
Abstract
A variety of developmental diseases of the cerebellum are associated with the dysregulation of proteins regulated by the ubiquitin–proteasome system (UPS). Dysfunction of the UPS is observed in several types of spinocerebellar ataxias associated with polyglutamine accumulation. Spinocerebellar ataxia type 3 is caused by a genetic defect in the Atxn3 gene, which codes for a deubiquitinase enzyme. Defects in the expression of a variety of ubiquitin ligases are associated with Freidrich’s ataxia, Ataxia-Telangiectasia, and cerebellar hemangioblastoma. Mutations in a number of genes for ubiquitin ligases are risk factors for autism. Subtypes of medulloblastoma are associated with specific defects in proteasome subunits and with deficiencies in components of the APC/C ubiquitin ligase complex regulating the cell cycle. Targeting various components of the UPS system may contribute to a future therapeutic approach that restores protein homeostasis in various cerebellar diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
In this chapter, we will discuss cerebellar diseases from the perspective of the ubiquitin–proteasome system. In some of these diseases, the ubiquitin–proteasome system (UPS) plays a key role in the disease, while in others the role of the ubiquitin–proteasome system, if any, is not clear. In at least three types of spinocerebellar ataxias, the protein product of the gene associated with the disease is an E3 ubiquitin ligase. We also discuss the role of the ubiquitin–proteasome system in cerebellar hemangioblastoma, in autism, and in medulloblastomas in terms of deficiencies of the ubiquitin–proteasome system.
The Ubiquitin–Proteasome System
The stability of most cellular proteins is controlled by the rate of their degradation through the proteasome, a catalytic chamber. Prior to degradation, the proteins are tagged with the ubiquitin molecule via a series of enzymes, a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3) [1]. An additional enzyme, a deubiquitinase (Dub), functions to remove ubiquitin [2,3,4]. This provides a way of recycling ubiquitin. Deubiquitinases can also, together with specific ubiquitin ligases, serve as on/off switch mechanisms for rapidly controlling proteins that are required for a short, or defined, period of time. The subunits and assembly of the proteasome have recently been described in detail [5]. Herein we discuss the role of the ubiquitin–proteasome system in various developmental diseases of the cerebellum.
Ataxia and Spinocerebellar Ataxia
Ataxia is a neurological condition in which a lack of coordination of muscle groups leads to abnormal gait. Such neurological conditions are often associated with the degeneration of parts of the cerebellum and the degeneration of neuronal pathways between the cerebellum and spinal cord; hence, they are called spinocerebellar ataxias (SCAs). As genes for various SCAs were identified, they were sequentially numbered. Currently there are over 40 subtypes of SCAs identified. SCA type 41, for example, is associated with a mutation in the TRPC3 gene [6]. A mouse model for this disease, the moonwalker mouse, has a mutation of this gene [7]. A number of SCAs are associated with defects in the ubiquitin–proteasome system. Tarlac and Storey [8] have noted that proteasome components and ubiquitin are often found co-localized with abnormal aggregates of proteins in neurons of SCA patients, particularly those with polyglutamine diseases.
Spinocerebellar Ataxia Type 1 (SCA1)
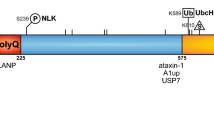
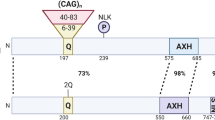
SCA1 is a polyglutamine disease [9]. It is associated with a CAG (cytosine–adenine–guanine) repeat in the Ataxin 1 gene (ATXN1) [10]. Loss of ATXN1 function is reported to contribute to the pathogenesis of SCA1 [11]. One protein to which the ataxin 1 protein binds is ubiquilin 4 (aka ataxin-1 ubiquitin-like interacting protein, A1UP) [12]. This protein also interacts with subunits of the proteasome, contributing to the mechanism by which misfolded proteins are degraded in this structure [13]. The E3 ubiquitin ligase CHIP can ubiquitinate wild-type ataxin 1, as well as its expanded polyQ form, and can protect against the toxicity of the expanded ataxin 1 protein [14]. Enhancing CHIP activity has been proposed as a therapy for polyQ diseases [15].
In a mouse model of SCA1 gene expression in the cerebellum of the ATXN1, polyQ mice were compared with wild type and ATXN1 knockout mice [11]. These investigators analyzed the genes altered in the strains of mice by Kegg analysis. Two sets of genes were expressed in opposite directions in ATXN1 knockout and ATN1 knockin mice, genes that could provide information concerning the mechanism of SCA1 pathogenesis. The two sets of genes, according to the Kegg analysis, were a group including three genes of the TCA cycle and a second group of five genes associated with the ubiquitin-mediated proteolysis (see their Supplemental Table 2). The five ubiquitin ligase genes in this table were ANAPC2, UBE2o, UBE3B, WWP2, and MID1 (aka Trim18). It should be noted that WWP2 is also known as atrophin interacting protein 2 (AIP2) (see below). Mutation of TRIM18 may result in a variety of genetic defects, including the Dandy–Walker malformation [16] and in some patients, agenesis of the cerebellar vermis [17].
Spinocerebellar Ataxia Type 2 (SCA2)
SCA2 is another polyglutamine disease. It is caused by a mutation in the ATXN2 gene [18, 19]. Mutation of this gene can also result in a Parkinson-like syndrome, as well as in amyotrophic lateral sclerosis [20]. Although the ubiquitin and ubiquitin-like conjugation database (UUCO) classifies the ATXN2 protein as an E3 ligase of the Ring family, most publications on SCA2 have not noted this.
Spinocerebellar Ataxia Type 3 (SCA3)/Machado–Joseph Disease and Ataxin 3
Machado–Joseph disease (MJD), although rare, is one of the most common spinocerebellar diseases. It was named after two individuals who first described it [21] . MJD is also referred to as spinocerebellar ataxia type 3 (SCA3); however, early investigators distinguished the two [22]. It is a progressive neurodegenerative disease leading to paralysis and death [23]. In addition to ataxia, the symptoms of this disease included memory deficits, dysarthria, alterations in saccadic eye movements, and dysphagia [24]. There is no current cure for this disease. SCA3/MJD, an autosomal dominant disease, is associated with a genetic abnormality (CAG trinucleotide repeats) of the ATXN3 (Ataxin 3) gene [25,26,27,28], a gene located on chromosome 14 (at 14q32.12). The Atxn3 gene codes for the protein ataxin-3. In SCA3/MJD, ataxin-3 accumulates in neurons as the disease progresses [29]. SCA3/MJD is one of the polyglutamine (polyQ; caused by expanded cytosine–adenine–guanine (CAG) repeats) neurodegenerative diseases associated with protein aggregates in neurons [30, 31]. The components of the ataxin-3 protein, including the polyQ region, have been described and illustrated by Matos et al. [28].
Ataxin-3 has been identified as a deubiquitinase enzyme [28]. It has several ubiquitin interacting regions, which account for its binding to polyubiquitinated protein chains [28]. Riess et al. [27] have illustrated a model of the normal function of ataxin-3. In this model, ataxin-3 facilitates the transport of ubiquitinated proteins to the proteasome for degradation. In SCA3/MJD, ubiquitinated proteins accumulate and proteasome activity is inhibited [32]. There is some data suggesting that in end-stage SCA3/MJD there is a defect preventing the assembly of the two major components of the proteasome, the proteolytic component and the regulatory component [33]. The presence of ubiquitin in neuronal inclusions in polyQ diseases has been taken as evidence for the role of the ubiquitin–proteasome system in the pathogenesis of these disorders [32].
Ataxin-3 functions as a polyubiquitin editing enzyme rather than simply as an enzyme that completely deubiquitinates its substrate [28, 34, 35]. According to Windborn et al., it binds to both Lys [48] and Lys [63] ubiquitin linkages but preferentially cleaves Lys [63] linkages [35].
In SCA3/MJD, the soluble polyglutamine proteins are toxic [28] and probably interfere with the normal function of ataxin-3 as a deubiquitinase. Ataxin-3 has been shown to interact with several ubiquitin ligases including CHIP and Parkin [28, 36]. It regulates the activity of these ligases by removing ubiquitin from them. It has been suggested that the activity of ataxin-3 is itself enhanced by ubiquitination [37, 38]. Its cellular role has been related to protein quality control [38]. Chai et al. [39] showed that the proteasome suppresses polyglutamine aggregation in neurons of SCA3/MJD patients and suggested that the ubiquitin–proteasome pathway has a key role in polyglutamine diseases including SCA3/MJD. However, data on the precise role of the proteasome, or its subunits, in SCA3/MJD is lacking. Rat and mouse models of SCA3/MJD have been developed [40, 41]. These models will contribute to determining the role of ataxin-3 and its polyglutamine form in the development and treatment of SCA3/MJD.
Spinocerebellar Ataxia 5 (SCA5)
Mutations in the SPTBN2 gene reportedly cause SCA5 [42]. This gene codes for one of the spectrin proteins, B-III-spectrin. Spectrin is an F-actin crosslinking protein composed of two chains, alpha and beta, making up a helix. It has a mechanical role in maintaining the shape of the cell but is also involved in cell signaling [43]. The alpha chain is reported to have E2 ubiquitin conjugase activity as well as E3 ubiquitin ligase activity [44,45,46]. The role of the alpha chain E2/E3 activity in the development of SCA5 has not been studied.
Spinocerebellar Ataxia 6 (SCA6)
SCA6 is a rare cerebellar ataxia with additional oculomotor symptoms. Both SCA6, which is progressive, and an episodic non-progressive ataxia, subtype 2 (see below) are associated with a mutation in the calcium channel subunit gene, CACNA1A. SCA6 is another polyglutamine disease caused by CAG repeats in the gene [47]. CACNA1A, also known as SCA6, is associated with the E3 ubiquitin ligase BCL6 [48]. This association is not well characterized.
Spinocerebellar Ataxia 7 (SCA7)
SCA7 is another disease caused by CAG nucleotide repeats. It is caused by a mutation in a gene on Chromosome 3, Atxn7. It is a progressive disease that results in ataxia and blindness. The ataxin-7 protein is part of a protein complex, the SAGA complex, that acts as a DUB, which regulates the transcription of a number of genes [49, 50]. The DUB protein that contributes to this complex is USP22 [51]. The polyglutamine expansion of the ataxin-7 protein apparently interferes with the function of the USP22 as a gene silencer [51, 52].
Spinocerebellar Ataxia 8 (SCA8)
SCA8 is associated with a trinucleotide repeat expansion in the two overlapping genes ATXN8 and ATXN8OS [53]. The latter codes for an antisense RNA for the ubiquitin ligase KLHL1 [54]. Both genes are highly expressed in the cerebellum and other brain tissues.
Spinocerebellar Ataxia 15 (SCA15)
SCA15 is a cerebellar ataxia in which atrophy of parts of the vermis is reported [55]. Mutations of the IPTR1 gene are associated with this disorder. Mutations of this gene are associated with abnormal regulation of calcium release by calmodulin and UBR4 (aka p600), a ubiquitin E3 ligase [56]. Mutations of UBR4 are associated with at least one subtype of episodic ataxias (see below).
Spinocerebellar Ataxia 17 (SCA17)
Sca17 is caused by a mutation in the gene (TBP) for the Tata-box binding protein. In a model of Sermwittayawong and Tan [57], the SAGA complex interacts with TBP to regulate transcription. These investigators, however, did not discuss the deubiquitinase activity of the SAGA complex in their model. If deubiquitinase activity is generally associated with the SAGA complex, as suggested by others [58, 59], it may also be important in regulating TBP in the cerebellum.
Spinocerebellar Ataxia Type 19 (SCA19) and Type 22 (SCA22)
SCA19 and SCA22 have been associated with mutations in the gene for a potassium channel component, KCND3 [60]. This protein has been classified as an E3 ubiquitin ligase of the BTB family in a supplemental table of a recent publication [61].
CHIP and Gordon Homes Syndrome
It has been shown that mutations in the Stub1 gene, which codes for the ubiquitin ligase CHIP, are associated with a number of autosomal recessive cerebellar ataxias [62, 63]. In Gordon Holmes syndrome (ataxia associated with hypogonadism), mutations in the Stub1 gene and loss of CHIP have been identified as the probable cause of this disorder [64]. Ronnebaum et al. [65] concluded that CHIP is required for the maintenance of normal cerebellar function.
Dentatorubropallidoluysian Atrophy
Dentatorubropallidoluysian atrophy (DRPLA) is another autosomal dominant neurodegenerative disease associated with cerebellar ataxia [66, 67]. It is also referred to Naito–Oyanagi disease [68]. Like SCA3/MJD, it is a genetic abnormality with trinucleotide repeats and polyglutamine proteins [69,70,71]. In DRPLA, there is an abnormality of the atrophin-1 gene (Atn1), expansion of a CAG repeat [72]. The abnormal form of the atrophin-1 protein accumulates in the brains of DRPLA patients [72].
Several atrophin-interacting proteins including AIP1, AIP2, AIP3, AIP4, and AIP5 have been identified [73]. Three of them are E3 ubiquitin ligases. AIP2 is also known as WWP2 (WW domain-containing protein ligase 2). AIP4 has been identified as a ubiquitin ligase homologous to the mouse E3 ligase Itch [74]. Among the substrates of this E3 ligase are the proteins Notch [75] and JunB [76]. AIP5 is also known as WWP1 (WW domain-containing protein ligase 1). AIP1 and AIP3 have not been described as E3 ligases. They are membrane-bound proteins with guanylate kinase-like regions [73].
Friedrich’s Ataxia and Ubiquitin-Competing Molecules
Friedrich’s ataxia (FRDA) is a hereditary protein disease. It is inherited as an autosomal recessive disease that initially presents itself in symptoms of gait disturbance and lack of coordination. FRDA is a disease that progressively impairs the muscular system. Other systems involved may include vision, hearing, speech, carbohydrate metabolism, and cardiac disorders. The pathology of FRDA has been reviewed by Koeppen AH 2011 [77]. FRDA results from failed transcription of the frataxin (FXN) gene [78, 79]. Gene silencing may contribute to this failure in transcription [77]. A deficiency in the FXN protein leads to the degenerative conditions characteristic of FRDA [80]. FXN has been located in the mitochondrial matrix. It is thought to play a significant role in maintaining adequate levels of iron in mitochondria [81].
Currently, there is no effective treatment for FRDA. However, the FRDA phenotype, in an in vitro mouse model, was partially reversed, using viral vectors encoding for the FXN gene [82]. This model provided an incentive to use this approach in humans. In humans, efforts have been made to reactivate the FXN gene using nicotinamide [83]. Underlying this research effort is the view that epigenetic regulation of the FXN gene is possible.
Another approach to increasing FXN is to manipulate its degradation. FXN is a protein that is degraded by the ubiquitin–proteasome system [84]. Theoretically, proteasome inhibitors could be used to increase tissue concentrations of FXN. However, this approach is limited to those inhibitors that cross the blood–brain barrier. Another limitation is that proteasome inhibitors are not specific enough. Rufini and colleagues have identified and tested a series of lead compounds capable of interfering with FXN ubiquitination and degradation [84]. Recently, they found that small molecules that bind to FXN compete with ubiquitin for binding to FXN (at a specific site on the molecule, lysine 147) and lead to the accumulation of FXN [85]. They named these molecules ubiquitin-competing molecules. Their results provided a rationale for a therapeutic use of ubiquitin-competing molecules in FRDA disease.
Episodic Ataxia and Ubiquitin Ligases
There are currently eight separate clinically recognized episodic ataxias (EA) [86]. In one form of EA, subtype 8, the UBR4 (Ubiquitin Protein Ligase E3 Component N-Recognin 4) gene on chromosome 1 was reported as the likely source of genetic variations causing this ataxia [56]. UBR4 (aka p600) is a ubiquitin E3 ligase [87, 88] that interacts with calmodulin, a calcium-binding protein. UBR4 also binds to ITPR1 (inositol trisphosphate receptor isoform 1), which regulates calcium release from the endoplasmic reticulum [56]. Conroy et al. [56] suggested the hypothesis that interference with the normal binding of calmodulin and/or ITPR1 to UBR4 resulted in a dysfunctional calcium sensing system leading to ataxia.
One of the most common types of EA (subtype 1) is reportedly caused by variations in the gene KCNA1, which codes for a potassium channel protein [89]. KCNA1 has been recently identified as having E3 ubiquitin ligase activity [61] (see Supplemental Table 4 in this reference). AS noted above, EA subtype 2 is caused by a mutation in the calcium channel subunit gene, CACNA1A.
Ataxia Telangiectasia (AT) and the ATM Protein
Ataxia telangiectasia (AT), also known as Louis–Bar’s syndrome [90], is an autosomal recessive disorder that results in various clinical symptoms including progressive ataxia. AT patients have a defect in a gene associated with the repair response to double-strand DNA breaks resulting from oxidative stress [91]. The ATM (ataxia telangiectasia mutated) protein, a serine–threonine protein kinase, phosphorylates several enzymes necessary for activation of the DNA damage checkpoint and repair response after double-strand DNA breaks [92].
Other proteins involved in ATM activation include the ubiquitin ligases RNF8 (Ring Finger 8) and CHFR (Checkpoint with forkhead and ring finger domains) [93]. Via phosphorylation, ATM can activate or inactivate many different proteins. Its effect on the ubiquitin–proteasome system during activation of the response to double-strand DNA breaks has been described by Shiloh and Ziv [93] as having several phases: 1. recruitment of ATM to the site of double-strand breaks (partially mediated by the E3 ubiquitin ligase SKP2); 2. a kinase cascade stimulating the phosphorylation of many proteins including other kinases; 3. recruitment of proteasomes to the site of DNA damage [94]; 4. modulation of ubiquitin ligases and DUBs (deubiquitinases) by phosphorylation; and 5. phosphorylation of substrates of E3 ligases preparing them for ubiquitination. Thus, E3 ligases control the stability of proteins such as p53 and NFκB. Among the E3 ligases listed by Shiloh and Ziv as influenced by ATM include Cop1 (aka RFWD2), MDM2, MDMX, and SIAH1. The deubiquitinase USP10 is also phosphorylated by ATM. Thus, it can be safely concluded that the ubiquitin–proteasome system plays an important role in the ATM response. Eventually, this information may be used to design therapeutic molecules that can be used in the management of AT.
Cerebellar Hemangioblastoma and the Von Hippel–Lindau Protein
Hemangioblastomas of the cerebellum are frequently associated with von Hippel–Lindau (VHL) disease [95, 96]. In this disease, there is a deficiency in the gene for the VHL tumor suppressor protein and overexpression of VEGF (vascular endothelial growth factor) [96]. Hemangioblastomas probably originate from hemangioblast progenitor cells [97].
The molecular mechanisms by which loss of the VHL gene or VHL protein leads to susceptibility to hemangioblastoma have been described [98]. The VHL protein has been shown to be a ubiquitin ligase [99]. One of its substrates is the transcription factor HIF-1α [100], a transcription factor for a number of proteins including VEGF [101]. Under normal conditions (normoxia), HIF-1α is ubiquitinated by VHL and degraded by the proteasome [102].
Autism-Associated Genes
The development of the cerebellum has been shown to differ in autistic patients compared to controls [103]. MRI studies showed hypoplasia of the cerebellum in autistic patients [104]. Postmortem studies showed significantly decreased numbers of Purkinje neurons in the cerebellum of patients with autism spectrum disorders (ASDs) [103]. Among the genes associated with ASD is the gene for the ubiquitin ligase UBE3A [105]. It was reported as upregulated in cells from individuals with autism [106]. The UBE3A gene is better known as the gene which, when deficient, causes Angelman syndrome [107]. It is a maternally expressed gene. The protein encoded by this gene is the E6-AP protein [108]. It is named for its association with the papillomavirus protein E6.
Recently, Louros and Osterweil [105] have noted that mutations in a number of genes of the ubiquitin–proteasome system have been identified as risk factors for ASD. In addition to UBE3A, ten other ubiquitin ligases were documented as risk factors for ASD (UBE3B, UBE3C, PARK2, FBXO40, RFWD2, Cullin 3, Cullin 7, HECW2, HERC2, and HUWE) in this review. The genes coding three deubiquitinases (USP9Y, USP45, and USP7) and the gene for the proteasome subunit PSMD10 were also listed as risk factors. The authors point out that these data provide strong evidence for the dysregulation of protein degradation in ASD. The number of ubiquitin–proteasome proteins listed as risk factors may reflect the heterogeneity of ASD diseases.
Medulloblastoma and Ubiquitin–Proteasome Components
Medulloblastoma, described as a malignancy of the cerebellum, actually describes a group of heterogeneous tumors, differing in histology, genetic expression, clinical outcome, and response to treatment. A consensus classification, however, was reported in 2012 [109]. In this classification, four major subtypes of medulloblastoma were recognized: the WNT group, the SHH group, and two additional groups simply referred to as Groups 3 and 4. In 2015, we suggested the possibility of classification of MBs according to their expression of ubiquitin ligases [110]. In support of this view are supplemental data of Thompson et al. [111] showing differential expression of at least 50 ubiquitin ligases among the various subtypes of MB. Since the Thompson data were reported before 2012, these investigators recognized five subgroups of MB rather than the four subgroups of the consensus classification. In a recent review [112], we identified these E3 ligases and indicated whether they were significantly upregulated or downregulated in the various subgroups of the Thompson supplementary dataset. We also noted the differential expression of 12 deubiquitinases among the various subtypes of MB in the Thompson dataset. Since the publication of our review, we noted that expression of the gene UBE3A, also an E3 ligase, was also differential expressed among some of the Thompson MB subgroups. We have noted above that the UBE3A gene is associated with Angelman syndrome and autism as well. Thus, in addition, this ubiquitin E3 ligase could be useful as a marker gene for a subtype of MB, the Thompson Group E MB (equivalent to MB consensus subtype 3). We noted above that the UBE3A gene codes for a protein, E6-AP. On further examination of the Thompson dataset, we note that several ubiquitin-conjugating enzymes are differently expressed among the various MB subtypes. In Table 1, we list the ubiquitin conjugases that were significantly upregulated or downregulated in the Thompson dataset compared to the other MB groups. Thus, E2 conjugases, E3 ligases, and deubiquitinases could all be useful as marker genes for the various subtypes of MB.
Another remarkable feature of the Thompson dataset [111] is that it shows differential expression of genes for proteasome subunits among the different subtypes of MB. The Wnt subgroup showed significant depression of expression of the PSMB1 gene; the SHH subgroup of MBs showed significant depression of expression of seven separate genes for proteasome subunits; the Thompson dataset also showed that their Group A MBs had significantly increased expression of genes for 13 separate proteasome subunits, including the genes for two catalytic subunits. The genes for eight subunits of the proteasome, including two catalytic subunits, were significantly decreased in their Group C tumors. The final group of Thompson MBs, Group E, also showed significant variations in several proteasome subunits. This was illustrated in the review of Vriend and Marzban [112] and reproduced as Fig. 1 (by permission). These results showed that genes for proteasome subunits are “signature” genes for subtypes of MBs and raise the possibility of targeting proteasome subunits therapeutically.

Differential expression of proteasome subunits in MB subtypes in Thompson supplemental dataset. (Used with permission, Springer, Cellular and Molecular Life Sciences, 2016)
Green, significantly increased; orange, significantly decreased compared to other groups; gray, not in the dataset or not significant
One ubiquitin ligase complex suggested in a therapeutic context for MB is casein kinase 1 delta, a substrate of the APC/C (anaphase promoting complex/cyclosome) complex [113]. The APC/C ubiquitin ligase is an important regulator of mitosis. Among the factors that regulate its activity is the human cytomegalovirus [114]. Many medulloblastomas are reportedly infected with this virus [115, 116]. Although a causative relationship between cytomegalovirus and MBs has not been definitively established, Baryawno et al. [116] have suggested an important role of this virus in the development of MB. This virus may be more significant in subgroups of MBs in which the activity of the APC/C ubiquitin ligase complex is impaired than in MBs in which this complex is fully functional. Further investigation on the interaction of viruses and ubiquitin ligases may provide information leading to new therapeutic approaches for several cerebellar diseases.
Prospective Expectations
Various developmental diseases of the cerebellum are associated with abnormal protein regulation [8]. It is becoming clear that a dysfunctional UPS has a key role in many of these disorders. As subsequent research identifies the specific components of the UPS that are dysfunctional, opportunities arise to target these constituents therapeutically. Inhibitors of ubiquitin ligases and ubiquitin conjugases, as well as inhibitors of deubiquitinases, may all be therapeutically significant in the treatment of some of these diseases. In cases in which disease is associated with proteasome dysfunction, proteasome inhibitors or proteasome-stimulating proteins may be clinically practical.
Abbreviations
- AIP:
-
Atrophin interacting protein
- AIUP:
-
Ataxin-1 ubiquitin-like interacting protein
- APC/C:
-
Anaphase promoting complex/cyclosome
- ASD:
-
Autism spectrum disorder
- AT:
-
Ataxia telangiectasia
- ATM:
-
Ataxia telangiectasia mutated
- ATXN1:
-
Ataxin 1
- ATXN3:
-
Ataxin 3
- CAG:
-
Cytosine–adenine–guanine repeat
- CHFR:
-
Checkpoint with forkhead and ring finger domains
- DRPLA:
-
Dentatorubropallidoluysian atrophy
- DUB:
-
Deubiquitinase
- E2:
-
Ubiquitin-conjugating enzyme
- E3:
-
Ubiquitin ligase
- EI:
-
Ubiquitin-activating enzyme
- FRDA:
-
Freidrich’s ataxia
- FXN:
-
Frataxin
- HIF-1:
-
Hypoxia-inducible factor 1
- ITPR:
-
Inositol triphosphate receptor isoform
- MB:
-
Medulloblastoma
- MJD:
-
Machado–Joseph disease
- RNF:
-
Ring finger protein
- SCA:
-
Spinocerebellar ataxia
- UBR:
-
Ubiquitin Protein Ligase E3 Component N-Recognin 4
- UPS:
-
Ubiquitin–proteasome system
- USP:
-
Ubiquitin-specific protease
- VEGF:
-
Vascular endothelial growth factor
- VHL:
-
Von Hippel–Lindau protein
References
Hershko A, Ciechanover A. The ubiquitin pathway for the degradation of intracellular proteins. Prog Nucleic Acid Res Mol Biol. 1986;33:19–56, 301.
Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–97.
Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695(1–3):189–207.
Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–63.
Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N, Ciechanover A. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 2016;26(8):869–85.
Fogel BL, Hanson SM, Becker EB. Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov Disord. 2015;30(2):284–6.
Becker EB. The Moonwalker mouse: new insights into TRPC3 function, cerebellar development, and ataxia. Cerebellum. 2014;13(5):628–36.
Tarlac V, Storey E. Role of proteolysis in polyglutamine disorders. J Neurosci Res. 2003;74(3):406–16.
Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4(3):221–6.
Sanchez I, Balague E, Matilla-Duenas A. Ataxin-1 regulates the cerebellar bioenergetics proteome through the GSK3beta-mTOR pathway which is altered in Spinocerebellar ataxia type 1 (SCA1). Hum Mol Genet. 2016;25:4021.
Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, Zoghbi HY. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010;6(7):e1001021.
Riley BE, Xu Y, Zoghbi HY, Orr HT. The effects of the polyglutamine repeat protein ataxin-1 on the UbL-UBA protein A1Up. J Biol Chem. 2004;279(40):42290–301.
Su V, Lau AF. Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell Mol Life Sci. 2009;66(17):2819–33.
Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Perez AM, et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281(36):26714–24.
Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL. In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol Dis. 2009;33(3):342–53.
Preiksaitiene E, Krasovskaja N, Utkus A, Kasnauskiene J, Meskiene R, Paulauskiene I, et al. R368X mutation in MID1 among recurrent mutations in patients with X-linked Opitz G/BBB syndrome. Clin Dysmorphol. 2015;24(1):7–12.
De Falco F, Cainarca S, Andolfi G, Ferrentino R, Berti C, Rodriguez Criado G, et al. X-linked Opitz syndrome: novel mutations in the MID1 gene and redefinition of the clinical spectrum. Am J Med Genet A. 2003;120A(2):222–8.
Lastres-Becker I, Rub U, Auburger G. Spinocerebellar ataxia 2 (SCA2). Cerebellum. 2008;7(2):115–24.
Li PP, Sun X, Xia G, Arbez N, Paul S, Zhu S, et al. ATXN2-AS, a gene antisense to ATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann Neurol. 2016;80(4):600–15.
Pulst SM. Degenerative ataxias, from genes to therapies: the 2015 Cotzias lecture. Neurology. 2016;86(24):2284–90.
Nakano KK, Dawson DM, Spence A, Machado disease. A hereditary ataxia in Portuguese emigrants to Massachusetts. Neurology. 1972;22(1):49–55.
Matilla T, McCall A, Subramony SH, Zoghbi HY. Molecular and clinical correlations in spinocerebellar ataxia type 3 and Machado-Joseph disease. Ann Neurol. 1995;38(1):68–72.
Bettencourt C, Lima M. Machado-Joseph Disease: from first descriptions to new perspectives. Orphanet J Rare Dis. 2011;6:35.
Seidel K, Siswanto S, Fredrich M, Bouzrou M, Brunt ER, van Leeuwen FW, et al. Polyglutamine aggregation in Huntington’s disease and spinocerebellar ataxia type 3: similar mechanisms in aggregate formation. Neuropathol Appl Neurobiol. 2016;42(2):153–66.
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8(3):221–8.
Limprasert P, Nouri N, Heyman RA, Nopparatana C, Kamonsilp M, Deininger PL, et al. Analysis of CAG repeat of the Machado-Joseph gene in human, chimpanzee and monkey populations: a variant nucleotide is associated with the number of CAG repeats. Hum Mol Genet. 1996;5(2):207–13.
Riess O, Rub U, Pastore A, Bauer P, Schols L. SCA3: neurological features, pathogenesis and animal models. Cerebellum. 2008;7(2):125–37.
Matos CA, de Macedo-Ribeiro S, Carvalho AL. Polyglutamine diseases: the special case of ataxin-3 and Machado-Joseph disease. Prog Neurobiol. 2011;95(1):26–48.
Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature. 2011;480(7378):543–6.
Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–47.
Schols L, Reimold M, Seidel K, Globas C, Brockmann K, Hauser TK, et al. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain. 2015;138(Pt 11):3316–26.
Jana NR, Nukina N. Recent advances in understanding the pathogenesis of polyglutamine diseases: involvement of molecular chaperones and ubiquitin-proteasome pathway. J Chem Neuroanat. 2003;26(2):95–101.
Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, et al. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol. 2002;51(3):302–10.
Burnett B, Li F, Pittman RN. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet. 2003;12(23):3195–205.
Winborn BJ, Travis SM, Todi SV, Scaglione KM, Xu P, Williams AJ, et al. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem. 2008;283(39):26436–43.
Durcan TM, Fon EA. Ataxin-3 and its e3 partners: implications for machado-joseph disease. Front Neurol. 2013;4:46.
Todi SV, Winborn BJ, Scaglione KM, Blount JR, Travis SM, Paulson HL. Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 2009;28(4):372–82.
Todi SV, Scaglione KM, Blount JR, Basrur V, Conlon KP, Pastore A, et al. Activity and cellular functions of the deubiquitinating enzyme and polyglutamine disease protein ataxin-3 are regulated by ubiquitination at lysine 117. J Biol Chem. 2010;285(50):39303–13.
Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL. Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet. 1999;8(4):673–82.
Cemal CK, Carroll CJ, Lawrence L, Lowrie MB, Ruddle P, Al-Mahdawi S, et al. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet. 2002;11(9):1075–94.
Alves S, Nascimento-Ferreira I, Dufour N, Hassig R, Auregan G, Nobrega C, et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3? Hum Mol Genet. 2010;19(12):2380–94.
Dick KA, Ikeda Y, Day JW, Ranum LP. Spinocerebellar ataxia type 5. Handb Clin Neurol. 2012;103:451–9.
Machnicka B, Grochowalska R, Boguslawska DM, Sikorski AF, Lecomte MC. Spectrin-based skeleton as an actor in cell signaling. Cell Mol Life Sci. 2012;69(2):191–201.
Chang TL, Cubillos FF, Kakhniashvili DG, Goodman SR. Ankyrin is a target of spectrin’s E2/E3 ubiquitin-conjugating/ligating activity. Cell Mol Biol (Noisy-le-Grand). 2004;50(1):59–66.
Hsu YJ, Goodman SR. Spectrin and ubiquitination: a review. Cell Mol Biol (Noisy-le-grand). 2005;Suppl 51:OL801-7.
Goodman SR, Petrofes Chapa R, Zimmer WE. Spectrin’s chimeric E2/E3 enzymatic activity. Exp Biol Med (Maywood). 2015;240(8):1039–49.
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15(1):62–9.
Miles RR, Crockett DK, Lim MS, Elenitoba-Johnson KS. Analysis of BCL6-interacting proteins by tandem mass spectrometry. Mol Cell Proteomics. 2005;4(12):1898–909.
Mohan RD, Abmayr SM, Workman JL. Pulling complexes out of complex diseases: spinocerebellar ataxia 7. Rare Dis. 2014;2:e28859.
Morgan MT, Haj-Yahya M, Ringel AE, Bandi P, Brik A, Wolberger C. Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science. 2016;351(6274):725–8.
Yang H, Liu S, He WT, Zhao J, Jiang LL, Hu HY. Aggregation of Polyglutamine-expanded Ataxin 7 protein specifically sequesters ubiquitin-specific protease 22 and deteriorates its deubiquitinating function in the Spt-Ada-Gcn5-acetyltransferase (SAGA) complex. J Biol Chem. 2015;290(36):21996–2004.
Li ZH, Yu Y, Du C, Fu H, Wang J, Tian Y. RNA interference-mediated USP22 gene silencing promotes human brain glioma apoptosis and induces cell cycle arrest. Oncol Lett. 2013;5(4):1290–4.
Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38(7):758–69.
Nemes JP, Benzow KA, Moseley ML, Ranum LP, Koob MD. The SCA8 transcript is an antisense RNA to a brain-specific transcript encoding a novel actin-binding protein (KLHL1). Hum Mol Genet. 2000;9(10):1543–51.
Storey E. Spinocerebellar ataxia type 15. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R). Seattle: University of Washington; 1993.
Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B, et al. A novel locus for episodic ataxia:UBR4 the likely candidate. Eur J Hum Genet. 2014;22(4):505–10.
Sermwittayawong D, Tan S. SAGA binds TBP via its Spt8 subunit in competition with DNA: implications for TBP recruitment. EMBO J. 2006;25(16):3791–800.
Wang L, Dent SY. Functions of SAGA in development and disease. Epigenomics. 2014;6(3):329–39.
Weake VM, Workman JL. SAGA function in tissue-specific gene expression. Trends Cell Biol. 2012;22(4):177–84.
Lee YC, Durr A, Majczenko K, Huang YH, Liu YC, Lien CC, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012;72(6):859–69.
Lehmann G, Udasin RG, Ciechanover A. On the linkage between the ubiquitin-proteasome system and the mitochondria. Biochem Biophys Res Commun. 2016;473(1):80–6.
Shi Y, Wang J, Li JD, Ren H, Guan W, He M, et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One. 2013;8(12):e81884.
Depondt C, Donatello S, Simonis N, Rai M, van Heurck R, Abramowicz M, et al. Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology. 2014;82(19):1749–50.
Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23(4):1013–24.
Ronnebaum SM, Patterson C, Schisler JC. Emerging evidence of coding mutations in the ubiquitin-proteasome system associated with cerebellar ataxias. Hum Genome Var. 2014;1:14018.
Yuasa T. Hereditary dentatorubro-pallidoluysian atrophy (DRPLA): clinical studies on 45 cases. Nihon Rinsho Jpn J Clin Med. 1993;51(11):3016–23.
Matilla-Duenas A. Machado-Joseph disease and other rare spinocerebellar ataxias. Adv Exp Med Biol. 2012;724:172–88.
Kanazawa I. Dentatorubral-pallidoluysian atrophy or Naito-Oyanagi disease. Neurogenetics. 1998;2(1):1–17.
Yamada M, Shimohata M, Sato T, Tsuji S, Takahashi H. Polyglutamine disease: recent advances in the neuropathology of dentatorubral-pallidoluysian atrophy. Neuropathology. 2006;26(4):346–51.
Tsuji S. Dentatorubral-pallidoluysian atrophy. Handb Clin Neurol. 2012;103:587–94.
Fan HC, Ho LI, Chi CS, Chen SJ, Peng GS, Chan TM, et al. Polyglutamine (PolyQ) diseases: genetics to treatments. Cell Transplant. 2014;23(4–5):441–58.
Suzuki Y, Yazawa I. Pathological accumulation of atrophin-1 in dentatorubralpallidoluysian atrophy. Int J Clin Exp Pathol. 2011;4(4):378–84.
Wood JD, Yuan J, Margolis RL, Colomer V, Duan K, Kushi J, et al. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol Cell Neurosci. 1998;11(3):149–60.
Feng L, Guedes S, Wang T. Atrophin-1-interacting protein 4/human Itch is a ubiquitin E3 ligase for human enhancer of filamentation 1 in transforming growth factor-beta signaling pathways. J Biol Chem. 2004;279(28):29681–90.
Qiu L, Joazeiro C, Fang N, Wang HY, Elly C, Altman Y, et al. Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J Biol Chem. 2000;275(46):35734–7.
Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol. 2002;3(3):281–7.
Koeppen AH. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci. 2011;303(1–2):1–12.
Chamberlain S, Shaw J, Rowland A, Wallis J, South S, Nakamura Y, et al. Mapping of mutation causing Friedreich’s ataxia to human chromosome 9. Nature. 1988;334(6179):248–50.
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271(5254):1423–7.
Busi MV, Gomez-Casati DF. Exploring frataxin function. IUBMB Life. 2012;64(1):56–63.
Patel PI, Isaya G. Friedreich ataxia: from GAA triplet-repeat expansion to frataxin deficiency. Am J Hum Genet. 2001;69(1):15–24.
Fleming J, Spinoulas A, Zheng M, Cunningham SC, Ginn SL, McQuilty RC, et al. Partial correction of sensitivity to oxidant stress in Friedreich ataxia patient fibroblasts by frataxin-encoding adeno-associated virus and lentivirus vectors. Hum Gene Ther. 2005;16(8):947–56.
Libri V, Yandim C, Athanasopoulos S, Loyse N, Natisvili T, Law PP, et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich’s ataxia: an exploratory, open-label, dose-escalation study. Lancet. 2014;384(9942):504–13.
Rufini A, Fortuni S, Arcuri G, Condo I, Serio D, Incani O, et al. Preventing the ubiquitin-proteasome-dependent degradation of frataxin, the protein defective in Friedreich’s ataxia. Hum Mol Genet. 2011;20(7):1253–61.
Rufini A, Cavallo F, Condo I, Fortuni S, De Martino G, Incani O, et al. Highly specific ubiquitin-competing molecules effectively promote frataxin accumulation and partially rescue the aconitase defect in Friedreich ataxia cells. Neurobiol Dis. 2015;75:91–9.
Choi KD, Choi JH. Episodic ataxias: clinical and genetic features. J Mov Disord. 2016;9(3):129–35.
Tasaki T, Mulder LC, Iwamatsu A, Lee MJ, Davydov IV, Varshavsky A, et al. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol. 2005;25(16):7120–36.
Parsons K, Nakatani Y, Nguyen MD. p600/UBR4 in the central nervous system. Cell Mol Life Sci. 2015;72(6):1149–60.
Brandt T, Strupp M. Episodic ataxia type 1 and 2 (familial periodic ataxia/vertigo). Audiol Neurootol. 1997;2(6):373–83.
Pelc S, Vis H. Familia ataxia with ocular telangiectasis (D. Louis-Bar syndrome). Acta Neurol Belg. 1960;60:905–22.
Subba RK. Mechanisms of disease: DNA repair defects and neurological disease. Nat Clin Pract Neurol. 2007;3(3):162–72.
Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26(56):7741–8.
Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210.
Levy-Barda A, Lerenthal Y, Davis AJ, Chung YM, Essers J, Shao Z, et al. Involvement of the nuclear proteasome activator PA28gamma in the cellular response to DNA double-strand breaks. Cell Cycle. 2011;10(24):4300–10.
Slater A, Moore NR, Huson SM. The natural history of cerebellar hemangioblastomas in von Hippel-Lindau disease. AJNR Am J Neuroradiol. 2003;24(8):1570–4.
Richard S, Campello C, Taillandier L, Parker F, Resche F. Haemangioblastoma of the central nervous system in von Hippel-Lindau disease. French VHL Study Group. J Intern Med. 1998;243(6):547–53.
Glasker S, Li J, Xia JB, Okamoto H, Zeng W, Lonser RR, et al. Hemangioblastomas share protein expression with embryonal hemangioblast progenitor cell. Cancer Res. 2006;66(8):4167–72.
Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19(6):617–23.
Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, et al. Identification of the von Hippel-Lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc Natl Acad Sci U S A. 1999;96(22):12436–41.
Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol. 2001;13(2):167–71.
Shih SC, Claffey KP. Hypoxia-mediated regulation of gene expression in mammalian cells. Int J Exp Pathol. 1998;79(6):347–57.
Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr, Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296(5574):1886–9.
Donovan AP, Basson MA. The neuroanatomy of autism – a developmental perspective. J Anat. 2016;230(1):4–15.
Hashimoto T, Tayama M, Murakawa K, Yoshimoto T, Miyazaki M, Harada M, et al. Development of the brainstem and cerebellum in autistic patients. J Autism Dev Disord. 1995;25(1):1–18.
Louros SR, Osterweil EK. Perturbed proteostasis in autism spectrum disorders. J Neurochem. 2016;139(6):1081–92.
Baron CA, Tepper CG, Liu SY, Davis RR, Wang NJ, Schanen NC, et al. Genomic and functional profiling of duplicated chromosome 15 cell lines reveal regulatory alterations in UBE3A-associated ubiquitin-proteasome pathway processes. Hum Mol Genet. 2006;15(6):853–69.
Buiting K, Williams C, Horsthemke B. Angelman syndrome – insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12(10):584–93.
Kishino T, Wagstaff J. Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics. 1998;47(1):101–7.
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–72.
Vriend J, Ghavami S, Marzban H. The role of the ubiquitin proteasome system in cerebellar development and medulloblastoma. Mol Brain. 2015;8(1):64.
Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924–31.
Vriend J, Marzban H. The ubiquitin-proteasome system and chromosome 17 in cerebellar granule cells and medulloblastoma subgroups. Cell Mol Life Sci. 2016;74(3):449–67.
Penas C, Govek EE, Fang Y, Ramachandran V, Daniel M, Wang W, et al. Casein kinase 1delta is an APC/C(Cdh1) substrate that regulates cerebellar granule cell neurogenesis. Cell Rep. 2015;11(2):249–60.
Wiebusch L, Bach M, Uecker R, Hagemeier C. Human cytomegalovirus inactivates the G0/G1-APC/C ubiquitin ligase by Cdh1 dissociation. Cell Cycle. 2005;4(10):1435–9.
Hawkins C, Croul S. Viruses and human brain tumors: cytomegalovirus enters the fray. J Clin Invest. 2011;121(10):3831–3.
Baryawno N, Rahbar A, Wolmer-Solberg N, Taher C, Odeberg J, Darabi A, et al. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J Clin Invest. 2011;121(10):4043–55.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vriend, J., Jiao, X. (2023). The Ubiquitin–Proteasome System and Cerebellar Developmental Disease. In: Marzban, H. (eds) Development of the Cerebellum from Molecular Aspects to Diseases. Contemporary Clinical Neuroscience. Springer, Cham. https://doi.org/10.1007/978-3-031-23104-9_12
Download citation
DOI: https://doi.org/10.1007/978-3-031-23104-9_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23103-2
Online ISBN: 978-3-031-23104-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)