
Abstract
The regular functioning of the human body is the result of interactions between numerous organs and biological systems, with the Renin-Angiotensin System (RAS) playing a crucial role in maintaining homeostasis for human existence. Though RAS is mainly known for regulation of blood pressure, however, its role in cancer development and progression has been emerging nowadays wherein this system was found to promote angiogenesis and inflammation in tumor niche. RAS comprises of numerous elements, of which angiotensin converting enzyme (ACE) is of utmost significance which converts angiotensin I (ATI) to angiotensin II (ATII), which then undergoes downstream signaling via binding to AT receptors. This signaling aids in hematopoiesis and the associated malignancies like leukemia, myeloma and lymphoma. ACE and the downstream signaling cascades upregulation could be seen in cancer models which are linked to the activation of multiple signaling events involving NF-κB, PI3K, MAPK, etc. The importance of RAS in hematological malignancies led to the exploration of RAS inhibitors for the cancer treatment. There are certain categories of RAS inhibitors which include ACE inhibitors, Angiotensin receptor blockers or renin inhibitors which have been tested in vitro either alone or in combination for therapeutics of various cancers including hematological malignancies. The local RAS and its association with cancer might opens up new avenues for investigation and development of novel therapies for hematological malignancies.
Nidhi Gupta and Shraddha Kapoor, equal contribution
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
The normal functioning of human body is interplay of the interactions among various organs and biological systems, out of which the role of Renin-Angiotensin system (RAS) is inevitably significant for maintaining homeostasis in human being’s survival [1, 2]. Since, from numerous years it has been well studied in context with heart-kidney axial system and also, as a key player for regulating blood pressure in the body [3]. In addition to systemic blood pressure regulation, it maintains tonicity of vascular tissues and extracelluar volumes, ion concentration gradient and vasomotor functions [1, 4]. Tissue specific expression of local RAS is responsible for diversity in functionality and processes [3]. Augmented levels of RAS activation have been evident in many organ specific tumors like kidney, pancreas, brain, cervix, etc. and also in hematopoietic cancers [5].
Apart from the role documented for “classical” RAS, the data has emerged discussing the revolutionary involvement of RAS in plethora of physiological as well as pathological conditions. One of the relevant areas of study is the implication of RAS in the biology of cancer. Cancer in itself is a broad area in focus for association of RAS, however, the scope of this chapter highlights the role of RAS in relation to Hematological Malignancies, mainly leukemia (Acute Myeloid Leukemia, Chronic Myeloid Leukemia, and Chronic Lymphoid Leukemia etc.), myeloma and lymphoma, and scope of treatment by targeting RAS in context to hematological malignancies which is less known till date.
Componential Organization of RAS
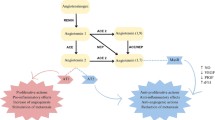
RAS system involves complex network of its multiple peptides, receptors and enzyme components; the key step of significance in circulating RAS is production of Angiotensin II (ATII) via two-step process involving Renin and Angiotensin-Converting Enzyme (ACE) [6] as shown in Fig. 20.1.

Componential organization of RAS involving numerous active peptide and receptor. Renin is secreted into the bloodstream by the juxtaglomerular cells of kidneys, where it interacts with angiotensinogen (AGN), which is produced by the liver. Renin catalyzes the conversion of angiotensinogen (AGN) to angiotensin I (ATI), which is then cleaved into ATII by angiotensin converting enzyme (ACE). The active biopeptide ATII elucidates its effect via AT1R and AT2R receptors. ACE2 also fragments ATII, resulting in AT (1–7) that bind with the MAS receptor (MASR). ATIII and ATIV are also created in the sequential processing of ATII. [RAS: Renin-Angiotensin system; ACEi: ACE inhibitor; ARB: Angiotensin Receptor Blockers; AP: Aminopeptidase]
Renin is produced by the kidneys in response to decreased arterial pressure, reduced sodium in the distal tubule or sympathetic nervous system activity via the β-adrenergic receptors. Renin is secreted into the bloodstream by juxtaglomerular cells, where it encounters angiotensinogen (AGN), which is ordinarily produced by the liver. Renin catalyses the conversion of angiotensinogen (AGN) to angiotensin I (ATI), which is rapidly cleaved by angiotensin converting enzyme (ACE) to create ATII. ATII causes the adrenal glands to release aldosterone, which accelerates sodium and water reabsorption, increasing blood volume and blood pressure. ATII also causes vasoconstriction of the arterioles in several organs of the body by acting on smooth muscle. Furthermore, ATII stimulates the posterior pituitary gland to release antidiuretic hormone, which causes water retention and activates the thirst reflex [7, 8].
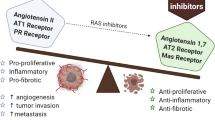
The active biopeptide ATII elucidate its effect via AT1R and AT2R receptor through downstream signaling. Also, ATII is fragmented by ACE2 to AT (1–7) which further interact through MAS receptor (MASR). Further ATIII and ATIV are also produced in stepwise processing of ATII [9].
RAS and Cancer: What’s Known Till Date?
There has been a recent upthrust of RAS in relation to cancer and malignancy implicated by various studies wherein the involvement of RAS in induction of cell proliferation, maintaining growth stimulatory signals, promoting angiogenesis and inflammation, combating apoptosis etc. were shown as the attributes of RAS functionality indicating its characteristics synchronous to cancer progression [10]. It has been found that impaired regulation of RAS in case of tumorigenesis might be crucial for studying its significance in cancer.
Carcinogenesis being a multi-factorial disease involves various hallmarks such as angiogenesis, inflammation, telomerase activity, etc. In addition, tumor microenvironment is a pre-requisite for the continuous proliferation of cancerous cells. Our group had reported the involvement of these factors in different cancers ranging from solid tumors to hematological malignancies [11,12,13,14,15]. In relation to RAS, the signaling promoted by peptide Angiotensin II is observed by studies to enhance angiogenesis mediated by vascular endothelial growth factor (VEGF) via growth of cellular vasculature [16]. Besides, other ways of stimulation of pathogenesis mechanism for evasion and growth of tumor mainly comprises of aggravation of ATII expression and comparative expression of Angiotensin converting enzymes-1 and 2 [17].
RAS Signaling: Role in Oncogenesis
Numerous findings have highlighted that the downstream regulation of cellular RAS signaling pathways may be associated with pace of tumor development or regression. Some evident molecule with anti-carcinogenic effect is AT2R receptor-interacting proteins (ATIP) that work in conjunction with AT2R receptors whose downstream function is generated by action of peptide ATII. These ATIP mainly ATIP-1 and ATIP-3 collectively promote tumor inhibitory actions by blocking receptor tyrosine kinase activation, thus, halting cellular proliferation [5, 18].
While interaction of ATII with AT1R has been contradictory to action with AT2R; tumor cells were found to have exacerbated expression levels of these AT1R receptors. ATII bio peptide also has cellular proliferation promoting effects in turn by ultimate increased levels of transcription factor like Nuclear Factor κB (NF-κB). This may be attributed by enhancement of apoptosis via AT2R receptor downstream signaling events supporting its anti-apoptotic activity while ATII binding to AT1R receptor in cancerous cells inhibits cell death and promotes cell growth and survival [19, 20]. Moreover, the MAPK or PI3K mitotic cascade events by AT1R activation via binding to ATII leading to NF-κB stimulation resulted in promotion of Bcl-xL and Bcl-2 anti-apoptotic molecules [6]. The amelioration of these mitotic effects by inhibiting molecules like ATII and NF-κB have been principally observed and utilized for RAS blockage in order to enhance apoptosis and decrease cell proliferation [5, 21].
Though multiple signaling pathways including PI-3 K, MAPK and JAK-STAT are deregulated in carcinogenesis and shown to be regulated by RAS to favour tumor progression. Another pathway such as adenosine monophosphate (AMP)-activated protein kinase (AMPK) signaling system is also reported to be relevant in oncogenesis. With the stability of p53 and the cyclin-dependent kinase inhibitors p21WAF1 and p27CIP1, AMPK pathway possess tumour suppressor role. This pathway also limits the production of mTOR-1 and hypoxia-inducible factor-1 (HIF-1), as well as fatty acids, triglycerides, cholesterol, glycogen, and proteins, resulting in cell growth inhibition. The association between RAS and AMPK signaling pathway was observed by in vivo studies in uninephrectomized (UNX) rat experimental model. AMPK expression reduced in carcinogenesis with increase in mutant p53 and Ki-67, however, antagonization of RAS by inhibitors resulted in elevated AMPK, hence, proves to be protective against malignant transformations.
Inhibition in levels of molecules like hypoxia-inducible factor-1α (HIF-1α) and mTOR-1 are also responsible for regulation of AMPK signaling pathway [5, 22].
Hematopoietic RAS
According to previous molecular biological investigations, the existence of local intrinsic bone marrow RAS was proved which was found to be regulatory in scenarios involving physiological and pathological hematopoiesis; one of earliest experimental evidence relating hematopoiesis production and differeniation in bone marrow to the components of RAS system [23]. The major effector peptide of RAS, angiotensin II (ATII), stimulates hematopoietic progenitor cell proliferation via binding to angiotensin type 1a (AT1a) receptors present on CD34+ hematopoietic stem cells. The existence of ACE in human primitive lympho-hematopoietic cells, embryonic, foetal and adult hematopoietic tissues has also been described [24, 25]. The elements of RAS system have been observed to be modulating in proliferative processes mainly; myelopoiesis, erythropoiesis, thrombopoiesis and even process involving formation of other immune cell linages [19].
The presence of a regulatory tissue RAS system has eased the investigations such as tracing the level of specific levels of glycoproteinaceous enzyme ACE would help in studying of pathophysiological aspect of many diseases [26].
RAS in Hematological Malignancies
Since RAS elements function in hematopoietic processes throughout the development, hence, there has been data suggesting the importance of this system in hematological malignancies which mainly comprises of leukemia, myeloma and lymphoma. The present chapter discussed the involvement of RAS in these types of hematological malignancies.
RAS and Leukemia
There are many studies elucidating association between presence of local bone marrow RAS with hematopoietic functioning and production of the neoplastic blood cells [23]. In the report by Aksu et al. the group evaluated the level of surface antigen marker ACE (CD143) isolated from bone marrow of Acute Myeloid Leukemia (AML) patients and found to be upregulated in leukemic cells of myeloid blast origin [27]. The levels of renin and ACE were investigated in both bone marrow aspirate and peripheral blood samples of leukemic and control patients. Unlike Renin, levels of ACE have shown to be increased significantly in bone marrow samples as compared to peripheral blood [28]. Regulatory role of an ACE-Acetyl-N-Ser-Asp-Lys-Pro (AcSDKP) system is also under scrutiny check in vicinity of bone marrow microenvironment for its role in involvement of hematopoiesis. The degradation of AcSDKP or goralatide is facilitated by ACE, hence, enhanced degradation could be observed due to increased production of ACE in leukemia [29,30,31,32]. The mechanism of AcSDKP to act as negative regulator of hematopoiesis is by keeping the S-phase population of hematopoietic and progenitor cells in non-dividing quiescent phase [19, 33] (Fig. 20.2). Role of analogs of AcSDKP are under study which remain unaltered in terms of degradation by ACE thus, playing important function in preventing hematopoiesis [34] (Fig. 20.3).

Scrutiny check by ACE-AcSDKP system on hematopoiesis and the associated malignancy. Thymosin β4 peptide is produced by various cell types whose degradation by oligopeptidase (OP) produce a smaller peptide AcSDKP (Acetyl-N-Ser-Asp-Lys-Pro) which inhibits hematopoiesis in bone marrow microenvironment. The degradation of AcSDKP by ACE promotes cellular proliferation and enhances the process of hematopoiesis and might also play a crucial role in hematological malignancy

Disease specific expression observed in hematological malignancies. The association between local bone marrow RAS, hematopoiesis and neoplastic conditions such as leukemia, lymphoma and myeloma have been elucidated as per the literature. Various inhibitors of RAS contribute have been tested in vitro for cancer therapeutics and hence, possess the potential of combating and managing the hematological malignancy
The evidence of disease specific expression and activation of local RAS such as a differentially altered activity in leukemia and lymphoma was indicated by study of Uz et al. The comparative expression monitoring in case of groups of myeloid or lymphoid blood malignancies for the important components of RAS mainly ACE1, ACE2, Renin and Angiotensin have been carried out [35]. The expression levels of Renin mRNA was found to be significantly elevated in lymphoid compared to myeloid classified disease, whereas for ACE1 and ACE2 expression elevation were found vice versa. The study also mentioned mRNA expression of RAS elements with active and without active disease status in patients [35].
The major RAS components’ (Renin, ACE and angiotensinogen) specific gene expression levels using real time quantitative PCR analysis method was studied in K562 experimental cell line to evaluate role of RAS in leukemia and further paving way for it to act as in vitro model system for finding effect of RAS system effecting drugs and association between respective leukemic cell proliferation pattern [36, 37].
While specific with quantitative expression of ACE, renin and angiotensinogen in case of AML, the patient had relevantly higher mRNA expression in the bone marrow samples [38]. A contemporary study to the work of Beyazit et al., traced the occurrence of AT receptors and other RAS components in cultured line of marrow stromal cells as well as in lineage of bone marrow cells, concluding the presence of local RAS supported autocrine and paracrine mechanism of hematopoiesis due to auto regulatory synthesis of marrow stromal cells mediated synthesis of Angiotensin II [39].
RAS and Lymphoma
Angiotensin Converting Enzyme (ACE) being an important enzyme for octapeptide ATII peptide production and RAS functioning, also associated with certain neoplastic conditions and while regulated by AcSDKP peptide negative regulator vis-à-vis affecting cellular proliferation in bone marrow environment [27, 36, 37, 40]. Thus, due to numerous significant role of this enzyme, ACE expression levels were mapped in macrophages of lymphoma linked proliferative disorder Hodgkin’s disease in lymph nodes to decipher its relationship in RAS functionality and thus, mechanism of tumorigenesis. This study demonstrated the ACE expression in intratumoral residual macrophages in lymph nodes of patients suffering from Hodgkin’s lymphoma (HL) versus negligible ACE expression obtained in case of controls and lymphoid hyperplasia. The study has prominent drawback of not demarcating in quantification of level of ACE expression in relation to the strength of staining and unable to differentiate between nodular sclerosing and mixed cellular sub categorized cases of Hodgkin’s lymphoma. ACE expression was reported to be directly proportional to macrophages infiltration is lymph node in HL which is linked to interaction between RAS system and thereby, enhancing evidence of lymphoma generation [41]. The other molecular and cellular evidences available with relevance to lymphoma (both Hodgkin’s and non-Hodgkin’s) generation in turn with tissue RAS system is least explored and further require experimental and clinical elucidation. The pronounced disease prevalence of Hodgkin’s Lymphoma in pediatrics population as compared to adult counterpart and treatable nature of these lymphoma provides an urge to be explored with greater scientific upthrust in the field to ensure disease free life to these individuals (Fig. 20.3) [42].
RAS and Multiple Myeloma
The levels of circulatory ACE enzyme important for investigation of systemic and portal disease was found to be augmented significantly in clinical cases of Multiple myeloma in bone marrow compared to controls. However, intra group staging of these MM patients according to International Staging System (ISS) criteria reported non-significant variation in ACE levels [24, 26]. Similarly other studies provide evidence of elevated RAS component expression levels directly relating to activation of local bone marrow RAS functioning in MM disease state (Fig. 20.3). Further reports indicate enhanced AT1R type receptor in MM and correlated with CD34 activation in bone marrow supporting the idea of AT1R receptor promotion of tumorigenic activity and angiogenesis (as mentioned before in the chapter under section RAS signaling) [43].
RAS Inhibitors as Cancer Therapy: Single or Combinatorial
The role of RAS in tumor progression and pathogenesis has been documented in numerous cancers. ATII signaling in RAS system drives neovascularization and boosts cell proliferation. An increasing amount of evidence demonstrates that ATII signalling increases VEGF-mediated angiogenesis in malignancy by directly impacting tumour and stromal cells as well as indirectly altering vascular cell development during angiogenesis. Hence, blocking of this system might be beneficial to combat the malignancy. Thus, the class of drugs that modulates the RAS functionally has been observed to be efficacious in in vitro and in vivo cancer models [6]. Suppressing the action of RAS by administration of Angiotensin Receptor Blockers (ARB), which acts through inhibiting the activity of AT1R; or ACE inhibitor (ACEi) which inhibits enzyme ACE, thus preventing synthesis of ATII; or directly renin inhibitor; all function as RAS antagonists, block the RAS influence on angiogenesis, reducing the growth of cancer and possibly lowering cancer risk over time and has proved to be preventive and helpful in treatment of certain types of cancers [6, 44]. However, these medications can impact additional potential pathways mediated by ACE internalisation and endonuclear localization in distinct cell types, which may be related to certain aspects of carcinogenesis. ACEi apparently inhibited tumour growth in both cancer patients and animal models, as well as the carcinogenesis process via inhibiting angiogenesis (Table 20.1) [16, 45, 46].
RAS inhibitors have been tested in hematological malignancies to evaluate their efficacy for treatment. ACEi captopril and trandolapril and ARB losartan decreased cell proliferation, increased apoptosis, and decreased c-myc expression in leukemic cell line in vitro [47, 48]. Captopril also reduced tumor burden in mice with colorectal liver metastasis following partial hepatectomy and hence, reported as an adjunct therapy [49]. Another ACEi Enalapril induced apoptosis in HL60 acute promyelocytic leukaemia cells via downregualtion of STAT5A [50]. Enalapril also significantly slowed the progression of pancreatic cancer precursor lesions by inhibiting NF-kB in tumour cells [51, 52]. Other in vivo studies have shown that perindopril, another ACEi has a potential inhibitory effect on tumor growth due to suppression of VEGF-induced angiogenesis in head and neck squamous cell carcinoma & renal cell carcinoma [53]. In addition, Perindopril or fosinopril or ARBs losartan inhibited the progression of diethylnitrosamine-induced hepatocellular carcinoma in mice via inactivation of NF-κB [21]. ARBs such as Telmisartan appears to downregulate Bcl-2, an anti-apoptotic protein, and activate caspase-3, causing cell death of renal cell carcinoma [54]. Moreover, the basic mechanism of action of ARB like Irbesartan may act by inhibiting the proliferating activity of bioactive angiotensin element which may in turn inhibits proliferating-cell nuclear antigen (PCNA) and cyclin D1 expression and modulate p53 expression via AT1R receptor in experimental MCF-7 breast cancer cell line [19, 55]. Furthermore, Aliskiren, a renin inhibitor, reduced cell proliferation via decreasing Notch1 and KRT6 expression, and regulating apoptosis in renal carcinoma cells [56].
Besides, the single treatment of RAS inhibitors, their combination with standard therapy has also been found effective in cancer treatment. ACEi Enalapril was found to prevent the cardiotoxic side effects associated with doxorubicin treatment in lymphoma patients [57]. Further, the combination treatment of ARB losartan and standard drug, doxorubicin resulted in an increase in sensitivity of drug treatment in AML cell lines [58].
Conclusion and Future Prospects
The multifactorial interplay of local tissue RAS concerning many peptides, enzymes, receptors and feedback regulation as well as its involvement in growth of tumor tissue, metastasizing tendency, autocrine and paracrine regulation of mechanism relevant to cellular proliferation, cell signaling, immune responses, extracellular matrix formation, apoptosis, angiogenesis are characteristics which favors the study of local RAS as a therapeutic target for its significant role in carcinogenesis. Most of the clinical reports correlated the levels of different local bone marrow RAS components in case of normal or altered hematopoiesis processes and neoplastic disorders to investigate its disease specific altered state.
The mechanism involving hematopoiesis regulation or dysregulation via local bone marrow RAS modulation in normal or pathological state may be characterized by changing functionality of transcriptional factors dependent signals involved in turn in regulation of gene expression; and BM microenvironment and stromal cells mediated growth factors signals. Moreover, targeting the components involved in hematopoietic regulation of local RAS might be of therapeutic significance in treatment modalities related to cancers. The local RAS and its relationship to pathogical conditions involving malignancy paves a new way for exploration and delineating into its molecular mechanism which could be investigated further to gain an in-depth insight into this area for utilization for novel therapeutics of hematological malignancies.
References
Patel S, Rauf A, Khan H, Abu- T (2017) Renin-angiotensin-aldosterone (RAAS): the ubiquitous system for homeostasis and pathologies. Biomed Pharmacother 94:317–325. https://doi.org/10.1016/j.biopha.2017.07.091
Atlas SA (2007) The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm 13(8 Suppl B):9–20. https://doi.org/10.18553/jmcp.2007.13.s8-b.9
Vargas Vargas RA, Varela Millán JM, Fajardo Bonilla E (2022) Renin-angiotensin system: Basic and clinical aspects-a general perspective. Endocrinol Diabetes Nutr 69(1):52–62. https://doi.org/10.1016/j.endinu.2021.05.012
Laghlam D, Jozwiak M, Nguyen LS (2021) Renin-angiotensin-aldosterone system and immunomodulation: a state-of-the-art review. Cells 10(7):1767. https://doi.org/10.3390/cells10071767
Afsar B et al (2021) Renin-angiotensin system and cancer: epidemiology, cell signaling, genetics and epigenetics. Clin Transl Oncol 23(4):682–696. https://doi.org/10.1007/s12094-020-02488-3
George AJ, Thomas WG, Hannan RD (2010) The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer 10(11):745–759. https://doi.org/10.1038/nrc2945
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM (2014) Classical Renin-Angiotensin system in kidney physiology. Compr Physiol 4(3):1201–1228. https://doi.org/10.1002/cphy.c130040
Fyhrquist F, Saijonmaa O (2008) Renin-angiotensin system revisited. J Intern Med 264(3):224–236. https://doi.org/10.1111/j.1365-2796.2008.01981.x
Yang J, Yang X, Gao L, Zhang J, Yi C, Huang Y (2021) The role of the renin-angiotensin system inhibitors in malignancy: a review. Am J Cancer Res 11(3):884–897
Wegman-Ostrosky T, Soto-Reyes E, Vidal- S, Sánchez-Corona J (2015) The renin-angiotensin system meets the hallmarks of cancer. J Renin-Angiotensin Aldosterone Syst 16(2):227–233. https://doi.org/10.1177/1470320313496858
Gupta N, Kumar R, Seth T, Garg B, Sharma A (2020) Targeting of stromal versican by miR-144/199 inhibits multiple myeloma by downregulating FAK/STAT3 signalling. RNA Biol 17(1):98–111. https://doi.org/10.1080/15476286.2019.1669405
Kumar R, Gupta N, null Himani, Sharma A (2018) Novel combination of tanshinone I and lenalidomide induces chemo-sensitivity in myeloma cells by modulating telomerase activity and expression of shelterin complex and its associated molecules. Mol Biol Rep 45(6):2429–2439. https://doi.org/10.1007/s11033-018-4409-z
Anand V et al (2019) CD44 splice variant (CD44v3) promotes progression of urothelial carcinoma of bladder through Akt/ERK/STAT3 pathways: novel therapeutic approach. J Cancer Res Clin Oncol 145(11):2649–2661. https://doi.org/10.1007/s00432-019-03024-9
Gupta N, Kumar R, Seth T, Garg B, Sati HC, Sharma A (2019) Clinical significance of circulatory microRNA-203 in serum as novel potential diagnostic marker for multiple myeloma. J Cancer Res Clin Oncol 145(6):1601–1611. https://doi.org/10.1007/s00432-019-02896-1
Khan R, Sharma M, Kumar L, Husain SA, Sharma A (2013) Interrelationship and expression profiling of cyclooxygenase and angiogenic factors in Indian patients with multiple myeloma. Ann Hematol 92(1):101–109. https://doi.org/10.1007/s00277-012-1572-5
Rosenthal T, Gavras I (2019) Renin-angiotensin inhibition in combating malignancy: a review. Anticancer Res 39(9):4597–4602. https://doi.org/10.21873/anticanres.13639
Dolomatov S, Zukow W, Novikov N, Markaryan A, Eremeeva E (2019) Expression of the renin-angiotensin system components in oncologic diseases. Acta Clin Croat 58(2):354–364. https://doi.org/10.20471/acc.2019.58.02.21
Nouet S et al (2004) Trans-inactivation of receptor tyrosine kinases by novel angiotensin II AT2 receptor-interacting protein, ATIP. J Biol Chem 279(28):28989–28997. https://doi.org/10.1074/jbc.M403880200
Haznedaroglu IC, Beyazit Y (2013) Local bone marrow renin-angiotensin system in primitive, definitive and neoplastic haematopoiesis. Clin Sci (Lond) 124(5):307–323. https://doi.org/10.1042/CS20120300
Velez Rueda JO, Palomeque J, Mattiazzi A (2012) Early apoptosis in different models of cardiac hypertrophy induced by high renin-angiotensin system activity involves CaMKII. J Appl Physiol (1985), 112(12):2110–2120. https://doi.org/10.1152/japplphysiol.01383.2011
Saber S, Mahmoud AAA, Goda R, Helal NS, El- E, Abdelghany RH (2018) Perindopril, fosinopril and losartan inhibited the progression of diethylnitrosamine-induced hepatocellular carcinoma in mice via the inactivation of nuclear transcription factor kappa-B. Toxicol Lett 295:32–40. https://doi.org/10.1016/j.toxlet.2018.05.036
Hardie DG, Alessi DR (2013) LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol 11:36. https://doi.org/10.1186/1741-7007-11-36
Haznedaroğlu IC, Tuncer S, Gürsoy M (1996) A local renin-angiotensin system in the bone marrow. Med Hypotheses 46(6):507–510. https://doi.org/10.1016/s0306-9877(96)90122-x
Haznedaroglu IC, Beyazit Y (2010) Pathobiological aspects of the local bone marrow renin-angiotensin system: a review. J Renin-Angiotensin Aldosterone Syst 11(4):205–213. https://doi.org/10.1177/1470320310379876
Rodgers KE, Xiong S, Steer R, diZerega GS (2000) Effect of angiotensin II on hematopoietic progenitor cell proliferation. Stem Cells 18(4):287–294. https://doi.org/10.1634/stemcells.18-4-287
Albayrak M et al (2012) Elevated serum angiotensin converting enzyme levels as a reflection of bone marrow renin-angiotensin system activation in multiple myeloma. J Renin-Angiotensin Aldosterone Syst 13(2):259–264. https://doi.org/10.1177/1470320312437070
Aksu S et al (2006) Over-expression of angiotensin-converting enzyme (CD 143) on leukemic blasts as a clue for the activated local bone marrow RAS in AML. Leuk Lymphoma 47(5):891–896. https://doi.org/10.1080/10428190500399250
Abali H et al (2002) Circulating and local bone marrow renin-angiotensin system in leukemic hematopoiesis: preliminary evidences. Hematology 7(2):75–82. https://doi.org/10.1080/10245330290022160
Rousseau- A et al (Oct.1998) Lisinopril, an angiotensin I-converting enzyme inhibitor, prevents entry of murine hematopoietic stem cells into the cell cycle after irradiation in vivo. Exp Hematol 26(11):1074–1079
Rieger KJ et al (1993) Involvement of human plasma angiotensin I-converting enzyme in the degradation of the haemoregulatory peptide N-acetyl-seryl-aspartyl-lysyl-proline. Biochem J 296(Pt 2):373–378. https://doi.org/10.1042/bj2960373
Rousseau A, Michaud A, Chauvet MT, Lenfant M, Corvol P (1995) The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem 270(8):3656–3661. https://doi.org/10.1074/jbc.270.8.3656
Rousseau- A, Lenfant M, Potier P (Jul.1996) Catabolism of the hemoregulatory peptide N-Acetyl-Ser-Asp-Lys-Pro: a new insight into the physiological role of the angiotensin-I-converting enzyme N-active site. Bioorg Med Chem 4(7):1113–1119. https://doi.org/10.1016/0968-0896(96)00104-6
Li J, Volkov L, Comte L, Herve P, Praloran V, Charbord P (1997) Production and consumption of the tetrapeptide AcSDKP, a negative regulator of hematopoietic stem cells, by hematopoietic microenvironmental cells. Exp Hematol 25(2):140–146
Gaudron S, Grillon C, Thierry J, Riches A, Wierenga PK, Wdzieczak-Bakala J (1999) In vitro effect of acetyl-N-Ser-Asp-Lys-Pro (AcSDKP) analogs resistant to angiotensin I-converting enzyme on hematopoietic stem cell and progenitor cell proliferation. Stem Cells 17(2):100–106. https://doi.org/10.1002/stem.170100
Uz B et al (2013) Local hematopoietic renin-angiotensin system in myeloid versus lymphoid hematological neoplastic disorders. J Renin-Angiotensin Aldosterone Syst 14(4):308–314. https://doi.org/10.1177/1470320312464677
Koca E et al (2007) Renin-angiotensin system expression in the K562 human erythroleukaemic cell line. J Renin-Angiotensin Aldosterone Syst 8(3):145–147. https://doi.org/10.3317/jraas.2007.019
Koca E et al (2007) Angiotensin-converting enzyme expression of the lymphoma-associated macrophages in the lymph nodes of Hodgkin’s disease. J Natl Med Assoc 99(11):1243–1244, 1246–1247
Beyazit Y et al (2007) Overexpression of the local bone marrow renin-angiotensin system in acute myeloid leukemia. J Natl Med Assoc 99(1):57–63
Strawn WB, Richmond RS, Ann Tallant E, Gallagher PE, Ferrario CM (2004) Renin-angiotensin system expression in rat bone marrow haematopoietic and stromal cells. Br J Haematol 126(1):120–126. https://doi.org/10.1111/j.1365-2141.2004.04998.x
Abali H, Güllü IH, Engin H, Haznedaroğlu IC, Erman M, Tekuzman G (2002) Old antihypertensives as novel antineoplastics: angiotensin-I-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists. Med Hypotheses 59(3):344–348. https://doi.org/10.1016/s0306-9877(02)00185-8
Mazur G, Haloń A, Wróbel T, Kuliczkowski K (2004) Macrophage/histiocytic antigen CD68 expression in neoplastic and reactive lymph nodes. Rocz Akad Med Bialymst 49(Suppl):73–75
Radhakrishnan V et al (2017) Management of hodgkins lymphoma: ICMR consensus document. Indian J Pediatr 84(5):371–381. https://doi.org/10.1007/s12098-017-2304-6
Saka B et al (2019) The role of the local bone marrow renin-angiotensin system in multiple myeloma. Turk J Haematol 36(3):178–185. https://doi.org/10.4274/tjh.galenos.2019.2018.0420
Lever AF et al (1998) Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 352(9123):179–184. https://doi.org/10.1016/S0140-6736(98)03228-0
Lucero HA, Kintsurashvili E, Marketou ME, Gavras H (2010) Cell signaling, internalization, and nuclear localization of the angiotensin converting enzyme in smooth muscle and endothelial cells. J Biol Chem 285(8):5555–5568. https://doi.org/10.1074/jbc.M109.074740
Yoshiji H, Kuriyama S, Noguchi R, Fukui H (2004) Angiotensin-I converting enzyme inhibitors as potential anti-angiogenic agents for cancer therapy. Curr Cancer Drug Targets 4(7):555–567. https://doi.org/10.2174/1568009043332790
De la Iglesia Iñigo S et al (2009) Induction of apoptosis in leukemic cell lines treated with captopril, trandolapril and losartan: a new role in the treatment of leukaemia for these agents. Leuk Res 33(6):810–816. https://doi.org/10.1016/j.leukres.2008.09.029=
Stanojkovic TP, Zizak Z, Mihailovic-Stanojevic N, Petrovic T, Juranic Z (2005) Inhibition of proliferation on some neoplastic cell lines-act of carvedilol and captopril. J Exp Clin Cancer Res 24(3):387–395
Riddiough GE et al (2022) Captopril, a renin-angiotensin system inhibitor, attenuates tumour progression in the regenerating liver following partial hepatectomy. Int J Mol Sci 23(9):5281. https://doi.org/10.3390/ijms23095281
Purclutepe O et al (2012) Enalapril-induced apoptosis of acute promyelocytic leukaemia cells involves STAT5A. Anticancer Res 32(7):2885–2893
Fendrich V et al (2014) Enalapril and ASS inhibit tumor growth in a transgenic mouse model of islet cell tumors. Endocr Relat Cancer 21(5):813–824. https://doi.org/10.1530/ERC-14-0175
Carlos-Escalante JA, de Jesús M, Rivas-Castro A, Pichardo PS, Arce C, Wegman-Ostrosky T (2021) The use of antihypertensive drugs as coadjuvant therapy in cancer. Front Oncol 11:660943. https://doi.org/10.3389/fonc.2021.660943
Fendrich V et al (2010) The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut 59(5):630–637. https://doi.org/10.1136/gut.2009.188961
de Araújo Júnior RF, Leitão Oliveira ALCS, de Melo Silveira RF, de Oliveira Rocha HA, de França Cavalcanti P, de Araújo AA (2015) Telmisartan induces apoptosis and regulates Bcl-2 in human renal cancer cells. Exp Biol Med (Maywood) 240(1):34–44. https://doi.org/10.1177/1535370214546267
Du N et al (2012) Angiotensin II receptor type 1 blockers suppress the cell proliferation effects of angiotensin II in breast cancer cells by inhibiting AT1R signaling. Oncol Rep 27(6):1893–1903. https://doi.org/10.3892/or.2012.1720
Hu J, Zhang L-C, Song X, Lu J-R, Jin Z (2015) KRT6 interacting with notch1 contributes to progression of renal cell carcinoma, and aliskiren inhibits renal carcinoma cell lines proliferation in vitro. Int J Clin Exp Pathol 8(8):9182–9188
Georgakopoulos P et al (2019) The role of metoprolol and enalapril in the prevention of doxorubicin-induced cardiotoxicity in lymphoma patients. Anticancer Res 39(10):5703–5707. https://doi.org/10.21873/anticanres.13769
Ghasemi M et al (2019) The impact of At1r inhibition via losartan on the anti-leukaemic effects of doxorubicin in acute myeloid leukaemia. J Renin-Angiotensin Aldosterone Syst 20(2):1470320319851310. https://doi.org/10.1177/1470320319851310
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gupta, N., Kapoor, S., Sharma, A., Sharma, A. (2023). Renin-Angiotensin System in Hematological Malignancies. In: Bhullar, S.K., Tappia, P.S., Dhalla, N.S. (eds) The Renin Angiotensin System in Cancer, Lung, Liver and Infectious Diseases. Advances in Biochemistry in Health and Disease, vol 25. Springer, Cham. https://doi.org/10.1007/978-3-031-23621-1_20
Download citation
DOI: https://doi.org/10.1007/978-3-031-23621-1_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23620-4
Online ISBN: 978-3-031-23621-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)