
Abstract
Micro RNAs (miRNAs) are small non coding RNA sequences consisting of 20–23 nucleotides that govern the post transcriptional expression of genes in both normal and disease condition of the cell. Thus, identification of most influencing miRNAs and the associated mRNAs becomes a research quest in diagnostic and prognostic application of cancer. In this study we conducted an integrated analysis of Next Generation Sequencing based miRNA-mRNA expression data to identify dysregulated miRNAs and their target mRNAs for Oral Cancer. A sensible combination of datamining tools such as Random Forest (RF), K-nearest Neighbour (KNN), Support Vector Machine (SVM), log-Fold Change, Adjusted p-values, Matthews coefficient correlation (MCC), Prediction accuracy was considered for this analysis. The prioritized cancer specific target genes obtained in this approach exhibited a MCC value of 0.9 and achieved a consistently higher prediction accuracy of 95% when subjected to classifiers RF, KNN and SVM. These target genes can be presented as predictive variables for early diagnosis of cancer. The selected miRNA-target genes can further be biologically validated to confirm their participation in disease specific pathways and biological processes.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Cancer has now ranked as leading cause of death worldwide [1]. Oral cancer (OC) is a category of head and neck squamous cell carcinoma, mostly developed on the lip, floor of the mouth, cheek lining, gingiva, palate or in the tongue. In India, OC is measured among top three types of cancers which accounts for more than 30% of all cancers [2]. Often OC is mostly diagnosed at its advanced stage i.e., when cancer has metastasized to another location, most likely the lymph nodes of the neck, which results in low treatment outcomes and leaves patient with significantly low survival rate [3]. Alcohol addiction, practice of tobacco products like cigarettes, smokeless tobacco and viral infection are the most common risk factors for oral cancer [4].
Cancer is a multi-step process which causes due to mutation in genes that controls cell behaviour. Mutated genes may result in uncontrolled growth of cells that invade and cause the adjacent tissue impairment [5]. Micro RNAs (miRNAs) are small non coding RNA sequences consisting of 20–23 nucleotides that are incriminated in numerous biological, anatomical processes including cell differentiation, cell signalling, apoptosis, metastasis and response to infection [6]. Dysregulated expression pattern of miRNA is an indicator for initiation and progression of various disease including cancer [23]. Hence, identification of dysregulated miRNAs becomes crucial towards understanding of the biological mechanism behind miRNAs. miRNA govern the post transcriptional expression of genes by complementary base pairing with target m-RNAs in both normal and disease condition of the cell [7]. Thus, prediction of the miRNA-mRNA target interactions becomes significant to elucidate the mechanism by which miRNA act in carcinogenesis process. However, it has become a current challenge to correctly characterize the course of action of miRNAs on their mRNA targets, because each miRNA has multiple mRNA targets and vice versa [8]. This association of miRNA-mRNA targets highlights the importance of integrating miRNA expression with downstream mRNA target genes [9].
2 Background Study
Several studies have been carried out in literature to identify novel miRNA signatures associated with cancer and elucidate miRNA-mRNA target interactions. Seo et al. applied an integrative approach of miRNA, mRNA and protein expression data for identifying cancer-related miRNAs and investigating the gene-miRNA association [10]. Modules of highly correrated miRNA, mRNA and proteins were constructed using SAMBA bi-clustering algorithm and a Bayesian network model. The regulatory relationship between these modules were then investigated for precise analysis of miRNA-target gene interactions. Another integrative approach was proposed in [11] to identify the mRNA targets of abnormally regulated miRNAs. Several aberrantly expressed miRNAs and the associated target mRNA signatures were identified in this approach across six different cancer types. Sathipati et al. proposed SVM-HCC model based on inheritable bi-objective combinatorial genetic algorithm for selecting novel miRNA signatures for predicting hepatocellular carcinoma stages [12]. A hierarchical integrative model was utilized in [13] to uncover the miRNA-mRNA associations utilizing the sequence data of miRNA and mRNA. The identified miRNA-mRNA pairs were observed to be involved in processes contributing to hepatocellular carcinoma progression. A biphasic technique of machine learning based feature selection followed by survival analysis was applied in [14] to identify the most significant miRNA biomarkers for breast cancer subtype prediction. There is a strong association of miRNAs in various oral carcinomatous process. Thus, the abnormal expression detected in samples obtained from oral cancer patients are clinically significant in prediction and the development of effective treatments [16]. Falzole et al. utilized miRNA expression data set from GEO and TCGA miRNA profiling datasets to identify miRNAs signatures specific to OC [15].
In this study we proposed an integrated computational approach for identification and analysis of dysregulated miRNAs and their target mRNAs for Oral Cancer. Dysregulated miRNAs were prioritized based on their contribution in predicting the diseased condition. Further, putative dysregulated target mRNAs specific to cancer were identified and their prediction ability in separating the clinical conditions was examined.
The paper is organized as follows: Sect. 3 briefly describes the steps of our proposed method. Section 4 presents and discuss the empirical results of this study. Finally, Sect. 5 presents the conclusions of our study.
3 Materials and Methods
3.1 Dataset Used
Next-generation Sequencing based miRNA and mRNA expression data for the same patient were utilized in this work. The dataset was taken from GDC data portal of TCGA (https://portal.gdc.cancer.gov/). The Cancer Genome Atlas (TCGA) is a consortium of cancer genomics spanning over 33 cancer types which applies high throughput genome analysis techniques for characterizing genetic mutations responsible for cancer [17]. The data set consists of expression values of 1881 miRNAs and 18283 genes for 120 tumor samples and 44 matched normal samples.
3.2 Proposed Model
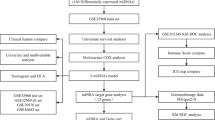
Figure 1 illustrates the workflow of our proposed model. The steps of our proposed work goes as follows:

Proposed model for identification of dysregulated miRNAs and associated target genes.
-
(A)
Data preparation: The data preparation step of our approach entailed removal of candidate miRNAs and genes with more than 30% of missing values followed by replacing the remaining missing values with mean of the sample [18]. Further, a logarithmic transformation base 2 [11] was applied on the resultant data in order to achieve normal distribution.
-
(B)
Identification of significant dysregulated miRNAs: A differential expression analysis of miRNAs and genes was done to find significant miRNAs and genes that show quantitative changes in expression levels between experimental groups normal and diseased. The candidate miRNAs and genes were investigated with the help of adjusted p-values and log-transformed fold change for identifying dysregulated miRNAs. A change in expression profile was considered as filtering criteria for identifying differentially expressed miRNAs. miRNAs with adjusted p-value \(\le \)0.05 and logFoldChange \(\ge \)2 [18] were considered to be significant in this study.
To evaluate the predictive ability of the differentially expressed miRNAs and to extract a handpicked of miRNAs, Random Forest (RF) classifier was adopted [11]. The RF is a learning method for classification, which works on the principle of ensemble learning by combining the solutions produced by multiple classifiers. The forest generated in the RF model consists of many decision trees of varying depths. RF method applies boot strap aggregating technique to train the decision trees. The samples left out during the training of each decision tree is referred as Out-Of-Bag (OOB) samples. For a new unseen sample, the learned RF model predicts by taking the average of the prediction outputs given by distinct decision trees. RF classifier can also be used to rank the features. Here we used Mean Decrease Accuracy (MDA) as the parameter for filtering significant miRNAs [24]. MDA is the proportion of observations that are incorrectly classified by removing the feature from the learned model [21]. The higher the MDA value, the more important the feature is.
-
(C)
Identification of miRNA-target genes using open source repository: The potential targets for the dysregulated miRNAs resulted in the previous step, were obtained using the TargetScan database [19]. For finding the target genes, we considered the conserved miRNAs only. Because more than 60% of human genes contain targets of conserved miRNAs across species [20]. Top predicted target genes with the highest aggregate probability of conserved targeting (Aggregate P\(_{CT}\)), irrespective of site conservation were considered for further analysis. Finally, a set of distinct miRNA-target genes were identified for the inputted dysregulated miRNAs.
-
(D)
Screening of statistically significant target genes: Differential expression analysis of the identified miRNA-target genes was done to screen the genes which express at different levels between clinical conditions. These genes are expected to offer precise biological insight into the processes affected by the condition(s) of interest. Furthermore, to unwrap the correspondence between the differentially expressed miRNA-target genes and disease of interest, a disease relatedness analysis was done by taking the data from NCBI (www.ncbi.nlm.nih.gov/geo/). Data of 8933 cancer-related (CR) genes and 316 oral cancer (OC) genes were obtained from NCBI. The screened target genes were investigated for the presence of cancer genes and oral cancer genes. Three different classifiers KNN (k = 3), SVM, RF were applied to examine the prognostic ability of the identified target genes against two clinical conditions. Parameters such as Specificity, Sensitivity, Precision, F-Score, Matthews coefficient correlation (MCC) and Prediction accuracy were used to measure the classification performance [22].
4 Results
We performed step wise analysis and selection of miRNA signatures and the associated target genes. The results obtained in this proposed work are presented on
-
(i)
Prioritizing miRNA signatures
-
(ii)
Identification of significant target genes specific to the disease
-
(iii)
Effectiveness of the final selected target genes.
-
(i)
Identification of prioritized miRNA signatures
The miRNA expression and gene expression data collected from TCGA produced were inputted to the data preparation step of our proposed model which resulted in 493 miRNA signatures and expression values of 16478 genes. The differential expression analysis of the resultant 493 miRNA signatures resulted in 244 differentially expressed miRNA signatures with adjusted p-value <0.05 and logFoldChange >2. To further identify the miRNA signatures which are significant in disease prognosis, RF classifier was used. miRNA signatures were ranked in decreasing order of MDA value. miRNA signatures with MDA value >1 were considered significant. This resulted in 72 significant dysregulated miRNA signatures out of 244. This ensures that these handpicked 72 miRNA signatures are differentially expressed and computationally proficient as well. Among all, the top 10 significant miRNA signatures are illustrated in Table 1.
-
(ii)
Identification of significant target genes
To obtain the potential targets for 72 notable miRNA signatures obtained in the previous step, a web-based target prediction tool: Target Scan was utilized. We systematically searched TargetScan for the identification of biological targets of the handpicked miRNAs. Target genes were queried for conserved miRNAs only. We obtained the top 50 targets for each conserved miRNAs with the highest aggregate probability of conserved targeting (PCT) value. It resulted in 1511 unique miRNA-target genes, which were finally mapped to pre-processed gene expression data obtained for OC. However, during mapping, a few miRNA-targets were dropped out because of its unavailability in the acquired TCGA gene expression data. This resulted in expression values of 1334 miRNA-target genes.
For selecting the target genes showing significant changes in different diseased conditions, the expression vector of 1334 target genes for tumor and normal samples were compared with respect to adjusted p-value and logarithmic fold change in expression levels. An adjusted p-value <0.05 and logFoldChange >2 was kept as cut off parameter. It resulted in 671 differentially expressed miRNA-target genes. The volcano plot in Fig. 2a clearly represents the differentially expressed miRNA-target genes marked with cyan colored dots.
Further, these target genes were reviewed for the existance of cancer-related genes and oral cancer genes using data collected from NCBI. Among 671 identified differentially expressed miRNA-targets, 331 genes were observed to be related to cancer whereas 19 genes were oral cancer genes. The proportion of CR and OC genes in the whole set of differentially expressed genes is demonstrated in Fig. 2b. These 350 (331 CR and 19 OC) genes were further validated in the following step with three different classifiers: KNN, RF and SVM.
-
(iii)
Effectiveness of the final selected target genes
To examine the predictive efficiency of the 350 target genes obtained in the previous step, we run the classifiers KNN (k = 3), RF and SVM. All the classifiers were run with 10-fold cross validation. Table 2 illustrates the results of classification. For all the three classifiers: KNN (k = 3), RF and SVM, the identified miRNA-target genes achieved an accuracy of 95.6%, 97.1% and 98.5% respectively. The acknowledged target features were observed with an average MCC value of 0.9 for the considered classifiers. A MCC value >0.5 is mostly considered to be significant in various machine learning platforms when there is an imbalanced ratio of input samples. The result shows that the distinguished target genes obtained in this approach can put a new light on OC prognosis with efficacy. These genes can further be biologically validated to confirm their participation in disease specific pathways and biological processes.

a) Volcano plot showing differentially expressed miRNA-target genes with cyan coloured dots.(b) Presence of cancer-related (CR) and oral cancer (OC) genes in the selected group of miRNA-target genes.
5 Conclusion
Identification of specific miRNAs and the associated target genes is crucial in characterizing the course of action of miRNAs in biological processes which lead towards cancer progression. The proposed work started with sample matched data of miRNA expression and gene expression to identify the dysregulated miRNAs and respective target genes. Up and down regulated miRNAs with high value for mean decrease in accuracy of the classifier were considered to be dysregulated. The specific top ranked target genes for the obtained dysregulated miRNAs were obtained from the online repository and further analyzed with respect to their differential expression and affinity towards the disease. The cancer specific target genes obtained in this approach were observed with significant prediction accuracy, which directs their use in prognostic application in diagnosis and treatment. These handpicked miRNA-target genes may further be biologically validated to confirm their role in biological processed and oncogenesis pathways.
References
Dikshit, R., et al.: Cancer mortality in India: a nationally representative survey. Lancet 379(9828), 1807–1816 (2012)
Borse, V., Konwar, A.N., Buragohain, P.: Oral cancer diagnosis and perspectives in India. Sens. Int. 1, 100046 (2020). https://doi.org/10.1016/j.sintl.2020.100046
Veluthattil, A.C., Sudha, S.P., Kandasamy, S., Chakkalakkoombil, S.V.: Effect of hypofractionated, palliative radiotherapy on quality of life in late-stage oral cavity cancer: a prospective clinical trial. Indian J. Palliat. Care 25(3), 383 (2019)
Lucenteforte, E., Garavello, W., Bosetti, C., La Vecchia, C.: Dietary factors and oral and pharyngeal cancer risk. Oral Oncol. 45(6), 461–467 (2009)
Fearon, E.R., Vogelstein, B.: A genetic model for colorectal tumorigenesis. Cell 61(5), 759–767 (1990)
Bartel, D.P.: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2), 281–297 (2004)
Jansson, M.D., Lund, A.H.: MicroRNA and cancer. Mol. Oncol. 6(6), 590–610 (2012)
Enerly, E., et al.: Correction: miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PloS one 8(9), e16915 (2013)
Lim, L.P., et al.: Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433(7027), 769–773 (2005)
Seo, J., Jin, D., Choi, C.H., Lee, H.: Integration of microRNA, mRNA, and protein expression data for the identification of cancer-related microRNAs. PLoS One 12(1), e0168412 (2017)
Bhowmick, S.S., Bhattacharjee, D., Rato, L.: Integrated analysis of the miRNA-mRNA next-generation sequencing data for finding their associations in different cancer types. Comput. Biol. Chem. 84, 107152 (2020)
Sathipati, S.Y., Ho, S.Y.: Novel miRNA signature for predicting the stage of hepatocellular carcinoma. Sci. Rep. 10(1), 1–12 (2020)
Varghese, R.S., et al.: Identification of miRNA-mRNA associations in hepatocellular carcinoma using hierarchical integrative model. BMC Med. Genom. 13(1), 1–14 (2020)
Sarkar, J.P., Saha, I., Sarkar, A., Maulik, U.: Machine learning integrated ensemble of feature selection methods followed by survival analysis for predicting breast cancer subtype specific miRNA biomarkers. Comput. Biol. Med. 131, 104244 (2021)
Falzone, L., et al.: Identification of novel MicroRNAs and their diagnostic and prognostic significance in oral cancer. Cancers 11(5), 610 (2019)
Fang, C., Li, Y.: Prospective applications of microRNAs in oral cancer. Oncol. Lett. 18(4), 3974–3984 (2019)
Tomczak, K., Czerwińska, P., Wiznerowicz, M.: The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp. Oncol. 19(1A), A68 (2015)
Mahapatra, S., Mandal, B., Swarnkar, T.: Biological networks integration based on dense module identification for gene prioritization from microarray data. Gene Rep. 12, 276–288 (2018)
Agarwal, V., Bell, G.W., Nam, J.W., Bartel, D.P.: Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005 (2015)
Xiong, P., Schneider, R.F., Hulsey, C.D., et al.: Conservation and novelty in the microRNA genomic landscape of hyperdiverse cichlid fishes. Sci. Rep. 9, 13848 (2019). https://doi.org/10.1038/s41598-019-50124-0
Hur, J.H., Ihm, S.Y., Park, Y.H.: A variable impacts measurement in random forest for mobile cloud computing. Wirel. Commun. Mob. Comput. (2017)
Mahapatra, S., Bhuyan, R., Das, J., Swarnkar, T.: Integrated multiplex network based approach for hub gene identification in oral cancer. Heliyon 7(7), e07418 (2021)
Ardekani, A.M., Naeini, M.M.: The role of MicroRNAs in human diseases. Avicenna J. Med. Biotechnol. 2(4), 161–79 (2010)
Han, H., Guo, X., Yu, H.: Variable selection using mean decrease accuracy and mean decrease gini based on random forest. In: 2016 7th IEEE International Conference on Software Engineering and Service Science (ICSESS). IEEE (2016)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this paper

Cite this paper
Mahapatra, S., Prasath, R., Swarnkar, T. (2022). Integrative Analysis of miRNA-mRNA Expression Data to Identify miRNA-Targets for Oral Cancer. In: Chbeir, R., Manolopoulos, Y., Prasath, R. (eds) Mining Intelligence and Knowledge Exploration. MIKE 2021. Lecture Notes in Computer Science(), vol 13119. Springer, Cham. https://doi.org/10.1007/978-3-031-21517-9_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-21517-9_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-21516-2
Online ISBN: 978-3-031-21517-9
eBook Packages: Computer ScienceComputer Science (R0)