
Abstract
Spinal muscular atrophy (SMA) is a rare genetic neuromuscular disease that affects individuals with a broad age range and spectrum of disease severity, characterized by progressive loss of spinal motor neurons leading to debilitating and sometimes fatal muscle weakness. Therapeutic options had been limited for this patient population, and to address this unmet medical need, a series of studies on risdiplam (EVRYSDI®) began in 2016. Risdiplam has now been approved in more than 90 countries, including the United States and EU for the treatment of SMA. The approvals were primarily based on two pivotal clinical studies, FIREFISH and SUNFISH, designed to represent a broad spectrum of people living with SMA. FIREFISH is an open-label, single-arm study in infants aged 1–7 months with type 1 SMA. SUNFISH is a randomized, double-blind, placebo-controlled study in patients aged 2–25 years with type 2 and 3 SMA. Real-world data (RWD) from a combination of peer-reviewed publications and individual patients were used to support data interpretation and registration. Natural history data from publications were used to define performance criteria for key clinical endpoints in FIREFISH. RWD from individual patients were also used to perform a robust statistical comparison and contextualize the results from SUNFISH. In this chapter, we present a case study describing how RWD from publications and individual patient data were used to support the development of risdiplam, a medicine to treat SMA.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- External control
- Natural history data
- Real-world data
- Real-world evidence
- Risdiplam
- Spinal muscular atrophy
1 Introduction
1.1 Spinal Muscular Atrophy
Spinal muscular atrophy (SMA) is a rare, debilitating genetic neuromuscular disease. It affects approximately 1 in 10,000 individuals and when untreated is the leading genetic cause of infant mortality [1]. SMA is characterized by progressive loss of motor neurons (nerve cells that control muscle movement). The disease is caused by mutations or deletions in the survival of motor neuron 1 (SMN1) gene, which leads to a deficiency in SMN protein [2]. SMN protein is found throughout the body and is essential for the function of nerves that control muscles and movement. Without SMN protein, motor neurons cannot function properly, which in turn leads to muscle wasting over time [3]. Depending on the type of SMA, an individual’s physical strength and their ability to walk, eat, or breathe can be significantly diminished or lost [4].
Patients with SMA are typically classified into types 1–4 based on the age of symptom onset and highest motor milestone achieved [5,6,7], with types 1, 2, and 3 SMA representing approximately 99% of the SMA population [8]. Table 1 shows a summary of the primary SMA types.
Since 2016, the US Food and Drug Administration (FDA) has approved three medications to treat SMA: nusinersen (SPINRAZA®), onasemnogene abeparvovec-xioi (ZOLGENSMA®), and risdiplam (EVRYSDI®). The goal of these disease-modifying treatments is to increase the availability of SMN protein, leading to clinically meaningful improvements in muscle function.
1.2 Risdiplam
At the time of initiating the risdiplam clinical development program in 2016, there was no approved treatment for SMA. Risdiplam was developed by Roche/Genentech (the sponsor) in partnership with PTC Therapeutics and the SMA Foundation to help address the unmet needs for children and adults with SMA. Risdiplam is a small molecule administered daily at home in liquid form by mouth or feeding tube. It is a selective SMN2 gene splicing modifier that increases the production of full-length SMN protein in the central nervous system and peripheral tissues [9]. It was hypothesized that increasing the amount of SMN protein would reduce motor neuron degeneration thereby limiting muscle atrophy.
A series of clinical studies on risdiplam were designed to represent a broad spectrum of people with SMA, from birth to 60 years of age.
-
FIREFISH (NCT02913482): an open-label, single-arm, two-part study in infants aged 1–7 months with type 1 SMA (N = 62).
-
SUNFISH (NCT02908685): a randomized, placebo-controlled, two-part study in children and young adults aged 2–25 years with type 2 or 3 SMA (N = 231).
-
JEWELFISH (NCT03032172): an open-label, single-arm study in children and adults aged 6 months to 60 years who have taken part in clinical trials for SMA or received other investigational or approved SMA therapies (N = 174).
-
RAINBOWFISH (NCT03779334): an open-label, single-arm study in infants genetically diagnosed with SMA and not yet presenting symptoms (N = 26).
Table 2 shows a summary of key milestones in the development of risdiplam.
As of September 2022, risdiplam has been approved in more than 90 countries and the dossier is under review in 18 countries. More than 7000 people have been treated with risdiplam across clinical trials, through the Compassionate Use Program/Pre-Approval Access and in the commercial setting.
The focus of this case study will be on the two pivotal studies in the risdiplam clinical development program, FIREFISH and SUNFISH. Both these studies had an operationally seamless design, with an exploratory dose-finding part (Part 1) and a confirmatory part (Part 2). Real-world data (RWD) were critical to support the clinical development planning, data interpretation, and registration of risdiplam. Here, we describe how RWD from publications were used to define performance criteria for key clinical endpoints in FIREFISH and benchmark the results for success in patients with type 1 SMA. In addition, RWD from individual patients were used to perform a robust statistical comparison and contextualize the results from SUNFISH in patients with type 2 and 3 SMA. In this chapter, we will discuss why we used RWD, how we used RWD and the impact of using RWD, including a summary of challenges and lessons learned.
2 FIREFISH Study: External Control Data from Publications
2.1 Design and Methods
2.1.1 Study Design
FIREFISH was an operationally seamless, two-part, open-label, multicenter Phase 2/3 study to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of risdiplam in infants with type 1 SMA aged 1–7 months at enrolment. Figure 1 shows a summary of the FIREFISH study design.

Study Design of Part 1 and Part 2 of FIREFISH. SMA spinal muscular atrophy
FIREFISH Part 1 was an exploratory, dose-finding study conducted in 21 infants with type 1 SMA, which determined the dose for use in Part 2 [10]. FIREFISH Part 2 was a confirmatory study conducted in 41 infants with type 1 SMA [11]. Part 1 and Part 2 enrolled different infants. The primary endpoint for the confirmatory Part 2 of the study was the proportion of infants sitting without support for at least 5 seconds (s) after 12 months of treatment, as assessed by Item 22 of the Bayley Scales of Infant and Toddler Development, third edition (BSID-III) Gross Motor Subscale [12]. Sitting without support was selected as the primary endpoint because achieving this milestone illustrates a divergence from the natural course of type 1 SMA as these infants would never achieve this motor milestone without treatment. Besides, sitting independently is clinically meaningful for infants, as it allows them to use their upper limbs to reach for objects, grasp objects, and feed themselves. In addition, event-free survival, defined as being alive without permanent ventilation, was a key secondary endpoint. The overall study design and choice of endpoints incorporated health authority advice from both the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) and the FDA. The efficacy outcome measures analyzed in FIREFISH Part 1 were consistent with those in Part 2, but no formal hypothesis testing was planned for Part 1.
2.1.1.1 RWD as a Component of Study Design in FIREFISH
The natural history of type 1 SMA is well defined and has been described in numerous studies [13, 14]. Untreated infants with type 1 SMA are never able to sit independently [15]. In addition, natural history shows that 50% of infants will have died or required permanent non-invasive ventilation support by 10.5 months of age and 92% of infants by 20 months of age [13].
Given the severity, rapid decline, and high mortality and morbidity in infants with type 1 SMA, a placebo comparator group was not included in the FIREFISH study. In the absence of a control arm, RWD played an important role in the design of the study and the analysis and interpretation of the study results. Natural history data were used to define thresholds of achievement, or “performance criteria,” against which to assess the efficacy of risdiplam treatment in Part 2 of the study. This approach can be acceptable in the development of new treatments for serious and rare diseases such as type 1 SMA if the natural course of the disease is well understood, the external comparator group is similar to the treatment group (e.g., with regard to patient characteristics and endpoints), and a large treatment effect is expected with the study drug. A treatment is considered to be effective if the threshold for success is crossed, and when the observed treatment effect is large, it is reasonable to exclude chance or bias as a possible explanation [16].
The FIREFISH study included objective endpoints that facilitated the comparison with RWD. The primary endpoint (sitting without support for at least 5 s) was assessed using strict criteria to minimize bias following the BSID-III manual, objectively measured by trained clinical evaluators, video recorded and scored by two independent reviewers. As sitting without support is never achieved in untreated infants with type 1 SMA [13], a large divergence from natural history would be expected when treated with an efficacious drug. Secondary endpoints included motor function, achievement of other developmental motor milestones, survival, and event-free survival. Performance criteria were defined for the primary and key secondary endpoints in Part 2 of the FIREFISH study.
2.1.1.2 Identification and Selection of Historical Data Sources for Defining Performance Criteria
First, a literature search was performed to identify publications in infants with type 1 SMA that included an endpoint reported in FIREFISH. A PubMed search was undertaken using the following search terms: (spinal muscular atrophy [Title/Abstract]) AND (observational OR cohort OR natural history OR registr* OR association OR describe OR description OR match* or control*) AND (“2000/01/01”[Date – Publication]: “3000/01/01”[Date – Publication]), which gave 938 hits (February 1, 2018). Titles and abstracts were reviewed to identify articles that appeared to report on the outcome measures included in the FIREFISH study in an observational setting, from both retrospective and prospective data collection, in infants with type 1 SMA. This left a total of 35 articles that could be used for comparison with the FIREFISH study. From these 35 articles, publications were excluded for the following reasons related to usability of the data, patient characteristics, and standard of care:
-
No data relating to any of the endpoints in the FIREFISH study
-
No extractable data to derive a performance criterion for any of the endpoints in the FIREFISH study, e.g., no longitudinal data were provided to calculate change from baseline values
-
No infants with a genetic confirmation of SMA
-
Standard of care not reflective of guidelines described in the consensus statement for standards of care in SMA [17], i.e., no consistent use of non-invasive ventilation or gastrostomy tube
Of the 35 observational studies identified, 18 were excluded for meeting one or more of these criteria. In addition to the 17 remaining observational studies, the untreated, matched cohort selected retrospectively as a comparator group in a Phase 1/2 study of valproic acid and carnitine in infants with type 1 SMA [18] was also included as a potential data source. Endpoints defined in the FIREFISH study were sometimes described in more than one of these studies. The identified studies were ranked based on the level of similarity of the patient population to the expected population in the FIREFISH study. When more than one source was available for an endpoint, the published study cohort with baseline characteristics most similar to those targeted by the FIREFISH study inclusion and exclusion criteria (i.e., the published study with the highest ranking) was selected to set the performance criterion. The following characteristics were considered when determining the similarity of the historical cohorts to the FIREFISH study population:
-
SMN2 copy number
-
Age at onset of symptoms
-
Age at enrolment (start of follow-up in study or presentation at treating center)
-
Type 1 SMA classification
-
Standard of care
-
Time period
-
Region
-
Type of treating center
Multicenter, prospective studies were given a higher ranking, while studies were given a lower ranking if the data collection occurred prior to the publication of the consensus statement for standards of care in SMA [17]; if the data were collected over a long period of time (e.g., 15 years) over which the standard of care would be expected to change; and if there was no information provided for important infant characteristics such as SMN2 copy number and age at onset of symptoms. Based on these criteria, the population in the NeuroNEXT SMA infant biomarker study [14, 19] was judged to be most similar to the expected population in the FIREFISH study. Whenever possible, the performance criterion derived from this study was selected as the benchmark to be used for hypothesis testing. When data for an endpoint were not available from the NeuroNEXT SMA infant biomarker study (e.g., for the Hammersmith Infant Neurological Examination, Module 2, which is a secondary endpoint not described in this book chapter), the benchmark was derived from the study conducted by De Sanctis et al. [20]. The NeuroNEXT SMA infant biomarker study included 16 patients with two copies of the SMN2 gene, and the study conducted by De Sanctis et al. [20] included 24 infants classified as type 1B. The demographic and baseline characteristics of these two cohorts and of the infants enrolled in FIREFISH Part 2 are presented in Table 3.
2.1.2 Statistical Methodology: Performance Criteria Approach
The performance criterion for the primary endpoint in Part 2 of the FIREFISH study (the proportion of infants sitting without support for at least 5 s after 12 months of treatment) was based on the natural history of the disease in which untreated patients with type 1 SMA never achieve sitting without support [13]. A threshold of 5% was chosen to provide sufficient confidence that any effect seen in the FIREFISH study would not otherwise have occurred in the natural history of infants enrolled in the study. An exact binomial test was performed to test the hypothesis that the proportion of infants who sit without support on treatment (p1) was:
If the one-sided p-value was ≤5%, then the null hypothesis would be rejected. If the lower limit of the two-sided 90% confidence interval (CI) (Clopper–Pearson) was above the 5% threshold, then the primary objective of the study would be considered achieved. With a sample size of 41 infants, a minimum of 6 infants sitting without support would be needed for a statistically significant result. Infants were classified as non-responders for the primary endpoint if they were not able to sit without support at Month 12, did not maintain sitting achieved at an earlier time-point, were withdrawn or died prior to Month 12, or had a missing assessment at Month 12.
The performance criterion for the key secondary endpoint of event-free survival was based on the NeuroNEXT SMA infant biomarker study [14]. The point estimate and 90% CI were obtained after selecting infants with two SMN2 gene copies, similar to the population in the FIREFISH study. The performance criterion was based on the upper limit of the 90% CI, derived using the complementary log-log transformation for the proportion of patients who were alive without permanent ventilation at 18 months of age. The benchmark was set at 18 months of age to reflect the expected average age of infants in Part 2 of the FIREFISH study after 12 months of treatment. The estimated proportion (90% CI) of patients alive without permanent ventilation at 18 months of age based on the available data was 20% (5–42), giving a performance criterion of 42%. Potential performance criteria were also calculated from other available data sources and documented in the appendix of the Statistical Analysis Plan, along with key selection criteria and rankings for transparency.
When a pre-defined benchmark could be determined for a secondary endpoint, hypothesis testing was performed. For the secondary endpoint of event-free survival, a z-test was performed to test the hypothesis that the proportion of infants alive without permanent ventilation at Month 12 on treatment (p2) was:
To control for multiplicity across the different endpoints, a hierarchical testing approach was implemented.
2.2 Results
The baseline characteristics of the patients enrolled in Part 1 and Part 2 of the FIREFISH study were representative of a population with well-established, symptomatic type 1 SMA. Of the 41 infants enrolled in Part 2, 22 (54%) were female. At enrolment, the median age of patients was 5.3 months (range: 2.2–6.9 months). The median age at onset of symptoms was 1.5 months (range: 1.0–3.0 months). No infants were able to sit without support at baseline. The results for sitting without support and event-free survival in FIREFISH Part 1 and Part 2 at Month 12 compared with the pre-defined performance criteria are shown in Table 4. The results are presented separately for Part 1 and Part 2 as they included different infants. In addition, the performance criteria were pre-defined for the confirmatory Part 2 only.
The primary efficacy endpoint of the study was successfully met. Twelve of 41 infants (29%; 90% CI 18–43) in Part 2 were able to sit without support for at least 5 s after 12 months of treatment. This proportion was significantly higher than the pre-defined performance criterion of 5% based on well-established natural history data (p < 0.0001). At 12 months of treatment, seven of 21 infants in Part 1 (33%; 90% CI 17–54) were able to sit without support for at least 5 s. All infants who were ongoing in the study had an assessment at Month 12. These results were clinically meaningful, as untreated patients with type 1 SMA are unable to sit without support at any age.
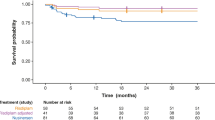
In Part 2, the proportion of infants alive without permanent ventilation at Month 12 was 85% (90% CI 73–92). Three infants died within the first 3 months following study enrolment, and three infants met the endpoint of permanent ventilation. One infant who attended the Month 12 visit a few days early and therefore had not yet reached 12 months from enrolment as of the data-cutoff date was censored in the analysis. The proportion of infants alive without permanent ventilation (85%) was significantly higher than the pre-defined performance criterion of 42% (p < 0.0001). Figure 2 shows a summary of the results for event-free survival.

FIREFISH Part 2: Event-free survival at Month 12 (Intent-to-Treat Population). (Source: Darras et al. [11])
In Part 1, the proportion of infants alive without permanent ventilation at Month 12 was 90% (90% CI 73–97). Two infants died prior to Month 12, and no infants met the definition of permanent ventilation. The median time to death or permanent ventilation was not estimable as few patients had an event. Clinically meaningful and statistically significant improvements were also observed for other key secondary endpoints in FIREFISH Part 2 [11]. The results of these analyses were used to confirm the benefits of risdiplam in type 1 SMA and thus to support the approval and registration of risdiplam in different countries around the world.
The results for Part 1 were used for the initial regulatory filing and are included in the United States prescribing information (EVRYSDI® prescribing information) [21]. This was because we filed early based on the Part 1 results, before the results from Part 2 were available due to the clear divergence shown from natural history and the high unmet medical need.
3 SUNFISH Study: External Control Data from Individual Patient Data
3.1 Design and Methods
3.1.1 Study Design
SUNFISH was an operationally seamless, two-part, multicenter, randomized, double-blind, placebo controlled, Phase 2/3 study, designed to assess the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of risdiplam in a broad patient population including children, teenagers, and adults aged 2–25 years with type 2 and 3 SMA. Figure 3 shows a summary of the SUNFISH study design.

Study design of Part 1 and Part 2 of SUNFISH [*Comprises two age groups (Cohort A: 2–11 years, two dose levels; Cohort B: 12–25 years, three dose levels). †Placebo-treated patients were switched to risdiplam in a blinded manner]
SUNFISH Part 1 was an exploratory, dose-finding study conducted in 51 patients with type 2 and ambulant or non-ambulant type 3 SMA, which determined the dose for use in Part 2. SUNFISH Part 2 was a confirmatory study conducted in 180 patients with type 2 or non-ambulant type 3 SMA [22]. Part 1 and Part 2 had different patients. The primary efficacy endpoint in Part 2 was the change in motor function assessed using the 32-item Motor Function Measure (MFM32) from baseline to Month 12. The MFM32 is a clinician-reported outcome measure that evaluates different levels of motor function in individuals with SMA, from distal fine motor movements of the hands such as using a touch-screen to more complex gross motor function activities such as standing and transfers [23]. The 32 items of this measure were scored using a 4-point Likert scale: 0: cannot initiate the task, 1: can perform the task partially, 2: can perform the task incompletely or completely but imperfectly; 3: can perform the task fully and “normally.” The raw score of the 32 items (range: 0–96) was converted to a 0–100 scale, where lower scores indicate poorer functional ability [24].
The overall study design and choice of endpoints incorporated health authority advice from both the EMA CHMP and the FDA.
3.1.1.1 RWD as a Component of Study Design in SUNFISH
RWD were important for determining the anticipated treatment effect for the primary endpoint (MFM32) in the SUNFISH study as natural history data demonstrated that patients with type 2 and 3 SMA had a decline in motor function over time. Patients with type 2 SMA are able to sit independently and occasionally stand or take a few steps, but are unable to walk independently [15]. Patients with type 3 SMA are able to sit, stand, and walk independently [15], though nearly a third of patients with type 3 SMA lose their ability to walk between the ages of 3 and 28 years [24]. Natural history studies show that patients with type 2 and 3 SMA have a decline in motor function over time, as reported in a number of publications with different validated motor function measures. For example, natural history data demonstrated that the overall slope of decline over time, using the MFM32 total score, is in the range of −0.9 points/year for patients with type 2 SMA and −0.6 points/year for patients with type 3 SMA [24]. In order to gain additional information on the Motor Function Measure (MFM) endpoint in a broad population, the sponsor co-funded a Natural History Study (NatHis-SMA; NCT02391831) in patients with type 2 and 3 SMA with the Institute of Myology, which was designed in collaboration with Patient Advisory Groups (SMA Europe, Cure SMA, SMA Foundation). This study was also critical to ensure access to good-quality data to perform a robust statistical analysis of the MFM endpoint in SUNFISH compared with RWD. RWD were used to generate an external comparator group of patients with type 2 and 3 SMA to give context to the SUNFISH Part 1 results before the placebo-controlled results from SUNFISH Part 2 were available.
3.1.1.2 Selection of External Comparator Sources
The external comparator group used to give context to the SUNFISH Part 1 results comprised of untreated patients with SMA from the NatHis-SMA study and the placebo arm of a Phase 2 trial of olesoxime for the treatment of SMA (NCT01302600).
-
The NatHis-SMA Study was a prospective, multicenter, longitudinal natural history study of patients with type 2 and 3 SMA. The primary objective of this study was to characterize the disease course in patients with type 2 and 3 SMA using standardized evaluations including the MFM. The study included 81 patients aged 2–29 years and was conducted in Europe between 2015 and 2018. The maximum duration of study participation for each patient was 24 months [25].
-
The Olesoxime Study was a Phase 2, parallel-group, placebo-controlled, randomized, double-blind, multicenter study, designed to assess the efficacy and safety of olesoxime over a 2-year period in patients with type 2 or non-ambulant type 3 SMA. The study included 165 patients aged 2–25 years, of whom 57 were randomized to placebo and was conducted in Europe between 2010 and 2013 [26]. The development of olesoxime has since been discontinued.
The NatHis-SMA study and olesoxime study were considered as appropriate sources for generating an external comparator group because of the following similarities to SUNFISH:
-
Similar patient population with type 2 and 3 SMA
-
All studies included the MFM scale as an outcome measure
-
Studies were conducted in Europe with an overlap of some study centers
-
SUNFISH and the olesoxime study were both conducted in the same controlled clinical setting. The olesoxime study was placebo controlled which provided a robust control arm
-
Investigators from SUNFISH and the NatHis-SMA study were trained in the same way with regard to the MFM scale application, hence the assessment was considered similar
-
The first year of follow-up in the NatHis-SMA study occurred just before study enrolment started for SUNFISH, hence patients had similar standards of care and calendar time bias (a bias associated with patients treated in the past progressing differently than those treated today due to changes in standard of care over time) was likely small.
3.1.1.3 Endpoint Used for Comparison with External Control Analysis
Although patient motor function was measured using the MFM in all three studies, the scale was not administered in the same way. In SUNFISH, all patients completed all 32 items (MFM32), whereas in the NatHis-SMA and olesoxime studies, patients aged less than 6 years completed 20 items (MFM20) while patients aged 6 years or older completed all 32 items. MFM total score was chosen to compare motor function between SUNFISH and the external comparator group. MFM total score was derived from the MFM20 total score for all patients aged less than 6 years and from the MFM32 total score for all patients aged 6 years or older. Both scales were transformed to 0–100%. Missing items on the MFM scale were recorded as 0 (i.e., cannot initiate) prior to calculation of total score. Only patients with an MFM assessment at baseline and at least one post-baseline assessment at Month 12 or Month 24 were included in the analysis.
3.1.2 Statistical Methodology
Patients in the external comparator group were weighted using Inverse Probability of Treatment Weighting based upon pre-selected prognostic factors at baseline: age at enrolment; SMA type; SMN2 copy number; ambulatory status; presence of scoliosis; MFM total score at baseline; and MFM scale used. This allowed a comparison of the treated and untreated groups with similar prognostic factors. A propensity score was estimated for each patient using logistic regression incorporating the pre-selected prognostic factors of treatment assignment (risdiplam vs no risdiplam) as independent variables. Patients with missing prognostic factors were excluded. Trimming, defined as removing extreme values and outliers [27], was applied to include only patients with an overlapping distribution of propensity scores. Inverse Probability of Treatment Weighting (IPTW) was applied to the propensity scores to derive weights only for the external comparator group based on the average effect for treated patients approach. The IPTW approach was chosen because it was considered to be an efficient approach where patients with unknown or missing prognostic factors were not included in the weighting procedure. A weight of 1 was given to each of the patients in the risdiplam-treated group and a weight of pj/(1 − pj) was given to the jth patient in the untreated external comparator group, where pj was the propensity score of the jth patient. In other words, the IPTW was applied to the propensity scores to derive weights only for the external control group based on the average effect for the treated patients (ATT) approach. To control for too much influence of patients with very low propensity scores, weights were truncated at the 99th percentile. The truncation was applied after trimming. The variance balance between the treated and untreated groups was assessed pre- and post-weighting. The standardized mean difference (SMD) was computed for each of the covariates to assess if adequate balance had been achieved between the treated and the untreated groups. Adequate balance was assumed if all SMDs were less than 0.25 [28].
The statistical analysis was performed after weighting was applied. Change from baseline in MFM total score was analyzed using a mixed model for repeated measures (MMRM) with treatment; time; time by treatment; MFM total score at baseline by time; and the prognostic factors (age at enrolment; SMA type; SMN2 copy number; ambulatory status; presence of scoliosis; MFM total score at baseline; and MFM scale used) as covariates. Estimated treatment differences in least squares mean change from baseline between patients treated with risdiplam in SUNFISH Part 1, and the external comparator were calculated with corresponding 95% CIs and p-values. The proportions of patients demonstrating improvement (≥3-points change from baseline in MFM total score) were analyzed via logistic regression. In the responder analyses, only patients with an MFM total score at baseline and the post-baseline time point (Month 12 or Month 24) of interest were included in the analysis. Supplemental analyses were performed on each of the external comparator data sources separately.
3.2 Results
After excluding patients with missing information on selected prognostic factors and trimming, 48 patients from the risdiplam arm of SUNFISH Part 1 and 109 patients from the external comparator group who had a valid MFM total score at baseline and Month 12 or Month 24 were included in this analysis. In particular, with the trimming, two treated patients from SUNFISH Part 1 were excluded due to extreme weights (i.e., the prognostic profile of these two patients was not similar to those in the external control group) and no patients from the external control group were excluded. In addition, no patients from either group were excluded due to truncation. After weighting was applied, weights were summed to generate an external comparator group of 49.3. All patients from SUNFISH Part 1 were given a weight of 1, to give a sum of weights of 48.0. The balance between the treated and untreated groups in terms of their baseline prognostic factors profile (covariate balance) was assessed using the SMDs. The SMDs are presented in Fig. 4 for each of the covariates prior to and after weighting.

Results of the covariate balance assessment (standardized mean difference values) of SUNFISH Part 1 compared with an external comparator group. “All” means based on the population from the external comparator group, “Region” means based on the patients included in the analysis and “ATT weighted region” means based on the patients after weighting. ATT average effect for treated patients, MFM motor function measure, SMA spinal muscular atrophy, SMN2 survival of motor neuron 2
Prior to weighting (for “All” and “Region”), except for the MFM total score, all covariates had already achieved balance between the treated patients in SUNFISH Part 1 and the untreated patients in the external comparator group, with their corresponding SMD values lying within the range of −0.25 to +0.25. After weighting (ATT weighted region), variance balance of each covariate was achieved with all SMDs close to 0 and lying within the −0.25 to +0.25 boundaries. Summary results for the baseline characteristics before and after weighting are shown in Table 5. After weighting, the baseline characteristics became more similar and more comparable between SUNFISH Part 1 and the external comparator group. For example, the mean age in the external comparator group was 10.5 years before weighting and 9.4 years after weighting, compared with 9.3 years in the SUNFISH Part 1 treated group. The proportion of patients in each age group also became more balanced between the two groups after weighting.
For the MMRM analysis, patients with a baseline and at least one post-baseline result at Month 12 or at Month 24 were included. In SUNFISH Part 1, all 48 patients had a result at baseline, Month 12 and Month 24. For the external comparator group, 109 patients had a result at baseline and at Month 12, and 79 patients had a result at Month 24. Under the MMRM analysis, for those with a result at Month 12 but not at Month 24, their missing results were assumed as missing at random, i.e., those with missing results behaved similarly to other patients with a similar covariate profile in the same treatment group. As shown in Table 6, at Month 12, the change from baseline in MFM total score was greater in the risdiplam group compared with the external comparator group, and this difference continued to increase at Month 24. The improvement in motor function in patients who received risdiplam treatment compared with the external comparator group was both clinically meaningful and highly statistically significant.
Figure 5 shows that risdiplam treatment in SUNFISH Part 1 led to an increase in mean MFM total score from baseline over 24 months, which was significantly different from the progressive decline observed in the untreated external comparator group [22].

Mean (LS means) change in motor function (as measured using the MFM) over 24 months in patients with Type 2 or 3 SMA in SUNFISH Part 1 compared with an external comparator group. Error bars represent the 95% CI. CI confidence interval, LS least squares, MFM motor function measure, SMA spinal muscular atrophy
After both 12 and 24 months of treatment, a significantly higher proportion of patients treated with risdiplam showed improvement (≥3-point change) in MFM total score compared with the untreated external comparator group (Fig. 6).

Percentage of patients whose MFM score increased by at least 3 points compared with their score at the start of the study, over 12 and 24 months in patients with Type 2 or 3 SMA in SUNFISH Part 1 compared with an external comparator group. MFM motor function measure
These results provided evidence of longer term efficacy of risdiplam in a broad population of patients with type 2 and 3 SMA compared with untreated patients. Supplemental analysis results from the weighted analysis comparing SUNFISH Part 1 with each of the two external comparator sources were generally in agreement with the above results, supporting the robustness of the conclusions. This retrospective weighted analysis comparing patients with type 2 and 3 SMA from SUNFISH Part 1 with two external comparator sources, the NatHis-SMA study and the placebo arm of the olesoxime study, helped to further understand the benefits of risdiplam. In summary, the improvement in motor function with risdiplam treatment in SUNFISH Part 1 was markedly different from the untreated external comparator groups, where the expected decline in function inherent to this progressive disease was observed. The difference observed at Month 12 continued to increase at Month 24, supporting and confirming the benefits of prolonged exposure to risdiplam. This analysis also complements recent results from the placebo-controlled Part 2 of the SUNFISH study, in which the primary endpoint of the change from baseline in MFM32 total score at Month 12 was met. At month 12, the least squares mean (95% CI) change from baseline in MFM32 was 1.36 (0.61–2.11) in the risdiplam group and −0.19 (−1.22–0.84) in the placebo group, with a treatment difference of 1.55 (0.30–2.81, p = 0.016) in favor of risdiplam [29].
4 Discussion
The use of RWD and comparisons with external controls are a rapidly evolving area. In this section, we summarize the benefits of using RWD, especially in a rare disease setting. We also discuss some of the challenges we faced and lessons learned.
4.1 Benefits of Using RWD
The use of RWD was critical in clinical development planning, contextualizing the results and providing substantial evidence of efficacy of a disease-modifying therapy in a rare disease setting. Randomized, double-blind, placebo-controlled (or active controlled if standard of care exists) clinical trials are the gold standard. Randomization avoids systematic differences between groups with respect to known or unknown baseline variables that could affect outcomes. Blinding minimizes the bias due to subject and investigator expectations, and a placebo arm provides internal evidence of assay sensitivity. However, placebo-controlled trials with blinding are not always feasible or appropriate. When an effective therapy that is known to prevent death or irreversible morbidity exists for a particular patient population that population cannot usually be ethically studied in placebo-controlled trials; the particular conditions and populations for which this is true may be controversial (ICH E10 [30]).
Given the severity, rapid decline, and high mortality and morbidity in infants with type 1 SMA described in natural history, a comparator placebo group was not included in the FIREFISH study. In addition, in 2016 when the study started, there were no approved disease-modifying therapies for SMA, which precluded an active comparator arm. In the absence of a control arm, comparisons with external comparators or available natural history data are a valid approach in the development of new treatments for serious and rare diseases. Such comparisons are possible if the natural history of the disease course is well understood, the external comparator group is similar to the treatment group (e.g., with regard to patient characteristics and endpoints), and a large treatment effect is expected to be seen with the study drug. In our case studies, the external comparators were carefully selected based on rigorous criteria described earlier.
RWD were also important in determining the anticipated treatment effect for the primary endpoint (MFM32) in the SUNFISH study as natural history data demonstrated that patients with type 2 and 3 SMA had a decline in motor function over time. In addition to clinical development planning, RWD were important in contextualizing study results for both FIREFISH and SUNFISH.
In FIREFISH, RWD from publications were used to define the performance criteria and benchmarks for success. Infants in FIREFISH attained motor milestones such as sitting without support that would never be achieved in infants with type 1 SMA without treatment. Infants in FIREFISH also achieved improved rates of event-free survival compared with those observed in natural history studies. These results confirmed that the disease course with risdiplam treatment substantially diverged from the natural history of the disease [11]. Infants treated with risdiplam also continued to benefit beyond Year 1. After 3 years of treatment in the FIREFISH study, event-free survival time was greatly improved compared with natural history [31].
In SUNFISH, the improvement in motor function with risdiplam treatment was markedly different from the untreated external comparator group, where the expected progressive decline in function inherent to SMA was observed. The comparison of SUNFISH data (Part 1 and later Part 2) with RWD also confirmed the longer term benefit of risdiplam over 24 months [32]. This was important because the placebo-controlled period in SUNFISH Part 2 was only for 1 year. RWD were used to provide substantial evidence of the efficacy of risdiplam, which was used to support registration and approval of risdiplam in different countries around the world.
The use of RWD for type 1 SMA was pivotal for regulatory decisions. The FDA stated, “…the study [FIREFISH] showed improvements in multiple clinical functional measures compared to the natural history of SMA, including motor function and developmental milestones as well as survival and ventilation free survival.” The RWD used to contextualize the FIREFISH study results were included in the US prescribing information, “Of the patients who were treated with the recommended dosage of EVRYSDI 0.2mg/kg/day, 41% (7/17) were able to sit independently for ≥5 s (BSID-III, item 22). These results indicate a clinically meaningful deviation from the natural history of untreated infantile-onset [type 1] SMA. As described in the natural history of untreated infantile-onset SMA, patients would not be expected to attain the ability to sit independently, and no more than 25% of these patients would be expected to survive without permanent ventilation beyond 14 months of age.” (EVRYSDI® prescribing information [21]).
The use of RWD for type 2 and 3 SMA was supplemental for regulatory decisions. In order to accelerate the regulatory review and approval timelines in the United States, the exploratory dose-finding SUNFISH Part 1 motor function results were compared with RWD to contextualize the study results before the confirmatory SUNFISH Part 2 placebo-controlled results were available. The RWD used to contextualize the results in patients with type 2 and 3 SMA showed clear divergence between patients treated with risdiplam and untreated patients from natural history. The use of RWD also accelerated the filing and approval timelines for risdiplam, which was crucial given the high unmet need and the severe nature of the disease. Approval in the United States was expedited by at least 7 months.
4.2 Challenges
The role of RWD in providing substantial evidence of efficacy was different across regulatory regions. All regions accepted RWD as the benchmark for success for type 1 SMA. However, for patients with type 2 and 3 SMA, the acceptance of RWD as substantial evidence of efficacy differed across regions. In the United States, the FDA accepted an early filing based on FIREFISH Part 1 and SUNFISH Part 1, supplemented with placebo-controlled data from SUNFISH Part 2 during the review. Approval by the FDA was granted in August 2020. In contrast, in Europe, the EMA requested all data from SUNFISH Part 1 and Part 2 at the time of filing. Approval by the EMA was granted in March 2021. To overcome this challenge, a flexible filing strategy was implemented across regions.
At the time of our submission to the FDA in 2019, the recent fit-for-purpose framework for RWD to support regulatory decision making was not in place. Statistical considerations for fit-for-use RWD to support regulatory decision making in drug development can be found in a recent publication [33].
The requirement to provide individual patient-level data also varied across regions. The FDA required all individual patient data in Clinical Data Interchange Standards Consortium (CDISC) format including RWD, whereas the EMA did not. In order to file in the United States, the legacy data from the NatHis-SMA study were converted to CDISC standards for submission to the FDA. A good understanding of CDISC standards and regulatory requirements, technical skills, and upfront planning for this activity were critical to avoid a delay in submission timelines.
Although every effort was made to derive the performance criteria from infants who were as similar as possible to the FIREFISH study population, there were some limitations and challenges associated with this approach. These included potential differences in patient characteristics between the historical cohorts and study population; the limited number of studies available for some endpoints or small sample sizes of the historical cohorts; and studies being conducted in a limited number of countries or sites. For some endpoints, a performance criterion could not be derived as no suitable natural history studies were available. The results observed for motor function and motor milestone endpoints were consistent across natural history studies, but for other endpoints such as event-free survival, the results were more variable. These endpoints are more dependent on individual clinician practice and caregiver preferences, in particular pulmonary and nutritional intervention strategies, and so may vary across countries and sites. Different definitions of permanent ventilation were also used in each study, including differences in the number of hours of ventilation per day, the number of consecutive days with this level of respiratory support, and the type of respiratory support provided. Despite these limitations, it should be noted that standard of care guidelines were considered during the selection of study sites, and the FIREFISH results were clearly differentiated from the natural course of type 1 SMA described in the literature.
Subtle differences in definitions of endpoints were also challenging for the SUNFISH and external comparator comparison. For example, MFM was administered differently across studies depending on a patient’s age. To overcome this issue, an MFM total score was derived using either the MFM32 or MFM20 items depending on the patient’s age. Missing data were also a challenge, with more missing data in the external comparator data sources. For example, for those included in the analysis, in the external comparator group, 73% of patients (i.e., 79 out of 109 patients) completed the Month 24 assessment, while in SUNFISH, all patients (i.e., all 48 patients included in the analysis) completed the Month 24 assessment. To deal with this, sensitivity analyses were performed to assess the robustness of the results using different methods for dealing with missing data.
4.3 Lessons Learned
The risdiplam clinical development program highlighted a number of key planning considerations that can be applied to other drug development programs in which RWD may play an important role, as follows:
-
1.
Incorporate RWD into the Clinical Development Plan
We engaged with regulators early and pushed regulatory boundaries with robust arguments. The FDA eventually accepted a single-arm design for FIREFISH despite a preference for a placebo-controlled study. The overall study designs and choice of endpoints for both FIREFISH and SUNFISH incorporated Health Authority advice from both the EMA CHMP and the FDA. We pre-planned the statistical analysis for the confirmatory parts, documented this in a statistical analysis plan, shared the statistical analysis plan with Health Authorities, and asked for feedback in advance of filing.
Collaborating with healthcare providers and Patient Advisory Groups (PAGs) was also important to the success of the program. In particular, PAGs were actively involved in designing the NatHis-SMA study, advising on the interpretation of the clinical study results from both FIREFISH and SUNFISH, and validating the meaningfulness of the results from both a patient and caregiver perspective. This feedback was further supported by data collected by PAGs (e.g., a treatment expectation survey conducted by SMA Europe [34]), which were incorporated into our regulatory dossiers to further contextualize our data.
-
2.
Take Steps to Reduce Bias When Using RWD
We selected data sources that reflected considerations from ICH E10 to minimize bias when using external controls [30], including selecting a control population as similar as possible to the study population and selecting more than one external control. In FIREFISH, the identified RWD studies were documented and ranked based on the level of similarity of the patient population to the expected population in the FIREFISH study. The endpoints were also robust and objectively measured.
In SUNFISH, two independent studies, the NatHis-SMA study and olesoxime study, were selected based on their similarities to SUNFISH, such as the same efficacy outcome measure (change in MFM total score) and similar patient population (type 2 and 3 SMA and age range). In addition, some of the study centers collecting the external comparator data also enrolled patients in SUNFISH, and the first year of follow-up in the NatHis-SMA study occurred just before trial enrolment started for SUNFISH. These common features mitigated potential biases relating to different endpoint bias; selection bias; regional bias; different standards of care; and calendar time bias. The RWD in the external comparator group were weighted based on key prognostic factors to ensure the populations were as similar as possible. The important prognostic factors that were used to perform the weighting were defined a priori and described in the statistical analysis plan. It was also important to ensure that these data were available. For example, if severity of scoliosis was an important prognostic factor and was not collected in the RWD, it could not be used for the calculation of propensity scores.
It is also recommended to perform sensitivity analyses. For SUNFISH, the comparison was performed on the pooled external comparator data and separately for each study to support the robustness of the conclusions. A compilation of the different sources of biases from various types of external controls, such as those used in this chapter, and the potential mitigation steps can be found in a recent publication [35].
5 Conclusion
Risdiplam has now been approved in more than 90 countries worldwide, including the United States and the EU for the treatment of SMA in a broad patient population. In this case study, the use of RWD was pivotal in clinical development planning, contextualizing the results and providing substantial evidence of the efficacy of risdiplam, a disease-modifying therapy in a rare disease setting.
The results from the comparison of SMA patients treated with risdiplam from both the FIREFISH and SUNFISH studies versus RWD clearly diverged from the natural history of the disease and were clinically meaningful.
RWD were also critical in our filing strategy and led to significantly reduced approval timelines in the United States in a rare disease setting with a high unmet medical need. RWD were more widely accepted for objective endpoints with well-defined natural history (e.g., motor milestones and survival).
References
Verhaart IE, Robertson A, Wilson I et al (2017) Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy–a literature review. Orphanet J Rare Dis 12(1): 1–15. https://doi.org/10.1186/s13023-017-0671-8.
Lefebvre S, Bürglen L, Reboullet S et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80(1):155–165. https://doi.org/10.1016/0092-8674(95)90460-3.
Hamilton G & Gillingwater TH (2013). Spinal muscular atrophy: going beyond the motor neurons. Trends Mol Med 19(1):40–50. https://doi.org/10.1016/j.molmed.2012.11.002.
Mercuri E, Finkel RS, Muntoni F et al (2018) Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 28(2):103–115. https://doi.org/10.1016/j.nmd.2017.11.005.
Munsat TL, Davies KE (1992) International SMA consortium meeting. Neuromuscul Disord 2(5–6):423–8. https://doi.org/10.1016/s0960-8966(06)80015-5.
Zerres K, Rudnik-Schöneborn S (1995) Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol 52(5):518–523. https://doi.org/10.1001/archneur.1995.00540290108025.
D’Amico A, Mercuri E, Tiziano FD et al (2011) Spinal muscular atrophy. Orphanet J Rare Dis 6:71. https://doi.org/10.1186/1750-1172-6-71.
Darras BT (2015) Spinal muscular atrophies. Pediatr Clin North Am 62:743–766. https://doi.org/10.1016/j.pcl.2015.03.010.
Ratni H, Ebeling M, Baird J et al (2018) Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J Med Chem 61(15):6501–6517. https://doi.org/10.1021/acs.jmedchem.8b00741.
Baranello G, Darras BT, Day JW et al (2021) Risdiplam in Type 1 Spinal Muscular Atrophy. N Engl J Med 384(10):915–923. https://doi.org/10.1056/NEJMoa2009965.
Darras BT, Masson R, Mazurkiewicz-Beldzinska M et al. (2021) Risdiplam-treated patients with Type 1 Spinal Muscular Atrophy versus historical controls. N Engl J Med 385(5):427–435. https://doi.org/10.1056/NEJMoa2102047.
Bayley N. (2006). Bayley Scales of Infant and Toddler Development– Third Edition. Psychological Corporation.
Finkel RS, McDermott MP, Kaufmann, P et al (2014) Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 83(9):810–817. https://doi.org/10.1212/WNL.0000000000000741.
Kolb SJ, Coffey CS, Yankey J et al (2017) Natural history of infantile-onset spinal muscular atrophy. Ann Neurol 82(6):883–891. https://doi.org/10.1002/ana.25101.
Finkel R, Bertini E, Muntoni F, Mercuri E; ENMC SMA Workshop Study Group. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord 2015 Jul;25(7):593–602.
Eichler HG, Bloechl-Daum B, Bauer P et al (2016) “Threshold-crossing”: A useful way to establish the counterfactual in clinical trials?, Clinical Pharmacology and Therapeutics, 100(6):699–712. https://doi.org/10.1002/cpt.515.
Wang CH, Finkel RS, Bertini ES (2007) Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 22(8):1027–1049. https://doi.org/10.1177/0883073807305788.
Krosschell KJ, Kissel JT, Townsend EL et al (2018) Clinical trial of L-Carnitine and valproic acid in spinal muscular atrophy type I. Muscle Nerve 57(2):193–199. https://doi.org/10.1002/mus.25776.
Kolb SJ, Coffey CS, Yankey J et al (2016) Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study. Ann Clin Transl Neurol 3(2):132–145. https://doi.org/10.1002/acn3.283.
De Sanctis R, Coratti G, Pasternak A et al (2016) Developmental milestones in type I spinal muscular atrophy. Neuromuscul Disord 26(11):754–759. https://doi.org/10.1016/j.nmd.2016.10.002.
EVRYSDI® prescribing information: https://www.gene.com/download/pdf/evrysdi_prescribing.pdf. Accessed July 2022.
Mercuri E, Baranello G, Boespflug-Tanguy O et al (2022) Risdiplam in Types 2 and 3 spinal muscular atrophy: a randomised, placebo-controlled, dose-finding trials followed by 24 months of treatment. Eur J Neurol [in press]. https://doi.org/10.1111/ene.15499.
Berard C, Payan C, Hodgkinson I and Fermanian J (2005) A motor function measure for neuromuscular diseases. Construction and validation study. Neuromuscul Disord 15(7):463–470. https://doi.org/10.1016/j.nmd.2005.03.004.
Vuillerot C, Payan C, Iwaz J et al (2013) Responsiveness of the motor function measure in patients with spinal muscular atrophy. Arch Phys Med Rehabil 94(8):1555–1561. https://doi.org/10.1016/j.apmr.2013.01.014.
Annoussamy M, Seferian AM, Daron A et al (2021) Natural history of Type 2 and 3 spinal muscular atrophy: 2-year NatHis-SMA study. Ann Clin Transl Neurol 8(2):359–373. https://doi.org/10.1002/acn3.51281.
Bertini E, Dessaud E, Mercuri E et al (2017) Safety and efficacy of olesoxime in patients with type 2 or non-ambulant type 3 spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 16(7):513–522. https://doi.org/10.1016/S1474-4422(17)30085-6.
Stürmer T, Webster-Clark M, Lund JL et al (2021) Propensity Score Weighting and Trimming Strategies for Reducing Variance and Bias of Treatment Effect Estimates: A Simulation Study. Am J Epidemiol 190(8):1659–1670. https://doi.org/10.1093/aje/kwab041.
Stuart EA (2010). Matching methods for causal inference: A review and a look forward. Stat Sci 25(1):1–21. https://doi.org/10.1214/09-STS313.
Mercuri E, Deconinck N, Mazzone E et al (2022) Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol 21(1):42–52. https://doi.org/10.1016/S1474-4422(21)00367-7.
European Medicines Agency. ICH-E10 Choice of control group in clinical trials. January 20, CPMP/ICH/364/96. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-10-choice-control-group-clinical-trials-step-5_en.pdf
Servais L et al (2022) FIREFISH Parts 1 and 2: Safety and efficacy of risdiplam in Type 1 spinal muscular atrophy (3-year data). Abstract presented at 14th European Paediatric Neurology Society (EPNS) Congress, Glasgow, UK, 28 April – 2 May 2022.
Servais L et al (2022) SUNFISH Part 2: 24-month efficacy of risdiplam compared with external control comparators. Abstract presented at Muscular Dystrophy Association (MDA) Clinical and Scientific Conference, Nashville, TN, USA, 13–16 March 2022.
Levenson M, He W, Dharmarajan S et al (2022) Statistical consideration for fit-for-use real-world data to support regulatory decision making in drug development, Journal of Biopharmaceutical Statistics [in press].
Gusset N, Stalens C, Stumpe E et al (2021) Understanding European patient expectations towards current therapeutic development in spinal muscular atrophy. Neuromuscul Disord 31(5):419–430. https://doi.org/10.1016/j.nmd.2021.01.012.
Burger HU, Gerlinger, C, Harbron C et al (2021) The use of external controls: To what extent can it currently be recommended? Pharm Stat 20(6):1002–1016. https://doi.org/10.1002/pst.2120.
Acknowledgments
The risdiplam clinical development program was funded by F. Hoffmann-La Roche Ltd in collaboration with the SMA Foundation and PTC Therapeutics. We thank all the patients who participated in the studies and the staff of clinical sites around the world. In addition, we thank Carol Reid who contributed to the statistical design and interpretation, Fani Petridis for valuable insights and Helena Bailes of Chrysalis Medical Communications for medical writing assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
McIver, T., El-Khairi, M., Yeung, W.Y., Pang, H. (2023). The Use of Real-World Data to Support the Assessment of the Benefit and Risk of a Medicine to Treat Spinal Muscular Atrophy. In: He, W., Fang, Y., Wang, H. (eds) Real-World Evidence in Medical Product Development . Springer, Cham. https://doi.org/10.1007/978-3-031-26328-6_20
Download citation
DOI: https://doi.org/10.1007/978-3-031-26328-6_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-26327-9
Online ISBN: 978-3-031-26328-6
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)