
Abstract
A number of human genetic disorders, including Huntington’s disease, myotonic dystrophy type 1, C9ORF72 form of amyotrophic lateral sclerosis and several spinocerebellar ataxias, are caused by the expansion of various microsatellite sequences in single implicated genes. The neurodegenerative and neuromuscular nature of the repeat expansion disorders considerably limits the access of researchers to appropriate cellular models of these diseases. This limitation, however, can be overcome by the application of induced pluripotent stem cell (iPSC) technology. In this paper, we review the current knowledge on the modeling of repeat expansion diseases with human iPSCs and iPSC-derived cells, focusing on the disease phenotypes recapitulated in these models. In subsequent sections, we provide basic practical knowledge regarding iPSC generation, characterization and differentiation into neurons. We also cover disease modeling in iPSCs, neuronal stem cells and specialized neuronal cultures. Furthermore, we also summarize the therapeutic potential of iPSC technology in repeat expansion diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
More than 30 incurable, neurological diseases are caused by an expansion of simple sequence repeats in human genomic DNA [1]. This atypical mutation occurs in coding or noncoding regions of involved genes and triggers protein gain of function, RNA gain of function or RNA loss of function pathogenic mechanisms [2]. A tract containing expanded trinucleotide CAG repeats is located in the open reading frame (ORF) of the causative genes for Huntington’s disease (HD); spinocerebellar ataxias (SCAs) type 1, 2, 3, 6, 7 and 17; spinobulbar muscular atrophy (SBMA); and dentatorubral-pallidoluysian atrophy (DRPLA) [2]. This expansion leads to the formation of toxic, elongated polyglutamine (polyQ) proteins. In polyQ disorders, abnormal proteins misfold, aggregate and alter protein–protein interactions and cell homeostasis [3]. Recent findings demonstrate that elongated repeats present in transcripts are also toxic to cells and are involved in the pathogenesis of polyQ diseases [4–6]. RNA toxicity is a hallmark of another group of repeat expansion diseases, in which the mutation is located in noncoding sequences [i.e., 5′ or 3′ untranslated region (UTR) or intron] of implicated genes. In myotonic dystrophies type 1 and type 2 (DM1, 2), Fragile X associated tremor/ataxia syndrome (FXTAS), and in Fuchs endothelial corneal dystrophy (FECD), the mutation triggers RNA toxicity via a gain of function mechanism [2, 7]. In these conditions, the mutant transcripts induce the formation of intranuclear ribonucleoprotein foci by sequestering RNA-binding proteins (RBPs) and other factors that are essential for cell function and survival [8–11]. More recently, toxic properties of transcripts with elongated GGGGCC tracts were demonstrated for amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) [12, 13]. Repeat-associated non-AUG (RAN) translation is another possible pathomechanism that leads to cellular dysfunction and death. RAN translation, initiated at structures formed by long CAG, CGG or GGGGCC repeats, leads to the production and accumulation of the potentially neurotoxic peptides (reviewed in [14]). In Friedreich’s ataxia (FRDA), a GAA repeat expansion in the intron of frataxin (FXN) leads to a substantial reduction in frataxin protein levels and a consequent loss of function of this important mitochondrial protein [15].
Most repeat expansion diseases are chronic, age-related neurological disorders (Table 1), and induced pluripotent stem cell (iPSC) technology allows for the possibility of modeling these conditions. iPSCs research has grown rapidly over the short time since Yamanaka’s discovery that somatic cells can be successfully reprogrammed into iPSCs with use of defined factors [16]. iPSCs are similar to embryonic stem cells in terms of self-renewal and the potential to differentiate into any cell type [17]. These properties make it possible to construct disease models that contain patient-specific genetic information. For this reason, iPSC technology has become an important basic research tool for disease pathomechanisms, including neurodegeneration.
In this review, we focus on specific phenotypes and the instability of repeats observed in human iPSCs and iPSC-derived neuronal cells, which allow for the modeling of repeat expansion diseases. Furthermore, we describe the use of human iPSCs and iPSC-derived cells in drug discovery and their potential for cell therapy.
Can iPSCs be considered as models of neurodegenerative diseases?
The generation and characterization of iPSCs derived from patients with repeat expansion diseases
The reprogramming process begins with obtaining somatic cells (e.g., fibroblasts) from patients. Then, reprogramming factors are delivered to restore the embryonic state. The “gold standard” method for somatic cell reprogramming is through the use of the four Yamanaka’s factors: Octamer-binding protein 4 (OCT4), Sex-determining region Y-box 2 (SOX2), Krüppel-like factor 4 (KLF4) and c-MYC; however, c-MYC can be omitted or replaced by other factors, such as NANOG and LIN28 (Fig. 1) [18]. It has also been shown that the silencing of TP53 expression can improve iPSC generation [19]. The reprogramming transcription factors are delivered into parental cells primarily via a retroviral system; however, other delivery methods that utilize non-integrating episomal vectors have been described more recently (Fig. 1) [20]. The biggest advantage of episomal vectors over retroviruses is the lower risk they pose to the integrity of the cell genome, although the reprogramming efficiency that can be obtained with episomal vectors is considerably lower compared to integrating methods [21].

The workflow of human iPSC technology in repeat expansion disease research. Patient fibroblasts obtained by skin biopsy are reprogrammed into iPSCs using defined reprogramming factors delivered by various integrating or non-integrating systems. iPSCs can be further differentiated through the intermediate step of neuronal precursors into heterogeneous or homogenous neuronal populations. At each differentiation step, the identity of specific cell types is verified by selected markers. iPSCs and iPSC-derived neurons are used in basic research to explore disease pathomechanisms and in therapy development to search for therapeutic compounds
iPSCs should demonstrate typical stem cells features, such as self-renewability and pluripotency, as evidenced by the expression of specific molecular markers and differentiation potential. Human iPSCs express specific surface markers, [e.g., stage-specific embryonic antigens (SSEAs) 3 and 4, as well as TRA antigens (TRA-1-60 and TRA-1-81)], and show the endogenous expression of the transcription factors NANOG, SOX2 and OCT4. These pluripotency markers are validated by immunocytofluorescence (ICF) staining and quantitative PCR (qPCR). Additionally, in vitro differentiation into the three embryonic germ layers and in vivo teratoma assays are the standard methods of iPSC pluripotency testing (Fig. 1). Because reprogramming requires global changes in the chromatin state and may cause extensive chromosome aberrations, all generated iPSC lines must be karyotyped [18]. Furthermore, in the case of repeat-associated diseases, the stability of the repeats (i.e., number of repeated units in consecutive passages) is an important feature to monitor.
Successful disease modeling with human iPSCs has been achieved for six polyQ disorders, including HD, SCA2, SCA3, SCA7, SBMA, and DRPLA, likewise for five non-polyQ diseases, namely, DM1, Fragile X syndrome (FXS), FXTAS, FRDA and ALS/FTD. Table 2 shows informations on the generation and characterization of iPSCs derived from patients suffering from these diseases.
Repeat expansion-associated phenotypes in patient-derived iPSCs
PolyQ diseases
There are no consistent results regarding the formation of insoluble protein inclusions, the hallmark of polyQ diseases, in patient-derived iPSCs. In differentiated iPSCs obtained by the HD iPSC Consortium, no aggregation of the expanded huntingtin (HTT) protein was observed, even after the addition of cellular stressors, such as H2O2, 3-methyladenine (3-MA), or repetitive exposure to glutamate [22]. However, it was reported that EM48-positive HTT aggregates were observed when an iPSC line carrying 72 CAG repeats was treated with the proteasome inhibitor MG132 [23, 24].
Gene microarray and proteomic analyses revealed that early disease phenotypes, including the deregulation of genes involved in cell death, oxidative stress and bioenergetics, can be detected in both, HD differentiated and undifferentiated iPSCs [22, 25]. It has been reported that HD iPSCs show alterations in intracellular signaling cascades [including mitogen-activated protein kinase (MAPK) and Wnt pathways], demonstrate altered levels of extracellular signal-regulated kinase (ERK), and exhibit enhanced lysosomal activity and deregulation of superoxide dismutase 1 (SOD1) expression [26–28].
Another pathomechanism frequently linked to neurodegeneration in polyQ diseases is disruption of mitochondrial integrity and function [29]. In the mitochondrial fraction of HD iPSCs, levels of proteins involved in mitochondrial dynamics, such as dynamin-related protein 1 (Drp1) and p53, were increased threefold to fourfold relative to control cells. This effect was reversed following treatment with P110-TAT, a selective Drp1 inhibitor that prevents Drp1/p53 complex translocation to the mitochondria and initiates mitochondrial damage and cell death [30]. Additional support for the role of the p53 pathway in HD pathogenesis is provided by the observation of decreased p53 protein levels in a juvenile HD iPSC line [27].
Non-polyQ diseases
Intranuclear RNA foci were detected in DM1 iPSCs and iPSC-derived neural stem cells (NSCs), neurons and astrocytes; no foci were found in the controls at any stage of cell differentiation [31, 32]. RNA foci were also detected in ALS/FTD iPSCs with long GGGGCC tracts of >1000 repeats [33]. These facts suggest that the nuclear foci phenotype is present only in cases of large repeat expansions in ALS and DM1 iPSC models.
In FXS, methylation of the fragile X mental retardation (FMR1) promoter induced by the presence of an elongated CGG tract leads to the transcriptional silencing of FMR1 and a loss of FMRP protein [34]. Although this methylation is developmentally regulated and is not detected in undifferentiated FXS embryonic stem (ES) cells, silencing of FMR1 was demonstrated in all FMR1 iPSC models, including naïve FXS iPSCs (i.e., without epigenetic memory) [35–39]. In the case of fibroblasts with unmethylated FMR1 promoter regions, reprogramming results in promoter methylation and epigenetic silencing of the mutated FMR1 [36, 40].
Because FXS and FXTAS are associated with the X chromosome, it is important to ask whether the inactive X chromosome is reactivated during reprogramming. To answer this question, isogenic, epi-isoautosomal (i.e., containing allelic differences elsewhere in the two X chromosomes), female iPSC models with pre-mutations in FMR1 were successfully established. All of the iPSC clones derived from fibroblast sub-clones exhibited active normal and pre-mutation alleles; no reactivation of the inactive X chromosome was observed. All of the iPSC clones maintained the same active X chromosome as the parental fibroblasts [41].
One of the primary molecular features of FRDA is the silencing of FXN expression. Significantly lower levels of FXN mRNA and protein were detected in several FRDA iPSC models compared to control iPSC lines [42–45]. FRDA iPSCs showed disturbances in gene expression, especially in genes related to mitochondrial function and DNA repair. In addition, the expression profile of many microRNAs was altered in these models [46].
Repeat instability in patient-derived iPSCs
Repeat instability is a dynamic type of mutation that is not only transmitted to offspring (parental transmission) but also generates somatic heterogeneity (mosaicism) [47]. Several properties of tandem repeats make iPSC generation and maintenance in culture challenging. First, longer tracts undergo expansion more often than shorter tracts [48]. Second, the nucleotide composition of the repeated tracts affects its stability [49]. Third, there is a correlation between the number of repeats and both the age of disease onset and the severity of the symptoms [50]. This effect is referred to as genetic anticipation and may substantially affect the molecular and cellular phenotypes observed in cell culture. For these reasons, simple repeated elements in cellular models should be carefully monitored during somatic cell reprogramming and subsequent subculture.
No expansion or contraction of CAG repeats was observed during reprogramming and long-term passaging in HD iPSC models. It has been shown that HD iPSCs carrying the same number of repeats as parental fibroblasts (72 CAG) [26, 51, 52] and iPSCs with 44 CAG repeats in HTT were stable for at least 40 passages [23, 28]. However, expansions were observed after iPSC differentiation in lines with long (109 CAG) repeats [22].
In the case of SCA2 and SCA3 iPSCs, the CAG repeat number was stable during iPSC reprogramming, long-term passaging (up to 26 and 20 passage, respectively), and differentiation into neuronal cells [53, 54]. Similarly, no expansion of the polyQ-coding sequence was observed in iPSCs obtained from SBMA and DRPLA patients [55]. However, in another study in which the SBMA iPSCs were generated by three different reprogramming methods, the authors identified some iPSC clones with elongated and shortened CAG repeat tracts compared to the parental fibroblasts. Observed differences might be caused by mosaicism within the fibroblast population [56]. In the case of SCA7 iPSCs, the number of CAG repeats has not been analyzed during reprogramming and proliferation [57].
In contrast to long CAG repeats, CTG repeats are highly unstable during both reprogramming and subsequent passages. CTG repeats expand more rapidly when the initial CTG tract in the parental cells is longer. In DM1 iPSCs, the interval between 57 and 126 CTG repeats appears to be an important range of lengths. When the tract is longer, the expansion rate increases dramatically [58]. Components of the mismatch repair (MMR) system were shown to be involved in the phenomenon of CTG repeat instability for the first time in E. coli [59]. In DM1 iPSCs, MutS homolog (MSH) 2 and MSH6 were up-regulated compared to the parental fibroblasts, and their expression levels resembled those observed in ESCs. Similar observations were made for MSH2, MSH3 and MSH6 proteins levels. MSH2 showed increased occupancy upstream and downstream of CTG repeats in DM1 iPSCs with a long pathogenic allele. The knockdown of MSH2 decreased the repeat expansion rate, indicating that elements of the MMR system can block CTG repeat instability [58]. Similarly to DM1, the MMR system has been shown to be responsible for the regulation of GAA repeat stability. Increased protein levels of MSH2, MSH3 and MSH6 were observed in FRDA iPSCs [44, 46, 60]. This effect related to considerable GAA repeat instability, was manifested by either expansion or contraction of the GAA tract in both alleles [43–46, 60]. The expansion of GAA repeats in FRDA iPSC models can be blocked by shRNA silencing of MSH2 and MSH6 genes [60]. Furthermore, the β-alanine-linked pyrrole-imidazole polyamides that specifically bind to the GAA triplet-repeat sequence impede GAA triplet repeat expansion by displacing MMR enzymes from the repeat region [60].
Full mutation in the FMR1 (over 200 CGG repeats, Table 1) shows instability in iPSC models. Interestingly, the length of the CGG repeat tract in FXS iPSCs appeared to be shorter than in parental fibroblasts [36, 37]. In pre-mutated FXTAS iPSCs, the length of CGG repeats was unchanged during reprogramming [41]. The expansion of pathogenic GGGGCC repeats during long-term passaging was not observed; however, repeat expansion during reprogramming was detected in some ALS iPSC clones due to somatic mosaicism of the parental fibroblasts [33, 61, 62].
iPSC-derived neuronal cells
Heterogeneous and homogeneous neuronal populations
Early phenotypes detected in iPSCs derived from patients with repeat expansion diseases show that even pluripotent cells can model certain aspects of neurodegenerative processes. iPSC technology is generally perceived as a tool that grants access to cells that would otherwise be extremely difficult to obtain human neurons. In a typical scenario, established and validated iPSCs are differentiated into the relevant neuronal subpopulation. The majority of available protocols result in heterogeneous cell cultures that contain only a fraction of expected cells. For some research purposes, pure cell cultures of not required as the desired neuronal populations can be easily detected with microscopy-based imaging techniques [63, 64]. However, despite the relative simplicity of the protocols, several limitations reduce the usefulness of heterogeneous neuronal cultures (see Ref. [65]) and may even lead to misinterpretation of the obtained results. First, heterogeneous cultures are not well suited for high-throughput analyses, including gene expression profiling using deep sequencing methods. Cells of different types and differentiation states, including NSCs, neurons, and glia, contribute considerably to culture variability and can mask the signal from cells of interest. Second, many of the morphological and physiological features of analyzed neurons that can be used to monitor degeneration are dependent on the cellular maturation status [66, 67]. Therefore, the contribution of progenitors at different stages of differentiation can greatly increase experimental variability and hinder the assessment of disease phenotypes. Lastly, long-term culture of heterogeneous neuronal populations is not trivial given that dividing cells in a culture can quickly overgrow post-mitotic neurons. It is, therefore, difficult to reproduce the exact composition of heterogeneous cultures in independent experiments. For this reason, typical experimental designs often require homogenous neuronal populations that can be obtained with additional purification steps. There is a steady flow of protocols for generating specific neuronal cell types [68], but iPSC and NSC differentiation into homogeneous neuronal populations is still a challenge. Differentiation to specialized neurons requires specific supplements, culture media, or extracellular matrices. When combined with the required rigorous purification procedures, these factors make such protocols expensive and time-consuming. Neurons generated using most of the currently available protocols resemble cells at an immature fetal stage, although a great deal of effort is now being dedicated to enhance the maturity and electrophysiological activity of neurons [63, 69–71]. Another limitation of homogenous cultures is the low quantity of material obtained from mature neurons for analysis. Despite these constraints, homogenous neuronal cultures approach should greatly reduce molecular, physiological and morphological variation and facilitate reliable interpretation of results. Available protocols can produce subtypes of neurons that are relevant to specific repeat expansion disease, including medium spiny neurons (MSN) for HD [22, 26, 30], motor neurons for ALS and SBMA [56, 62, 72], dorsal root ganglion peripheral neurons for FRDA [42, 45] and forebrain neurons for FXS [39].
Differentiation of iPSCs into neuronal subtypes affected in repeat expansion diseases
NSCs derived from iPSCs can be obtained through the formation of embryoid bodies (EBs), rosettes, stromal feeder layer co-cultures or EZ spheres [i.e., free floating cell aggregates in medium with epidermal growth factor (EGF) and fibroblast growth factor (FGF)-2] [73–76]. In the next step, NSCs are exposed to growth factors involved in cell fate choices that induce specific neuronal subtype differentiation [77]. The process of iPSCs differentiation into mature neurons usually requires induction or inhibition of the cellular pathways that are normally involved in the maturation of neurons affected in particular repeat expansion diseases.
The activation of the sonic hedgehog (SHH) pathway appears to be essential for gamma-aminobutyric acid (GABA)ergic striatal neurons and motor neurons [78, 79]. SHH is the best-known morphogen of the central nervous system (CNS) that regulates the regionalization the subpallial region, from which the striatum develops. SHH also promotes axon guidance and neuronal specialization at later stages of CNS development [80]. It has been shown that the use of SHH agonists in vitro (e.g., purmorphamine) promotes differentiation into (1) HD-associated GABAergic cells that express striatal MSN markers [e.g., dopamine- and cAMP-regulated phosphoprotein (DARPP)-32, CTIP2, and microtubule associated protein 2 (MAP-2)] [78] and (2) ALS-associated motor neurons that express ChAT, SEMI32, and homeobox 9 (HB9) [61, 62, 72]. In the case of iPSC differentiation into peripheral sensory neurons involved in FRDA, activators of SHH, such as bone morphogenic proteins, are used to obtain PRPH-positive cells [45]. Moreover, methods that apply neurotrophin-3 and dbcAMP were used to generate cells expressing BRN3A, PRPH and ISL1 [42].
Coordination of SHH and Wnt signaling is important in striatal development. These proteins induce PAX6 expression, which stimulates cortical neuron development [78]. During MSN formation, the Wnt pathway is blocked. One commonly used antagonist of Wnt signaling is Dickopff-1 (Dkk-1) [81]. Wnt pathway inhibition in combination with SHH pathway activation is essential for striatal neuron specialization [78]; however, other factors may also be required, such as valproic and retinoic acids. Valproic acid increases the number of GABAergic neurons and enhances neurite outgrowth [82]. Retinoic acid is a morphogen that promotes striatal and motor neuron differentiation through the regulation of NolzI/GAD67 and cooperation with neurogenin 2, respectively [83, 84]. Moreover, the GAD67 enzyme is involved in GABA neurotransmitter synthesis [83]. The different supplements that were used for iPSC differentiation to specialized neurons affected in repeat expansion disorders are shown in Table 3.
Repeat expansion-associated phenotypes in iPSC-derived neuronal cells
Neuronal stem cells
Characteristic pathophysiological markers that are associated with repeat expansion disorders can be observed even at the early stages of neuronal differentiation. For example, abnormal neural rosette formation was observed during the differentiation of SCA2 iPSCs into NSCs, and ataxin-2 expression in SCA2 NSCs was lower than in fibroblasts. Although ataxin-2 was expressed more abundantly in SCA2-derived neuronal cells than in glial cells, it was not form cytoplasmic or nuclear polyglutamine protein inclusions [53]. Another example of early pathophysiological markers associated with repeat expansion is the formation of RNA foci, which were observed in the nuclei of iPSC-derived NSC models of DM1 [32], ALS [62] and HD [85]. RNA foci exert their cytotoxicity by sequestering and reducing the functional levels of important cellular proteins (reviewed in [12, 85–87]).
Compared to NSCs with no CAG tract expansion, NSCs derived from HD iPSCs showed changed gene expression pattern, disturbances in cytoskeletal structure, cellular adhesion and energy metabolism, as well as increased caspase 3/7 activity [22, 26, 88]. Moreover, some HD NSC-related phenotypes are associated with neuronal cell development. Mattis et al. [89] showed that after differentiation of iPSCs into mixed population of astrocytes and neuronal cells, HD-derived cultures maintained significantly more nestin-expressing neural progenitors compared to control cells. These persistent progenitors were selectively susceptible to brain-derived neurotrophic factor (BDNF) withdrawal due to a loss of signaling from the TrkB receptor. The resulting cell death was connected with increased sensitivity to glutamate-induced cellular toxicity [89].
The length of CAG expansions was stable during both iPSC differentiation into NSCs [26, 53] and long-term passaging [28, 54]; however, mild expansion (~3 CAG repeats per passage) in NSC lines with more than 100 CAG repeats in HTT was observed [22]. On the other hand, CTG and GAA repeats, which are highly unstable in iPSC lines, exhibit no length instability for 6 and 10 weeks in culture, respectively, when differentiated into NSCs [58, 60]. Thus far, no studies regarding the stability of CGG and GGGGCC repeats in neuronal precursors have been published.
Mature neurons
Morphological and functional changes The presence of expanded repeats can cause morphological changes or alter the electrophysiology of neurons. Compared to control cells, morphological alterations of neurites were observed in HD neuron-like cells obtained by direct conversion of fibroblast cells. Some of the neurites were degenerated and exhibited abnormal branching [90]. Reduced neurite outgrowth was also observed in HD MAP2-positive cells [25]. Fewer, considerably shorter, less branched and flatter Tuj1-positive neurons were observed in FXS iPSC-derived cells [37]. Fewer and shorter neurites were also observed in forebrain neurons differentiated from FXS iPSCs, and that effect was explained by defects in both neurite initiation and extension [39]. In FXTAS, the pre-mutation led to shorter neurite extensions and a reduced postsynaptic density of protein 95 expression [41]. An increased number of degenerating DARPP-32- and GABA-positive cells was reported in HD neuron-like cultures compared to control cells [90]. This result is consistent with the selective loss of GABAergic neurons in the striatum of HD patients [91]. Similarly, a higher rate of cell death was observed in HD neurons [22]. Following BDNF withdrawal, the death rate was significantly higher for cells with expanded CAG repeats compared to control lines [22, 89, 92]. However, the addition the ataxia-telangiectasia mutated (ATM) protein inhibitors, e.g., KU-60019, protected against cell death [92]. It was also shown that the neuronal loss and caspase-3 activation observed in HD iPSC-derived DARPP-32-positive neurons following BDNF withdrawal can be suppressed by silencing Gpr52. This protein is a neural surface receptor that stabilizes the HTT protein in vitro and in vivo [93]. These observations confirmed that BDNF plays important role in striatal degeneration observed in HD patients.
Increased glutamate receptor-dependent cell death was observed in HD iPSC-derived neurons [22] and in ALS iPSC-derived neurons [61]. In these experiments, cells with expanded repeat tracts were 100-fold more sensitive to glutamate treatment than control cells [61]. Higher sensitivity to cellular stressors (e.g., 3-MA, chloroquine and H2O2) was also observed in these cells compared to control neurons [22, 33, 94]. Stress induced by H2O2 caused changes in the expression of genes involved in the repair of DNA double strand breaks. This effect can explain why HD iPSC-derived neurons are more susceptible to H2O2-induced DNA damage than normal cells [22]. However, the activation of A2AR can prevent DNA damage and apoptosis via a protein kinase cAMP (PKA)-dependent pathway [94]. A shorter life span was also observed in SCA2 iPSC-derived neurons [53].
Electrophysiological alterations of iPSC-derived neurons are commonly caused by expanded repeats. ALS iPSC-derived motor neurons demonstrated electrical excitability. Compared to control motor neurons, these cells produced fewer spikes upon depolarization, exhibited a progressive loss of action potential output, and showed spontaneous synaptic activity and ionic conductance [62, 72]. These results indicate that early dysfunction or loss of ion channels (e.g., channels conducting Na+ or K+) may contribute to the initiation of downstream degenerative pathways that lead to a loss of motor neurons in ALS. FRDA iPSC-derived neuron cells were capable of firing action potentials and demonstrated Na+ and K+ current activity. However, these cells also showed delayed functional maturation compared to control cells, as evidenced by poor excitability of young neurons (~35 days of differentiation) and the acquisition of normal electrophysiology by older cells at ~60 days after differentiation [44]. FXTAS iPSC-derived neurons showed aberrant calcium activity, with a higher number of spontaneous Ca2+ transients that had significantly larger mean amplitudes compared to control neurons. Additionally, a glutamate challenge caused a profound and sustained elevation of intracellular Ca2+ levels in neurons containing CGG expansions, whereas normal cells quickly recovered to baseline after a transient rise in Ca2+ levels [41]. These data demonstrate that different pathogenic mechanisms and disturbances in signaling pathways occur in neuronal in vitro cultures derived from patient iPSCs.
Protein aggregation Protein aggregation is the most common hallmark of polyQ diseases. It has been shown that excitation-induced Ca2+ influx into iPSC-derived neurons containing CAG expansions in the ATXN3 induces calpain-dependent proteolysis of the mutant ataxin-3 protein. As a consequence, excitation led to the generation of expanded polyQ fragments that formed SDS-insoluble aggregates. This aggregation of ataxin-3 involved a neuron-specific cascade and depended on functional Na+, K+ and Ca+ channels. Excitation-induced aggregation was not observed in non-neuronal cells, including iPSCs, fibroblasts or glia [54]. Two publications described experiments in which GABAergic NSCs differentiated from HD iPSCs were injected into the striata of YAC128 transgenic mice and QUIN-lesioned rats. In these conditions, no EM48-positive HTT aggregates were observed 12 weeks after transplantation [23, 95]. However, clear EM48-positive signals were observed 33 weeks after transplantation of HD iPSC-derived NSCs into the lateral ventricle of neonatal CF-1 mouse brains [23]. These results may suggest that HTT aggregates develop at later time points after transplantation. In neuron-like cells that were directly reprogrammed from HD fibroblasts, mutant HTT aggregates formed in both the nucleus and non-nuclear regions, including cell soma and the neuropils [90]. In neurons derived from SBMA iPSCs, the aggregation of androgen receptor (AR) protein following dihydrotestosterone (DHT) treatment increased compared to control cells [55, 56]. AR aggregates were not observed in iPSCs, which may indicate that aggregation is suppressed in the pluripotent state [55]. Moreover, motor neurons from patient iPSCs containing >60 CAG repeats had increased acetylated α-tubulin levels compared to controls. Additionally, these cells showed alterations in lysosomal localization and post-translational modification, which may be a consequence of reduced histone deacetylase 6 (HDAC6) activity and a disruption of HDAC6-mediated microtubule transport [56].
Toxic RNA mechanism RNA foci containing mutant transcripts with expanded repeats, which have been observed in iPSCs, were also detected in different types of neurons. Small, discrete and punctate nuclear inclusions of transcripts containing GGGGCC repeats were found in neurons derived from ALS iPSCs [33]. In addition, GGGGCC repeats were shown to form a G-quadruplex structure [96, 97], which sequestered different RBPs. As many as 19 proteins that bind to GGGGCC repeat mRNA were identified in a heterogeneous neuronal population derived from ALS iPSCs [61]. One of these proteins, ADARB2, was experimentally validated. ADAR proteins comprise a family of CNS-enriched adenosine deaminases that not only co-localize with GGGGCC RNA but also mediate A-to-I RNA editing in non-specialized neurons [61, 98]. Neurons treated with siRNA against ADARB2 showed a reduced number of RNA foci, confirming an interaction between ADARB2 and GGGGCC mRNA. Similarly, GGGGCC-containing foci were observed in ALS iPSC-derived motor neurons. These foci co-localized with hnRNPA1 and PUR-α RBPs, supporting the hypothesis that RNA toxicity contributes to C9ORF72 repeat expansion diseases [62]. It has also been shown that GGGGCC RNA co-localizes with RanGAP1, which regulates nucleocytoplasmic transport mechanisms that are related to neurodegeneration [99]. GGGGCC RNA foci occur both in the nucleus as well as neurites, where branching defects were observed [100]. An analysis of RNA-seq datasets from ALS iPSC-derived neurons showed that a wide range of extracellular matrix proteins and matrix metalloproteinases was reduced in ALS iPSC-derived motor neurons [101]. RAN-translation products may sporadically co-localize with C9ORF72 RNA foci in the same cell [14]. The cytoplasmic accumulation of dipeptide poly-(Gly-Pro) RAN proteins and large, cytoplasmic RNA foci were observed in heterogeneous neuronal cultures [33, 61]. Notably, the RAN translation products were not detected in homogeneous cultures of motor neurons [62]. Foci containing mutant DM1 mRNA and sequestered splicing factors, such as muscleblind-like (MBNL) family proteins, were found in terminally differentiated iPSC-derived neurons and astrocytes [32].
Therapeutic potential of iPSC technology
Different compounds can reverse negative disease phenotypes
iPSCs and their derived cells can be used for high-throughput screening of compounds that may have a therapeutic potential. For example, NSCs derived from FXS iPSCs were used to screen approximately 5000 compounds, including approved drugs, in a highly sensitive, time-resolved Förster resonance energy transfer (FRET) assay. Six of these compounds modestly increased FMR1 expression and FMRP levels [102]. Other authors demonstrated that decreased levels of FMR1 mRNA could be specifically rescued after 5-azaC treatment in both FXS iPSCs and derived neurons [38].
In neurons derived from SBMA iPSCs, the aggregation of mutant AR with an expanded polyQ tract was decreased by 17-allylaminogeldanamycin (17-AAG) treatment [55]. 17-AAG is a HSP90 inhibitor and a candidate drug for SBMA therapy. This compound specifically binds to the ATP-binding site of HSP90, shifting the HSP90 complex to the proteasome-targeted form, resulting in enhanced degradation of mutant AR [103].
In fibroblasts from ALS patients and ALS iPSC-derived neurons, antisense oligonucleotides designed to activate RNase H-mediated C9ORF72 RNA degradation or block the toxic GGGGCC expansion rescued RBP aggregation, aberrant gene expression and neurotoxicity, but not the formation of RAN-translation products [61]. RNA foci formation was also suppressed following treatment with antisense oligonucleotides targeting the C9ORF72 transcript, with no observed toxicity to the iPSC-derived cultured neurons [62].
iPSC technology allows preclinical testing of new therapeutic strategies for repeat expansion diseases. Soragni et al. [104] described a phase I clinical trial in which the authors determined the efficacy of 2-aminobenzamide HDACi (109) in FXN upregulation and histone modification using FRDA iPSC-derived neurons. The patients were monitored for increases in FXN expression and chromatin modification in peripheral blood mononuclear cells, as well as for adverse effects. In iPSC-derived neurons, HDACi (109) had no effect on GAA repeat stability or the induction of GAA RNA foci formation [104]. Moreover, NSCs differentiated from FRDA-derived iPSCs provided insight into the mechanism by which the FXN upregulation is achieved by 2-aminobenzamide class HDAC inhibitors. Quantitative proteomic methods showed that targets of these HDAC inhibitors are involved not only in transcriptional regulation but also in mRNA translation [104, 105]. However, a large number of observed targets raised concerns regarding the use of 2-aminobenzamides as human therapeutics for FRDA [105].
Correction of repeat length for regenerative medicine and diseases modeling
The combination of two powerful technologies, genome engineering and human iPSC technology, has opened a new era for disease modeling and regenerative medicine. Zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and more recently described clustered, regularly interspaced, short, palindromic repeats/Cas9 (CRISPR/Cas9) systems all support homologous recombination. These systems are promising strategies that have the potential to correct genes associated with repeat expansion diseases [106]. The correction of the mutated gene in iPSCs or their derived differentiated cells is a long-awaited strategy that gives hope for replacement therapies and may also be used to generate human isogenic cell line models without pronounced inter-individual variability due to diverse genetic and epigenetic backgrounds.
Homologous recombination was used for CAG repeat correction in HD iPSCs [52]. The mutant HTT allele (with 73 CAG repeats) was successfully altered to have the normal CAG repeat length of 21. Corrected iPSCs showed rescued disease phenotypes, such as elimination of the expanded mutant HTT protein, reduced mitochondrial bioenergetic disturbances and cell death. Importantly, corrected cells maintained pluripotency characteristics. Repeat length correction also normalized altered cellular signaling, including the cadherin, TGF-β and BDNF pathways, as well as caspase activation. More importantly, the transplantation of corrected HD iPSC-derived NSCs into the mouse striatum resulted in not only cell survival but also in their ability to differentiate into GABAergic and DARPP-32-positive neurons [52]. To improve recombination efficiency, screening and the generation of isogenic cell line models of HD, the same group recently used the CRISPR/Cas9 system [107]. CRISPR/Cas9 was also used for successful CGG correction in FXS iPSC models, after which the CGG repeats were completely removed. The authors observed extensive demethylation, an open chromatin state and restored FMR1 mRNA and FMRP expression; these effects persisted in NSCs and mature neurons [108].
ZFNs were used for genome editing of fibroblasts obtained from patients with FRDA. Following GAA tract cutting, the fibroblasts were reprogrammed to iPSCs and differentiated to neurons. Reduction in the length of the expanded GAA repeats ameliorated FRDA-associated phenotypes in iPSC-derived neurons. Corrected cells expressed three fold higher levels of FXN mRNA and protein as well as 30 % increased aconitase activity and 25 % higher ATP levels compared to non-corrected neurons [109]. Interestingly, correction of only one mutant allele causes increased FXN expression and rescues several FRDA biomarkes, indicating that even limited genetic intervention in FRDA homozygous patients might be curative.
Genome editing was also performed in DM1 NSCs derived from iPSCs, taking advantage of homologous recombination mediated by a pair of site-specific TALENs. This method successfully prevented the production of expanded CUG transcripts by introducing polyA signals (PASs) upstream of the CTG expansion. The insertion of exogenous PASs led to the ablation of nuclear RNA foci and a return of aberrant alternative splicing to the normal pattern [110].
Conclusions
Localized pathology, often limited to specific subtypes of neurons, makes modeling repeat expansion diseases a challenging task. iPSC technology is expected to help develop new and necessary cellular model systems that will broaden our understanding of neurodegeneration and be suitable for medical research, including drug discovery and regenerative medicine.
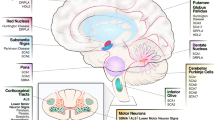
Although iPSCs are usually considered as an intermediate step in generating specialized cells that could serve as an appropriate disease model, a growing body of evidence demonstrates that phenotypes characteristic of repeat expansion diseases can be detected even in pluripotent cells (Fig. 2). In particular, iPSCs can be a good model for detecting and analyzing molecular changes related to the early phenotypes of repeat expansion diseases, including alterations in gene expression, changes in cellular signaling, and mutant RNA/protein aggregation. These studies address important issues because there is still little known regarding the transition from the subtle molecular processes of the pre-symptomatic stages of neurodegenerative diseases to the subsequent and pronounced alterations that characterize symptomatic phases.

Disease hallmarks observed in iPSCs and iPSC-derived cells. iPSC technology provides a platform to study cellular phenotypes of repeat expansion diseases at each step step of differentiation. iPSCs and their derivative cells can be used to generate isogenic cell lines by genome editing techniques and to test compounds with therapeutic potential
One of the biggest advantages of iPSC technology is that it allows for the generation of virtually any cell type for more accurate disease modeling. In the case of repeat expansion diseases, iPSCs allow for the generation of human neurons, which could otherwise only be obtained from scarce and ethically problematic sources, including human embryos and post-mortem fetal tissues. Numerous protocols enable the generation of neurons that express markers characteristic of neurons undergoing disease-specific selective degeneration. These cellular models facilitate studies of neuropathological processes by providing an opportunity to analyze phenotypes that are limited to the affected tissue (Fig. 2). Moreover, the advent of genome editing technology provides new possibilities for the production of improved and more reliable isogenic iPSC-based neuronal models for both basic research and therapy development.
Abbreviations
- 17-AAG:
-
17-Allylaminogeldanamycin
- 3-MA:
-
3-Methyladenine
- ALS:
-
Amyotrophic lateral sclerosis
- AR:
-
Androgen receptor
- ATM:
-
Ataxia-telangiectasia mutated protein
- ATXN3:
-
Ataxin-3
- BDNF:
-
Brain-derived neurotrophic factor
- CNS:
-
Central nervous system
- CRISPR/Cas9:
-
Clustered, regularly interspaced, short, palindromic repeats/Cas9 system
- DARPP-32:
-
Dopamine- and cAMP-regulated phosphoprotein
- DHT:
-
Dihydrotestosterone
- DKK-1:
-
Dickopff-1
- DM1:
-
Myotonic dystrophy type 1
- DRP1:
-
Dynamin-related protein 1
- DRPLA:
-
Dentatorubral-pallidoluysian atrophy
- EB:
-
Embryoid body
- EGF:
-
Epidermal growth factor
- ERK:
-
Extracellular signal-regulated kinase
- ESC:
-
Embryonic stem cell
- FECD:
-
Fuchs endothelial corneal dystrophy
- FGF:
-
Fibroblast growth factor
- FMR1:
-
Fragile X mental retardation 1
- FTD:
-
Frontotemporal dementia
- FXN:
-
Frataxin
- FXS:
-
Fragile X syndrome
- FXTAS:
-
Fragile X associated tremor/ataxia syndrome
- GABA:
-
Gamma-aminobutyric acid
- HB9:
-
Homeobox 9
- HD:
-
Huntington’s disease
- HDAC:
-
Histone deacetylase
- HTT:
-
Huntingtin
- ICF:
-
Immunocytofluorescence
- iPSC:
-
Induced pluripotent stem cell
- KLF4:
-
Krüppel-like factor 4
- MAP-2:
-
Microtubule-associated protein 2
- MAPK:
-
Mitogen-activated protein kinase
- MMR:
-
Mismatch repair system
- MSH:
-
MutS homolog
- MSN:
-
Medium spiny neuron
- NSC:
-
Neural stem cell
- OCT4:
-
Octamer-binding protein 4
- ORF:
-
Open reading frame
- PAS:
-
PolyA signals
- PKA:
-
Protein kinase cAMP
- polyQ:
-
Polyglutamine
- qPCR:
-
Quantitative PCR
- RAN:
-
Repeat associated non-AUG translation
- RBP:
-
RNA binding protein
- SBMA:
-
Spinobulbar muscular atrophy
- SCA:
-
Spinocerebellar ataxia
- SHH:
-
Sonic hedgehog
- SOD1:
-
Superoxide dismutase 1
- SOX2:
-
Sex-determining region Y-box 2
- SSEA:
-
Stage-specific embryonic antigen
- TALEN:
-
Transcription activator-like effector nuclease
- UTR:
-
Untranslated region
- ZFN:
-
Zinc finger nuclease
References
Mirkin SM (2007) Expandable DNA repeats and human disease. Nature 447:932–940. doi:10.1038/nature05977
Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621
Williams AJ, Paulson HL (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31:521–528. doi:10.1016/j.tins.2008.07.004
Fiszer A, Krzyzosiak WJ (2013) RNA toxicity in polyglutamine disorders: concepts, models, and progress of research. J Mol Med 91(6):683–691
Chan HY (2014) RNA-mediated pathogenic mechanisms in polyglutamine diseases and amyotrophic lateral sclerosis. Front Cell Neurosci 8:431. doi:10.3389/fncel.2014.00431
Nalavade R, Griesche N, Ryan DP, Hildebrand S, Krauss S (2013) Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis 4:e752. doi:10.1038/cddis.2013.276
Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, Baratz KH (2012) A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One 7:e49083. doi:10.1371/journal.pone.0049083
Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19(17):4439–4448. doi:10.1093/emboj/19.17.4439
Mohan A, Goodwin M, Swanson MS (2014) RNA–protein interactions in unstable microsatellite diseases. Brain Res 1584:3–14. doi:10.1016/j.brainres.2014.03.039
Echeverria GV, Cooper TA (2012) RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res 1462:100–111. doi:10.1016/j.brainres.2012.02.030
Du J, Aleff RA, Soragni E, Kalari K, Nie J, Tang X, Davila J, Kocher JP, Patel SV, Gottesfeld JM, Baratz KH, Wieben ED (2015) RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J Biol Chem 290:5979–5990. doi:10.1074/jbc.M114.621607
Gendron TF, Belzil VV, Zhang YJ, Petrucelli L (2014) Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol 127(3):359–376. doi:10.1007/s00401-013-1237-z
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA 110(19):7778–7783
Wojciechowska M, Olejniczak M, Galka-Marciniak P, Jazurek M, Krzyzosiak WJ (2014) RAN translation and frameshifting as translational challenges at simple repeats of human neurodegenerative disorders. Nucleic Acids Res 42:11849–11864. doi:10.1093/nar/gku794
Butler JS, Napierala M (2015) Friedreich’s ataxia—a case of aberrant transcription termination? Transcription 6(2):33–36
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. doi:10.1016/j.cell.2006.07.024
Takahashi K, Yamanaka S (2016) A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 17(3):183–193. doi:10.1038/nrm.2016.8
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. doi:10.1016/j.cell.2007.11.019
Rasmussen MA, Holst B, Tumer Z, Johnsen MG, Zhou S, Stummann TC, Hyttel P, Clausen C (2014) Transient p53 suppression increases reprogramming of human fibroblasts without affecting apoptosis and DNA damage. Stem Cell Rep 3(3):404–413. doi:10.1016/j.stemcr.2014.07.006
Malik N, Rao MS (2013) A review of the methods for human iPSC derivation. Methods Mol Biol 997:23–33. doi:10.1007/978-1-62703-348-0_3
Kang X, Yu Q, Huang Y, Song B, Chen Y, Gao X, He W, Sun X, Fan Y (2015) Effects of integrating and non-integrating reprogramming methods on copy number variation and genomic stability of human induced pluripotent stem cells. PLoS One 10(7):e0131128
Consortium THi (2012) Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11(2):264–278
Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, Shim SH, Choi C, Chang DJ, Kwon J, Oh SH, Shin DA, Kim HS, Do JT, Lee DR, Kim M, Kang KS, Daley GQ, Brundin P, Song J (2012) Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 30(9):2054–2062. doi:10.1002/stem.1135
Cheng PH, Li CL, Chang YF, Tsai SJ, Lai YY, Chan AW, Chen CM, Yang SH (2013) miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am J Hum Genet 93(2):306–312. doi:10.1016/j.ajhg.2013.05.025
Chae JI, Kim DW, Lee N, Jeon YJ, Jeon I, Kwon J, Kim J, Soh Y, Lee DS, Seo KS, Choi NJ, Park BC, Kang SH, Ryu J, Oh SH, Shin DA, Lee DR, Do JT, Park IH, Daley GQ, Song J (2012) Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem J 446:359–371. doi:10.1042/BJ20111495
Zhang N, An MC, Montoro D, Ellerby LM (2010) Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr 2:RRN1193. doi:10.1371/currents.RRN1193
Szlachcic WJ, Switonski PM, Krzyzosiak WJ, Figlerowicz M, Figiel M (2015) Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis Model Mech 8(9):1047–1057. doi:10.1242/dmm.019406
Camnasio S, Delli Carri A, Lombardo A, Grad I, Mariotti C, Castucci A, Rozell B, Lo Riso P, Castiglioni V, Zuccato C, Rochon C, Takashima Y, Diaferia G, Biunno I, Gellera C, Jaconi M, Smith A, Hovatta O, Naldini L, Di Donato S, Feki A, Cattaneo E (2012) The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis 46:41–51. doi:10.1016/j.nbd.2011.12.042
Cha MY, Kim DK, Mook-Jung I (2015) The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med 47:e150
Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X (2013) Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Investig 123(12):5371–5388. doi:10.1172/JCI70911
Xia G, Ashizawa T (2015) Dynamic changes of nuclear RNA foci in proliferating DM1 cells. Histochem Cell Biol. doi:10.1007/s00418-015-1315-5
Xia G, Santostefano KE, Goodwin M, Liu J, Subramony SH, Swanson MS, Terada N, Ashizawa T (2013) Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell Reprogram 15(2):166–177. doi:10.1089/cell.2012.0086
Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126(3):385–399. doi:10.1007/s00401-013-1149-y
Hansen RS, Gartler SM, Scott CR, Chen SH, Laird CD (1992) Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum Mol Genet 1(8):571–578
Urbach A, Bar-Nur O, Daley GQ, Benvenisty N (2010) Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6:407–411. doi:10.1016/j.stem.2010.04.005
de Esch CE, Ghazvini M, Loos F, Schelling-Kazaryan N, Widagdo W, Munshi ST, van der Wal E, Douben H, Gunhanlar N, Kushner SA, Pijnappel WW, de Vrij FM, Geijsen N, Gribnau J, Willemsen R (2014) Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem cell reports 3(4):548–555. doi:10.1016/j.stemcr.2014.07.013
Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ (2011) Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 6:e26203. doi:10.1371/journal.pone.0026203
Bar-Nur O, Caspi I, Benvenisty N (2012) Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J Mol Cell Biol 4:180–183. doi:10.1093/jmcb/mjs007
Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, Bhattacharyya A (2014) iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev 23(15):1777–1787. doi:10.1089/scd.2014.0030
Smeets HJ, Smits AP, Verheij CE, Theelen JP, Willemsen R, van de Burgt I, Hoogeveen AT, Oosterwijk JC, Oostra BA (1995) Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet 4(11):2103–2108
Liu J, Koscielska KA, Cao Z, Hulsizer S, Grace N, Mitchell G, Nacey C, Githinji J, McGee J, Garcia-Arocena D, Hagerman RJ, Nolta J, Pessah IN, Hagerman PJ (2012) Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet 21:3795–3805. doi:10.1093/hmg/dds207
Eigentler A, Boesch S, Schneider R, Dechant G, Nat R (2013) Induced pluripotent stem cells from friedreich ataxia patients fail to upregulate frataxin during in vitro differentiation to peripheral sensory neurons. Stem Cells Dev 22(24):3271–3282. doi:10.1089/scd.2013.0126
Lee YK, Ho PW, Schick R, Lau YM, Lai WH, Zhou T, Li Y, Ng KM, Ho SL, Esteban MA, Binah O, Tse HF, Siu CW (2013) Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflugers Arch 466(9):1831–1844. doi:10.1007/s00424-013-1414-x
Hick A, Wattenhofer-Donze M, Chintawar S, Tropel P, Simard JP, Vaucamps N, Gall D, Lambot L, Andre C, Reutenauer L, Rai M, Teletin M, Messaddeq N, Schiffmann SN, Viville S, Pearson CE, Pandolfo M, Puccio H (2013) Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis Model Mech 6:608–621. doi:10.1242/dmm.010900
Liu J, Verma PJ, Evans-Galea MV, Delatycki MB, Michalska A, Leung J, Crombie D, Sarsero JP, Williamson R, Dottori M, Pebay A (2011) Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem cell reviews 7(3):703–713. doi:10.1007/s12015-010-9210-x
Ku S, Soragni E, Campau E, Thomas EA, Altun G, Laurent LC, Loring JF, Napierala M, Gottesfeld JM (2010) Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAATTC triplet repeat instability. Cell Stem Cell 7:631–637. doi:10.1016/j.stem.2010.09.014
Wells RD, Dere R, Hebert ML, Napierala M, Son LS (2005) Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucleic Acids Res 33:3785–3798. doi:10.1093/nar/gki697
Figura G, Koscianska E, Krzyzosiak WJ (2015) In vitro expansion of CAG, CAA, and mixed CAG/CAA repeats. Int J Mol Sci 16(8):18741–18751
Lopez Castel A, Cleary JD, Pearson CE (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11:165–170. doi:10.1038/nrm2854
Pearson CE, Nichol Edamura K, Cleary JD (2005) Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 6:729–742. doi:10.1038/nrg1689
Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ (2008) Disease-specific induced pluripotent stem cells. Cell 134:877–886. doi:10.1016/j.cell.2008.07.041
An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM (2012) Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11:253–263. doi:10.1016/j.stem.2012.04.026
Xia G, Santostefano K, Hamazaki T, Liu J, Subramony SH, Terada N, Ashizawa T (2013) Generation of human-induced pluripotent stem cells to model spinocerebellar ataxia type 2 in vitro. J Mol Neurosci MN 51(2):237–248. doi:10.1007/s12031-012-9930-2
Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, Doerr J, Ladewig J, Mertens J, Tuting T, Hoffmann P, Klockgether T, Evert BO, Wullner U, Brustle O (2011) Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 480:543–546. doi:10.1038/nature10671
Nihei Y, Ito D, Okada Y, Akamatsu W, Yagi T, Yoshizaki T, Okano H, Suzuki N (2013) Enhanced aggregation of androgen receptor in induced pluripotent stem cell-derived neurons from spinal and bulbar muscular atrophy. J Biol Chem 288:8043–8052. doi:10.1074/jbc.M112.408211
Grunseich C, Zukosky K, Kats IR, Ghosh L, Harmison GG, Bott LC, Rinaldi C, Chen KL, Chen G, Boehm M, Fischbeck KH (2014) Stem cell-derived motor neurons from spinal and bulbar muscular atrophy patients. Neurobiol Dis 70:12–20. doi:10.1016/j.nbd.2014.05.038
Luo Y, Fan Y, Zhou B, Xu Z, Chen Y, Sun X (2012) Generation of induced pluripotent stem cells from skin fibroblasts of a patient with olivopontocerebellar atrophy. Tohoku J Exp Med 226:151–159
Du J, Campau E, Soragni E, Jespersen C, Gottesfeld JM (2013) Length-dependent CTG.CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum Mol Genet 22:5276–5287. doi:10.1093/hmg/ddt386
Jaworski A, Rosche WA, Gellibolian R, Kang S, Shimizu M, Bowater RP, Sinden RR, Wells RD (1995) Mismatch repair in Escherichia coli enhances instability of (CTG)n triplet repeats from human hereditary diseases. Proc Natl Acad Sci USA 92(24):11019–11023
Du J, Campau E, Soragni E, Ku S, Puckett JW, Dervan PB, Gottesfeld JM (2012) Role of mismatch repair enzymes in GAA.TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J Biol Chem 287:29861–29872. doi:10.1074/jbc.M112.391961
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80(2):415–428. doi:10.1016/j.neuron.2013.10.015
Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5(208):208ra149
Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, Zhang SC (2010) Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci 107:4335–4340. doi:10.1073/pnas.0910012107
Murry CE, Keller G (2008) Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 132(4):661–680
Sandoe J, Eggan K (2013) Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat Neurosci 16(7):780–789
Takazawa T, Croft GF, Amoroso MW, Studer L, Wichterle H, Macdermott AB (2012) Maturation of spinal motor neurons derived from human embryonic stem cells. PLoS One 7:e40154. doi:10.1371/journal.pone.0040154
Hernandez F, Perez M, de Barreda EG, Goni-Oliver P, Avila J (2008) Tau as a molecular marker of development, aging and neurodegenerative disorders. Curr Aging Sci 1(1):56–61
Compagnucci C, Nizzardo M, Corti S, Zanni G, Bertini E (2013) In vitro neurogenesis: development and functional implications of iPSC technology. Cell Mol Life Sci CMLS 71(9):1623–1639. doi:10.1007/s00018-013-1511-1
Velasco I, Salazar P, Giorgetti A, Ramos-Mejia V, Castano J, Romero-Moya D, Menendez P (2014) Concise review: generation of neurons from somatic cells of healthy individuals and neurological patients through induced pluripotency or direct conversion. Stem Cells 32(11):2811–2817. doi:10.1002/stem.1782
Pre D, Nestor MW, Sproul AA, Jacob S, Koppensteiner P, Chinchalongporn V, Zimmer M, Yamamoto A, Noggle SA, Arancio O (2014) A time course analysis of the electrophysiological properties of neurons differentiated from human induced pluripotent stem cells (iPSCs). PLoS One 9:e103418. doi:10.1371/journal.pone.0103418
Koehler KR, Tropel P, Theile JW, Kondo T, Cummins TR, Viville S, Hashino E (2011) Extended passaging increases the efficiency of neural differentiation from induced pluripotent stem cells. BMC Neurosci 12(1):82. doi:10.1186/1471-2202-12-82
Devlin AC, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, Vallier L, Shaw CE, Chandran S, Miles GB (2014) Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun 6:5999
Ebert AD, Shelley BC, Hurley AM, Onorati M, Castiglioni V, Patitucci TN, Svendsen SP, Mattis VB, McGivern JV, Schwab AJ, Sareen D, Kim HW, Cattaneo E, Svendsen CN (2013) EZ spheres: a stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res 10(3):417–427. doi:10.1016/j.scr.2013.01.009
Kurosawa H (2007) Methods for inducing embryoid body formation: in vitro differentiation system of embryonic stem cells. J Biosci Bioeng 103(5):389–398
Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L (2008) Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev 22(2):152–165
Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, Nishikawa SI, Sasai Y (2000) Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 28(1):31–40
Yang N, Ng YH, Pang ZP, Sudhof TC, Wernig M (2011) Induced neuronal cells: how to make and define a neuron. Cell Stem Cell 9(6):517–525
Li XJ, Zhang X, Johnson MA, Wang ZB, Lavaute T, Zhang SC (2009) Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development 136(23):4055–4063. doi:10.1242/dev.036624
Ma X, Turnbull P, Peterson R, Turnbull J (2013) Trophic and proliferative effects of Shh on motor neurons in embryonic spinal cord culture from wildtype and G93A SOD1 mice. BMC Neurosci 14:119
Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, Lipton SA, Ding S (2011) Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9(2):113–118. doi:10.1016/j.stem.2011.07.002
Purro SA, Dickins EM, Salinas PC (2012) The secreted Wnt antagonist Dickkopf-1 is required for amyloid beta-mediated synaptic loss. J Neurosci 32(10):3492–3498
Laeng P, Pitts RL, Lemire AL, Drabik CE, Weiner A, Tang H, Thyagarajan R, Mallon BS, Altar CA (2004) The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J Neurochem 91(1):238–251. doi:10.1111/j.1471-4159.2004.02725.x
Chatzi C, Brade T, Duester G (2011) Retinoic acid functions as a key GABAergic differentiation signal in the basal ganglia. PLoS Biol 9(4):e1000609. doi:10.1371/journal.pbio.1000609
Lee S, Lee B, Lee JW, Lee SK (2009) Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron 62(5):641–654. doi:10.1016/j.neuron.2009.04.025
Urbanek MO, Krzyzosiak WJ (2016) RNA FISH for detecting expanded repeats in human diseases. Methods 98:115–123. doi:10.1016/j.ymeth.2015.11.017
Wojciechowska M, Krzyzosiak WJ (2011) Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet 20:3811–3821. doi:10.1093/hmg/ddr299
Pettersson OJ, Aagaard L, Jensen TG, Damgaard CK (2015) Molecular mechanisms in DM1—a focus on foci. Nucleic Acids Res. doi:10.1093/nar/gkv029
Ring KL, An MC, Zhang N, O’Brien RN, Ramos EM, Gao F, Atwood R, Bailus BJ, Melov S, Mooney SD, Coppola G, Ellerby LM (2015) Genomic analysis reveals disruption of striatal neuronal development and therapeutic targets in human Huntington’s disease neural stem cells. Stem Cell Rep 5(6):1023–1038
Mattis VB, Tom C, Akimov S, Saeedian J, Ostergaard ME, Southwell AL, Doty CN, Ornelas L, Sahabian A, Lenaeus L, Mandefro B, Sareen D, Arjomand J, Hayden MR, Ross CA, Svendsen CN (2015) HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum Mol Genet 24(11):3257–3271
Liu Y, Xue Y, Ridley S, Zhang D, Rezvani K, Fu XD, Wang H (2014) Direct reprogramming of Huntington’s disease patient fibroblasts into neuron-like cells leads to abnormal neurite outgrowth, increased cell death, and aggregate formation. PLoS One 9:e109621. doi:10.1371/journal.pone.0109621
Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57(5):369–384
Lu XH, Mattis VB, Wang N, Al-Ramahi I, van den Berg N, Fratantoni SA, Waldvogel H, Greiner E, Osmand A, Elzein K, Xiao J, Dijkstra S, de Pril R, Vinters HV, Faull R, Signer E, Kwak S, Marugan JJ, Botas J, Fischer DF, Svendsen CN, Munoz-Sanjuan I, Yang XW (2014) Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med 6(268):268ra178. doi:10.1126/scitranslmed.3010523
Yao Y, Cui X, Al-Ramahi I, Sun X, Li B, Hou J, Difiglia M, Palacino J, Wu ZY, Ma L, Botas J, Lu B (2015) A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife 4:e05449. doi:10.7554/eLife.05449
Chiu FL, Lin JT, Chuang CY, Chien T, Chen CM, Chen KH, Hsiao HY, Lin YS, Chern Y, Kuo HC (2015) Elucidating the role of the A2A adenosine receptor in neurodegeneration using neurons derived from Huntington’s disease iPSCs. Hum Mol Genet 24(21):6066–6079
Jeon I, Choi C, Lee N, Im W, Kim M, Oh SH, Park IH, Kim HS, Song J (2014) In vivo roles of a patient-derived induced pluripotent stem cell line (HD72-iPSC) in the YAC128 model of Huntington’s disease. Int J Stem Cells 7:43–47. doi:10.15283/ijsc.2014.7.1.43
Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM (2012) C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep 2:1016
Reddy K, Zamiri B, Stanley SY, Macgregor RB Jr, Pearson CE (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem 288(14):9860–9866
Hideyama T, Yamashita T, Suzuki T, Tsuji S, Higuchi M, Seeburg PH, Takahashi R, Misawa H, Kwak S (2010) Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci 30(36):11917–11925
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525(7567):56–61
Schweizer Burguete A, Almeida S, Gao FB, Kalb R, Akins MR, Bonini NM (2015) GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. eLife 4:e08881. doi:10.7554/eLife.08881
Satoh J, Yamamoto Y, Kitano S, Takitani M, Asahina N, Kino Y (2014) Molecular network analysis suggests a logical hypothesis for the pathological role of c9orf72 in amyotrophic lateral sclerosis/frontotemporal dementia. J Cent Nerv Syst Dis 6:69–78. doi:10.4137/JCNSD.S18103
Kumari D, Swaroop M, Southall N, Huang W, Zheng W, Usdin K (2015) High-throughput screening to identify compounds that increase fragile X mental retardation protein expression in neural stem cells differentiated from fragile X syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl Med 4(7):800–808
Rusmini P, Simonini F, Crippa V, Bolzoni E, Onesto E, Cagnin M, Sau D, Ferri N, Poletti A (2011) 17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy. Neurobiol Dis 41:83–95. doi:10.1016/j.nbd.2010.08.023
Soragni E, Miao W, Iudicello M, Jacoby D, De Mercanti S, Clerico M, Longo F, Piga A, Ku S, Campau E, Du J, Penalver P, Rai M, Madara JC, Nazor K, O’Connor M, Maximov A, Loring JF, Pandolfo M, Durelli L, Gottesfeld JM, Rusche JR (2014) Epigenetic therapy for Friedreich ataxia. Ann Neurol 76(4):489–508. doi:10.1002/ana.24260
Shan B, Xu C, Zhang Y, Xu T, Gottesfeld JM, Yates JR 3rd (2014) Quantitative proteomic analysis identifies targets and pathways of a 2-aminobenzamide HDAC inhibitor in friedreich’s ataxia patient iPSC-derived neural stem cells. J Proteome Res 13(11):4558–4566. doi:10.1021/pr500514r
Singh AM, Adjan Steffey VV, Yeshi T, Allison DW (2015) Gene editing in human pluripotent stem cells: choosing the correct path. m-Cells—choosing-the-correct-path/630 [pii]. J Stem Cell Regen Biol 1:(1)
An MC, O’Brien RN, Zhang N, Patra BN, De La Cruz M, Ray A, Ellerby LM (2014) Polyglutamine disease modeling: epitope based screen for homologous recombination using CRISPR/Cas9 system. PLoS Curr. doi:10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a
Park CY, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, Benvenisty N, Kim DW (2015) Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep 13(2):234–241
Li Y, Polak U, Bhalla A, Rozwadowska N, Butler JS, Lynch D, Dent SY, Napierala M (2015) Excision of expanded GAA repeats alleviates the molecular phenotype of Friedreich’s ataxia. Mol Ther 23(6):1055–1065. doi:10.1038/mt.2015.41
Xia G, Gao Y, Jin S, Subramony S, Terada N, Ranum LP, Swanson MS, Ashizawa T (2014) Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 iPS-cell derived neural stem cells. Stem Cells 33(6):1829–1838. doi:10.1002/stem.1970
Juopperi TA, Kim WR, Chiang CH, Yu H, Margolis RL, Ross CA, Ming GL, Song H (2012) Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington's disease patient cells. Mol Brain 5:17. doi:10.1186/1756-6606-5-17
Kaufmann M, Schuffenhauer A, Fruh I, Klein J, Thiemeyer A, Rigo P, Gomez-Mancilla B, Heidinger-Millot V, Bouwmeester T, Schopfer U, Mueller M, Fodor BD, Cobos-Correa A (2015) High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J biomol screen 20(9):1101–1111. doi:10.1177/1087057115588287
Acknowledgments
This work was supported by a Grant from National Science Center (2012/06/A/NZ1/00094 to Wlodzimierz J. Krzyzosiak) and by the Polish Ministry of Science and Higher Education, under the KNOW program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
E. Jaworska, E. Kozlowska and P. M. Switonski contributed equally to this work.
Rights and permissions
About this article

Cite this article
Jaworska, E., Kozlowska, E., Switonski, P.M. et al. Modeling simple repeat expansion diseases with iPSC technology. Cell. Mol. Life Sci. 73, 4085–4100 (2016). https://doi.org/10.1007/s00018-016-2284-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2284-0