years; however, younger and older patients have been reported [72]. Anti-GM1 antibodies, a class of IgM, are frequently found in sera of MMN patients, which provides an explanation for motor nerve involvement. Motor nerve myelin contains a high amount of GM1, especially around the Ranvier nodes and in comparison to sensory nerves [73]. Initially, muscle weakness affects one of the upper limbs and spreads to the contralateral limb, which is followed by lower limb involvement. Brachial involvement is common. Some nerves are severely affected, while others are totally spared or minimally involved. Dropped wrist and weakness of the hand or distal foot muscles are common symptoms. Atrophy can be observed, but it is not well correlated with weakness. Progression is expected in both treated and untreated patients, but the level of progression may vary between individuals [74]. Sensory findings are usually not present but the vibration sense can be minimally impaired. Electrophysiological examinations show motor neuropathy with conduction blocks and normal sensory NCVs. However, in some patients, conduction block cannot be shown, probably due to the proximal location of the blocks. Proximal stimulation of nerves (Erb points), F-wave studies and examinations of the asymptomatic nerves must be performed. Previously, nerve conduction blocks were thought to be caused by severe demyelination, but accumulated data suggest that anti-GM1 antibodies are responsible for the blocks and reduced motor NCVs. The functions of sodium and potassium channels, located around Ranvier nodes, are disrupted by anti-GM1 antibodies, resulting in the failure of action potential propagation. Therefore, MMN can be considered part of the nodo-paranodopathy group of neuropathies. However, there is no consensus regarding the function of anti-GM1 antibodies in the pathogenesis of MMN [74].
Radiological examination with MRI and ultrasound can be performed, but there are no certain criteria for the diagnosis of MMN. Diagnosis is made on clinical grounds and electrodiagnostic parameters. A diagnostic criteria for MMN has been proposed by the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) [75].
The two core criteria for MMN (both must be present) are the following:
-
1.
Slowly progressive or stepwise progressive, focal, asymmetric limb weakness, that is, motor involvement in the motor nerve distribution of at least two nerves for more than 1 month. If symptoms and signs are present only in the distribution of one nerve, only a possible diagnosis can be made.
-
2.
No objective sensory abnormalities except for minor vibration sense abnormalities in the lower limbs.
The supporting criteria and exclusion criteria are also defined. The differential diagnosis is important for treatment planning and predicting prognosis (Table 10.6). The main concern is to distinguish MMN from amyotrophic lateral sclerosis (ALS). The absence of pyramidal signs and bulbar involvement in MMN are important clues. The progression of MMN is relatively slower than ALS. Flail leg syndrome (FLS) is an atypical variant of amyotrophic lateral sclerosis (ALS) characterised by progressive weakness and atrophy of lower limbs alone. Contrarily, flail arm syndrome (FAS), an atypical presentation of ALS, is characterised by upper limbs involvement [75]. The pure motor variant of CIDP is also an important disorder to consider for the differential diagnosis because this variant also has conduction block. CSF findings may help to exclude a variant of CIDP. Treatment of MMN with IVIG is effective. Patients with impairments affecting activities of daily living must be treated as soon as the diagnosis made. Plasmapheresis and corticosteroid treatment are not recommended because the worsening of symptoms has been reported [73].
Inflammatory Neuropathies
Inflammatory neuropathies are members of autoimmune peripheral nerve disorders. The temporal course of these disorders can be acute or chronic. The exact aetiology or pathophysiological mechanisms are unknown; however, it is widely agreed that an aberrant immune response causes demyelination and axonal degeneration. This abnormal immune response involves both humoral and cellular elements of immune system. Understanding of the underlying etiological and pathophysiological mechanisms is essential to choose appropriate treatments and estimate the prognosis. Acute forms of immune neuropathies are grouped under the Guillain-Barré syndrome (GBS), whereas chronic forms constitute chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a rare disease with a reported prevalence of 0.67–10.6 cases per 100,000 people [76]. A meta-analysis reported an estimated incidence of 0.33 per 100,000 and prevalence of 2.81 per 100,000 in 2019 [77]. The discrepancy between reported prevalence rates is probably due to the use of different diagnostic criteria [78]. The risk of CIDP increases with age and males are more likely to be affected than females. No risk factors for CIDP have not been reported yet. CIDP is an autoimmune demyelinating disorder which affects nerve roots and peripheral nerves. Large, myelinated fibres are most severely involved. The clinical course may be monophasic, chronic progressive or relapsing remitting [77]. The symptoms usually show a progressive course for more than 8 weeks. Muscle weakness, sensory loss and areflexia are the characteristic findings. Severe postural tremors may be an accompanying feature that can be resistant to CIDP treatment. Both proximal and distal parts of the limbs are affected symmetrically. Distal muscle weakness is more prominent; however, the presence of proximal weakness is a useful clue for CIDP diagnosis. Patients often have difficulty walking and performing usual activities of daily living. Cranial and respiratory muscle involvement has been reported but is very rare in comparison to GBS. Systemic symptoms such as fever, weight loss, autonomic dysfunction and severe pain are not expected in typical CIDP patients. The presence of these symptoms should suggest other possible diagnoses. A diagnosis of CIDP requires the exclusion of other possible aetiologies.
Diagnosis is based on history, clinical findings, electrophysiological examinations and nerve pathology because there is no available diagnostic marker yet. Different diagnostic criteria have been proposed, but the definition of atypical forms and CIDP-mimics complicates the already complicated diagnosis [77, 79]. Identification of immunoglobulin G4 (IgG4) antibodies in some patients with CIDP-like clinical findings is a notable advancement. Patients with IgG4 antibodies against nodal and paranodal proteins usually do not respond to standard CIDP therapies, but rituximab is an effective treatment [76, 80].
The typical CIDP phenotype has sensory-motor peripheral nerve involvement with a progressive course for longer than 8 weeks. It has a symmetric distribution and is predominantly proximal. At least 50% of patients are in this group. A careful evaluation should be performed to exclude more common types of sensory-motor neuropathies, such as paraproteinemic and hereditary neuropathies. As opposed to the slow progressing group of patients, around 18% of patients are known as acute onset A-CIDP cases. The distinct features of A-CIDP are acute onset of the disease and rapid progression that reaches maximal involvement before 8 weeks. It is also symmetrically distributed but with proximal and distal involvement. The course of A-CIDP is either with relapses or slow progressive. A-CIDP should be differentiated from subacute inflammatory demyelinating polyneuropathy and acute inflammatory demyelinating neuropathy, which have overlapping features. Electrophysiological examination is needed for CIDP diagnosis and differential diagnosis. Electrophysiological findings of demyelination are slowed conduction velocities, long distal sensory and motor latencies, partial conduction block in motor nerves (Fig. 10.6) and increased temporal dispersion. F-response and blink reflex latencies may also be prolonged. Sural saving and more prominent involvement of upper limb sensory nerves are characteristic findings for both acute and chronic acquired demyelinating neuropathies. The only needle EMG finding may be decreased recruitment activity due to slow conduction velocity or conduction block. In the chronic phase, due to secondary axonal degeneration, positive sharp waves and fibrillation potentials appear, as well as reinnervation potentials such as large polyphasic motor unit potentials. Strict criteria are difficult to achieve, and in some patients, when clinical findings strongly indicate CIDP diagnosis, electrophysiological findings are insufficient to fulfil the defined criteria [81]. When standard methods do not reveal the expected abnormalities, magnetic or electrical stimulation of nerve roots might yield rewarding results [82]. Magnetic resonance imaging of the proximal parts of nerves, namely, roots and plexuses, are useful to show thickening and enlargement of the affected structures, especially in the treatment of new patients. Contrast enhancements of the roots are informative. However, the MRI technique is difficult, time-consuming, expensive and sometimes unavailable. Instead of MRI, ultrasound examination can be used to show the t proximal median nerve thickening and brachial plexus enlargement [21, 22]. Nerve biopsy is not required when the presentation is one of typical CIDP. However, nerve biopsy should be considered in rapidly progressive or treatment-refractory neuropathy, in multifocal cases, or when vasculitis, amyloidosis or a neoplastic process is suspected. Both forms of typical CIDP benefit from IVIG, corticosteroids and plasma exchange. Subcutaneous immunoglobulin treatment is also approved for CIDP treatment by the FDA.

Motor conduction block in an ulnar nerve and common peroneal nerve. Ulnar nerve conduction velocity: 27.6 m/s and 38.4 m/s; peroneal nerve conduction velocity: 27.4 m/s
CIDP Variants
The European Academy of Neurology/Peripheral Nerve Society published a revision of the 2010 guidelines for the diagnosis and treatment of CIDP. The use of the term ‘CIDP variant’ is proposed instead of the term ‘atypical CIDP’. CIDP variants consist of distal CIDP, multifocal CIDP, focal CIDP, motor CIDP and sensory CIDP [79].
Distal CIDP, also called distal acquired demyelinating symmetric neuropathy (DADS), is a CIDP variant which often presents with weakness and sensory deficits which are symmetrical and more pronounced in the distal parts of limbs [79]. Gait instability due to ataxia and action tremor are other symptoms of distal CIDP. Nerve conduction studies show disproportionate prolongation of distal motor latencies compared to nerve conduction slowing. The terminal latency index is equal to or less than 0.25 in motor nerves. A lack of conduction block and absent sural nerve sensory potentials are noteworthy differences [77]. In this group of patients, monoclonal gammopathy of undetermined significance (MGUS) may be present with IgA, IgG or, more commonly, IgM isotypes of immunoglobulins. Around 50% of patients with IgM monoclonal gammopathy have antibodies against myelin-associated glycoprotein (MAG) [76]. It is suggested that DADS with anti-MAG antibodies lies outside of the CIDP spectrum as there is no response to IVIG, corticosteroids or plasma exchange. It is reported that around 50% of anti-MAG positive patients are responsive to rituximab in some series [83]. Monoclonal gammopathy may be associated with a haematological malignancy in some patients with distal symmetric polyneuropathy [76]. Distal CIDP patients without anti-MAG antibodies have a more favourable response to classical CIDP treatments [76, 79, 84].
Multifocal CIDP is also known as many other names such as Lewis-Sumner syndrome, multifocal acquired demyelinating sensory and motor neuropathy (MADSAM) and multifocal demyelinating neuropathy with persistent conduction block [79]. In their original article, Lewis and Sumner reported the findings of five cases with asymmetric sensorimotor neuropathy having a chronic course [84]. Findings were more prominent in the upper limbs with focal nerve involvement. Electrophysiologic manifestations of persistent multifocal conduction block were present. Histopathological examinations of sural nerve biopsies revealed primary demyelinating disorders. Two of five patients had subacute optic neuritis. Two patients were treated with prednisone. Those patients showed improvement with the corticosteroid; however they relapsed when doses were reduced. Untreated patients were reported to have progressed. They suggested that chronic multifocal demyelinating neuropathy with persistent conduction block seems to be a variant of chronic acquired demyelinating polyneuropathy and it may be immunologically mediated [84]. Furthermore, 16 patients with similar characteristics were later reported, and beneficial effects of IVIG treatment were noted. The term MADSAM was proposed to point out the differences between multifocal motor neuropathies. The definition of multifocal CIDP variant is almost the same except for the name in the European Academy of Neurology/Peripheral Nerve Society guideline on the diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy [85].
Focal CIDP is rare and usually affects the brachial or lumbosacral plexus but can affect individual peripheral nerves as well [85].
Motor CIDP affects motor nerves in a rather symmetrical fashion with proximal and distal weakness mostly in young males. Clinical and electrophysiological examination show no sensory abnormalities. If two or more nerves have low sensory nerve action potential amplitudes in electrophysiology studies while clinical findings suggest motor CIDP, then it is called motor-predominant CIDP. The most common electrophysiology findings are F-wave abnormalities and conduction blocks. IVIG treatment has beneficial effects, while corticosteroid treatment in motor CIDP is a controversial subject. Patients with motor CIDP may get worse with corticosteroid therapy, but motor predominant cases may benefit. On the other hand, some reports suggest using corticosteroids in IVIG-resistant cases [86].
Sensory CIDP is characterised by the damage of sensory fibres which results in an impairment of the vibration, position and cutaneous senses. An ataxic gait is observed due to the loss of proprioceptive senses. As the name implies, muscle weakness is not an expected finding in sensory CIDP [85]. However, in some cases, motor nerve conduction studies reveal slow velocity and a conduction block. The term sensory-dominant CIDP is used for this type. Treatment with IVIG and corticosteroids yields results similar to typical CIDP [87, 88].
Disorders Not Classified as CIDP
Patients who have clinically suspected CIDP with normal nerve conduction examinations may have chronic immune sensory polyradiculopathy (CISP). When the dorsal spinal roots that are proximal to the dorsal root ganglia are damaged, neurons in the dorsal root ganglia remain intact [85]. Therefore, sensory nerve conduction velocity and SNAPs remain normal. Patients who meet the criteria for CIDP may have IgG4 antibodies against nodal NF140/186 and paranodal proteins such as neurofascin isoforms, neurofascin-155 (NF155), contactin-1 (CNTN1) and contactin-associated protein 1 (CASPR1) [74, 79]. The organisation of myelinated axons allows the transmission of impulses in a saltatory fashion, saving energy. Ranvier nodes with paranodal regions are an important part of this organisation. The site of damage in nodal and paranodal neuropathies is at the paranodal section of myelinated axons [74]. Due to the absence of inflammation and true demyelination, the group with IgG4 antibodies is excluded from CIDP classification [74, 79]. Patients who have antibodies often present with specific clinical features. Corticosteroids have a partial effect, while response to IVIG is poor. Treatment with rituximab and plasma exchange appear to be more effective [74, 89]. Different pathophysiological mechanisms may underly the different responses to treatments [74, 79, 87]. The term autoimmune nodopathies is proposed for these conditions [85]. Nodal and paranodal antibodies are also mentioned in the acute inflammatory neuropathies section.
CIDP Diagnosis
There are numerous proposed diagnostic criteria for CIDP [90]. The European Academy of Neurology/Peripheral Nerve Society published a guideline on the diagnosis and treatment of CIDP in 2021. This guideline is a comprehensive and useful tool for the diagnosis, classification and treatment of CIDP [79]. The complexity of neuropathic disorders forces clinicians to use the guidelines. In this section, diagnostic parameters are briefly discussed. Clinical, electrodiagnostic and supportive criteria are defined as well as criteria for immunological testing in patients with clinically suspected CIDP. A recent proposal suggests clinically classifying CIDP as typical CIDP and CIDP variants. Electrodiagnostic certainty of CIDP is defined as CIDP and possible CIDP. Previously defined definite and probable CIDP criteria are found to be neither sensitive nor specific enough to differentiate the two types [79, 81]. Clinical diagnosis of typical CIDP requires the presence of all the symptoms listed which includes distal and proximal involvement of both the upper and lower limbs in progressive or relapsing forms, in addition to sensory impairment in at least two limbs, symptom evolution for at least 8 weeks and absence or reduction of tendon reflexes in all limbs [79]. Electrophysiological findings of demyelinating lesions include motor conduction velocity slowing, increased distal motor latency, increased F wave latency, partial motor conduction block, increased distal compound muscle activity potential duration and reduced SNAP amplitude/increased latency SNAP/decreased sensory nerve conduction velocity. Different combinations of these abnormalities, out of certain defined limits, are sought during diagnosis. Supportive criteria include CSF analysis, nerve biopsy and imaging of the nerve’s radices using MRI and ultrasound techniques [77, 79].
Differential Diagnosis
Clinical and electrophysiological findings of CIDP may be similar to findings of genetic neuropathies such as CMT type 1 or transthyretin familial amyloid polyneuropathy. The haematological malignancies, diabetes mellitus and recently described nodopathies and paranodopathies are also important diagnoses which should be excluded to achieve an optimum treatment response. Vitamin B toxicity and chemotherapeutic agents may cause CIDP-like features, and so exposure to them should be questioned during history taking. CSF examination results with elevated protein levels and a normal leucocyte count support CIDP diagnosis. Radiological evidence of enlarged nerves and nerve roots are also helpful findings (Table 10.7). In the presence of atypical findings for CIDP such as prominent pain, tremor, autonomic dysfunction ataxia or muscle atrophy at the onset of disease, painless injuries and respiratory muscle involvement CIDP diagnosis should be reevaluated [91].
Treatment
The aim of treatment for CIDP is to stop the immune attack of peripheral nerves which causes demyelination and secondary axonal degeneration. If early and effective treatments are administered, symptoms and long-term prognosis improve. Treatment decisions are made according to symptom severity and disease course. The presence of disabling symptoms which affects quality of life is a predictive factor for treatment. Severe symptoms and findings are difficult to manage. Switches between the treatment types and combination therapies may be required before reaching an optimum treatment point. After symptoms stabilise, the target of treatment is then sustained improvement and further remission. In difficult cases, referral to a neuromuscular specialist may be beneficial for treatment optimisation. As a first step, corticosteroids and IVIG treatment are considered for typical CIDP and CIDP variants. Plasma exchange is also effective; however it is more difficult to administer and requires an intensive care unit. Patients may not tolerate this treatment. Subcutaneous immunoglobulin treatment is approved for maintenance therapy by the FDA. Additionally, some immunosuppressive drugs may be used; however there is no evidence-based information detailing the appropriate drugs or doses that can be used. Symptomatic treatment for pain and motor impairments are also important factors for improving quality of life. Each treatment has its own pros and cons. Both availability and patients’ characteristics are important factors that influence the choice of treatment. Treatments can be evaluated as induction therapies and maintenance therapies. Glucocorticoids are more effective at accomplishing long-term remission. However, there is not yet a consensus about the best corticosteroid regimen [79, 92]. Other treatments like azathioprine, mycophenolate mofetil and ciclosporin are suggested as IVIG or glucocorticoid sparing agents for maintenance treatment. Patients who are resistant to the abovementioned proven effective treatments can be treated with rituximab, ciclosporin or cyclophosphamide [79, 92, 93].
The prognosis of CIDP is variable. Most patients respond to any of the standard treatments; however 10–15% of patients are refractory to IVIG, glucocorticoids and plasma exchange. The cure or remission can be accomplished in about 30% of patients [94].
Guillain-Barré Syndrome
Guillain-Barré syndrome (GBS) was first described by Guillain, Barré and Strohl in 1916. Previously, Landry published an article regarding ascending paralysis but did not mention the absence of tendon reflexes and albuminocytologic dissociation. GBS is the most frequent reason of acute flaccid paralysis. Most epidemiological information is gathered from developed countries, and therefore, data from other parts of the world are lacking. GBS has a range of incidence of 1.81–1.91/100,000 person years. Incidence increases with age, and males are more susceptible than females (M/F = 1.5). Usually, an antecedent infection is reported before acute limb weakness develops which may progress for up to 4 weeks until reaching a plateau phase. Sensory symptoms may precede motor symptoms; however electrophysiological evidence of sensory involvement is less than that of motor findings. Weakness usually follows an ascending route, but it can start in the proximal muscles. The need for mechanical ventilation arises in approximately 25–30% of patients due to respiratory muscle weakness [95,96,97]. The most common infection is gastroenteritis due to C. jejuni. Investigations disclosed molecular mimicry occurring between nerve structures and antigens of bacteria in C. jejuni-related GBS. Zika virus-associated GBS and recently described severe acute respiratory syndrome corona virus-2 (SARS-CoV-2)-related GBS cases are also reported. Determining a clinical diagnosis of GBS is not difficult when classical findings are present, which consist of ascending paralysis and areflexia, and additionally albuminocytologic dissociation is present with an infection history of 2 weeks prior. Although historically, GBS is a single disorder, but now several variant forms have been reported (Table 10.8). The presence of variant forms and overlap between different forms causes a diagnostic challenge. Treatment with IVIG and plasma exchange improves the prognosis of GBS, and rates of 5% mortality and 20% severe disability are reported. Therefore, it is important to recognise the large spectrum of clinical patterns which constitute GBS for correct and timely diagnosis.
Variant Forms of Guillain-Barré Syndrome
Acute inflammatory demyelinating polyneuropathy (AIDP) is the most frequently seen form of GBS which constitutes around 85–90% of cases. During the Zika epidemic, an increased incidence of AIDP was observed in French Polynesia and South America. Even though no causative antibodies have been identified in infection-related AIDP, molecular mimicry between the viruses and human proteins is suggested in recent studies as an underlying mechanism [98]. Classically, AIDP presents with progressive, ascending and symmetrical muscle weakness associated with reduced or absent deep tendon reflexes. Electrophysiological evidence of demyelination such as slow NCV, prolonged distal motor latency and partial conduction block are characteristic findings. However, in the early phase, an absence of F waves or the H-reflex may be the only pathological findings. When peripheral nerve examinations are normal, lumbar root stimulation is a useful method to identify proximal demyelinating lesions which cannot be demonstrated with standard nerve conduction studies [82]. Reduced recruitment activity may be the sole needle EMG finding in the early phase. If secondary axonal degeneration occurs, spontaneous activity with reduced recruitment is observed.
Acute axonal neuropathies consist of acute motor axonal neuropathy (AMAN) and acute motor and sensory axonal neuropathy (AMSAN). These primary axonal forms are often encountered in China, Japan and Mexico. In Western countries, axonal forms comprise approximately 5–10% of GBS cases [95, 99]. AMAN was first reported in 1986. AMAN particularly affects young people following C. jejuni infection. As the name AMAN implies, in this axonal form of GBS, motor nerves are affected, while sensory nerves remain intact. In some patients, deep tendon reflexes may be spared or even increased. An electrophysiological study demonstrated motor neuron excitability due to dysfunction of the intraneuronal inhibitory circuits in the spinal cord. The proposed mechanism of the dysfunction is immune-mediated damage of the spinal inhibitory intraneuronal network [100]. This finding is associated with C. jejuni infection [101]. Although, an absence or decrease of deep tendon reflexes is required for GBS diagnosis, this group of patients may be an exception. Hyperreflexia can be seen in patients with AMAN which is possibly a nodopathy [101]. Electrophysiological findings of AMAN include decreased compound muscle action potentials (CMAPs) with slightly slow or normal motor NCVs. Needle EMG shows reduced recruitment patterns and spontaneous activity approximately 3 weeks after disease onset. Antibodies against the gangliosides GM1, GD1a, GalNac-GD1a and GD1b are associated with the development of AMAN. Molecular mimicry may be the underlying mechanism responsible for the production of antibodies against gangliosides. Antibodies and complement activity cause axonal damage with variable severity or reversible conduction block at the nodal/paranodal parts of the nodes of Ranvier. Both abnormalities can be present on the same nerve. In some patients, conduction block can be resolved rapidly without causing axonal damage. This temporary conduction block is called reversible conduction failure (RCF) which possibly occurs due to loss of voltage-gated sodium channels. RCF is a different condition than classical demyelinating conduction block. It is a part of a nodo-/paranodopathy concept [74].
AMSAN affects both sensory and motor nerves with prominent axonal degeneration. Clinically, AMSAN is like AMAN; however, it is more severe and has a delayed and incomplete recovery with sensory involvement. GM1, GD1a, GalNac-GD1a and GD1b antibodies are involved in the pathophysiological process. Electrophysiological investigations demonstrate axonal damage with low or absent CMAPs and SNAP amplitudes. NCVs are slightly low or near normal. Due to axonal degeneration, active denervation potentials and reduced recruitment activity are demonstrated by needle EMG examination [95].
GQ1b syndromes are forms of GBS which are frequently associated with antibodies against GQ1b. The external ocular muscles, muscle spindles and most likely the reticular formation in the brainstem contains GQ1b antigens. In susceptible patients, infection with microorganisms with GQ1b epitopes may stimulate anti-GQ1b antibodies which result in a continuous spectrum. Miller Fisher syndrome (MFS), Bickerstaff brainstem encephalitis (BBE) and pharyngeal-cervical-brachial (PCB) variants are included in this spectrum [102]. Haemophilus influenzae and C. jejuni infection is frequent before GQ1b syndromes. It is reported that MFS and BBE relapse more frequently than other forms of GBS [103]. Relapsing MFS may be associated with HLADR2 positivity [104]. MFS is characterised by a triad of ophthalmoplegia, ataxia and areflexia which was first described by Fisher in three male patients in 1956 [105]. MFS is more frequent in Asia and occurs in around 20% of the cases in Asia and in 5–10% of the cases in the United States and Europe. In addition to the characteristic triad, approximately 25% of cases will have some limb weakness during the disease. Autonomic involvement of the pupil is present, with fixed dilated pupils in some patients. Incomplete forms are reported such as ophthalmoplegia without ataxia, ophthalmoplegia and ptosis or ataxia without ophthalmoplegia [106]. Unilateral ophthalmoplegia is also a reported finding. Approximately 80–90% of patients have GQ1b antibodies. Electrophysiological examinations may demonstrate absent SNAPs or reduced SNAP amplitudes without slowing of sensory NCVs which indicates sensory neuronopathy. Motor NCVs are normal in patients without paresis, but abnormalities compatible with axonal involvement can be found in patients with muscle weakness [104]. MFS prognosis is quite favourable in general since it is usually self-limiting and patients recover fully by 6 months [107].
BBE presents with ataxia, ophthalmoplegia and encephalopathy with GQ1b positivity. Altered consciousness in patients with BBE suggests involvement of the ascendant reticular-activating system. The blood-brain barrier (BBB) prevents the entry of large molecules into the brain parenchyma. However, there are some areas like the area postrema where the BBB is relatively more permeable for large molecules. Even though it has not been proven experimentally, there is a possibility that anti-GQ1b antibodies travel through the BBB at this site. In addition, facial weakness, bulbar symptoms and pupillary abnormalities can be found. According to a review of 53 BBE patients, mild limb weakness was found almost in half of the patients, and deep tendon reflexes were normal or brisk in approximately 40% of the patients. The treatment of BBE is like that of classical GBS including IVIG and plasmapheresis. Rare variants are also reported such as paraparesis, acute pandysautonomia, pure sensory GBS, facial diplegia and distal limb paraesthesia and acute bulbar palsy [95, 96, 108].
Differential diagnosis is relatively easy when characteristic findings are present. However, a long list of other diagnoses must be considered depending on the subtypes, patient characteristics and geographical localisation. Spinal cord and cranium imaging help to differentiate central nervous system disorders. Electrodiagnostic studies are used for excluding acute myopathies, neuromuscular junction diseases, anterior horn diseases and polyradiculopathic conditions. A change in the state of consciousness is not an expected finding in GBS, except for BBE. A severe neurological deficit that persists without a significant recovery or relapsing pattern suggests another diagnosis other than GBS. On the other hand, sural sparing is in favour of GBS and helps to discriminate GBS from other neuropathies [109, 110]. Treatment with IVIG and plasmapheresis is effective, and studies show no significant difference regarding their effectiveness. Patient characteristics and availability are the factors which affect the choice of treatment [111].
All About the Pathology of Neuropathies
Neuropathy Muscle Biopsy Findings
Muscle biopsy is a method that is used more frequently than nerve biopsy because it is easier to sample and has much less morbidity. Especially in hereditary neuropathies, diagnosing a significant proportion of the cases with genetic examination has greatly reduced the need for nerve biopsy examination. During the course of many diseases that can cause neuropathy, particularly diabetes or systemic vasculitis, nerve biopsy is not often considered necessary. However, for certain diseases, especially for localised vasculitis in the nerves, nerve biopsy examination may be deemed essential. Simultaneous muscle biopsy is recommended during nerve biopsy examination. Muscle biopsy examination is also not uncommon for the differential diagnosis of myopathy in some cases where muscle or nerve involvement cannot be clearly differentiated. Regardless of the indication, it is possible to diagnose any disease that affects nerves from a muscle biopsy specimen because maintaining the nerve supply is directly related to the health of the muscle fibres. The detrimental dysvoluminal alterations in muscle fibres will result from any disruption of these neurotrophic influences. Neuropathies affect the muscles innervated by the involved nerves, and some of these changes are pathognomonic for neuropathies. These myopathies due to neurogenic diseases are generally classified as neurogenic myopathies or neurogenic (denervation) atrophy of the muscle. Neurogenic atrophy may stem from diseases of the anterior horn cell or its myelinated axons [112,113,114].
The pathological effects of denervation are generally the same in almost all neurogenic atrophies except in SMA type 1. One of the most important diagnostic findings of neurogenic myopathies is the unusual clustering of cell nuclei called nuclear clumping. The presence of nuclear clumping alone in a muscle biopsy suggests a disease of neurogenic origin (Fig. 10.7). Except for small clusters of specialised nuclei at the neuromuscular junction, each skeletal muscle fibre includes hundreds of nuclei just below the plasma membrane scattered along the fibre. Like all other cell types, skeletal muscle fibres have dynamically controlled nuclear positions in both space and time. Nuclear movements are mediated by the cytoskeleton, which transfers pushing or pulling forces onto the nuclear membrane. The cytoskeleton plays a major role in this activity, and various molecular connections between the nuclear membrane and cytoplasmic components such as actin and other intermediary filaments have been found for various cell types. The mechanisms governing nuclear localisation inside cells have been partly identified in recent studies. However, it is not yet understood exactly how nuclei arrive at positions along fibres of skeletal muscle. Recent studies show that nuclei preferentially localise near blood vessels, especially in slow-twitch-oxidised fibres. In addition, it has been determined that desmin deficiency significantly alters the distribution of nuclei along fibres but does not prevent their close association with vessels. Consistent with the role of desmin in nuclear spacing, denervation has been shown to affect desmin filament organisation and nuclear distribution [115,116,117].

Nuclear clumping in neurogenic myopathy (H&E × 100)
Denervation is an important cause of nuclear misplacement. During permanent denervation, up to the 90% of severely atrophic muscle fibres present a reorganisation of their nuclear distribution. The permanent injury of lower motor neurons causes skeletal muscle fibre atrophy which mainly occurs during the first few weeks or months following injury. In the later phases of denervation, muscle fibre atrophy consistently progresses. Still, many severely atrophied muscle fibres remain present in the denervated muscle at this late stage, some of which have lost all contractile proteins and the helical distribution of myonuclei, which are aggregated in the core of the muscle fibre (nuclear clumps). Adipocytes and collagen sheets fill the empty areas of muscle tissues, and finally fibrosis replaces muscle fibres (Fig. 10.8). Nuclear clumping, a pathognomonic change due to denervation, becomes increasingly evident, especially in the later stages of denervation. In the first months, or more so in the later months of the first year, there may be a mild myopathic appearance during the histopathological examination of muscle [115].

Fibrosis in a later phase of neurogenic myopathy (Masson’s trichrome × 200)
In cross-sections of muscle biopsy, normal muscle cells are polygonal in shape and similar in size. Each muscle fibre is surrounded by a very thin layer of connective tissue called the endomysium that cannot be discriminated without special connective tissue staining such as trichrome staining. Muscle fibres usually become irregular in size and shape during any neuromuscular disease. After repetitive injuries, the size and shape of the muscle fibres change. However, in the neuropathies, sizes of muscle fibres vary greatly. A few fibres remain as almost normal size, while most fibres are very small, with some fibres disappearing, leaving only the nuclear clumps behind [112,113,114].
Some atrophic fibres are angular in shape, and these angular fibres are very specific for neurogenic myopathies (Fig. 10.9). During acute denervation, atrophic fibres randomly scatter. At this stage, small fibres are flattened and angular, and most of them can be glycolytic in type (type 2). If the denervation continues, the population of type 1 and type 2 fibres becomes a more irregular mixture. Advanced denervation demonstrates a pattern of atrophy that progresses from a random distribution to a grouping of affected fibres. Rarely, some atrophic fibres appear as regenerated fibres with more basophilic cytoplasm and internally located nuclei (Fig. 10.10). In addition, there is no inflammatory infiltration like that of the muscular dystrophies. In the early phases of neuropathies, muscle biopsy findings are like that of a noninflammatory myopathy. In these phases, histopathological differential diagnosis may be impossible, as the differences muscle fibres’ sizes are minimal (Fig. 10.11), and there are no/scarce nuclear clumps or angulated fibres. On the other hand, advanced fibrosis observed in a muscle biopsy in late-stage neuropathies can be interpreted as the last stage of muscular dystrophy. In this situation, the occasional presence of muscle fibres with almost normal size is in favour of neuropathy. For example, a 23-year-old male patient, who started at 18 years of age and had proximal muscle weakness more prominent in the lower extremities, had creatine kinase levels around 4000 IU/L. Clinically, muscular dystrophy was suspected, and genetic testing was completed. For Becker muscular dystrophy and limb-girdle muscular dystrophies, there were no genetic abnormalities. In the neuromuscular disease panel, a heterozygous c.2263G > A (p.Glu755Lys) variant was detected in the kinesin family member 5A (KIF5A) gene localised on chromosome 12q.13, which was associated with spastic paraplegia type 10 and Charcot-Marie-Tooth type 2 diseases. The same variant was also present in the heterozygous form in his older brother, who had signs of proximal myopathy. The patient had been evaluated as having end-stage muscular dystrophy due to the presence of severe atrophy and interstitial fibrosis in the muscle biopsy (Fig. 10.12). However, when the biopsy was re-examined in the light of genetic tests, it was decided that it would be more accurate to evaluate the pathology as neurogenic muscle atrophy due to the presence of myofibers that remained relatively unchanged and almost normal in size (Fig. 10.13) [112,113,114].

Atrophic fibres with angular shape in neurogenic myopathy (H&E × 100)

Some atrophic fibres resemble regenerated fibres with more basophilic sarcoplasm (H&E × 200)

Mild myopathic appearance in a muscle biopsy of a 19-year-old male with vasculitic neuropathy (H&E × 200)

Small atrophic fibres between the adipose and fibrous tissues in a muscle biopsy of a 23-year-old male patient with KIF5A gene variation (H&E × 100)

There are normal-sized myofibers with atrophic ones in a patient with KIF5A gene variation (Gomori’s trichrome × 200)
However, angular-shaped atrophic myofibers are not seen in infantile denervation, and almost all muscle fibres are round. In these cases, the most pathognomonic appearance in the differential diagnosis is the presence of muscle cell groups composed of larger myofibers between small round atrophic muscle fibres. Immunohistochemical analysis of sarcolemmal proteins enables us to better select cell groups with significant size differences (Fig. 10.14). In summary, denervation atrophy of muscle fibres can be defined by the presence of atrophic fibres with an angular shape, significantly decreased myofiber size and markedly increased myofibrillary disorganisation highlighted with nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) staining (Fig. 10.15). The occurrence of target fibres due to myofibrillary disorganisation is suggestive of reinnervation [118]. The target fibres which have a central pallor area surrounded by an internal very darkly stained rim and external normal-stained sarcoplasm are seen in about 20–30% of cases with neurogenic myopathy. They are more common in peripheral neuropathies than in amyotrophic lateral sclerosis (ALS). Fibres which have a central pallor area without a darkly stained rim are referred to as targetoid fibres [112,113,114].

Myofiber groups consist of small or large round fibres which are specific for infantile denervation. Severe size differences of myofibers are highlighted by sarcolemmal merosin expression (DAB × 100)

Myofibrillary disorganisation is suggestive of reinnervation. (a) Target fibre, (b) spiral-like appearance in a fibre due to myofibrillary disorganisation
Neurogenic myopathies are generally seen in diseases affecting lower motor neurons or peripheral nerves. Before deciding whether to undertake a muscle biopsy, neurogenic disorders can be diagnosed by the presence of sensory deficiency, a distal-dominant distribution of muscle weakness and from the results of electrophysiological tests. In this situation, a muscle biopsy is not performed. However, it can be challenging to distinguish between neurogenic and myopathic weaknesses. During the early denervation phases, there is random atrophy of both fibre types. The atrophic angulated fibres can also be seen as type 1 or type 2 fibres with oxidative enzyme stains except for the NADH-TR because all the atrophied fibres, regardless of the type, are stained darkly like type 1 fibres by NADH-TR stain [113]. During peripheral neuropathies, type 2 fibres can be more atrophied, and atrophic fibres can become angulated in muscle biopsy. The most striking finding of neurogenic changes in muscle is fibre-type grouping, where the same type of muscle fibres group together (Fig. 10.16). Repeated cycles of denervation and reinnervation cause the grouping of atrophied myofibers in neuropathies (Fig. 10.17). Fibre-type grouping is present if more than nine consequent myofibers of the same fibre type are seen together (Fig. 10.18). The formation of groups of atrophied myofibers is one of the most specific changes of denervation. Examples include inherited or chronic polyneuropathies, like Charcot-Marie-Tooth disease, and motor neuron illnesses, like spinal muscular atrophy and amyotrophic lateral sclerosis. Due to reinnervation from the collateral sprouting of surviving axons after denervation, the typical checkerboard pattern of muscle fibre types is lost [114,115,116,117].

Schematic of the typical motor unit and three steps of denervation are shown

Myofiber grouping with fast myosin staining is suggestive of neurogenic myopathy (DAB × 100)

Myofiber grouping can be highlighted with enzyme staining (NADH-TR × 200)
If neurogenic diseases are chronic and long-standing, myofiber hypertrophy is also seen in the muscle biopsy. It is thought that these hypertrophic fibres represent a compensatory reaction in response to the inefficiency of the atrophied fibres. As this process is not type selective, hypertrophic fibres may be oxidative or glycolytic. The muscle may also exhibit fibre splitting and even necrotic fibres after long-standing denervation. This appearance may create a pseudo-myopathic image. Very little muscle may be seen in a biopsy when there is advanced neurogenic atrophy. The tiny, undetectable bundles of atrophic fibres may be found mingled with adipose tissue (Fig. 10.19). These advanced atrophic myofibers, which cannot be seen in the fibroadipose tissue, can be identified with special dyes (Fig. 10.20) [114,115,116,117,118,119].

Unnoticeable atrophic fibres among fibroadipose tissue (H&E × 100)

Atrophied fibres are highlighted with immunohistochemical staining with anti-alpha sarcoglycan antibody (DAB × 100)
Neuropathy Nerve Biopsy Findings
It is not uncommon for inflammatory processes which occur in peripheral nerves to cause difficulty in reaching a differential diagnosis regarding noninflammatory or hereditary neuropathies. This is particularly relevant if vasculitides are localised to peripheral nerves or if they are systemic, as this means reaching a differential diagnosis will not be possible without a nerve biopsy examination (Fig. 10.21). It has been reported that concomitant muscle biopsy examination increases the chance of diagnosing vasculitis, even with isolated peripheral nerve vasculitides because there may be damaged or inflamed vessels involved in the neighbouring muscles. On the other hand, the diagnosis of vasculitis cannot be made from time to time because the diagnostic necrotising vascular lesions are not observed homogeneously along the nerve (Fig. 10.22). A 19-year-old male patient, who presented with progressive, ascending, symmetrical muscle weakness associated with reduced or absent deep tendon reflexes, was thought to be at the early stages of Guillain-Barre syndrome. However, electrophysiological evidence of demyelination such as slow nerve conduction velocity, prolonged distal motor latency and partial conduction block was absent. As the course of the disease was atypical, nerve biopsy was also performed. There was no denervation or axonal degeneration in the biopsy. Severe vascular attack targeting sural nerve vessels, including endoneurial capillaries, were observed (Fig. 10.23). The clinician was informed, and emergency treatment was commenced. It should not be forgotten that vasculitis is a pathology of the peripheral nerves that requires urgent intervention. If vascular destruction is inhibited in the early period, axonal damage and demyelination secondary to ischaemia can be prevented or delayed. As an early diagnosis is very essential for the prevention of ischaemic axonal damage, searching for changes which suggest vasculitis is important. Such changes that supported the presence of neighbour vasculitides include luminal narrowing/thrombosis, intimal hyperplasia, disorganisation of the media, breakup of the elastic lamina, vessel sclerosis, recanalization, proliferation of epineurial capillaries and focal perineurial damage. Multifocal axonal degeneration is also a very important finding suggesting remote vasculitides. Identifying vascular immunoglobulin and complement deposition may also help to diagnosis vasculitis (Fig. 10.24). The presence of hemosiderin, due to old haemorrhages in nerves (Fig. 10.25), or focal calcification of vessel walls is important evidence suggesting neighbour vasculitides [120,121,122].

Perivascular inflammatory cells in the perineurium of a sural nerve biopsy taken for the differential diagnosis of peripheral neuropathy (HE × 200)

Similar perivascular inflammatory cuff of vessels in the perimysium of the adjacent muscle (H&E × 200)

(a) Vasculitis in the sural nerve of a 19-year-old adolescent which can be seen in the panoramic picture (H&E × 40). (b) A perineurial vessel is entirely infiltrated and damaged with inflammatory cells (H&E × 400)

Vascular deposits of immunoglobulin G, observed using direct immunofluorescent (DIF) examination, in a large epineurial vessel of a sural nerve (fluorescein × 200)

Hemosiderin deposition in the vascular wall is highlighted by Prussian blue (Perl’s Prussian blue × 200)
Nerve biopsy offers much more limited diagnostic clues compared to muscle biopsy. Furthermore, since the way in which genes cause hereditary neuropathies has been further understood and markers of immune system-mediated processes can be detected via blood tests, the indications for nerve biopsy have diminished. For neuropathies which develop in systemic vasculitis conditions such as polyarteritis nodosa (PAN) or Wegener’s granulomatosis (WG), for typical distal sensory neuropathies which develop in diabetic patients or for neuropathies which develop due to an infectious agent, the diagnosis is often confirmed using blood tests and electrophysiological studies; thus, the need for nerve biopsy is eliminated. However, nerve biopsy examination is still essential, especially in atypical and suspicious cases. Clinicians generally have three basic expectations from pathologists who examine a nerve biopsy which include grading the presence of inflammatory cells or vessel damage, grading any deficiency of myelin and grading any axon degeneration. The main question is whether basic pathology is in the production of myelin or in the axon. However, it should not be forgotten that in the advanced stages of demyelination, even in pure myelination defects, pathology is not limited to conduction defects due to the deterioration of the isolation of the axon, and the demyelinated axon also begins to degenerate and eventually disappear. Therefore, in an end-stage nerve disease, differentiating between primary myelinopathy and primary axonopathy may not be impossible.
Electron microscopic examination highlights endothelial cell necrosis and basal lamina disruption and identifies the presence of hypertrophic endothelial cells with prominent intraluminal projections which is a useful clue regarding vasculitis. Similarly, areas of focal axonal degeneration due to ischaemia can be observed in detail with electron microscopic examination. On the other hand, nonspecific axonal degenerative changes and consequent segmental demyelination can be seen with both light and electron microscopic examination. Although focal demyelination can be observed in neuropathy associated with necrotising vasculitis, focal axonal damage is often accompanied by ischaemia, unlike that seen in acute or chronic inflammatory polyneuropathies. With ischaemic injury due to vasculitis, all fibres may be affected, or unmyelinated fibres may be preserved. Detailed discrimination of fibre type in a nerve plexus cannot be ascertained with light microscopic examination. In summary, with electron microscopic examination of ultrathin sections and of semithin sections of resin blocks that can only be stained with toluidine blue, the state of vessels, myelin sheaths and axons can be examined in much more detail. In these examinations, it is even possible to tell which group of nerve fibres the pathology particularly affects. However, the most important part of the nerve biopsy examination relates to the presence of inflammation. For this reason, signs of vascular damage should be investigated first, and then inflammatory cells should be examined. As endoneurial lymphocytes resemble the nuclei of Schwann cells, immunohistochemical examination is required for the differential diagnosis. The majority of inflammatory cells in any vasculitis are T lymphocytes (95%) and macrophages. Depending on the severity and stage of the inflammatory process, varying numbers of neutrophils and eosinophils may also be present [121].
It is suggested that direct immunofluorescence (DIF) examination of combined peroneal nerve/peroneus brevis biopsies reveals immunoglobulin or complement deposits in epineural vessel walls in 70–80% of nerve biopsies in patients with suspected isolated peripheral nervous system (PNS) vasculitis and diabetic neuropathy. In summary, immunohistochemical and immunofluorescent examinations are very important for identifying the presence of inflammatory cells and their immune phenotype in a nerve biopsy, and these examinations can only be performed on frozen or paraffin tissue sections. For this reason, examination of paraffin and frozen sections using different staining methods is indispensable for nerve biopsy examination. Additionally, different histochemical and immunohistochemical examinations can also detail the status of myelin and axons (Fig. 10.26). Nerve dysfunction in vasculitis is assumed to occur based on ischaemia secondary to vessel destruction. Patients who have symmetrical sensorimotor neuropathy, which is an atypical finding for vasculitis, are likely to be affected by several unrelated, mild lesions that together will have a greater overall impact on the longest nerve fibres. Although segmental demyelination caused by ischaemia rarely presents as a conduction block in electrophysiological examinations, axonal destruction is always the dominant defect in vasculitides. The diameter range of epineurial arterioles is 75–350 μm, while that of perineurial and endoneurial arteries is much less than 75 μm. Most vasculitic processes are vessel size-specific, but the much more common injuries are due to disease of epineurial vessels of the peripheral nerves. Epineurial vascular involvement is typical of PAN, Churg-Strauss syndrome (CSS) and WG, while there is a tendency towards the involvement of small vessels of the epineurium in isolated PNS vasculitis. Involvement of veins is more common in WG and CSS than in PAN. Endoneurial vessels are usually <30 micrometres in size, and arterioles, venules and capillaries of this calibre, in both the endoneurium and epineurium, are the typical site of injury in systemic lupus erythematosus (SLE), hypersensitivity vasculitis, Henoch-Schoenlein purpura (HSP) and essential mixed cryoglobulinemia. Both small- and large-sized vessels may be involved in the collagen diseases. It must be noted that overlap syndromes, in which vessels of all sizes are involved, are not uncommon. Activated complement and immunoglobulin deposits are found in almost all inflamed vessel walls, regardless of the aetiology. Even though perivascular immune complexes are seen in vasculitis, they do not cause it; rather, they develop after a cell-mediated attack. Interaction with foreign antigen results in the activation of complement, the adhesion of neutrophils and other inflammatory cells to the blood vessels and the release of toxic substances that can cause vessel wall necrosis. Vasculitis likely involves a variety of disease and tissue-specific underlying mechanisms. Hepatitis B antigen found in PAN is an example of where a causal antigen source has been identified. Regardless of the underlying disease, activated T cells make up a significant portion of the inflammatory infiltrate. The observation that they are primarily of the CD8 subtype points to an important role for cytotoxic T-cell-mediated damage, possibly directed at a vascular antigen or an antigen presented by endothelial cells [120, 121].

Minimal myelin defect is highlighted using special stains in a patient with systemic lupus erythematosus. (a) Myelin is seen as red globes (modified trichrome × 100) and (b) immunohistochemical examination by myelin basic protein (DAB × 100)
Many infectious diseases such as acquired immunodeficiency syndrome (AIDS), leprosy, syphilis and Lyme disease can lead to peripheral neuropathy during their course. Different clinical manifestations such as distal symmetrical sensory polyneuropathy, acute or chronic inflammatory demyelinating polyneuropathy, human immunodeficiency virus (HIV)-associated mononeuropathy multiplex (MNM) syndromes, cytomegalovirus-associated lumbosacral polyradiculomyelopathy, autonomic neuropathy and even paraneoplastic neuropathy secondary to the developing lymphoproliferative disease may be encountered in patients with AIDS. In all infectious diseases, histopathological findings can be very variable, depending on the direct effect of the infectious agent or the reactions that develop due to the immune response. Among these agents, the most specific histological finding is perhaps the granulomas observed in leprosy. Similarly, the detection of granulomas in sarcoidosis nerve biopsies is a pathognomonic and diagnostic finding (Fig. 10.27). However, with infections that have peripheral nerve involvement, pathological findings may not be observed in biopsy specimens because these diseases do not cause diffuse involvement of the nerve, like the vasculitides. Therefore, today nerve biopsy examination is not performed in patients with neuropathy if diagnosed with a specific infectious disease [112,113,114, 119,120,121,122].

A granuloma with two giant cells between two nerve plexi in a patient with sarcoidosis (H&E × 40)
Amyloidosis is another cause of neuropathy involving primary axons. Amyloid storage is highly diagnostic if observed with nerve biopsy examination. It is one of the most well-known diseases associated with protein misfolding due to gain-of-toxic function. It is characterised by the build-up and aggregation of toxic and dysfunctional proteins that harm tissues and cells. Under optical and electron microscopes, amyloid appears amorphous and fibrillar, respectively, and is recognised as an extracellular proteinaceous material. When stained with Congo red and observed under polarised light, amyloid fibrils exhibit apple green-yellow birefringence, setting them apart from other protein aggregates. Three patterns of amyloid deposits can be seen in the peripheral nerves. Areas of amyloid accumulation, seen according to these patterns, are found in extraneural connective tissue, widely in the endoneurium (Fig. 10.28), or in the walls of vessels in the nerve. Amyloidosis is generally associated with paraproteinaemia, for example, multiple myeloma, or chronic inflammatory disorders such as familial Mediterranean fever (FMF). Stored materials in different tissues in these disorders are a part of special amyloid types such as light chain amyloid (AL) and serum A amyloid (AA), respectively. In addition, there are hereditary familial types of amyloidosis, and up until today several distinct familial amyloid polyneuropathies (FAP), all of which are autosomal dominant, have been described. Mutations of the transthyretin (TTR), apolipoprotein A1 (APOA1) and gelsolin (GSN) genes cause these disorders which can potentially fatal. Transthyretin (TTR), encoded on chromosome 18, is a serum transport protein previously called pre-albumin, which serves as a carrier for several substances, including thyroxine and vitamin A. Apolipoprotein A1, encoded on chromosome 11, is a plasma protein with an extensive α-helical structure synthesised by the liver and the small intestine. The neuropathic pattern of symptoms is associated with the Gly26Arg mutation. Gelsolin is an actin-modulating protein encoded on chromosome 9. An accumulation of fibril aggregates of amyloid precursor proteins in the peripheral nerves and other systemic organs results in early autonomic symptoms, unexplained cardiomyopathy, bilateral carpal tunnel syndrome and progressive course in patients with a family history. The predominant nerve degeneration in amyloidosis is axonal degeneration which involves smaller fibres. The predilection of damage to a special nerve area is very diagnostic for some disorders. Alcohol and diabetic neuropathies also predominantly affect the smaller nerve fibres, like amyloidosis, and they cause neuropathy of smaller nerves [2, 28, 114, 121].

Apple green-yellow birefringence localised to the nerve plexus is observed under polarised light in a patient with amyloid neuropathy (HE × 40)
In some biopsies, granular material accumulations consisting of ground substance, extracellular microfilaments and scarce fibroblasts are seen adjacent to the inner perineurium (Fig. 10.29). These cushion-like accumulations are called Renaut bodies. They appear as large, loosely spiralled, elongated connective tissue accumulations in somatic and occasionally autonomic nerves. They generally involve more than one nerve fascicule. They are not seen in foetal life and their numbers increase during age. It is suggested that repetitive trauma or pressure may play a role in their pathogenesis due to their presence at the entrapped site of nerves. The collection of fluid, which may also be slightly granular, all around and under the perineurium is called sub-perineurial oedema. It is better visualised with plastic or frozen sections than fixed sections, and it has scattered fibroblasts, mast cells and macrophages (Fig. 10.30). It may be stained with alcian blue because of the mucopolysaccharides it contains. Sub-perineurial oedema may be seen in different disorders such as thiamine deficiency, leprosy, ischaemic (atherosclerotic) diseases, vasculitides, immune-mediated/hereditary demyelinating diseases, lead toxicity and ipilimumab-associated neuropathy [119,120,121,122].

(a) Renaut bodies (arrows) in several plexi of a sural nerve biopsy (H&E × 40). (b) Close-up view of a Renaut body (H&E × 200)

(a) Sub-perineurial oedema in a sural nerve biopsy (Gomori’s trichrome × 400). (b) Sub-perineurial oedema can be seen in several nerve plexi of a muscle biopsy (Gomori’s trichrome × 100)
Acquired inflammatory neuropathies are the main peripheral neuropathy group that constitute the most common indication for nerve biopsy examination today. Nerve biopsy examination was widely used to differentiate hereditary neuropathies from acquired inflammatory or immune-mediated neuropathies, especially in times when genetically diagnosing hereditary demyelinating neuropathies was not so widespread. Differentiating primary demyelinating neuropathy from primary axonal neuropathy can significantly narrow the differential diagnosis (Fig. 10.31). This distinction also has profound implications for prognosis and treatment because many demyelinating neuropathies are inflammatory. Electrophysiological tests are always necessary in this context, as it is often not easy to distinguish clinically. On physical examination, clues of a demyelinating process include early loss of reflexes disproportionate to weakness, greater motor than sensory defects and palpably enlarged nerves. If peripheral neuropathies are classified as diseases that mainly affect axons or myelin production, it is seen that most infectious, toxic, nutritional and metabolic disorders, including vasculitis, mostly cause axonal degeneration (Table 10.9). On the other hand, diseases characterised by myelinisation defects comprise the acquired demyelinating neuropathies (Fig. 10.32), rare toxic neuropathies due to perhexiline, amiodarone, solvents or chloroquine, most subtypes of Charcot-Marie-Tooth disease and a few metabolic diseases such as Krabbe, Tangier or Niemann-Pick diseases [121].

Severe myelination defect in transverse semithin section of a sural nerve biopsy (toluidine blue × 1000)

Myelination defect in longitudinal semithin section of a sural nerve biopsy (toluidine blue × 1000)
Due to their chronic and fluctuating course, chronic idiopathic inflammatory polyneuropathies (CIDPs) are one of the most common group of diseases for which nerve biopsy is performed for the differential diagnosis of many disorders, including hereditary diseases. Two important reasons for performing a nerve biopsy are to confirm the demyelinating nature of the disease and to distinguish it from other diseases that may give similar clinical findings (Fig. 10.33). The histopathological hallmark of CIDP is primary demyelination. This feature is the most constant and important finding in the sural nerve biopsy. The presence of thinly myelinated fibres in the biopsy confirms a disturbance of myelination with repetitive demyelination and remyelination processes (Fig. 10.34). Repeated primary demyelination and remyelination eventually lead to the formation of onion bulbs (Fig. 10.35). These structures are layers of supernumerary Schwann cells with intervening collagen arranged in rings around longitudinal nerve fibres (Fig. 10.36). Although hypertrophic neuropathy is a prominent feature of several genetically determined neuropathies, onion bulbs can also occur in CIDP and, to a lesser extent, in diabetic neuropathy and other acquired polyneuropathies. On the other hand, the widespread presence of onion bulbs distributed diffusely among nerve fascicles is the hallmark of most familial hypertrophic neuropathies. Onion bulbs can be visualised in paraffin-embedded material using haematoxylin and eosin (H&E) sections, and their presence may be confirmed with collagen IV staining of Schwann cell basal laminae. Onion bulbs may have a central myelinated or demyelinated axon. While the presence of axons can be highlighted by neurofilament immunoreactivity (Fig. 10.37), axons can even be seen in H&E-stained sections (Fig. 10.38). In some types of inherited demyelinating neuropathies, especially CMT-1, onion bulbs may be seen in 30–100% of the visible myelinated and demyelinated nerve fibres. Axonal degeneration is not an expected finding in hereditary and acquired demyelinating polyneuropathies. However, some reactive changes like axonal swelling and degeneration can be observed in demyelinated axons, especially in the advanced stages of the diseases (Fig. 10.39). Nonuniform involvement within and between fascicles, macrophage-mediated myelin stripping, perivascular lymphocytic infiltrates, specific endoneurial signs of active demyelination such as numerous naked axons, scattered endoneurial macrophages and Schwann cell mitosis are histological features that support CIDP more so than CMT-1. The presence of giant axons is a pathognomonic feature of giant axonal neuropathy (GAN) which is a rare hereditary disease. However, in some atypical CIDPs or toxic neuropathies, including glue and lacquer thinner sniffing toxic neuropathies, there may be giant axonal swellings that can make them difficult to differentiate from GAN. Especially in CIDP, the presence of many myelin-digestion chambers (Fig. 10.40) is indicative of axonal degeneration and the increased demyelination gap between Ranvier nodes [120,121,122].

Severe myelination defect with myelin basic protein immune staining (DAB × 400)

Note the presence of thinly myelinated large nerve fibres in a transverse semithin section of sural nerve biopsy (toluidine blue × 1000)

Severe myelination defect with occurrence of onion bulbs (Gomori’s trichrome × 200)

Patchy myelination defect with fibrosis in the sural nerve of a patient with CIDP (modified trichrome × 100)

Axons can be highlighted by neurofilament immune reactivity (DAB × 100)

The presence of axons can be discriminated in most fibres (modified trichrome × 200)

A few giant axons can be highlighted by neurofilament immune reactivity (DAB × 200)

Axonal swelling can be discriminated (Holmes stain × 400)
Identification of inflammatory cell infiltration is diagnostic, especially in immune-mediated or inflammatory polyneuropathies such as AIDP and CIDP because in hereditary disorders, inflammation is almost never detected. On the other hand, inflammation is not very evident in CIDP, which is an inflammatory process. Therefore, immunohistochemical studies are necessary for the diagnosis of CIDP. The presence of more than three T lymphocytes per plexus (Fig. 10.41), or the presence of T lymphocytes, albeit in small numbers, on the walls of the perineurial and endoneurial vessels, makes the diagnosis of inflammatory neuropathy certain (Fig. 10.42). This is because normally, inflammatory cells can only be seen in small numbers around epineurial vessels [121].

A small collections of T lymphocytes around the endoneurial vessel highlighted by CD3 immune reactivity (DAB × 100)

T lymphocytes around the perineurial vessel highlighted by CD3 immune reactivity (DAB × 400)
Axonal degeneration of the nerves is a very important clue for the diagnosis of axonal neuropathy. The features of axonal degeneration include the presence of myelin-digestion chambers (Fig. 10.43) and myelin ovoids (Fig. 10.44). Axonal degeneration can be indirectly diagnosed by the presence of giant axons and the presence of axonal regeneration. Axonal regeneration, like Wallerian degeneration, is diagnosed by the presence of small clusters of small axons surrounding small myelin sheaths (Fig. 10.45). In some patients, axonal degeneration is so silent that axonal atrophy may be the only evidence of axonal degeneration (Fig. 10.46). Except for GAN and vasculitic neuropathy, most axonal neuropathies do not have specific histopathological findings in the nerve biopsy that indicate aetiology [120,121,122]. On the other hand, some nerve biopsies can be normal. For example, in a sural nerve biopsy of a 33-year-old male with a 1-year history of distal numbness and muscle wasting, no pathological features were determined (Fig. 10.47 and Table 10.10).

The myelin-digestion chambers occur due to axonal degeneration (modified trichrome × 200)

Axonal degeneration and myelin ovoids can be discriminated in a superficial peroneal nerve biopsy (toluidine blue and basic fuchsin stain × 1000). This photograph is from the archive of Rahul Phadke, MD, PhD, Associate Professor of Neuropathology, Dubowitz Neuromuscular Centre, UCL QS Institute of Neurology, London

The myelin-digestion chambers can be highlighted by myelin basic protein immune reactivity (DAB × 100)

The presence of small clusters of small axons can be discriminated by neurofilament immune reactivity (DAB × 100)

Normal sural nerve with resin semithin section (toluidine blue and basic fuchsin stain × 200). This photograph is from the archive of Rahul Phadke, MD, PhD, Associate Professor of Neuropathology, Dubowitz Neuromuscular Centre, UCL QS Institute of Neurology, London
Nerve biopsy is rarely used when determining the differential diagnosis of a large spectrum of miscellaneous diseases such as mitochondrial, metabolic and toxic pathologies that may cause peripheral neuropathy. This is because most of these pathologies have no diagnostic findings or it is very difficult to encounter the pathognomonic findings in nerve biopsy. For example, peripheral nerves are frequently affected in mitochondrial diseases. However, it is not possible to diagnose neural mitochondrial pathologies with oxidative enzyme stains. It has been reported that the detection of proliferating mitochondria in bizarre shapes in the cytoplasm of Schwann cells is diagnostic if they can be detected in electron microscopic examinations. Similarly, the presence of intra-axonal periodic acid-Schiff (PAS) positive hyaline concentric lamellar bodies in the nerve plexi in polyglucosan body disease, giant lysosomes in Schwann cells in Chediak-Higashi disease and banana bodies in the cytoplasm of Schwann cells in Farber disease are diagnostic. However, these changes, which can only be detected by electron microscopic examination, are extremely unlikely to be seen in these small tissue sections. For this reason, patients with these rare diseases in which pathognomonic findings can be exhibited in nerve biopsy, have been published as case reports in the literature [121].
Almost all neuropathies mentioned above affect both motor and sensory nerves equally, or in other words, the sural nerve, totally composed of sensory nerves which is sampled for nerve biopsies is almost always involved. However, some disorders predominantly or only involve motor nerves. Motor neuron diseases like amyotrophic lateral sclerosis (ALS), which is the most common form, are neurological disorders characterised by the degeneration of motor neurons. ALS generally effects both lower motor neurons (LMN) and upper motor neurons (UMN). Despite the classical clinical presentation of ALS which is usually very diagnostic, diagnosis may be challenging in patients presenting with sporadic progressive disease of the LMNs. Furthermore, motor neuropathy (MN) primarily affects the motor nerves. In most cases, nerve conduction studies differentiate between these situations. While UMN signs are absent, demyelinating features are present in MN. However, demyelination may not always be identifiable, and with electrophysiological examination, purely axonal findings may be found in some cases. As early differentiation between ALS and MN is important for the prognosis and therapeutic approach, a motor nerve biopsy should be acquired. Therefore, in this situation, obturator nerve biopsy with a muscle biopsy from the gracilis muscle should be considered as a potential differential diagnostic tool [120,121,122].
References
Hammi C, Yeung B. Neuropathy. In: StatPearls. Treasure Island, FL: StatPearls; 2005. p. 1137–61. ISBN 9780721694917. https://doi.org/10.1016/B978-0-7216-9491-7.50048-X.
Barrell K, Smith AG. Peripheral neuropathy. Med Clin North Am. 2019;103(2):383–97. https://doi.org/10.1016/j.mcna.2018.10.006.
Hanewinckel R, van Oijen M, Ikram MA, van Doorn PA. The epidemiology and risk factors of chronic polyneuropathy. Eur J Epidemiol. 2016;31(1):5–20. https://doi.org/10.1007/s10654-015-0094-6.
Thompson PD, Thomas PK. Clinical patterns of peripheral neuropathy. In: Peripheral neuropathy. Philadelphia: WB Saunders; 2005. p. 1137–61.
Vij N, Traube B, Bisht R, Singleton I, Cornett EM, Kaye AD, Imani F, Mohammadian EA, Varrassi G, Viswanath O, Urits I. An update on treatment modalities for ulnar nerve entrapment: a literature review. Anesthesiol Pain Med. 2020;10(6):e112070. https://doi.org/10.5812/aapm.112070.
Merriam-Webster Medical Dictionary. https://www.merriam-webster.com/dictionary/neuropathy.
Rutkove SB. Overview of upper extremity peripheral nerve syndromes. Waltham, MA: UpToDate.
Padua L, Coraci D, Erra C, Pazzaglia C, Paolasso I, Loreti C, Caliandro P, Hobson-Webb LD. Carpal tunnel syndrome: clinical features, diagnosis, and management. Lancet Neurol. 2016;15(12):1273–84. https://doi.org/10.1016/S1474-4422(16)30231-9.
Oh SJ, Slaughter R, Harrell L. Paraneoplastic vasculitic neuropathy: a treatable neuropathy. Muscle Nerve. 1991;14(2):152–6. https://doi.org/10.1002/mus.880140210.
Levinson N, Price RS. Mononeuropathy multiplex. In: Cucchiara BL BL, Price RS, editors. Decision-making in adult neurology. Amsterdam: Elsevier; 2021. p. 188–189.e2.
Kincaid JC. Upper extremity neuropathies. Handb Clin Neurol. 2019;161:197–205. https://doi.org/10.1016/B978-0-444-64142-7.00049-7.
David WS, Sadjadi R. Chapter 13. Clinical neurophysiology of lower extremity focal neuropathies. In: Levin KH, Chauvel P, editors. Handbook of clinical neurology, vol. 161. Amsterdam: Elsevier; 2019. p. 207–16.
Rutkove SB. Overview of lower extremity peripheral nerve syndromes. https://www.uptodate.com/contents/overview-of-lower-extremity-peripheral-nerve-syndromes?search=mononeuropathy&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1.
Bromberg MB. Brachial plexus syndromes. Waltham, MA: UpToDate.
Jspeert J, Janssen RMJ, van Alfen N. Neuralgic amyotrophy. Curr Opin Neurol. 2021;34(5):605–12. https://doi.org/10.1097/WCO.0000000000000968.
Klein CJ, Dyck PJ, Friedenberg SM, Burns TM, Windebank AJ, Dyck PJ. Inflammation and neuropathic attacks in hereditary brachial plexus neuropathy. J Neurol Neurosurg Psychiatry. 2002;73(1):45–50. https://doi.org/10.1136/jnnp.73.1.45.
Sasaki H, Kawamura N, Dyck PJ, Dyck PJB, Kihara M, Low PA. Spectrum of diabetic neuropathies. Diabetol Int. 2020;11(2):87–96. Published 2020 Jan 8. https://doi.org/10.1007/s13340-019-00424-7.
Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173–93. Published 2020 Aug 13. https://doi.org/10.1016/j.cnp.2020.07.005.
Twydell TP. Lumbosacral plexus syndromes. Waltham, MA: UpToDate.
Kothari MJ. Treatment and prognosis of cervical radiculopathy. Waltham, MA: UpToDate.
Hsu PS, Armon C, Levin K. Acute lumbosacral radiculopathy: Pathophysiology, clinical features, and diagnosis. Waltham, MA: UpToDate.
Callaghan BC, Price RS, Chen KS, Feldman EL. The importance of rare subtypes in diagnosis and treatment of peripheral neuropathy: a review. JAMA Neurol. 2015;72(12):1510–8. https://doi.org/10.1001/jamaneurol.2015.2347.
Samson M, Puéchal X, Devilliers H, et al. Mononeuritis multiplex predicts the need for immunosuppressive or immunomodulatory drugs for EGPA, PAN and MPA patients without poor-prognosis factors. Autoimmun Rev. 2014;13(9):945–53.
Torvik A, Berntzen AE. Necrotizing vasculitis without visceral involvement. Postmortem examination of three cases with affection of skeletal muscles and peripheral nerves. Acta Med Scand. 1968;184(1–2):69–77.
Abdelhakim S, Klapholz JD, Roy B, Weiss SA, McGuone D, Corbin ZA. Mononeuritis multiplex as a rare and severe neurological complication of immune checkpoint inhibitors: a case report. J Med Case Reports. 2022;16(1):81. Published 2022 Feb 24. https://doi.org/10.1186/s13256-022-03290-1.
Kelkar P, Parry GJ. Mononeuritis multiplex in diabetes mellitus: evidence for underlying immune pathogenesis. J Neurol Neurosurg Psychiatry. 2003;74(6):803–6. https://doi.org/10.1136/jnnp.74.6.803.
Karam C. Peripheral neuropathies associated with Vasculitis and autoimmune connective tissue disease. Continuum (Minn). 2020;26(5):1257–79. PMID: 33003001. https://doi.org/10.1212/CON.0000000000000917.
Klein CJ. Charcot-Marie-tooth disease and other hereditary neuropathies [published correction appears in Continuum (Minneap Minn). 2021 Feb 1;27(1):289]. Continuum (Minn). 2020;26(5):1224–56. https://doi.org/10.1212/CON.0000000000000927.
Russell JA. General approach to peripheral nerve disorders. Continuum (Minn). 2017;23:1241–62. https://doi.org/10.1212/CON.0000000000000519.
Rutkove SB. Overview of polyneuropathy. https://www.uptodate.com/contents/overview-of-polyneuropathy/print?search=peripheral%20neuropathy&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1.
Siao P, Kaku M. A clinician’s approach to peripheral neuropathy. Semin Neurol. 2019;39(5):519–30. https://doi.org/10.1055/s-0039-1694747.
Wang W, Litchy WJ, Mandrekar J, et al. Blink reflex role in algorithmic genetic testing of inherited polyneuropathies. Muscle Nerve. 2017;55(3):316–22. https://doi.org/10.1002/mus.25250.
Watson JC, Dyck PJ. Peripheral neuropathy: a practical approach to diagnosis and symptom management. Mayo Clin Proc. 2015;90(7):940–51. https://doi.org/10.1016/j.mayocp.2015.05.004.
Bird TD. Charcot-Marie-tooth hereditary neuropathy overview. In: Adam MP, et al., editors. GeneReviews®. Seattle: University of Washington; 1998. Accessed 24 Feb 2022.
Eichler FS. Hereditary sensory and autonomic neuropathies. Waltham, MA: UpToDate.
Edvardson S, Cinnamon Y, Jalas C, Shaag A, Maayan C, Axelrod FB, Elpeleg O. Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann Neurol. 2012;71(4):569–72. https://doi.org/10.1002/ana.23524.
Kang PB. Charcot-Marie-Tooth disease: genetics, clinical features, and diagnosis. Waltham, MA: UpToDate.
Taşdelen E, Calame DG, Akay G, et al. Novel RETREG1 (FAM134B) founder allele is linked to HSAN2B and renal disease in a Turkish family. Am J Med Genet A. 2022;188(7):2153–61. https://doi.org/10.1002/ajmg.a.62727.
Conceição I, González-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9. https://doi.org/10.1111/jns.12153.
Grisold W, Carozzi VA. Toxicity in peripheral nerves: an overview. Toxics. 2021;9(9):218. Published 2021 Sep 11. https://doi.org/10.3390/toxics909021.
Valentine WM. Toxic peripheral neuropathies: agents and mechanisms. Toxicol Pathol. 2020;48(1):152–73. https://doi.org/10.1177/019262331985432.
Yamamoto S, Egashira N. Pathological mechanisms of bortezomib-induced peripheral neuropathy. Int J Mol Sci. 2021;22(2):888. Published 2021 Jan 17. https://doi.org/10.3390/ijms22020888.
Hadtstein F, Vrolijk M. Vitamin B-6-induced neuropathy: exploring the mechanisms of pyridoxine toxicity. Adv Nutr. 2021;12(5):1911–29. https://doi.org/10.1093/advances/nmab033.
Gwathmey KG, Grogan J. Nutritional neuropathies. Muscle Nerve. 2020;62(1):13–29. https://doi.org/10.1002/mus.26783.
Attaluri P, Castillo A, Edriss H, Nugent K. Thiamine deficiency: an important consideration in critically ill patients. Am J Med Sci. 2018;356(4):382–90. https://doi.org/10.1016/j.amjms.2018.06.015.
Kulkantrakorn K. Pyridoxine-induced sensory ataxic neuronopathy and neuropathy: revisited. Neurol Sci. 2014;35(11):1827–30. https://doi.org/10.1007/s10072-014-1902-6.
Aroda VR, Edelstein SL, Goldberg RB, et al. Long-term metformin use and vitamin B12 deficiency in the diabetes prevention program outcomes study. J Clin Endocrinol Metab. 2016;101(4):1754–61. https://doi.org/10.1210/jc.2015-3754.
Palmer BF. Uremic polyneuropathy. Available at: Uremic polyneuropathy - UpToDate. Orbach H, Tishler M, Shoenfeld Y. intravenous immunoglobulin and the kidney—a two-edged sword. Semin Arthritis Rheum. 2004;34(3):593–601. https://doi.org/10.1016/j.semarthrit.2004.06.003.
Gandhi Mehta RK, Caress JB, Rudnick SR, Bonkovsky HL. Porphyric neuropathy. Muscle Nerve. 2021;64(2):140–52. https://doi.org/10.1002/mus.27232.
Castelli G, Desai KM, Cantone RE. Peripheral neuropathy: evaluation and differential diagnosis. Am Fam Physician. 2020;102(12):732–9.
Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, Shaw JE, Bright D, Williams R, IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843. Epub 2019 Sep 10. https://doi.org/10.1016/j.diabres.2019.107843.
Tesfaye S, Selvarajah D, Gandhi R, Greig M, Shillo P, Fang F, Wilkinson ID. Diabetic peripheral neuropathy may not be as its name suggests: evidence from magnetic resonance imaging. Pain. 2016;157(Suppl 1):S72–80. https://doi.org/10.1097/j.pain.0000000000000465.
Liu X, Xu Y, An M, Zeng Q. The risk factors for diabetic peripheral neuropathy: a meta-analysis. PloS One. 2019;14(2):e0212574. PMID: 30785930; PMCID: PMC6382168. https://doi.org/10.1371/journal.pone.0212574.
Boulton AJ, Gries FA, Jervell JA. Guidelines for the diagnosis and outpatient management of diabetic peripheral neuropathy. Diabet Med. 1998;15(6):508–14. https://doi.org/10.1002/(SICI)1096-9136(199806)15:6<508::AID-DIA613>3.0.CO;2-L.
Sinnreich M, Taylor BV, Dyck PJ. Diabetic neuropathies. Classification, clinical features, and pathophysiological basis. Neurologist. 2005;11(2):63–79. https://doi.org/10.1097/01.nrl.0000156314.24508.ed.
Hagedorn JM, Engle AM, George TK, et al. An overview of painful diabetic peripheral neuropathy: diagnosis and treatment advancements. Diabetes Res Clin Pract. 2022;188:109928. https://doi.org/10.1016/j.diabres.2022.109928.
Sloan G, Selvarajah D, Tesfaye S. Pathogenesis, diagnosis and clinical management of diabetic sensorimotor peripheral neuropathy. Nat Rev Endocrinol. 2021;17(7):400–20. https://doi.org/10.1038/s41574-021-00496-z.
Ziegler D, Keller J, Maier C, Pannek J. Diabetic neuropathy. Exp Clin Endocrinol Diabetes. 2021;129(S01):S70–81. https://doi.org/10.1055/a-1284-6245.
Ziegler D, Papanas N, Schnell O, et al. Current concepts in the management of diabetic polyneuropathy. J Diabetes Investig. 2021;12(4):464–75. https://doi.org/10.1111/jdi.13401.
Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetic autonomic neuropathy. Diabetes Care. 2003;26(5):1553–79. https://doi.org/10.2337/diacare.26.5.1553.
Cheshire WP, Freeman R, Gibbons CH, et al. Electrodiagnostic assessment of the autonomic nervous system: a consensus statement endorsed by the American autonomic society, American Academy of Neurology, and the International Federation of Clinical Neurophysiology [published correction appears in Clin Neurophysiol. 2021 May;132(5):1194]. Clin Neurophysiol. 2021;132(2):666–82. https://doi.org/10.1016/j.clinph.2020.11.024.
Seçil Y, Ozdedeli K, Altay B, Aydoğdu I, Yilmaz C, Ertekin C. Sympathetic skin response recorded from the genital region in normal and diabetic women. Neurophysiol Clin. 2005;35(1):11–7. https://doi.org/10.1016/j.neucli.2004.12.001.
Scollard D, Barbara Stryjewska B, Dacso M. Leprosy: epidemiology, microbiology, clinical manifestations, and diagnosis. Waltham, MA: UpToDate.
Halperin JJ. Lyme neuroborreliosis. Curr Opin Infect Dis. 2019;32(3):259–64. https://doi.org/10.1097/QCO.0000000000000545.
Kaku MC, Simpson DM. Epidemiology, clinical manifestations, diagnosis, and treatment of HIV-associated distal symmetric polyneuropathy (HIV-DSPN). Waltham, MA: UpToDate.
Finsterer J, Scorza FA, Scorza CA, Fiorini AC. Peripheral neuropathy in COVID-19 is due to immune-mechanisms, pre-existing risk factors, anti-viral drugs, or bedding in the intensive care unit. Arq Neuropsiquiatr. 2021;79(10):924–8. https://doi.org/10.1590/0004-282X-ANP-2021-0030.
Dalmau J, Rosenfeld M. Paraneoplastic syndromes affecting spinal cord, peripheral nerve, and muscle. Waltham, MA: UpToDate.
Antoine JC, Camdessanché JP. Paraneoplastic neuropathies. Curr Opin Neurol. 2017;30(5):513–20. https://doi.org/10.1097/WCO.0000000000000475.
Sancho Saldaña A, Mahdi-Rogers M, Hadden RD. Sensory neuronopathies: A case series and literature review [published correction appears in J Peripher Nerv Syst. 2021 Jun;26(2):238]. J Peripher Nerv Syst. 2021;26(1):66–74. https://doi.org/10.1111/jns.12433.
Koike H, Katsuno M. Paraproteinemia and neuropathy. Neurol Sci. 2021;42(11):4489–501. https://doi.org/10.1007/s10072-021-05583-7.
Katz JS, Saperstein DS. Asymmetric acquired demyelinating polyneuropathies: MMN and MADSAM. Curr Treat Options Neurol. 2001;3(2):119–25. https://doi.org/10.1007/s11940-001-0046-1.
Hameed S, Cascella M. Multifocal motor neuropathy. In: StatPearls. Treasure Island, FL: StatPearls; 2022. https://www.ncbi.nlm.nih.gov/books/NBK554524/. Accessed 5 Feb 2022.
Lange DJ, Robinson-Papp J. Multifocal motor neuropathy. Waltham, MA: UpToDate.
Uncini A, Vallat JM. Autoimmune nodo-paranodopathies of peripheral nerve: the concept is gaining ground. J Neurol Neurosurg Psychiatry. 2018;89(6):627–35. https://doi.org/10.1136/jnnp-2017-317192.
Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society--first revision. J Peripher Nerv Syst. 2010;15(4):295–301. https://doi.org/10.1111/j.1529-8027.2010.00290.x.
Bunschoten C, Jacobs BC, Van den Bergh P, Cornblath DR, van Doorn PA. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol. 2019;18(8):784–94. https://doi.org/10.1016/S1474-4422(19)30144-9.
Gwathmey KG, Smith AG. Immune-mediated neuropathies. Neurol Clin. 2020;38(3):711–35. https://doi.org/10.1016/j.ncl.2020.03.008.
Stino AM, Naddaf E, Dyck PJ, Dyck P. Chronic inflammatory demyelinating polyradiculoneuropathy-diagnostic pitfalls and treatment approach. Muscle Nerve. 2021;63(2):157–69. https://doi.org/10.1002/mus.27046.
Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force-Second revision [published correction appears in Eur J Neurol 2022 Apr;29(4):1288]. Eur J Neurol. 2021;28(11):3556–83. https://doi.org/10.1111/ene.14959.
Querol L, Devaux J, Rojas-Garcia R, Illa I. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol. 2017;13(9):533–47. https://doi.org/10.1038/nrneurol.2017.84.
Van Schaik IN, Bril V, van Geloven N, et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (PATH): a randomised, double-blind, placebo-controlled, phase 3 trial [published correction appears in lancet Neurol. 2018 Jan;17 (1):26] [published correction appears in lancet Neurol. 2018 Aug;17(8):661]. Lancet Neurol. 2018;17(1):35–46. https://doi.org/10.1016/S1474-4422(17)30378-2.
Kurt Incesu T, Secil Y, Tokucoglu F, et al. Diagnostic value of lumbar root stimulation at the early stage of Guillain-Barré syndrome. Clin Neurophysiol. 2013;124(1):197–203. https://doi.org/10.1016/j.clinph.2012.07.004.
Bunschoten C, Blomkwist-Markens PH, Horemans A, van Doorn PA, Jacobs BC. Clinical factors, diagnostic delay, and residual deficits in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2019;24(3):253–9. https://doi.org/10.1111/jns.12344.
Lewis RA, Sumner AJ, Brown MJ, Asbury AK. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32(9):958–64. https://doi.org/10.1212/wnl.32.9.958.
Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force-Second revision [published correction appears in J Peripher Nerv Syst. 2022 Mar;27(1):94] [published correction appears in Eur J Neurol. 2022 Apr;29(4):1288]. J Peripher Nerv Syst. 2021;26(3):242–68. https://doi.org/10.1111/jns.12455.
Pegat A, Boisseau W, Maisonobe T, et al. Motor chronic inflammatory demyelinating polyneuropathy (CIDP) in 17 patients: clinical characteristics, electrophysiological study, and response to treatment. J Peripher Nerv Syst. 2020;25(2):162–70. https://doi.org/10.1111/jns.12380.
Arends S, Drenthen J, Van den Bergh PYK, et al. Electrodiagnostic subtyping in Guillain-Barré syndrome: Use of criteria in practice based on a survey study in IGOS [published online ahead of print, 2022 Jun 14]. J Peripher Nerv Syst. 2022;27:197. https://doi.org/10.1111/jns.12504.
Doneddu PE, Cocito D, Manganelli F, et al. Atypical CIDP: diagnostic criteria, progression and treatment response. Data from the Italian CIDP database. J Neurol Neurosurg Psychiatry. 2019;90(2):125–32. https://doi.org/10.1136/jnnp-2018-318714j.
Illa I. ARTHUR ASBURY LECTURE: chronic inflammatory demyelinating polyradiculoneuropathy: clinical aspects and new animal models of auto-immunity to nodal components. J Peripher Nerv Syst. 2017;22(4):418–24. https://doi.org/10.1111/jns.12237.
Abraham A, Breiner A, Katzberg HD, Lovblom LE, Perkins BA, Bril V. Treatment responsiveness in CIDP patients with diabetes is associated with unique electrophysiological characteristics, and not with common criteria for CIDP. Expert Rev Clin Immunol. 2015;11(4):537–46. https://doi.org/10.1586/1744666X.2015.1018891.
Allen JA. The misdiagnosis of CIDP: a review. Neurol Therapy. 2020;9(1):43–54. https://doi.org/10.1007/s40120-020-00184-6l.
Lewis RA, Muley SA. Chronic inflammatory demyelinating polyneuropathy: treatment and prognosis. Waltham, MA: UpToDate.
Eftimov F, Winer JB, Vermeulen M, de Haan R, van Schaik IN. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2013;12:CD001797. Published 2013 Dec 30. https://doi.org/10.1002/14651858.CD001797.pub3.
Kaplan AA, Fridey JL. Therapeutic apheresis (plasma exchange or cytapheresis): complications. UpToDate, Waltham, MA
Chandrashekhar S, Dimachkie M. Guillain-Barré syndrome in adults: pathogenesis, clinical features, and diagnosis. Waltham, MA: UpToDate.
Shahrizaila N, Lehmann HC, Kuwabara S. Guillain-Barré syndrome. Lancet. 2021;397(10280):1214–28. https://doi.org/10.1016/S0140-6736(21)00517-1.
Sheikh KA. Guillain-Barré Syndrome. Continuum (Minn). 2020;26(5):1184–204. https://doi.org/10.1212/CON.0000000000000929.
Koike H, Katsuno M. Emerging infectious diseases, vaccines and Guillain-Barré syndrome. Clin Exp Neuroimmunol. 2021;12(3):165–70. https://doi.org/10.1111/cen3.12644.
Berciano J. Axonal degeneration in Guillain-Barré syndrome: a reappraisal. J Neurol. 2021;268(10):3728–43. https://doi.org/10.1007/s00415-020-10034-y.
Versace V, Campostrini S, Rastelli E, et al. Understanding hyper-reflexia in acute motor axonal neuropathy (AMAN). Neurophysiol Clin. 2020;50(3):139–44. https://doi.org/10.1016/j.neucli.2020.05.004.
Uncini A, Notturno F, Kuwabara S. Hyper-reflexia in Guillain-Barré syndrome: systematic review. J Neurol Neurosurg Psychiatry. 2020;91(3):278–84. https://doi.org/10.1136/jnnp-2019-321890.
Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry. 2013;84(5):576–83. https://doi.org/10.1136/jnnp-2012-302824.
Cabrero FR, Morrison EH. Miller Fisher syndrome. In: StatPearls [Internet]. Treasure Island, FL: StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK430685/.
Morena J, Elsheikh B, Hoyle JC. Recurrent miller Fisher: a case report along with a literature and an EMG/NCS review. Neurohospitalist. 2021;11(3):263–6. https://doi.org/10.1177/1941874420987053.
Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med. 1956;255(2):57–65. https://doi.org/10.1056/NEJM195607122550201.
Tan CY, Razali SNO, Goh KJ, Shahrizaila N. Determining the utility of the Guillain-Barré syndrome classification criteria. J Clin Neurol. 2021;17(2):273–82. https://doi.org/10.3988/jcn.2021.17.2.273.
Sejvar JJ, Kohl KS, Gidudu J, et al. Guillain-Barré syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2011;29(3):599–612. https://doi.org/10.1016/j.vaccine.2010.06.003.
Wakerley BR, Yuki N. Pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2014;85(3):339–44. https://doi.org/10.1136/jnnp-2013-305397.
Tham SL, Prasad K, Umapathi T. Guillain-Barré syndrome mimics. Brain Behav. 2018;8(5):e00960. Published 2018 Apr 10. https://doi.org/10.1002/brb3.960.
Umapathi T, Koh JS, Cheng YJ, Goh EJH, Lim CSJ. The utility of sural-sparing pattern in the electrodiagnosis of regional subtypes of Guillain-Barré syndrome. Clin Neurophysiol Pract. 2020;5:43–5. PMID: 32140628; PMCID: PMC7044466. https://doi.org/10.1016/j.cnp.2019.12.002.
Querol L, Lleixà C. Novel immunological and therapeutic insights in Guillain-Barré syndrome and CIDP. Neurotherapeutics. 2021;18(4):2222–35. https://doi.org/10.1007/s13311-021-01117-3.
Diniz G, Tosun Yildirim H, Ünalp A, Barutçuoğlu M, Güzel O, Polat M, Türe S, Özgönül F, Serdaroğlu G. The evaluation of muscle biopsy findings in children with neuromuscular disorders. J Behcet Uz Child Hosp. 2012;2(2):62–7. https://doi.org/10.5222/buchd.2012.062.
Ang LC. Skeletal muscle. In: Rosai J, editor. Ackerman and Rosai surgical pathology. 9th ed. Amsterdam: Elsevier; 2004. p. 2663–81.
Dubowitz V, Sewry C, Oldfors A. Muscle biopsy: a practical approach. Philadelphia, PA: Saunders Elsevier; 2013. p. 1–27.
Carraro U, Kern H. Severely atrophic human muscle fibers with nuclear misplacement survive many years of permanent denervation. Eur J Transl Myol. 2016;26(2):5894. https://doi.org/10.4081/ejtm.2016.5894.
Dupin I, Etienne-Manneville S. Nuclear positioning: mechanisms and functions. Int J Biochem Cell Biol. 2011;43(12):1698–707. https://doi.org/10.1016/j.biocel.2011.09.004.
Ralston E, Lu Z, Biscocho N, Soumaka E, Mavroidis M, Prats C, Lømo T, Capetanaki Y, Ploug T. Blood vessels and desmin control the positioning of nuclei in skeletal muscle fibers. J Cell Physiol. 2006;209(3):874–82. https://doi.org/10.1002/jcp.20780.
Lu JQ, Mubaraki A, Yan C, Provias J, Tarnopolsky MA. Neurogenic muscle biopsy findings are common in mitochondrial myopathy. J Neuropathol Exp Neurol. 2019;78(6):508–14. https://doi.org/10.1093/jnen/nlz029.
Park YE, ShinJH KDS. Muscle pathology in neuromuscular disorders. Ann Clin Neurophysiol. 2020;22(2):51–60. https://doi.org/10.14253/acn.2020.22.2.51.
Oh SJ. Color atlas of nerve biopsy pathology. 1st ed. London: CRC; 2001. https://doi.org/10.1201/9781420039801.
Bilbao JM, Schmidt RE. Biopsy diagnosis of peripheral neuropathy. 2nd ed. Cham: Springer Nature; 2015.
Lanigan LG, Russell DS, Woolard KD, Pardo ID, Godfrey V, Jortner BS, Butt MT, Bolon B. Comparative pathology of the peripheral nervous system. Vet Pathol. 2021;58(1):10–33. https://doi.org/10.1177/0300985820959231.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tokuçoğlu, F., Diniz, G. (2023). Peripheral Neuropathies. In: Diniz, G. (eds) Clues for Differential Diagnosis of Neuromuscular Disorders. Springer, Cham. https://doi.org/10.1007/978-3-031-33924-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-031-33924-0_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-33923-3
Online ISBN: 978-3-031-33924-0
eBook Packages: MedicineMedicine (R0)