
Abstract
Recent progress has led to nucleotide-based gene silencing strategies as a potential therapy for spinocerebellar ataxia type 3 (SCA3) and other hereditary ataxias. But recent setbacks for antisense oligonucleotide (ASO) therapy in another CAG repeat disease, Huntington’s disease (HD), remind us of the importance of broadening the search for potential routes to disease-modifying therapies. Here we review alternative approaches. We begin by introducing some of the complexities and nuances of SCA3 that can help guide therapeutic strategies. We then review the myriad biological pathways that are potentially druggable in SCA3, and the current use of genetic and small molecule screens to identify targets and possible therapeutic agents. Given the importance of employing model systems in which the disease gene and protein are assessed at physiological concentrations in the human genomic context, we discuss the emerging importance and challenges of using human stem cells to study disease mechanisms and test therapeutic targets and novel pharmacologic agents. We conclude by considering SCA3 and related SCAs beyond biochemical pathways and as diseases of impaired connectivity, emphasizing the largely untapped potential of modulating neuronal activity and brain connectivity for possible disease-modifying therapies.
For inclusion in: Trials of Cerebellar Ataxia: From Disease Models to Therapeutics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Now is an exciting time in SCA3 research. Decades of hard work by many groups to understand disease mechanisms and develop treatments could soon lead to rationally based disease-modifying therapies for this progressive and fatal neurodegenerative disease. As we write this in 2022, at least one company is performing a human clinical trial of nucleotide-based gene silencing therapy as a potential disease-modifying treatment in SCA3. As contributors to the preclinical work establishing gene silencing as a treatment strategy (McLoughlin et al. 2018), we fervently hope these gene-directed therapies find their way into the clinic. The role of nucleotide-based gene silencing therapies in SCA3 is well covered in Dr. McLoughlin’s chapter in this collection (McLoughlin 2022). Here, we focus instead on other approaches to therapy. We do not know where the best therapies will come from in SCA3, and researchers must adopt a broad perspective in order to capture the most effective agents (Costa 2020; Da Silva et al. 2019). Doing so requires that scientists recognize the complexities of SCA3 disease pathogenesis and consider alternative approaches to ameliorating symptoms and altering disease course. It is also worth considering, even at the basic science stage, how addressing these alternative mechanisms may be tracked to develop biomarkers of disease and target engagement.
What follows represents a selective review of the current state of therapeutic development for SCA3, emphasizing research published in the past 5 years. We begin by introducing some of the complexities and nuances of SCA3 that can help guide therapeutic strategies. We then review the use of genetic and small molecule screens to identify targets and potential therapies, discuss the importance and challenges of using human stem cells to study disease mechanisms and test therapeutic targets, and conclude by considering the SCAs as dysfunctional circuitry and reviewing the largely untapped potential of modulating neuronal activity and brain connectivity.
2 Complex Issues in SCA3 to Consider as Therapies Are Sought
2.1 SCA3 Is Dominantly Inherited, but Elements Beyond Toxic Gain of Function Likely Contribute
Like other SCAs, SCA3 is an autosomal dominant disorder in which a single copy of the disease gene harboring the CAG repeat expansion causes disease (Paulson and Shakkottai 2020; Costa and Paulson 2012). In rare individuals carrying two expanded alleles, the disease is more severe with symptoms beginning early in life (Shang et al. 2018). An overwhelming preponderance of evidence from model systems and human disease tissue supports the view that the polyglutamine expansion in the disease protein ATXN3 drives one or more toxic effects in the nervous system (Paulson et al. 2017; McLoughlin et al. 2020). In other words, toxic properties conferred on the gene and encoded protein (so called “gain of function”) drive the propagation of the disease. Moreover, the gene itself does not appear to be essential since mice lacking Atxn3 appear essentially normal. It should be noted that while the exact interactions and dependencies of wild-type ATXN3 and polyglutamine-expanded ATXN3 are not fully understood, there is little evidence that loss of gene function is a major contributor to human disease. Thus, unlike in Huntington’s disease (HD), where the gene protein is essential for early brain development, current antisense nucleotide-based approaches to reduce ATXN3 expression likely do not suffer from concerns that marked reduction in the ATXN3 protein itself will be deleterious.
And yet a contribution of loss of gene function to disease has not been formally excluded in SCA3. In a diseased brain, ATXN3 tends to concentrate in the nucleus of neurons, disappearing from the cytoplasm where it normally is more abundant. Potentially, this “loss” of ATXN3 from the cytoplasm contributes to disease pathogenesis. Moreover, recent studies reveal retinal involvement in SCA3 and a role for ATXN3 in the retina (Toulis et al. 2020, 2022), where the loss of the protein perturbs retinal structure and highlights a role for ATXN3 in regulating cilia and phagocytosis, both of which are fundamental to photoreceptor function. Further data indicate that ATXN3 modulates molecular features in certain cancers and participates in DNA repair (Chakraborty et al. 2020; Gong et al. 2021; Herzog et al. 2020; Zhuang et al. 2021). Accordingly, we should be mindful of the potential for deleterious effects of gene-silencing strategies that reduce ATXN3 levels too effectively and therapeutics targeting ATXN3 itself should be developed and analyzed with careful consideration of function of the native protein as well.
2.2 SCA3 Is a Neurodegenerative Disease, but Neurons Are Not the Only Involved Cell Type
The accumulation of ATXN3 nuclear inclusions in neurons of SCA3 disease brain, coupled with the fact that specific brain regions undergo profound atrophy in SCA3, placed attention on neurons as the principal cell type affected in SCA3. Recent studies, however, point to oligodendrocytes as an unexpected major contributor as well (Schuster et al. 2022). In that work, transcriptional changes in a mouse model of SCA3 revealed oligodendrocytes as the cell population most affected early on. While it remains to be seen whether ATXN3-mediated effects in oligodendrocytes drive disease processes, it will be important that therapeutic strategies not be focused solely on neurons lest nonneuronal populations prove paramount in targeting. Lessons might be learned from Alzheimer’s disease, where an intense early focus on neurons delayed recognition of the critical disease contributions of astrocytes and microglia. In SCA3, we now know that oligodendrocytes are likely important, and recent data suggest that microglia are involved as well (Campos et al. 2022). As inflammation is proving to be an important contributor to other neurodegenerative diseases, it is critical that we define potential direct roles in SCA3 for microglia and astrocytes in disease pathogenesis. Finally, as we consider the roles of polyglutamine-expanded ATXN3 in various cell types, it behooves us to evaluate the levels and function of wild-type ATXN3 in response to the disease state, as this may provide insight into future treatment effects.
2.3 Proteotoxicity of the ATXN3 Disease Protein Is Important, but Not the Only Contributor to Disease
The CAG repeat expansion in SCA3 encodes an elongated stretch of the amino acid glutamine in ATXN3. Studies ranging from recombinant protein in vitro to cell-based models and various animal models of disease consistently have shown that expanded polyglutamine-containing ATXN3 is prone to aggregate and form intracellular inclusions in select brain regions and cell populations (reviewed in Costa and Paulson 2012; Paulson et al. 2017). Similarly, polyglutamine-containing disease proteins in SCAs 1, 2, 6, 7, and 17 also aggregate when the disease protein contains a polyglutamine expansion (Paulson et al. 2017). Accordingly, the prevailing view is that the primary toxic species in SCA3 is the ATXN3 protein containing the expansion. Understandably, then, mutant ATXN3 has been the target of many strategies to define disease-modifying therapies. That said, some evidence in model systems suggest toxicity at the RNA level occurs with higher expansion sizes (Li et al. 2008), and GC-rich repeat expansions are prone to undergo repeat-associated non-methionine (RAN) translation, in which protein can be translated in all three reading frames across the repeat leading to polyserine and polyalanine RAN products as well as the “expected” polyglutamine (Cleary et al. 2018). Evidence is building for RAN translation in Huntington’s disease (Cleary et al. 2018) and SCA8 (Perez et al. 2021) and is hinted at in SCA31 (Ishiguro et al. 2021), but remains limited in the more common CAG repeat expansion SCAs. One advantage of nucleotide-based gene silencing strategies is that they will reduce levels of both the RNA transcript and the disease protein. As a result, potential toxicity from RNA effects or RAN translation would also be mitigated. Therapies focused on the ATXN3 protein, or affected pathways downstream of the polyglutamine-expanded protein, would fail to address these potential upstream toxic effects.
2.4 SCA3 Affects the Brain, but Little Is Known About Disease in Other Organs
SCA3 is unquestionably a neurodegenerative disorder. Hence, effective therapies must penetrate the brain or be directly delivered to the nervous system. Clinically, the evidence for significant organ involvement beyond the central and peripheral nervous system is limited. The disease protein, however, is expressed ubiquitously. Thus, there may be subtler subclinical effects associated with the expression of the disease gene that have been missed to date. Retinal involvement in SCA3 is a recently described example (Toulis et al. 2022). And many SCA3 patients develop significant peripheral neuropathy that might not be fully addressed by a CNS-directed therapy. Accordingly, therapies such as orally delivered small molecules that can act throughout the body have a potential advantage over therapies that are directly delivered to the CNS, provided that the orally delivered compound displays favorable pharmacokinetics in the nervous system.
2.5 The ATXN3 Disease Protein Maybe Small, but Its Function Is Complex and Far-Reaching
ATXN3 is a specialized deubiquitinase that preferentially cleaves longer poly-ubiquitin chains (reviewed in Costa and Paulson 2012). A relatively small protein, ATXN3 readily moves in and out of the nucleus and participates in diverse ubiquitin-dependent processes at many places in the cell. Its various functions include working with ubiquitin ligases to modulate ubiquitin chain composition, regulating aggresome production, participating in DNA repair processes, and modulating autophagy and endocytic processes (Costa and Paulson 2012; Chakraborty et al. 2020; Dantuma and Herzog 2020; Rosselli-Murai et al. 2020; Zeng et al. 2020). Some properties of the protein suggest that ATXN3 is a unique deubiquitinase that participates in protein quality control pathways (e.g., ubiquitin-proteasome system and autophagy) that are themselves perturbed in SCA3. Precisely how the deubiquitinase activity and diverse functions of ATXN3 are perturbed in disease remains a work in progress. In principle, its deubiquitinase activity represents a therapeutic target, but currently, we do not know if the enzyme activity of polyglutamine-expanded (mutant) ATXN3 is deleterious or beneficial compared to its native function and may have wide-reaching ramifications in cellular proteostasis. A potential negative consequence of inhibiting the enzyme activity of ATXN3 is that it could produce an avid ubiquitin-binding protein that lacks the ability to cleave the ubiquitin chains it binds; essentially the protein could act like a dominant-negative isoform on client proteins with potentially deleterious effects.
2.6 Studies of Overexpressed or Transgenic ATXN3 Have Shed Important Light on Disease Mechanisms but May Not Mirror the Human Disease State
Most of the experiments providing insight into SCA3 disease mechanisms have relied on high-level expression of the disease protein in cellular and animal models, and often not from the endogenous locus. Overexpression of the disease protein accelerates the appearance of molecular and neuropathological phenotypes in short-lived model systems, whereas expression of the protein at endogenous levels has more subtle effects that can become impractical in a laboratory setting. But as helpful as overexpression systems have been, they run the risk of leading to artifacts and spurious findings that do not mirror the physiological state of cells in humans with SCA3. Similarly, relying on transgenic expression models poses possible confounding factors of altered regulation. Particularly as scientists search for genes or compounds that regulate levels of SCA disease proteins, it will be important to screen for such regulatory factors in model systems that mimic the native physiological levels and actions of the disease gene and protein. Doing so poses its own challenges, but it avoids the hazard of focusing on regulatory factors that might simply work to mitigate an overexpression artifact. The emergence of human stem cells derived from SCA3 patients or embryos (discussed further below) allows for the examination of the disease protein and its effects under the most germane physiological conditions.
2.7 ATXN3 Maybe the Obvious Target in SCA3, but Targets Beyond and Downstream of ATXN3 Also Need to Be Explored
Observed effects beyond the polyglutamine expansion suggest additional, potentially targetable pathways in SCA3. For example, single- and double-strand DNA breaks accumulate in cellular models of HD (Illuzi et al. 2008), and overexpression of DNA repair enzymes can ameliorate the disease phenotype in mouse models (Enokido et al. 2010). In SCA3, ATXN3 promotes the activity of the 3′ phosphatase and 5′ kinase PNKP (Chatterjee et al. 2014) and has been linked to nonhomologous end-joining (NHEJ) repair of double-strand DNA breaks (Chakraborty et al. 2020). The DNA repair pathway may also offer a route through which to target repeat expansion instability as small molecules that contract CAG repeats have been published (Nakamori et al. 2018). Epigenetic mechanisms have also been implicated in SCA3 as DNA methylation within the ATXN3 promoter inversely correlates with the age of onset and intergenerational repeat instability (Wang et al. 2016).
2.8 Most Research in SCA3 Has Focused on Disease Effects at the Cellular Level, but Network-Level Effects Remain Understudied
Addressing the widespread reaches of polyglutamine-expanded ATXN3 demands a comprehensive approach to improve patient care. While SCA3 is clearly a systemic disease with extra-CNS complications, even within the nervous system we must expand our disease framework. We have greatly improved our understanding of the biochemical mechanisms at play in SCA3 and the other SCAs, but this has not led to novel drugs beyond gene-silencing therapies, as mentioned above. It is therefore equally valuable to consider SCAs as diseases of impaired network connectivity. We are in a unique position to learn from the success of deep brain stimulation (DBS) in Parkinson’s disease (PD) and essential tremor (ET) as a demonstration of the viability of electrophysiological perturbation for symptomatic management. A pharmacological approach with specificity against individual ion channels offers the opportunity to elicit similar therapeutic effects without surgical intervention.
3 Screens to Identify Targetable Pathways for Potential Therapy
On the one hand, the dominant-toxic nature of disease pathogenesis in SCA3 conceptually simplifies the route to therapy: agents that can reduce identified toxic species have strong therapeutic potential. On the other hand, many factors conspire to make the search for SCA3 therapies more complicated, including the chronicity of disease; diverse functions of the disease protein, including participation in stress pathways that are themselves implicated in disease; involvement of multiple cell types; and, the dynamic nature of the underlying mutation, a variably sized repeat expansion that could have deleterious effects at the DNA, RNA, and protein levels. Many researchers have successfully interrogated specific cellular pathways (e.g., molecular chaperones and autophagy) as potential routes to therapy, and nucleotide-based strategies that directly target the SCA3 disease gene have seen tremendous progress recently (see McLoughlin 2022). Here we focus instead on small-molecule and genetic screens that have taken a more unbiased approach to identify compelling therapeutic agents. The number of such screens to date is limited, partly because the development of efficient and reproducible screening platforms takes considerable time and effort. With the advent of clustered regularly interspaced short palindromic repeat (CRISPR)-based approaches that enable rapid full-genome screens to find regulators of predefined readouts (say, ATXN3 levels) and the emergence of human stem cell lines harboring trackable epitope tags engineered into an endogenous gene of interest, we expect that the range, depth, and quality of screens for potential SCA3 therapies will ramp up.
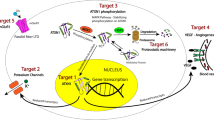
Figure 1 highlights the many biological factors that could be modulated to alter levels of ATXN3, specific functions, or downstream effects tied to the protein. Arguably all are worth pursuing, and in many cases, promising initial data already support further investigations. These include boosting autophagy, enhancing chaperone activity, inhibiting specific protein-protein interactions, and blocking the proteolytic cleavage of ATXN3 that produces putative toxic fragments (recent examples include Lee et al. 2021; Robinson et al. 2021; Vasconcelos-Ferreira et al. 2022). Many such studies were driven by a candidate target assessment and not linked to larger-scale screens. As outlined below, published screens to date have been performed to find compounds or genes that reduce levels of ATXN3 or reduce the toxic effects of ATXN3. We again stress the need to evaluate wild-type and polyglutamine-expanded ATXN3 separately and together to better understand potential treatment effects. Ideal screens will incorporate assays by which total and mutant ATXN3 levels can be measured in response to any cellular perturbation.

Biological routes to potential disease-modifying treatment in SCA3. Cellular levels and activity of the disease protein ATXN3 are likely regulated by numerous biological pathways, illustrated here. Unbiased genetic and small molecule screens can identify genes or compounds that alter ATXN3 concentration, subcellular localization, or deubiquitinase activity, each of which may influence the toxicity of the disease protein. Alternatively, existing compounds or candidate genes implicated in specific pathways can be directly tested for disease-modifying effects. Current nucleotide-based gene silencing approaches act by reducing ATXN3 transcript levels
Using an immortalized nonneuronal cell line that stably expresses luciferase-tagged ATXN3, Costa and colleagues (Costa et al. 2016) screened a small-molecule library largely containing FDA-approved drugs. A series of secondary screens in other cell lines and in cerebellar brain slices from a SCA3 mouse model identified the atypical antipsychotic aripiprazole as an ATXN3 modulator. Short-term exposure of SCA3 mice to this drug resulted in a marked reduction of aggregated ATXN3. The mechanism by which aripiprazole acted on ATXN3 was unclear, particularly since the starting cell line, derived from HEK293 cells, was not known to express neurotransmitter receptors by which the drug might work. A recent follow-up study in a nematode model of SCA3 suggests that aripiprazole acts through dopamine and serotonin receptors to reduce the motor effects of ATXN3 (Jalles et al. 2022). To date, aripiprazole has not been tested in a clinical trial of patients with SCA3. Because some SCA3 patients develop parkinsonism and long-term exposure to aripiprazole can elicit a movement disorder among other side effects, aripiprazole usage in SCA3 will have to be monitored closely.
This same cell-based screening platform was used in a druggable genome screen to identify genes that regulate levels of ATXN3 (Ashraf et al. 2020). In this screen, 317 candidate genes were identified and 100 were selected for validation, 33 of which were confirmed in multiple secondary assays. Of these, 15 were independently validated in separate cell lines as modulators of ATXN3 levels. Further analysis in a Drosophila model of disease confirmed the effects of several genes in vivo, and one gene, FBXL3, was shown to reduce ATXN3 levels through ubiquitin-dependent processes. Additional analyses revealed a molecular network linked to tumor necrosis factor/nuclear factor-Kappa B and to the extracellular signal-regulated kinases 1 and 2 (ERK1/2), which are pharmacologically targetable. Further analysis of these identified genes in mouse models and human disease tissue will be required to confirm their relevance to human disease. Limitations to this screen include the fact that an immortalized nonneural cell line was used that overexpresses ATXN3. Despite these limitations, the screen demonstrated the feasibility of further genetic screens in more physiologically relevant cellular and animal models.
Maciel and colleagues performed a small molecule screen in a nematode (Caenorhabditis elegans) model of SCA3 that displays ATXN3-mediated immotility (Teixeira-Castro et al. 2015). From this screen, the antidepressant citalopram surfaced as a drug that could rescue motility. Moreover, a follow-up study in a mouse model of SCA3 showed that citalopram reduced ATXN3 aggregation (Ashraf et al. 2019). Thus, citalopram, a widely prescribed antidepressant, holds promise as a potential disease-modifying therapy in SCA3. The small size, short life cycle, and genetic tractability of the nematode make it a strong organismal platform for genetic and compound screening. Leveraging these advantages, Maciel and colleagues conducted further studies in the nematode that highlight the importance of serotonin signaling as a modulator of ATXN3 proteotoxicity, and support future human clinical trials of serotonin-acting agents such as specific 5-HT1A agonists (Pereira-Sousa et al. 2021).
The nematode model of SCA3 has proved useful in other genetic and compound screens. For example, a screen of nearly 4000 compounds, many FDA-approved, revealed five lead compounds that restored motility in the model (Fardghassemi and Parker 2021). Three of these compounds were found to modulate a key transcriptional regulator of autophagy, TF EB/HLH-30. This screen not only highlights the relevance of autophagy to SCA3 (Fig. 1), in part as a potential clearance mechanism for mutant ATXN3, but also supports the use of specific autophagy-regulating small molecules as possible therapeutic agents. In another recent study employing this nematode model, an RNA interference (RNAi) screen of nearly 400 transcription factor genes identified one with apparent neuroprotective activities, FKH-2/FOXG1 (Fardghassemi et al. 2021). The mechanism of neuroprotection by this transcription factor has not yet been defined.
The above studies underscore the need for additional large-scale screens to identify therapeutic targets and possible therapeutic agents. What is currently lacking are human stem cell-based screening platforms that can capture changes in ATXN3 when expressed at endogenous levels. CRISPR-based genetic screens, coupled with automated cell sorting that employs a robust, sensitive anti-ATXN3 antibody or ligand to quantify ATXN3 levels, could capture genes that up- or down-regulate ATXN3. A suitably tagged form of ATXN3, genetically engineered into the endogenous ATXN3 locus of human stem cells, might enable a physiologically relevant screening platform for small molecules, provided that the signal to detect ATXN3 is sensitive and specific.
4 Human Stem Cells as a New Tool for Mechanistic and Translational Studies
Animal models have proven to be an excellent platform for assessing pathogenic mechanisms and testing candidate therapeutics for SCA3 and other CAG repeat expansion diseases including HD. Nevertheless, these models have limitations. Human stem cells provide a compelling opportunity to expand basic and translational investigations of SCA3 and other repeat expansion diseases. Favorable characteristics include the fact that the human disease gene is expressed at endogenous levels from its native locus within the full human genomic context. Here we review the use of stem cells in SCA3. Because HD has been more extensively examined using stem cells than any of the SCAs, we also refer to the more robust HD literature to highlight the potential of stem cells.
Both human induced pluripotent stem cells (iPSCs) and human embryonic stem cells (hESCs) are beginning to play significant roles in the study of various repeat expansion diseases. iPSCs are generated by reprogramming adult somatic cells (e.g., skin fibroblasts and peripheral blood mononuclear cells) to an embryonic-like state. Yamanaka originally demonstrated that iPSCs could be derived from fibroblasts by transducing various transcription factors (Oct4, Sox2, Klf4, and c-Myc) into murine embryonic and adult fibroblasts (Takahashi and Yamanaka 2006). More recently, nonintegrative gene delivery approaches have also been established (Al Abbar et al. 2020). The resultant iPSCs, in turn, can be differentiated into various cell types, permitting cell type-specific analyses. The intermediate pluripotent state can also be skipped through direct reprogramming, which allows adult somatic cells to transition directly from one lineage to another. For example, Ambasudhan et al. (2011) used this approach to generate functional neurons directly from adult human fibroblasts. In contrast, hESCs are not generated through reprogramming. As the name implies, hESCs are derived from the inner cell mass of blastocyst-stage embryos and can give rise to all somatic cell types in the embryo (Lee and Lee 2011).
Stem cells hold promise as model systems in which to discover potential therapeutic targets. For both SCA3 and HD, various nucleotide-based approaches (e.g., siRNAs, shRNAs, microRNAs, and ASOs) and small molecules have successfully reduced the mutant transcript and/or disease protein levels (Alves et al. 2008, 2010; Ashraf et al. 2019; Carroll et al. 2011; Costa et al. 2016; Estevez-Fraga et al. 2020; Komatsu 2021; McLoughlin et al. 2018; Moore et al. 2019; Sun et al. 2014; Tabrizi et al. 2019). While nucleotide-based gene silencing may lead to disease-modifying therapies for SCA3 and other dominantly inherited ataxias, ataxia researchers need to continue seeking and developing alternative therapeutic strategies. A promising avenue for uncovering such strategies is to identify the molecular mechanisms regulating mutant protein production, stability, and/or clearance as highlighted in Fig. 1.
Stem cells as a therapeutic strategy may also hold promise. In their review, Sivandzade and Cucullo (2021) explain that stem cells could either replace damaged cells with differentiated ones or promote an environment conducive to regeneration through neurotrophic support. Alterations to the CNS environment elicited by stem cells also might prevent damage to the remaining healthy neurons and glia. We caution that the field of regenerative medicine is relatively young and further advances are needed for stem cell therapy to enter the clinical setting as a treatment option for SCA3 and other polyglutamine expansion diseases.
For a variety of reasons, both iPSCs and hESCs are excellent tools for seeking and developing alternative therapeutic strategies. First, stem cells obtained from patients can recapitulate many disease-associated phenotypes, including transcriptional dysregulation, mutant protein aggregation, lysosomal dysfunction, and neuronal vulnerability (Cheng et al. 2013; Hansen et al. 2016; He et al. 2021; Jaworska et al. 2016; Jeon et al. 2012; Koch et al. 2011; Lu and Palacino 2013; Moore et al. 2019; Niclis et al. 2013; Tousley and Kegel-Gleason 2016). Second, stem cells can be differentiated into various somatic cell types, which enables the examination of disease-specific cell type vulnerability. Third, stem cells express the disease gene of interest (e.g., ATXN3 or HTT) at endogenous levels from the native locus, eliminating nonphysiological effects and spurious results that may be observed when disease genes are overexpressed. Fourth, in stem cells, signal transduction pathways that contribute to disease development and progression may be uncovered more readily. Finally, isogenic cell lines can be established as critical controls for stem cell lines and their generation is currently underway in SCA3. Previous research has revealed that single nucleotide polymorphisms in the genome of different patient samples can affect research results. The generation of multiple isogenic cell lines for a disease enables one to distinguish critical disease gene-associated findings from effects due simply to variations in genetic background. Two approaches can be used to generate isogenic pairs of cell lines. The first approach removes the mutation from patient-specific cells, while the second introduces the mutation into wild-type cells. Transcription activator-like effector nucleases (TALENs) and CRISPR/Cas9 are frequently used as gene-editing tools and have successfully been employed to generate isogenic lines for SCA3 and HD (Dabrowska et al. 2020; He et al. 2021; Lu and Palacino 2013; Malankhanova et al. 2017, 2020; Ooi et al. 2019). These control lines are essential when performing compound screens or identifying differentially expressed genes and proteins that are associated with a disease. Not only can isogenic lines enable understanding of the contribution of the ATXN3 CAG expansion to disease-linked cellular phenotypes compared to otherwise identical cells harboring a normal CAG repeat, scientists can also genetically increase the CAG repeat expansion to enhance the disease phenotype and better understand clinical heterogeneity and even the phenomenon of anticipation.
To date, numerous scientific questions pertaining to HD have been addressed in iPSCs and hESCs. Malankhanova et al. (2020) generated isogenic lines using HD-iPSCs reprogrammed from fibroblast clones. The resulting cells were differentiated into striatal medium spiny neurons, which are known to be selectively vulnerable in HD. While iPSC-derived neurons harboring the CAG-expanded HTT allele did not develop huntingtin protein aggregates, they did accumulate ultrastructural defects detectable by electron microscopy. The authors suggested these defects may occur early in the pathogenesis of HD, before aggregate formation. Niclis et al. (2013) compared two HD-hESC lines to a wild-type control line both in the undifferentiated state and during differentiation into forebrain neurons. The two HD-hESC lines had CAG-expanded HTT alleles of 37 repeats and 51 repeats, respectively. Whether as undifferentiated or differentiating cells, HD-hESC and wild-type lines were indistinguishable with respect to growth, viability, pluripotent gene expression, mitochondrial activity, and capacity to differentiate into neurons. Furthermore, the expression levels of genes known to be perturbed in HD were similar across the hESC lines. The authors did note, however, that neurons derived from HD-hESCs with the larger repeat expansion (51 CAG repeats) displayed elevated glutamate-evoked responses. These studies confirm that stem cells with a CAG-expanded HTT allele maintain pluripotent parameters and can differentiate into various somatic cells. In addition, the neuronal progeny may display phenotypes associated with HD.
In studies of SCA3, similar questions have been tackled using iPSCs and hESCs derived from patients. Koch et al. (2011) reported the first SCA3-iPSC line, which paved the way for the development of additional lines. For example, He et al. (2021) used CRISPR/Cas9-mediated homologous recombination to correct SCA3-iPSC lines; for each line, the abnormal CAG repeat expansion was replaced with a normal repeat length. Neurons derived from the SCA3-iPSC lines exhibited several phenotypic abnormalities: polyglutamine protein aggregates; decreased mitochondrial membrane potential and glutathione expression; and, increased reactive oxygen species, intracellular Ca2+ concentrations, and lipid peroxidase malondialdehyde levels. Importantly, neurons generated from genetically corrected SCA3-iPSC lines did not display these abnormal phenotypes. This study highlights the ability of isogenic lines to unveil phenotypes associated with the CAG-expanded ATXN3 allele.
Moore et al. (2019) reported the first NIH-approved SCA3-hESC line, which recapitulated certain molecular features of human disease, most notably the production of aggregates. Their study highlighted the potential for SCA3-hESCs to function as a cell-based disease model. When SCA3-hESC cells were exposed to a validated anti-ATXN3 antisense oligonucleotide (ASO), the ASO reduced the expression of mutant ATXN3, reversing ATXN3 aggregation and aggresome formation in SCA3-hESCs. As discussed further below, whereas the phenotype of ATXN3 aggregation was spontaneously observed in the SCA3-hESC line, similar aggregation in differentiated SCA3-iPSC lines required that the cells be stressed through depolarization (Koch et al. 2011). Whether this discrepancy reflects a difference in the ability of iPSC versus hESC lines to mirror molecular features of disease will require further head-to-head comparisons of multiple iPSC and hESC lines harboring the same mutation.
There are numerous discrepancies with respect to protein aggregation in stem cell models of CAG repeat diseases. Most of the HD literature, for example, suggests that HD-iPSCs do not exhibit protein aggregation spontaneously or even after exposure to cellular stressors (e.g., hydrogen peroxide, 3-methyladenine, and repetitive exposure to glutamate) (Jaworska et al. 2016). Both Jeon et al. (2012) and Cheng et al. (2013), however, were able to trigger aggregate formation in stem cells by exposing them to a proteasome inhibitor. It is also unclear whether neurons derived from HD-iPSCs contain aggregates (Jaworska et al. 2016; Jeon et al. 2012; Malankhanova et al. 2020; Zhang et al. 2010). With respect to SCA3, several groups have reported excitation-induced aggregation in neurons, but not in iPSCs and other non-neuronal cells (Hansen et al. 2016; He et al. 2021; Jaworska et al. 2016; Koch et al. 2011). The literature pertaining to HD-hESCs is inconsistent (Lu and Palacino 2013; Niclis et al. 2009; Ooi et al. 2019). Lu and Palacino (2013) were able to trigger the formation of aggregates after transfecting cells with cDNA encoding HTT exon1 fragments with various polyglutamine lengths (Q23 for wild-type; Q73 and Q145 for HD). Otherwise, the literature suggests that HD-hESCs and neuronal progeny often do not develop HTT aggregates. In contrast, Moore et al. (2019) found that SCA3-hESCs developed protein aggregates in the absence of cellular stressors. The undifferentiated state and early passage number (or young age) associated with stem cells and neuronal progeny, respectively, may account for the absence of protein aggregation in many of these studies (Ooi et al. 2019). Nekrasov et al. (2016) observed HTT inclusions in older (6-month-old) neurons derived from HD-iPSCs. Following intracerebral transplantation in mice, Jeon et al. (2012) found that neurons derived from HD-iPSCs contained aggregates when assessed 33 or 40 weeks later.
Stem cell-based research on HD and SCA3 over the past decade has not shed much light on somatic CAG repeat instability, which recently has surfaced as a potential contributor to disease pathogenesis (Paulson 2018). With respect to HD, most literature suggests that iPSCs do not exhibit repeat instability during long-term passaging (Camnasio et al. 2012; Jaworska et al. 2016; Jeon et al. 2012). Interestingly, Mattis et al. (2012) found that iPSC-derived neural stem cells (NSCs) displayed mild repeat instability following long-term passaging: by passage 26, the CAG-expanded HTT allele in one of the lines contained 118 repeats rather than 110. The CAG repeat length in SCA3-iPSCs appears to remain stable following long-term passaging and differentiation into neurons (Jaworska et al. 2016; Ou et al. 2016). Limited information is available for repeat stability in hESCs. Most studies suggest that, like HD-iPSCs, HD-hESCs do not exhibit repeat instability. Neuronal progeny, on the other hand, often have minor repeat instability (Niclis et al. 2009; Ooi et al. 2019). To date, repeat length instability in SCA3-hESCs following long-term passaging and differentiation has not been assessed rigorously. Potentially, high fidelity of DNA replication in stem cells may prevent repeat instability from occurring. Alternatively, the process of somatic repeat instability might require greater time and more accumulated cell divisions than is typically assessed in stem cell studies employing undifferentiated or differentiated cell populations (Ooi et al. 2019).
Evidence to date raises the intriguing possibility that iPSC and hESC lines differ in ways that would favor one versus the other for mechanistic and translational studies. Epigenetic differences are the most likely reason why the two cell types may differ (Narsinh et al. 2011). Direct comparisons of disease-relevant phenotypes (e.g., repeat instability, protein aggregation, and transcriptional changes) are needed to determine the relative value of each stem cell type as a disease model for SCA3 or other repeat expansion ataxias.
As two-dimensional cell culture is somewhat limited, three-dimensional (3D) organoids may offer new insights into the pathogenesis of SCA3 and other CAG repeat diseases. Organoids are generated through the aggregation of stem cells. When exposed to various signaling molecules, aggregated stem cells differentiate into multiple cell types. Through self-organization and self-renewal, these 3D structures mimic organ-specific cellular patterns and functions. In their review, Hou and Kuo (2022) highlight the strengths associated with CNS organoids, including their organ-like spatial cell arrangements and microenvironment. Unlike 2D cultures, the presence of multiple cell types with proper orientation and adjacency in CNS organoids promotes the occurrence of paracrine- and direct contact-mediated interactions similar to those that occur in vivo. Research involving 2D culture facilitated the discovery of cell-intrinsic mechanisms that promote disease development and progression. CNS organoids provide an opportunity to build on this understanding by examining pathological events in the context of neuronal networks. This system could reveal how repeat instability, aggregation propensity, and cellular vulnerability vary between cell types. The use of organoids could also uncover early neurodevelopmental aspects of HD, SCA3, and other CAG repeat diseases, which remain understudied. Lastly, as the HD ASOs are thought to have been toxic due to high concentrations required to reach deep brain tissue, organoids offer an early screening step to evaluate such possible adverse effects.
Recent developments have enhanced the utility of CNS organoids as a model for neurodegenerative diseases. For example, distinct types of CNS organoids have successfully been established: cortical, striatal, midbrain, cerebellar, and motor neuron. The process of generating 3D structures can be divided into two sequential, induction steps. The first and second steps result in the formation of neuroepithelium and region-specific lineages, respectively. By modifying the signaling molecules present during the second step, the developmental patterning characteristic to a specific CNS region can be initiated (Bang et al. 2021; Hou and Kuo 2022). Fused region-specific brain organoids are also becoming more prevalent. For example, Chang et al. (2020) highlighted multiple studies that examine fused dorsal–ventral-patterned organoids as models for brain development. They point out that a caveat associated with CNS organoids is the lack of a circulatory system. Scientists have attempted to resolve this issue by transplanting CNS organoids into an in vivo environment. Another approach entails engineering hESCs that ectopically express erythroblast transformation specific (ETS) variant transcription factor 2 (ETV2) and therefore can form vascular-like structures. As summarized by Tidball et al. (2022), most methods for establishing CNS organoids result in 3D structures with multiple neural rosettes. These rosettes correlate with neural tube formation during embryonic development and, unfortunately, promote structural heterogeneity between organoids. To reduce this heterogeneity and enhance their biological relevance, Tidball et al. (2022) developed a protocol to establish CNS organoids with a single neural rosette organizing center.
The use of CNS organoids to study both SCA3 and HD is in its infancy. Depla et al. (2020) generated iPSC-derived cerebral organoids from a healthy control patient and used them to examine the viral transduction efficiency and distribution of rAAV5, a commonly used AAV serotype. The rAAV5 was engineered to deliver microRNA targeting ATXN3 mRNA and was able to lower the expression of wild-type ATXN3 protein by 30%. The organoid literature pertaining to HD is more extensive. For example, Conforti et al. (2018) generated cerebral organoids using iPSCs derived from healthy and HD patients. When compared to the control organoids, the HD organoids exhibited defects in striatal and cortical fate differentiation, cytoarchitecture, and neuronal maturation.
Overall, as is true for mouse models, no one stem cell model system is likely sufficient when studying the underlying molecular mechanisms of HD, SCA3, or other SCAs. Each model system has strengths and weaknesses. For example, the process of generating iPSCs from somatic cells may result in the loss of epigenetic modifications that are critical for disease development and progression (Narsinh et al. 2011). Due to ethical reasons, it is more difficult to acquire hESCs compared to iPSCs. With respect to CNS organoids, the process of generating them is expensive and time intensive. Furthermore, this model system is still relatively new and in the process of being fine-tuned.
5 Impaired Connectivity as a Druggable SCA Target: Insight into Systems-Based Approach
While many therapeutic and mechanistic efforts remain focused on the root cause of SCAs, it has become apparent that analyzing downstream consequences of the genetic perturbation represents an important and unique opportunity for therapeutic intervention. Simply put, it makes physiological sense that correcting the genetic mutation in monogenic diseases like the SCAs should prevent future pathogenesis. This approach, however, is tempered by several caveats. The recent failure of two trials in HD using ASO therapy to reduce the expression of the disease gene gives insight into potential pitfalls (Kingwell 2021; Kwon 2021). Both trials against HD were stopped prematurely due to failure to meet primary endpoints, with one trial (Generation HD1) actually resulting in worsened patient outcomes. Which patients are most likely to benefit from genetic corrective therapy? And is targeting both wild-type and mutant isoforms ideal or prohibitive? In HD, it is clear that targeting both isoforms may be deleterious, though, as above, ATXN3 knockout models do not suffer similar phenotypes as HD models do. Preclinical models using anti-ATXN3 ASOs improve the motor phenotype, but it remains unclear if this will translate to human benefits. When is the best time to administer a gene-targeting intervention? What about patients that are already symptomatic? What about clinical heterogeneity in patients with similar ATXN3 repeat lengths? While these outstanding questions are being explored in ongoing clinical research, it is worth considering how common downstream pathways might be exploited for therapeutic intervention. Such a strategy would ideally complement ongoing efforts at gene-based therapies. In this section, we examine SCA3 and other SCAs as diseases of impaired network connectivity that offer novel targets for intervention.
One major hallmark of SCAs is cerebellar Purkinje cell (PC) degeneration (Durr 2010; Paulson et al. 2017). It is also clear that impaired cerebellar Purkinje neuron firing is a shared feature in several SCA models. Moreover, alterations in PC spiking due to ion channel dysfunction occur concurrently with motor impairment and well before cell loss (Shakkottai et al. 2011; Hansen et al. 2013; Chopra et al. 2018). Recent work tied these changes in PC membrane excitability to reduced expression and activity of large-conductance calcium-activated potassium (BK) channels in SCAs 1, 2, and 7 (Dell’Orco et al. 2015, 2017; Chopra et al. 2018; Stoyas et al. 2020). In SCA1 transgenic mice, BK expression is transcriptionally repressed. Restoring BK channel expression or function rescues membrane hyperexcitability, improves motor dysfunction, and reduces PC degeneration (Dell’Orco et al. 2015; Chopra et al. 2018). These data suggest that ion channel dysfunction may drive neurotoxicity. Therefore, augmenting BK activity represents an attractive therapeutic strategy in SCA1 and possibly other SCAs, and is supported by recently published work (Srinivasan et al. 2022). An alternative strategy to restore appropriate PC spiking is combining chlorzoxazone and baclofen, which has proven successful in both a SCA1 mouse model and a limited open-label trial of SCA1 patients (Bushart et al. 2021a). Despite not modulating BK function directly, this dual compound treatment strategy is expected to move into a Phase 2 Clinical Trial soon.
These findings raise several unanswered questions. For example, what specific channel should be targeted? As described above, BK (KCNMA1) channel activation is a compelling strategy for SCAs 1, 2, and 7. In contrast, SCA3 PCs are hyperexcitable and exhibit depolarization block (Shakkottai et al. 2011), and thus the irregular firing in SCA3 could theoretically be rectified by either a sodium channel blocker or a potassium channel activator. Recordings from Purkinje neurons in SCA3 transgenic mice, though, have demonstrated a clear reduction in voltage-gated potassium channel activation, specifically Kv1.6 (Kcna6) and Kv3.3 (Kcnc3) (Shakkottai et al. 2011; Bushart et al. 2021b) (Fig. 2a). BK (KCNMA1) is transcriptionally repressed in SCA1, but the mechanisms driving Kcna6 and Kcnc3 dysfunction in SCA3 are less clear. Intraventricular delivery of an ASO against the ATXN3 gene in SCA3 transgenic mice restored proper Purkinje excitability and recovered expression levels of Kcna6 and Kcnc3 (Bushart et al. 2021b), suggesting that regulation of channel expression lies downstream of ATXN3 function and is perturbed by the polyglutamine expansion.

Electrophysiological phenotyping in spinocerebellar ataxia. (a) The aberrant cerebellar spiking activity in SCAs can be traced to dysfunction or repressed expression of voltage-gated potassium channels. In SCAs 1, 2, and 7, decreased expression and function of Kcnma1 (BK) seems to be the root driver of slow and irregular spiking and therefore represents an ideal target. In SCA3, however, Kcnma1 expression remains unchanged while Kcna6 and Kcnc3 levels and activity are diminished. (b) Although the cerebellar cortex, and particularly the Purkinje cell layer, is predominantly affected in SCAs, SCA3 and several other SCAs can show marked involvement of the deep cerebellar nuclei. Gradient shading from lighter to darker indicates an increasing degree of involvement. (Image created with Biorender and Kenhub)
Although the majority of ion channel dysfunction in SCAs has focused on Purkinje neurons since they may be the most affected cell type (Kasumu and Bezprozvanny 2012), there is variability among SCAs in which cell populations are affected within the cerebellum. While mouse models of SCAs 1, 2, and 7 show irregular and slow PC spiking, underscoring the selective vulnerability of PCs in these CAG/polyglutamine repeat SCAs, SCA3 mouse models show irregular spiking not only in PCs (Shakkottai et al. 2011) but also in neurons of the deep cerebellar nuclei (DCN) (Mayoral-Palarz et al. 2022) (Fig. 2b). Interestingly, while irregular spiking in PCs and DCN neurons correlates with motor symptom onset, there is no worsening of this electrophysiological perturbation as clinical ataxia symptoms progress in a mouse model of the disease (Bushart et al. 2021a, b; Mayoral-Palarz et al. 2022). This lack of correlation suggests that the progressive ataxia seen in SCA3 is due, at least in part, to dysfunction outside the cerebellum. While neuropathological and imaging studies have confirmed atrophy of the brainstem, cerebral cortex, thalamus, and basal ganglia in SCA3 patients (Rüb et al. 2008; Rezende et al. 2018), the degree of electrophysiological perturbations in these regions remains unknown.
Many questions remain unanswered about the range and relevance of electrophysiological perturbations in SCA3 and other SCAs. That said, targeting neuronal dysfunction in the SCAs through specific ion channel modulation is a viable therapeutic strategy that should be pursued both as independent symptomatic and disease-modifying therapy, and as a complement to approaches aimed at the root genetic cause and other biochemical pathways.
More globally, addressing neuronal circuitry is important to consider for other SCAs as we move beyond a biochemical view of disease to a more systems-wide view. Examining synaptic transmission and neurotransmitter levels may uncover additional targets for SCA3 and other SCAs. For example, glutamate receptor activity is perturbed in multiple SCAs (Meera et al. 2016, 2017) and is thought to lead to aberrant calcium signaling, potential depolarization block, and cytotoxicity. This mechanism has led to a Phase 3 Trial of troriluzole, a glutamate reuptake activator, the results of which are imminently pending.
6 Conclusion and the Future of SCA3 Therapeutics
The last few decades have seen an explosion in our understanding of the biological forces driving SCA3 pathogenesis. With this new knowledge and the recent advent of patient-derived biomarkers, the field is primed to move more therapeutics into the clinical phase than ever. For effective SCA3 therapeutics to advance to the next stage, we expect numerous leaps and shifts to occur. As mentioned above, patient-derived stem cell and organoid models will need to move to the main stage as platforms to understand disease biology and test novel therapeutics. The amenability of stem cells to genetic modification also opens the door to genetic therapies beyond ASOs including CRISPR-mediated correction of the CAG/polyglutamine expansion. Such efforts, already underway in cellular models, will prove challenging in patients. As CRISPR-based delivery and genetic manipulation techniques improve, however, we hope that such strategies will emerge as a viable option for patient intervention. While targeting the root cause of SCA3—the CAG/polyglutamine expansion—will likely serve as the base of many SCA3 therapeutic regimens, it is also clear that monotherapy may not be sufficient. Pharmacological agents focused on protein quality control machinery, bioenergetics, protein-protein interactions, and network connectivity will serve as valuable complements to further probe disease biology and improve patient outcomes.
References
Al Abbar A, Ngai SC, Nograles N, Alhaji SY, Abdullah S. Induced pluripotent stem cells: reprogramming platforms and applications in cell replacement therapy. Biores Open Access. 2020;9(1):121–36.
Alves S, Nascimento-Ferreira I, Auregan G, Hassig R, Dufour N, Brouillet E, Pedroso de Lima MC, Hantraye P, de Almeida LP, Déglon N. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS One. 2008;3(10):e3341.
Alves S, Nascimento-Ferreira I, Dufour N, Hassig R, Auregan G, Nóbrega C, Brouillet E, Hantraye P, Pedroso de Lima MC, Déglon N, de Almeida LP. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3? Hum Mol Genet. 2010;19(12):2380–94.
Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, Lipton SA, Ding S. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 2011;9(2):113–8.
Ashraf NS, Duarte-Silva S, Shaw ED, Maciel P, Paulson HL, Teixeira-Castro A, Costa M d C. Citalopram reduces aggregation of ATXN3 in a YAC transgenic mouse model of Machado-Joseph disease. Mol Neurobiol. 2019;56(5):3690–701.
Ashraf NS, Sutton JR, Yang Y, Ranxhi B, Libohova K, Shaw ED, Barget AJ, Todi S v, Paulson HL, Costa M d C. Druggable genome screen identifies new regulators of the abundance and toxicity of ATXN3, the spinocerebellar ataxia type 3 disease protein. Neurobiol Dis. 2020;137:1–13.
Bang S, Lee S, Choi N, Kim HN. Emerging brain-pathophysiology-mimetic platforms for studying neurodegenerative diseases: brain organoids and brains-on-a-chip. Adv Healthc Mater. 2021;10(12):1–27.
Bushart DD, Huang H, Man LJ, Morrison LM, Shakkottai VG. A chlorzoxazone-baclofen combination improves cerebellar impairment in spinocerebellar ataxia type 1. Mov Disord. 2021a;36:622–31.
Bushart DD, Zalon AJ, Zhang H, Morrison LM, Guan Y, Paulson HL, Shakkottai VG, McLoughlin HS. Antisense oligonucleotide therapy targeted against ATXN3 improves potassium channel–mediated Purkinje neuron dysfunction in spinocerebellar ataxia type 3. Cerebellum. 2021b;20:41–53.
Camnasio S, Carri AD, Lombardo A, Grad I, Mariotti C, Castucci A, Rozell B, Riso PI, Castiglioni V, Zuccato C, Rochon C, Takashima Y, Diaferia G, Biunno I, Gellera C, Jaconi M, Smith A, Hovatta O, Naldini L, et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis. 2012;46(1):41–51.
Campos AB, Duarte-Silva S, Fernandes B, das Neves SP, Marques F, Teixeira-Castro A, Neves-Carvalho A, Monteiro-Fernandes D, Portugal CC, Socodato R, Summavielle T, Ambrósio AF, Relvas JB, Maciel P. Profiling microglia in a mouse model of Machado-Joseph disease. Biomedicine. 2022;10(2):237.
Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, Hayden MR. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–85.
Chakraborty A, Tapryal N, Venkova T, Mitra J, Vasquez V, Sarker AH, Duarte-Silva S, Huai W, Ashizawa T, Ghosh G, MacIel P, Sarkar PS, Hegde ML, Chen X, Hazra TK. Deficiency in classical nonhomologous end-joining-mediated repair of transcribed genes is linked to SCA3 pathogenesis. Proc Natl Acad Sci U S A. 2020;117(14):8154–65.
Chang Y, Kim J, Park H, Choi H, Kim J. Modelling neurodegenerative diseases with 3D brain organoids. Biol Rev. 2020;95(5):1497–509. https://doi.org/10.1111/BRV.12626.
Chatterjee A, Saha S, Chakraborty A, Silva-Fernandes A, Mandal SM, Neves-Carvalho A, Liu Y, Pandita RK, Hegde ML, Hegde PM, Boldogh I, Ashizawa T, Keoppen AH, Pandita TK, Maciel P, Sarkar PS, Hazra TK. The role of mammalian DNA end-processing enzyme polynucleotide kinase 3′-phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet. 2014;11:e1004749.
Cheng PH, Li CL, Chang YF, Tsai SJ, Lai YY, Chan AWS, Chen CM, Yang SH. miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am J Hum Genet. 2013;93(2):306–12.
Chopra R, Bushart DD, Shakkottai VG. Dendritic potassium channel dysfunction may contribute to dendrite degeneration in spinocerebellar ataxia type 1. PLoS One. 2018;13:e0198040.
Cleary JD, Pattamatta A, Ranum LPW. Repeat-associated non-ATG (RAN)translation. J Biol Chem. 2018;293(42):16127–41.
Conforti P, Besusso D, Bocchi VD, Faedo A, Cesana E, Rossetti G, Ranzani V, Svendsen CN, Thompson LM, Toselli M, Biella G, Pagani M, Cattaneo E. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc Natl Acad Sci U S A. 2018;115(4):E762–71.
Costa M d C. Recent therapeutic prospects for Machado-Joseph disease. Curr Opin Neurol. 2020;33(4):519–26.
Costa M d C, Paulson H. Toward understanding Machado-Joseph disease. Prog Neurobiol. 2012;97(2):239–57.
Costa M, Ashraf NS, Fischer S, Yang Y, Schapka E, Joshi G, Mcquade TJ, Dharia RM, Dulchavsky M, Ouyang M, Cook D, Sun D, Larsen MJ, Gestwicki JE, Todi S v, Ivanova MI, Paulson HL. Unbiased screen identifies aripiprazole as a modulator of abundance of the polyglutamine disease protein, ataxin-3. Brain. 2016;139(11):2891–908.
da Silva JD, Teixeira-Castro A, Maciel P. From pathogenesis to novel therapeutics for spinocerebellar ataxia type 3: evading potholes on the way to translation. Neurotherapeutics. 2019;16(4):1009–31.
Dabrowska M, Ciolak A, Kozlowska E, Fiszer A, Olejniczak M. Generation of new isogenic models of Huntington’s disease using CRISPR-Cas9 technology. Int J Mol Sci. 2020;21(5):1–13.
Dantuma NP, Herzog LK. Machado-Joseph disease: a stress combating deubiquitylating enzyme changing sides. Adv Exp Med Biol. 2020;1233:237–60.
Dell’Orco JM, Wasserman AH, Chopra R, Ingram MAC, Hu Y-S, Singh V, Wulff H, Opal P, Orr HT, Shakkottai VG. Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J Neurosci. 2015;35:11292–307.
Dell’Orco JM, Pulst SM, Shakkottai VG. Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum Mol Genet. 2017;26:3935–45.
Depla JA, Sogorb-Gonzalez M, Mulder LA, Heine VM, Konstantinova P, van Deventer SJ, Wolthers KC, Pajkrt D, Sridhar A, Evers MM. Cerebral organoids: a human model for AAV capsid selection and therapeutic transgene efficacy in the brain. Mol Ther Methods Clin Dev. 2020;18:167–75.
Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9(9):885–94.
Enokido Y, Tamura T, Ito H, Arumughan A, Komuro A, Shiwaku H, Sone M, Foulle R, Sawada H, Ishiguro H, Ono T, Murata M, Kanazawa I, Tomilin N, Tagawa K, Wanker EE, Okazawa H. Mutant huntingtin impairs Ku70-mediated DNA repair. J Cell Biol. 2010;189:425–43.
Estevez-Fraga C, Flower MD, Tabrizi SJ. Therapeutic strategies for Huntington’s disease. Curr Opin Neurol. 2020;33(4):508–18.
Fardghassemi Y, Parker JA. Overexpression of FKH-2/FOXG1 is neuroprotective in a C. elegans model of Machado-Joseph disease. Exp Neurol. 2021;337:1–10.
Fardghassemi Y, Maios C, Parker JA. Small molecule rescue of ATXN3 toxicity in C. elegans via TFEB/HLH-30. Neurotherapeutics. 2021;18(2):1151–65.
Gong B, Zhang J, Hua Z, Liu Z, Thiele CJ, Li Z. Downregulation of ATXN3 enhances the sensitivity to AKT inhibitors (perifosine or MK-2206) but decreases the sensitivity to chemotherapeutic drugs (etoposide or cisplatin) in neuroblastoma cells. Front Oncol. 2021;11:1–15.
Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–83.
Hansen SK, Stummann TC, Borland H, Hasholt LF, Tümer Z, Nielsen JE, Rasmussen MA, Nielsen TT, Daechsel JCA, Fog K, Hyttel P. Induced pluripotent stem cell – derived neurons for the study of spinocerebellar ataxia type 3. Stem Cell Res. 2016;17(2):306–17.
He L, Wang S, Peng L, Zhao H, Li S, Han X, Habimana J d D, Chen Z, Wang C, Peng Y, Peng H, Xie Y, Lei L, Deng Q, Wan L, Wan N, Yuan H, Gong Y, Zou G, et al. CRISPR/Cas9 mediated gene correction ameliorates abnormal phenotypes in spinocerebellar ataxia type 3 patient-derived induced pluripotent stem cells. Transl Psychiatry. 2021;11(1):1–13.
Herzog LK, Kevei É, Marchante R, Böttcher C, Bindesbøll C, Lystad AH, Pfeiffer A, Gierisch ME, Salomons FA, Simonsen A, Hoppe T, Dantuma NP. The Machado–Joseph disease deubiquitylase ataxin-3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell. 2020;19(1):1–15.
Hou PS, Kuo HC. Central nervous system organoids for modeling neurodegenerative diseases. IUBMB Life. 2022;74:812–25. https://doi.org/10.1002/iub.2595.
Illuzi J, Yerkes S, Parekh-Olmedo H, Kmiec EB. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res. 2008;87(3):733–47.
Ishiguro T, Nagai Y, Ishikawa K. Insight into spinocerebellar ataxia type 31 (SCA31) from drosophila model. Front Neurosci. 2021;15:648133.
Jalles A, Vieira C, Pereira-Sousa J, Vilasboas-Campos D, Mota AF, Vasconcelos S, Ferreira-Lomba B, Costa MD, da Silva JD, Maciel P, Teixeira-Castro A. Aripiprazole offsets mutant ATXN3-induced motor dysfunction by targeting dopamine D2 and serotonin 1A and 2A receptors in C. elegans. Biomedicine. 2022;10(2):1–24.
Jaworska E, Kozlowska E, Switonski PM, Krzyzosiak WJ. Modeling simple repeat expansion diseases with iPSC technology. Cell Mol Life Sci. 2016;73(21):4085–100.
Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, Shim SH, Choi C, Chang DJ, Kwon J, Oh SH, Shin DA, Kim HS, Do JT, Lee DR, Kim M, Kang KS, Daley GQ, Brundin P, Song J. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30(9):2054–62.
Kasumu A, Bezprozvanny I. Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum. 2012;11(3):630–9.
Kingwell K. Double setback for ASO trials in Huntington disease. Nat Rev Drug Discov. 2021;20:412–3.
Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, Doerr J, Ladewig J, Mertens J, Tüting T, Hoffmann P, Klockgether T, Evert BO, Wüllner U, Brüstle O. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature. 2011;480(7378):543–6.
Komatsu H. Innovative therapeutic approaches for Huntington’s disease: from nucleic acids to GPCR-targeting small molecules. Front Cell Neurosci. 2021;15:785703.
Kwon D. Failure of genetic therapies for Huntington’s devastates community. Nature. 2021;593(7858):180.
Lee JE, Lee DR. Human embryonic stem cells: derivation, maintenance and cryopreservation. Int J Stem Cells. 2011;4(1):9–17.
Lee JH, Lin SY, Liu JW, Lin SZ, Harn HJ, Chiou TW. n-Butylidenephthalide modulates autophagy to ameliorate neuropathological progress of spinocerebellar ataxia type 3 through mTOR pathway. Int J Mol Sci. 2021;22(12):1–21.
Li LB, Yu Z, Teng X, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453(7198):1107–11.
Lu B, Palacino J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013;27(5):1820–9.
Malankhanova TB, Malakhova AA, Medvedev SP, Zakian SM. Modern genome editing technologies in Huntington’s disease research. J Huntingtons Dis. 2017;6(1):19–31.
Malankhanova T, Suldina L, Grigor’eva E, Medvedev S, Minina J, Morozova K, Kiseleva E, Zakian S, Malakhova A. A human induced pluripotent stem cell-derived isogenic model of Huntington’s disease based on neuronal cells has several relevant phenotypic abnormalities. J Pers Med. 2020;10(4):1–26.
Mattis VB, Svendsen SP, Ebert A, Svendsen CN, King AR, Casale M, Winokur ST, Batugedara G, Vawter M, Donovan PJ, Lock LF, Thompson LM, Zhu Y, Fossale E, Atwal RS, Gillis T, Mysore J, Li JH, Seong I, et al. Induced pluripotent stem cells from patients with Huntington’s disease show CAG repeat expansion associated phenotypes. Cell Stem Cell. 2012;11(2):264–78.
Mayoral-Palarz K, Neves-Carvalho A, Maciel P, Khodakhah K. Cerebellar neuronal dysfunction accompanies early motor symptoms in spinocerebellar ataxia type 3. Dis Model Mech. 2022;15(8):dmm049514.
McLoughlin HS. Antisense oligonucleotide therapy against SCA3. In: Trials for cerebellar ataxias: from cellular models to human therapies. Springer; 2022. (in press).
McLoughlin HS, Moore LR, Chopra R, Komlo R, McKenzie M, Blumenstein KG, Zhao H, Kordasiewicz HB, Shakkottai VG, Paulson HL. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann Neurol. 2018;84(1):64–77.
McLoughlin HS, Moore LR, Paulson HL. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol Dis. 2020;134:1–11.
Meera P, Pulst SM, Otis TS. Cellular and circuit mechanisms underlying spinocerebellar ataxias. J Physiol. 2016;594:4653–60.
Meera P, Pulst S, Otis T. A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. eLife. 2017;6:e26377.
Moore LR, Keller L, Bushart DD, Delatorre RG, Li D, McLoughlin HS, Costa M d C, Shakkottai VG, Smith GD, Paulson HL. Antisense oligonucleotide therapy rescues aggresome formation in a novel spinocerebellar ataxia type 3 human embryonic stem cell line. Stem Cell Res. 2019;39:1–13.
Nakamori M, Panigrahi GB, Lanni S, Gall-Duncan T, Hayakawa H, Tanaka H, Luo J, Otabe T, Li J, Sakata A, Caron M-C, Joshi N, Prasolava T, Chiang K, Masson J-Y, Wold MS, Wang X, Lee MYWT, Huddleston J, Munson KM, Davidson S, Layeghifard M, Edward L-M, Gallon R, Santibanez-Koref M, Murata A, Takahashi MP, Eichler EE, Shlien A, Nakatani K, Mochizuki H, Pearson CE. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat Genet. 2018;52(2):146–59.
Narsinh KH, Plews J, Wu JC. Comparison of human induced pluripotent and embryonic stem cells: fraternal or identical twins? Mol Ther. 2011;19(4):635–8.
Nekrasov ED, Vigont VA, Klyushnikov SA, Lebedeva OS, Vassina EM, Bogomazova AN, Chestkov I v, Semashko TA, Kiseleva E, Suldina LA, Bobrovsky PA, Zimina OA, Ryazantseva MA, Skopin AY, Illarioshkin SN, Kaznacheyeva E v, Lagarkova MA, Kiselev SL. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol Neurodegener. 2016;11(1):1–15.
Niclis JC, Trounson AO, Dottori M, Ellisdon AM, Bottomley SP, Verlinsky Y, Cram DS. Human embryonic stem cell models of Huntington disease. Reprod Biomed Online. 2009;19(1):106–13.
Niclis JC, Pinar A, Haynes JM, Alsanie W, Jenny R, Dottori M, Cram DS. Characterization of forebrain neurons derived from late-onset Huntington’s disease human embryonic stem cell lines. Front Cell Neurosci. 2013;7(37):1–13.
Ooi J, Langley SR, Xu X, Utami KH, Sim B, Huang Y, Harmston NP, Tay YL, Ziaei A, Zeng R, Low D, Aminkeng F, Sobota RM, Ginhoux F, Petretto E, Pouladi MA. Unbiased profiling of isogenic Huntington disease hPSC-derived CNS and peripheral cells reveals strong cell-type specificity of CAG length effects. Cell Rep. 2019;26(9):2494–2508.e7.
Ou Z, Luo M, Niu X, Chen Y, Xie Y, He W, Song B, Xian Y, Fan D, Ouyang S, Sun X. Autophagy promoted the degradation of mutant ATXN3 in neurally differentiated spinocerebellar ataxia-3 human induced pluripotent stem cells. Biomed Res Int. 2016;2016:6701793.
Paulson H. Repeat expansion diseases. Handb Clin Neurol. 2018;147:105–23.
Paulson H, Shakkottai V. Spinocerebellar ataxia type 3. In: GeneReviews. Seattle: University of Washington, Seattle; 2020. https://www.ncbi.nlm.nih.gov/books/NBK1196/.
Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias – from genes to potential treatments. Nat Rev Neurosci. 2017;18:613–26.
Pereira-Sousa J, Ferreira-Lomba B, Bellver-Sanchis A, Vilasboas-Campos D, Fernandes JH, Costa MD, Varney MA, Newman-Tancredi A, Maciel P, Teixeira-Castro A. Identification of the 5-HT1A serotonin receptor as a novel therapeutic target in a C. elegans model of Machado-Joseph disease. Neurobiol Dis. 2021;152:1–14.
Perez BA, Shorrock HK, Banez-Coronel M, Zu T, Romano LE, Laboissonniere LA, Reid T, Ikeda Y, Reddy K, Gomez CM, Bird T, Ashizawa T, Schut LJ, Brusco A, Berglund JA, Hasholt LF, Nielsen JE, Subramony SH, Ranum LP. CCG⋅CGG interruptions in high-penetrance SCA8 families increase RAN translation and protein toxicity. EMBO Mol Med. 2021;13(11):e14095. https://doi.org/10.15252/emmm.202114095.
Rezende TJR, de Paiva JLR, Martinez ARM, Lopes-Cendes I, Pedroso JL, Barsottini OGP, Cendes F, França MC. Structural signature of SCA3: from presymptomatic to late disease stages: brain damage stages in SCA3/MJD patients. Ann Neurol. 2018;84:401–8.
Robinson KJ, Yuan K, Plenderleith SK, Watchon M, Laird AS. A novel calpain inhibitor compound has protective effects on a zebrafish model of spinocerebellar ataxia type 3. Cell. 2021;10(10):1–15.
Rosselli-Murai LK, Joseph JG, Lopes-Cendes I, Liu AP, Murai MJ. The Machado–Joseph disease-associated form of ataxin-3 impacts dynamics of clathrin-coated pits. Cell Biol Int. 2020;44(5):1252–9.
Rüb U, Brunt ER, Deller T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado–Joseph disease). Curr Opin Neurol. 2008;21:111–6.
Schuster KH, Zalon AJ, Zhang H, DiFranco DM, Stec NR, Haque Z, Blumenstein KG, Pierce AM, Guan Y, Paulson HL, McLoughlin HS. Impaired oligodendrocyte maturation is an early feature in SCA3 disease pathogenesis. J Neurosci. 2022;42(8):1604–17.
Shakkottai VG, Costa M d C, Dell’Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31:13002–14.
Shang XJ, Xu HL, Yang JS, Chen PP, Lin MT, Qian MZ, Lin HX, Chen XP, Chen YC, Jiang B, Chen YJ, Chen WJ, Wang N, Zhou ZM, Gan SR. Homozygote of spinocerebellar ataxia type 3 correlating with severe phenotype based on analyses of clinical features. J Neurol Sci. 2018;390:111–4.
Sivandzade F, Cucullo L. Regenerative stem cell therapy for neurodegenerative diseases: an overview. Int J Mol Sci. 2021;22(4):1–21.
Srinivasan SR, Huang H, Chang W-C, Nasburg JA, Nguyen HM, Strassmaier T, Wulff H, Shakkottai VG. Discovery of novel activators of large-conductance calcium-activated potassium channels for the treatment of cerebellar ataxia. Mol Pharmacol. 2022;102(1):438–49.
Stoyas CA, Bushart DD, Switonski PM, Ward JM, Alaghatta A, Tang M-B, Niu C, Wadhwa M, Huang H, Savchenko A, Gariani K, Xie F, Delaney JR, Gaasterland T, Auwerx J, Shakkottai VG, Spada ARL. Nicotinamide pathway-dependent Sirt1 activation restores calcium homeostasis to achieve neuroprotection in spinocerebellar ataxia type 7. Neuron. 2020;105:630–644.e9.
Sun X, Marque LO, Cordner Z, Pruitt JL, Bhat M, Li PP, Kannan G, Ladenheim EE, Moran TH, Margolis RL, Rudnicki DD. Phosphorodiamidate morpholino oligomers suppress mutant huntingtin expression and attenuate neurotoxicity. Hum Mol Genet. 2014;23(23):6302–17.
Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron. 2019;101(5):801–19.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Teixeira-Castro A, Jalles A, Esteves S, Kang S, da Silva Santos L, Silva-Fernandes A, Neto MF, Brielmann RM, Bessa C, Duarte-Silva S, Miranda A, Oliveira S, Neves-Carvalho A, Bessa J, Summavielle T, Silverman RB, Oliveira P, Morimoto RI, Maciel P. Serotonergic signalling suppresses ataxin 3 aggregation and neurotoxicity in animal models of Machado-Joseph disease. Brain. 2015;138(Pt11):3221–37.
Tidball AM, Niu W, Ma Q, Takla TN, Walker JC, Margolis JL, Mojica-Perez SP, Sudyk R, Moore SJ, Chopra R, Shakkottai VG, Murphy GG, Li JZ, Parent JM. Self-organizing single-rosette brain organoids from human pluripotent stem cells. BioRxiv. 2022:1–36. https://doi.org/10.1101/2022.02.28.482350.
Toulis V, García-Monclús S, de la Peña-Ramírez C, Arenas-Galnares R, Abril JF, Todi S v, Khan N, Garanto A, Costa M d C, Marfany G. The deubiquitinating enzyme ataxin-3 regulates ciliogenesis and phagocytosis in the retina. Cell Rep. 2020;33(6):1–19.
Toulis V, Casaroli-Marano R, Camós-Carreras A, Figueras-Roca M, Sánchez-Dalmau B, Muñoz E, Ashraf NS, Ferreira AF, Khan N, Marfany G, Costa M d C. Altered retinal structure and function in spinocerebellar ataxia type 3. Neurobiol Dis. 2022;170:1–15.
Tousley A, Kegel-Gleason KB. Induced pluripotent stem cells in Huntington’s disease research: progress and opportunity. J Huntingtons Dis. 2016;5(2):99–131.
Vasconcelos-Ferreira A, Carmo-Silva S, Codêsso JM, Silva P, Martinez ARM, França MC, Nóbrega C, Pereira de Almeida L. The autophagy-enhancing drug carbamazepine improves neuropathology and motor impairment in mouse models of Machado–Joseph disease. Neuropathol Appl Neurobiol. 2022;48(1):1–14.
Wang C, Peng H, Li J, Ding D, Chen Z, Long Z, Peng Y, Zhou X, Ye W, Li K, Xu Q, Ai S, Song C, Weng L, Qiu R, Xia K, Tang B, Jiang H. Alteration of methylation status in the ATXN3 gene promoter region is linked to the SCA3/MJD. Neurobiol Aging. 2016;53:192.e5–192.e10.
Zeng C, Zhao C, Ge F, Li Y, Cao J, Ying M, Lu J, He Q, Yang B, Dai X, Zhu H. Machado-Joseph deubiquitinases: from cellular functions to potential therapy targets. Front Pharmacol. 2020;11:1311.
Zhang N, An MC, Montoro D, Ellerby LM. Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr. 2010;2:1–11.
Zhuang S, Xie J, Zhen J, Guo L, Hong Z, Li F, Xu D. The deubiquitinating enzyme ATXN3 promotes the progression of anaplastic thyroid carcinoma by stabilizing EIF5A2. Mol Cell Endocrinol. 2021;537:1–9.
Acknowledgments
Our research and the preparation of this article are supported by NIH grants R25NS089450 (SS) R35NS122302 (HP), U01NS106670 (HP), and NIH T32 GM145470 (RP).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Powers, R., Paulson, H., Srinivasan, S. (2023). Seeking Therapies for Spinocerebellar Ataxia: From Gene Silencing to Systems-Based Approaches. In: Soong, Bw., Manto, M., Brice, A., Pulst, S.M. (eds) Trials for Cerebellar Ataxias. Contemporary Clinical Neuroscience. Springer, Cham. https://doi.org/10.1007/978-3-031-24345-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-031-24345-5_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-24344-8
Online ISBN: 978-3-031-24345-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)