
Abstract
Cellular senescence has become a subject of great interest within the ageing research field over the last 60 years, from the first observation in vitro by Leonard Hayflick and Paul Moorhead in 1961, to novel findings of phenotypic sub-types and senescence-like phenotype in post-mitotic cells. It has essential roles in wound healing, tumour suppression and the very first stages of human development, while causing widespread damage and dysfunction with age leading to a raft of age-related diseases. This chapter discusses these roles and their interlinking pathways, and how the observed accumulation of senescent cells with age has initiated a whole new field of ageing research, covering pathologies in the heart, liver, kidneys, muscles, brain and bone. This chapter will also examine how senescent cell accumulation presents in these different tissues, along with their roles in disease development. Finally, there is much focus on developing treatments for senescent cell accumulation in advanced age as a method of alleviating age-related disease. We will discuss here the various senolytic and senostatic treatment approaches and their successes and limitations, and the innovative new strategies being developed to address the differing effects of cellular senescence in ageing and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
What Is Cellular Senescence?
Cellular senescence is a state of irreversible growth arrest in which cells cease to proliferate but do not undergo apoptosis. Instead, these cells experience significant changes in morphology, metabolism and enzyme activity, as well as developing a pro-inflammatory phenotype with wide-ranging implications for the surrounding microenvironment. These phenotypic changes are responsible for both beneficial and detrimental roles of senescent cells in human development. While such cells are key to embryonic development, wound healing and tumour suppression in early life, they have also been associated with ageing and age-related disease. In each of these circumstances, senescent cells accumulate as a result of upregulated anti-apoptotic pathways and downregulated pro-apoptotic pathways, promoting cell survival and persistent cell cycle arrest.
One of the major determining factors of a senescent phenotype is the activation of a persistent DNA damage response (DDR). Double- and single-strand DNA breaks are detected by DNA damage kinases ataxia-telangiectasis mutated (ATM), ataxia-telangiectasis and rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK) (Ciccia and Elledge 2010), which initiate a cascade of downstream signalling to recruit DNA repair proteins. Factors involved in this DNA repair process localise at the site of damage and can form foci identifiable by immunofluorescent staining and used in the detection of senescent cells (d'Adda di Fagagna et al. 2003; Jackson and Bartek 2009). Such factors include ATM (Dupre et al. 2006), p53 binding protein (53BP1) (Eliezer et al. 2009) and phosphorylated histone 2AX (yH2AX) (Burma et al. 2001). H2AX is also responsible for chromatin remodelling protein recruitment, which allows alterations to chromatin structure to permit DDR protein access to the site of damage (Nakamura et al. 2010).
ATM and the checkpoint kinase Chk2 also promote activation of the transcriptional regulator p53 (Turenne et al. 2001), a major regulator of both senescent and apoptotic pathways (Seluanov et al. 2001). p53 in turn stimulates activation of the cyclin-dependent kinase (CDK) inhibitor p21 (Jackson and Pereira-Smith 2006) leading to cell cycle arrest (Dutto et al. 2015) via retinoblastoma protein (pRb) hypophosphorylation and stabilised binding to E2F transcription factor 1 (E2F1) (Georgakilas et al. 2017). Chk2 is also responsible for the degradation of CDC25 phosphatase, which prevents G1-S phase transition and supports growth arrest (Mailand et al. 2000). An additional barrier to cell cycle progression is established by double strand break (DSB) activation of the CDK inhibitor p16, a positive regulator of the pRb pathway and able to induce cell cycle arrest by inhibiting CDK4/6 and reinforcing pRb inhibition by E2F1 (Beausejour et al. 2003).
The major beneficial role of cellular senescence is as a tumour suppressor mechanism. By preventing damaged and pre-malignant cells from dividing, cellular senescence acts as a barrier to tumour development. This is largely achieved by modifying levels of cell cycle regulators, primarily p53, p21 and p16, which induce a permanent halt to cell cycle progression. Secretion of SASP factors IL-6 and IL-8 can also interact with specific cell-surface receptors to trigger signalling pathways such as the DDR. This is discussed in further detail below.
During mammalian embryonic development, senescent cells have been found in the apical ectodermal ridge (AER) and neural roof plate. It is here that senescent cells are thought to influence growth and patterning through p21, p15 and SASP signalling. Indeed, studies have found that mice lacking p21 exhibit dysfunctional embryonic senescence (Storer et al. 2013). Notably, two fibroblast growth factor genes (FGF4 and FGF8) involved in AER signalling for proliferation and pattern formation were also reduced in p21-deficient mice, suggesting that this aspect of AER function is compromised when senescence is prevented. Senescent markers have also been linked to placental formation and function. In particular, the known SASP components matrix metalloproteinases 2 and 9 MMP2 and MMP9 are important in achieving trophoblast invasion in early pregnancy, demonstrating a link between SASP and placental development. Moreover, activity of these MMPs is decreased in murine placentas with deficient senescent signalling (Gal et al. 2019). Other reports have detailed a p21-dependent senescence in the mesonephros and endolymphatic sac in the inner ear (Munoz-Espin et al. 2013).
Cellular senescence is also important to the process of wound healing, both by limiting proliferation of damaged tissue and recruiting immune cells to eliminate the accumulated senescent cells. This is demonstrated in liver fibrosis, where senescent hepatic stellate cells are known to accumulate and secrete greater levels of extracellular matrix (ECM) degrading enzymes and are the subject of enhanced immune surveillance. This promotes immune-mediated clearance of the senescent cells, alleviating fibrosis progression and liver scarring (Krizhanovsky et al. 2008). Similar findings have also been reported in the pancreas, in which accumulated senescent pancreatic stellate cells correlate with greater inflammation and fibrosis, which is limited by immune-mediated clearance of senescent cells (Fitzner et al. 2012). Furthermore, in vivo studies have demonstrated that cellular senescence is involved in tissue regeneration in both zebrafish (Da Silva-Alvarez et al. 2020) and salamanders (Yun et al. 2015).
In addition to mediating immune clearance of senescent cells, pro-inflammatory SASP factors are also involved in numerous other processes within wound healing. For example, platelet-derived growth factor AA (PDGF-AA) accelerates wound closure by promoting myofibroblast differentiation (Demaria et al. 2014), while cellular communication network factor 1 (CCN1) limits skin fibrosis by inducing fibroblast senescence via cell-surface receptor interaction in cutaneous wound healing (Jun and Lau 2010). These senescent cells exhibit a persistent DDR, p53 and p16 activation, and express antifibrotic genes. This, along with SASP-mediated clearance of the senescent cells, restricts fibrosis in cutaneous injury. The pro-inflammatory senescence-associated secretory phenotype (SASP) is responsible for triggering immune-mediated clearance of senescent cells by recruiting key immune cells including macrophages, B cells, T cells and NK cells (Coppe et al. 2008; Krizhanovsky et al. 2008; Storer et al. 2013). This clearance of senescent cells is key to both wound healing, by limiting scarring and fibrosis, and tumour suppression, by eliminating pre-malignant and damaged cancerous cells (Xue et al. 2007; Krizhanovsky et al. 2008; Ruscetti et al. 2018).
In contrast to these positive effects in early life and development, it is well established that senescent cells are also associated with the ageing process and age-related disease, discussed in greater detail in the section on Senescence and Age-Related Disease, below. Alongside this, the SASP has been shown to possess a paradoxical role in cancer development over time. While SASP factors are key to immune-mediated clearance of senescent cells, contributing to tumour suppression, it has also been noted that the chronic inflammation generated by SASP production may contribute to diseases such as diabetes (Xu et al. 2015; Prattichizzo et al. 2018), COPD (Birch et al. 2015), osteoarthritis (Livshits et al. 2009; Xu et al. 2017) and cancer (Bavik et al. 2006; Coppe et al. 2010; Ortiz-Montero et al. 2017). It has also been observed that accumulation of senescent cells can promote cancer development, and can be accompanied by SASP-mediated paracrine effects on cancer progression (Eggert et al. 2016; Lau et al. 2019) and treatment failure (Demaria et al. 2017). Notably, Demaria and colleagues reported that selective elimination of senescent cells by navitoclax treatment alleviated a number of chemotherapy-induced side effects, including cancer recurrence and bone marrow suppression. Similar observations of increased chemotherapy-induced fatigue in patients with greater senescent marker presence were reported in patients with breast cancer (Demaria et al. 2017).
History
Cellular senescence was first described in 1961 by Hayflick and Moorhead, who determined that normal proliferating fibroblasts in culture were limited to a finite number of divisions before halting cell cycle progression (Hayflick and Moorhead 1961). This is now referred to as the ‘Hayflick limit’ (Hayflick 1965). This form of senescence resulting from extended cell culture is termed replicative senescence, discussed in more detail in the section on Replicative Mechanisms, below. Other forms of senescent phenotype are triggered by different stimuli, but share a large proportion of common markers and characteristics are also described below. One central feature of all of these phenotypes is the choice between apoptosis and senescence induction. How this choice is made remains unclear, but there are many possible determining factors, such as damage severity, stimulus type and cell type. It is also possible that a shared mediator may be involved in this choice, as both apoptotic and senescent pathways are regulated by the p53 pathway (Kirschner et al. 2015).
The contrasting roles of senescence in embryonic development and early life versus ageing and disease have also caused much debate within the ageing research field. It is striking that cells initially so beneficial to human development and survival should have such detrimental effects later in life. One prominent hypothesis is the antagonistic pleiotropy theory of ageing, which proposes a preferential selection of genes with reproductive fitness advantages during early life, but which may come with negative consequences in later life as an unintended consequence. Though to date there is little evidence to support this theory.
Characteristics and Markers
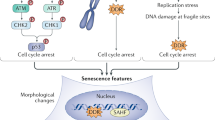
Cellular senescence can result from numerous stress-inducing triggers including telomere attrition, persistent DNA damage, oncogene activation, mitochondrial dysfunction and oxidative stress. The specific senescent profiles produced by each of these triggers is discussed in further detail below, but there are a number of common characteristics and markers that are observed in multiple models of senescence (Fig. 7.1)

The senescent phenotype: Cellular senescence can be triggered by a number of different stressors, but a selection of common markers have been identified which can be used to determine the presence of senescent cells. (a) elevated senescence-associated beta-galactosidase (SA-β-Gal) activity identified by blue histochemical stain at pH 6, (b) elevated reactive oxygen species (ROS) production by the mitochondria, (c) DNA damage caused by a range of stressors can be identified as foci of DNA repair machinery components, (d) telomere shortening, (e) senescence-associated heterochromatin foci (SAHF) and F) production of pro-inflammatory cytokines, chemokines and growth factors that form the senescence-associated secretory phenotype (SASP)
.
Telomere Shortening
Telomeres are end chromosome structures formed by repetitive DNA sequences and capped with proteins that protect them from being mistakenly identified as damage or double strand breaks by DNA repair processes (Blackburn 1991; d'Adda di Fagagna et al. 2004). However, DNA polymerase activity during cell division cannot fully copy these telomeric regions, leading to progressive shortening of telomeres with each cell division. Eventually, a critical length is reached at which the protective telomeric caps are lost during replication, and the newly uncapped single-stranded telomere ends are recognised by DNA repair machinery as double strand breaks (DSBs) (d'Adda di Fagagna et al. 2003). This triggers a DNA damage response (DDR) which attempts to repair the ‘damage’; however, telomeres are inaccessible to DNA repair machinery as components of the protective shelterin complex inhibit the non-homologous end joining repair pathway (Bae and Baumann 2007). This inability of DNA repair processes to fix the perceived damage leads to a persistent DDR (Galbiati et al. 2017), which is required for cellular senescence (Fumagalli et al. 2014).
Some DNA repair components translocate to the site of the damage and accumulate into identifiable foci. These foci are also present at the sites of non-telomeric DNA damage, and can be used as quantifiable markers of senescent cells (Takai et al. 2003). In the case of telomere attrition, these foci localise at the telomeres and are termed telomere-associated foci, or TAFs. It is also worth noting that a persistent DDR can also be triggered at telomeres following DNA damage, irrespective of telomere length, such as that resulting from ROS-induced oxidative damage (von Zglinicki 2002; Reichert and Stier 2017). The reduced efficiency of DNA repair at telomeres promotes a persistent DDR regardless of the initial cause of damage (Bae and Baumann 2007; Hewitt et al. 2012; Fumagalli et al. 2014).
The role for telomere shortening in cellular senescence is further supported by the observation that overexpression of telomerase prevented senescence in a study of telomerase-negative human retinal pigment epithelial cells and foreskin fibroblasts, along with extending the replicative lifespan of such cells (Bodnar et al. 1998).
DNA Damage
As discussed, a persistent DDR can be triggered by DNA damage at sites other than telomeres, culminating in permanent cell cycle arrest (Fumagalli et al. 2014). Often, a persistent DDR is triggered by double strand DNA breaks (DSBs), though single-strand breaks (SSBs) can also trigger a DDR. At these points of damage, key components of the DNA repair machinery can accumulate and provide identifiable markers of a persistent DDR and senescent cells. Such markers include phosphorylated histone 2AX (γH2AX), mediator of DNA damage checkpoint protein 1 (MDC1), 53BP1 and ATM (Burma et al. 2001; Eliezer et al. 2009; Lavin et al. 2015). These foci of DNA repair machinery components are referred to as DNA damage foci (DDF), and when present at telomeric sequences, telomere-associated foci (TAF) which represent a robust marker of senescence (Hewitt et al. 2012). Work by Galbiati and colleagues provided evidence of prolonged checkpoint activation in senescence by demonstrating the presence of long-term unrepaired DSBs in persistent DDFs (Galbiati et al. 2017).
Notably, inhibition of the key DDR factors ATM, ATR and CHK1/2 enables senescent cells to re-enter the cell cycle, confirming the role of a persistent DDR in maintaining senescent growth arrest (d'Adda di Fagagna et al. 2003; Di Micco et al. 2006; Mallette and Ferbeyre 2007). The induction of senescence in response to severe DNA damage also demonstrates its key beneficial role in tumour suppression. By limiting the proliferation of cells which have accumulated excessive DNA damage, cellular senescence restricts the ability of pre-cancerous and cancerous cells to continue to multiply and impact on tissue function.
SA-B-Gal
A common marker of cellular senescence is increased β-galactosidase activity (Dimri et al. 1995) that is not observed in quiescent or immortalised cells. Though β-galactosidase activity has previously been observed in confluent quiescent cells, this was reversible, distinguishing senescent cells from these phenotypes (Yang and Hu 2005). Similarly, elevated β-galactosidase activity has been shown in immortalised cells, but was dependent on high confluency, and in cultures at low cell density β-Gal staining was not observed (Severino et al. 2000). This increase in β-galactosidase activity occurs in response to elevated lysosomal activity (Kurz et al. 2000) following induction of senescence (senescence-associated β-galactosidase, or SA-β-Gal). This reliable marker has also been observed in vivo (Dimri et al. 1995). However, while a common marker of senescence it is not unique to the phenotype, and can also be present in serum-starved or highly confluent cell cultures (Severino et al. 2000; Yang and Hu 2005). As such, the presence of SA-β-Gal activity is not sufficient alone to identify cellular senescence.
Cyclin-Dependent Kinase Inhibitors
Senescent cells also exhibit greater upregulation of cyclin-dependent kinase inhibitors. Specifically, p21 and p16 signalling is key to maintaining the senescent growth arrest, as inhibitors of cell cycle progression. Serrano and colleagues identified that mice lacking p16 were more prone to tumour formation (Serrano et al. 1996) and that oncogene-induced senescence involved elevated p16 expression (Serrano et al. 1997). p16 has also been used in a number of studies to identify and selectively eliminate senescent cells in vivo (Baker et al. 2011), which has led to improvements in age-related pathologies (Baker et al. 2016; Bussian et al. 2018; Patil et al. 2019; Ogrodnik et al. 2021). These two markers are generally robust identifiers of senescence; however, not all forms of senescence include elevated p16 levels (Munoz-Espin et al. 2013; Storer et al. 2013), and some aspects of senescence are independent of p16 expression. Moreover, in senescent cells lacking p16, an inhibition of p53 activity can cause a dramatic increase in SASP production along with re-entry into the cell cycle, producing dangerous pro-inflammatory cells prone to malignant transformation (Beausejour et al. 2003; Coppe et al. 2008; Rodier et al. 2009). Therefore these CDK inhibitors cannot be used individually to confirm the presence of senescence (Coppe et al. 2010).
Morphology
One of the most noticeable markers of senescence is a change in cell morphology. Upon the induction of a senescent phenotype cells become significantly enlarged, with a flattened cell body and a greater cytoplasm: nucleus ratio in vitro. The nucleus itself also becomes enlarged, and can be used as a marker of senescent cells both in vitro and in vivo (Mitsui and Schneider 1976; Yoon et al. 2016; Fielder et al. 2022). Some studies have even suggested that this increase in cell size may in fact drive the permanent growth arrest, as a result of a cell’s inability to appropriately scale up nuclear and protein component synthesis (Neurohr et al. 2019).
Chromatin Alterations
Senescent cells also display significant changes to chromatin architecture, including loss of nuclear lamina allowing release of cytosolic chromatin fragments (CCFs) (Ivanov et al. 2013; Dou et al. 2017), the formation of heterochromatin foci (Narita et al. 2003), and altered DNA methylation (Hanzelmann et al. 2015).
Senescence-associated heterochromatin foci (SAHF) can be used as markers of senescent cells and can be identified as dense DAPI-positive foci within the nucleus. These condensed chromatin structures are associated with the silencing of E2F activity and dependent upon the Rb tumour suppressor pathway (Narita et al. 2003). Notably, SAHF do not develop in cells where growth arrest is reversible. However, some have noted that they appear to be specific to oncogene-induced senescence and senescence induced by stressors of DNA replication. SAHF are also thought to lessen persistent DDR signalling, as HDAC inhibitor-induced chromatin relaxation results in elevated DDR signalling and promotion of apoptosis (Di Micco et al. 2011).
CCFs have been found to trigger the cGAS-STING pathway (Chang et al. 2017; Dou et al. 2017). Treatment with HDAC inhibitors has also been shown to reduce CCF formation and subsequent SASP production (Vizioli et al. 2020).
Lack of DNA Replication/Cell Proliferation
Another major marker of cellular senescence is the lack of cell proliferation and DNA replication. These can be identified by immunostaining for the proliferation marker Ki67 (Lawless et al. 2010), though this is not unique to senescent cells.
SASP
The SASP is a major part of the senescent phenotype and responsible for much of both the beneficial and detrimental effects of senescent cell accumulation. The secretome is composed primarily of pro-inflammatory cytokines, chemokines, growth factors, matrix metalloproteinases and ECM-degrading proteins (Coppe et al. 2008, 2010). Most commonly, IL-6 and IL-8 are used as identifiers of senescent cells and are regularly used in cell culture models. These cytokines are also known to both reinforce and induce senescence via autocrine and paracrine mechanisms (Acosta et al. 2008; Nelson et al. 2012). However, it should be noted that some forms of senescence do not possess the IL-1 arm of the SASP (Wiley et al. 2016) or exhibit an otherwise altered SASP not inclusive of these two markers, as in the case of p16 overexpression (Coppe et al. 2011). Moreover, SASP can also vary between cell type and senescence trigger. Therefore, while valuable markers of senescence, IL-6 and IL-8 are not sufficient on their own. Nevertheless, there are common SASP factors observed across most forms of senescence and are widely used together to monitor the presence of senescent cells and effectiveness of senolytic and senomorphic drug treatments (discussed below in: Senescence and age-related disease).
This diverse and variable pro-inflammatory phenotype is primarily controlled via the transcription regulator NFκB, which regulates the activation of SASP genes (Chien et al. 2011; Ohanna et al. 2011). In multiple models of senescence activation of the cGAS-STING pathway is known to regulate SASP production via NFκB, providing a link between CCF formation and SASP production (West et al. 2015). Though the relationship between cGAS-STING is not straightforward, as downregulation reduces SASP production in vivo, while activation has also been shown to lead to accumulation of nuclear DNA in the cytosol (Takahashi et al. 2018).
Numerous other components of the DDR signalling pathway influence SASP production, and studies have demonstrated that a persistent DDR is required in order for a full SASP to be established (Rodier et al. 2009). Indeed, p38MAPK is known to be both necessary and sufficient to induce a SASP via NFκB activation (Freund et al. 2011). Moreover, inhibition of key DDR components ATM, Chk2 and p53 has been found to suppress the SASP, while heterochromatin reorganisation and SAHF formation has been linked to greater nuclear pore density in OIS, which can influence SASP gene expression (Boumendil et al. 2019). High mobility group box 1 and 2 (HMGB1/2) proteins have also been identified as regulators of the SASP, by inducing the expression of SASP genes in OIS (Huang et al. 2015; Aird et al. 2016) and signalling the immune system to senescent cells (Davalos et al. 2013). In contrast, cells which enter senescent growth arrest as a result of p21 or p16 overexpression do not produce a SASP (Rodier et al. 2009; Coppe et al. 2011), meaning that SASP production alone cannot be used as a universal marker of senescence.
Though multiple robust markers of cellular senescence have been identified, no one marker is unique to senescence, and the heterogeneity of the senescent phenotype between cell type, senescent stimuli and resulting phenotypic sub-type show that not all markers are present in all forms of senescence. Individual markers are therefore insufficient to identify senescence cells alone, and multiple markers must be used to determine the presence of cells in a senescent state (Gorgoulis et al. 2019).
Types and Mechanisms of Cellular Senescence
Replicative
The first description of cellular senescence in normal cultured human fibroblasts by Hayflick and Moorhead in 1961 pertained primarily to what is known as replicative senescence; the circumstance in which cells have reached their replicative limit as a result of critically short telomeres leading to growth arrest. The role of telomere shortening in senescence has been confirmed by reports of senescence prevention and extended replicative lifespan in cells overexpressing telomerase (Bodnar et al. 1998). Following extended proliferation and the arrival at this critical telomere length (Harley et al. 1990)—and triggering a persistent DDR as discussed previously—these cells arrest in the G1 phase of the cell cycle (Stein and Dulic 1995). However, despite being unable to continue proliferating, such cells are also resistant to apoptotic cell death processes. This is thought to be a result of upregulated anti-apoptotic members of the BCL-2 family (Yosef et al. 2016). It should also be noted, however, that there are numerous determining factors that impact the response to extreme stress, including cell type, stimuli strength, and regulation of pro- and anti-apoptotic proteins (Rebbaa et al. 2003; Hampel et al. 2004).
Stress-Induced
Accumulation of DNA damage at sites other than telomeres and at telomeres that are not critically shortened can also trigger induction of a senescent phenotype (Di Leonardo et al. 1994). Such damage is most often caused by stimuli such as ionising radiation, oxidative stress (Parrinello et al. 2003), or genotoxic chemicals including chemotherapeutic drug treatments (Roninson 2003). Under these circumstances senescent cells have been known to accumulate in young animals, showing that senescence is not strictly an age-related phenotype (Le et al. 2010; Shao et al. 2014). This stress-induced senescence primarily involves elevated p16 expression and pRb signalling pathways in response to DNA damage (Parrinello et al. 2003). Upregulation of pro-inflammatory SASP factors has also been shown to induce senescent changes in otherwise healthy cells through a process known as bystander senescence, discussed in further detail later.
Oncogene-Induced
Oncogene-induced senescence (OIS) was first described by Serrano and colleagues in 1997, following the observation that oncogenic RAS protein induced premature cellular senescence characterised by elevated p53 and p16 activity. This was later supported by others who found a similar senescence induction in the presence of oncogenic BRAF (Michaloglou et al. 2005), E2F (Lazzerini Denchi et al. 2005), PTEN and p53 (Chen et al. 2005). It is also thought that OIS may be triggered by excessive mitogenic stimulation (Mathon et al. 2001), supported by the observation that cells relieved of mitogenic pressure by culture in serum-free medium do not undergo OIS (Woo and Poon 2004). It is important to note, however, that the extension of replicative capacity by telomerase does not promote cancerous transformation, demonstrating a key difference between immortalised and tumorigenic cells (Morales et al. 1999).
The presentation of this oncogene-induced senescent phenotype is largely similar to other forms of senescence, with chromatin alterations and formation of heterochromatin foci (Zhang et al. 2005), markers of a persistent DDR (Di Micco et al. 2006), and establishment of a SASP (Coppe et al. 2008). One notable difference, however, is the induction of a hyperproliferative phase caused by oncogene expression. This period of hyperproliferation is accompanied by altered DNA replication and triggers a persistent DDR, as in other senescent sub-types (Bartkova et al. 2006; Di Micco et al. 2006; Halazonetis et al. 2008).
A role for oncogene-induced ROS production in OIS establishment has been identified, in which excessive ROS generated by NADPH oxidases promote hyperproliferation and DNA damage (Ogrunc et al. 2014). Though this is not universal for all OIS triggers. For example, activation of the PI3K-AKT pathway induces a p53-dependent senescence, but lacks the hyperproliferation and DDR observed in other forms of OIS (Astle et al. 2012). OIS has also been found to exhibit a more amplified SASP compared to other forms of senescence (Coppe et al. 2008).
Bystander-Induced
It is also now well established that senescent cells can promote senescence induction in neighbouring healthy cells though SASP-mediated paracrine effects (Nelson et al. 2012). Among the known SASP factors, the transforming growth factor beta (TGF-β) family, vascular endothelial growth factor (VEGF) and chemokines CC motif chemokine ligand 2 (CCL2) and chemokine ligand 20 (CCL20) have all been found to trigger major senescent pathways including a persistent DDR following interaction with cell-surface receptors (Acosta et al. 2013). High levels of IGFβ can also cause elevated ROS production and p53-dependent DDR (Moiseeva et al. 2006). In vivo studies have further demonstrated an increase in senescent cell accumulation and tissue ageing in a mouse model with shortened telomeres following extended exposure to interferon gamma (IFNγ) (Katlinskaya et al. 2016), supporting the role of SASP factors in senescence induction in animal models.
TGF-β signalling demonstrates a paradoxical effect in bystander senescence, as it can both cause p16-pRb signalling and SAHF formation (Vijayachandra et al. 2003; Zhang and Cohen 2004), and has also been shown to prevent the spread of bystander-induced senescence in liver, improving regeneration (Bird et al. 2018). The ability of some SASP factors to improve regenerative capacity over short exposure times has been demonstrated in mouse keratinocytes (Ritschka et al. 2017) and skeletal muscle (Chiche et al. 2017). Comparatively, following long-term exposure paracrine senescence is induced, suggesting that this relationship may be more complicated than first thought and may be dependent on numerous factors.
The ability of the SASP to promote further induction of senescence likely contributes to the accumulation of senescent cells and deterioration of tissue function. It is thought that dysfunctional mitochondria produce excessive amounts of toxic reactive oxygen species (ROS), which can trigger ROS-dependent NFκB signalling and subsequently senescence in bystander cells (Nelson et al. 2018).
MiDAS
It has been well established that mitochondria are required for the development of a senescent phenotype (Correia-Melo et al. 2016) and that manipulation of mitochondrial function can impact senescence development (Wiley et al. 2016). Moreover, mitochondrial dysfunction has been shown to promote senescence, and vice versa (Correia-Melo et al. 2016; Wiley et al. 2016; Nelson et al. 2018). A major regulator of this relationship is the production of mitochondrial reactive oxygen species. Notable changes in mitochondrial morphology during senescence have also been described, including a hyperfused network with increased mitochondrial mass (Yoon et al. 2006; Dalle Pezze et al. 2014).
Work by Wiley and colleagues demonstrated that mitochondrial dysfunction-induced senescence (MiDAS) exhibits a unique SASP relative to other forms of senescence, which lacks the pro-inflammatory IL-1 arm of the phenotype. This is accompanied by loss of LaminB1, a reduced NAD+/NADH ratio, activation of AMPK and p53, and—surprisingly—a reduction in NFκB signalling. Inhibition of sirtuin 3 (SIRT3) specifically resulted in a senescent phenotype with a distinct secretome lacking IL-1β, IL-6 and IL-8. This led to the hypothesis that a functional mitochondrial network is essential for a complete SASP to occur. Similar mitochondrial function-modifying interventions include mtDNA depletion, pharmacological inhibition of respiratory complexes, and heat shock protein family A member 9 (HSPA9) depletion, a key mitochondrial chaperone, although these are supraphysiological conditions. Each of these interventions also demonstrated a unique senescent phenotype lacking the IL-1 arm of the SASP.
MiDAS also appears to be independent of DNA damage and ROS production as the phenotype is reversible in the presence of pyruvate, but not antioxidants. It is thought that MiDAS can be driven by a decline in the NAD+/NADH ratio as a result of mitochondrial dysfunction. A low NAD+/NADH ratio then causes activation of AMPK and subsequently p53 activation. Notably, elevated p53 activity suppresses NFκB signalling and is therefore a likely cause of a SASP lacking the IL-1 arm.
Table 7.1 is a summary of some of the well-established markers of senescence and their presence in specific sub-types of senescent phenotype. It should also be noted, however, that this list is not exhaustive and that these phenotypes demonstrate heterogeneity between different cell types, for example. Moreover, there remains some debate in the literature as to the presence or relevance of these markers (Mirzayans et al. 2012).
In Vivo Relevance of Senescent Cells
The well-known roles for senescent cells in human development and disease can be observed in a variety of tissues, and the development of a robust profile of senescent markers across different tissue types has been a valuable goal within the ageing field for many years. The heterogeneity of the phenotype makes this more difficult, but over the years senescent cells have been observed in many tissue types and organisms, providing the basis for an overarching picture of senescence in vivo. One major contributor to this work has been the establishment of key animal models, namely INK-ATTAC (Baker et al. 2011) and P16-3MR mice (Demaria et al. 2014). These models of premature ageing have allowed researchers to examine the progress and impact of senescent cell accumulation, as well as investigate potential therapeutics targeted specifically at senescent cells (discussed below, in Anti-senescence interventions). Importantly, Baker and colleagues demonstrated that selective removal of p16-expressing senescent cells produced significant improvement in numerous age-related phenotypes.
Another commonly used model of in vivo ageing is the BubR1 hypomorphic mouse model, in which mice express ~90% less of the BubR1 protein, responsible for ensuring chromosome segregation into daughter cells during mitosis. This leads to a phenotype of shortened lifespan, infertility, and cataracts, among others (Baker et al. 2004) and is associated with greater p16 expression (Baker et al. 2008a, b). Subsequent investigations of this rapid ageing model incorporated the Cdkn2ap16-knockout mice, which produced BubR1 mice lacking p16. These mice consequently did not develop the ageing-related phenotype of poor muscle, eye and adipose tissue function (Baker et al. 2008a, b), demonstrating the requirement for p16 in the pro-ageing phenotype. This was further confirmed in a study of naturally aged mice and the INK-ATTAC system, where an extended lifespan and healthspan was observed (Baker et al. 2011).
It is also worth noting that our understanding of which cells can senesce is also expanding, with the recognition of senescent markers (Table 7.2). In the past it was thought that cellular senescence was restricted only to cells capable of proliferation. That has now been called into question by the observation of senescent-like phenotypes in neurons, skeletal myofibers and cardiomyocytes, among others (Jurk et al. 2012; Anderson et al. 2019; da Silva et al. 2019; Matias et al. 2022). Though these findings are still in the early stages of exploration, it presents an exciting new avenue for our understanding of cellular senescence as a whole.
Cell Types and Tissues in Which Senescence Has Been Observed
Age-Dependent Changes in the Abundance
Senescent cells are known to accumulate with age (Dimri et al. 1995; Wang et al. 2009; Jurk et al. 2014). Indeed, ageing is associated with greater prevalence of DDR signalling in both proliferating and non-proliferating cells (Fumagalli et al. 2012; Rossiello et al. 2017). Moreover, the accumulation of senescent cells has been linked to impaired immune-mediated clearance (Ovadya et al. 2018) resulting from a weakened immune system with age.
In addition to a decline in tissue function, there has also been an observed reduction in regenerative capacity with greater senescent cell abundance (Krishnamurthy et al. 2006; Sousa-Victor et al. 2015). This is thought to result from an increase in senescent stem and progenitor cells, which subsequently are unavailable to provide new cells for tissue growth and regeneration. This is also true of muscle progenitor cells such as satellite cells, which have important roles in wound healing and regeneration and can become senescent with age, leading to a decline in wound healing capabilities (Zwetsloot et al. 2013; Sousa-Victor et al. 2015), as well as a paracrine SASP-mediated impairment of tissue regeneration by HSCs (Gnani et al. 2019).
Senescence in Age-Related Disease
The accumulation of senescent cells with age can contribute to overall tissue dysfunction (Janzen et al. 2006; Molofsky et al. 2006) and development of disease. This link between cellular senescence and disorders of ageing has received further support from recent genome-wide association studies (GWAS) which confirmed the INK4/ARF locus as a major genomic determinant of susceptibility to diseases such as cardiovascular disease, neurodegenerative disease and cancer (Jeck et al. 2012). Moreover, in vivo studies have demonstrated significant tissue dysfunction and shortened lifespan in healthy mice when administered a quantity of senescent cells via transplantation (Xu et al. 2017). The stresses associated with disease can also trigger greater conversion of cells to senescence, particularly as aged cells are less capable of combating stress and damage (Weyand and Goronzy 2016; Chen et al. 2020).
The presence of numerous senescent markers has been identified in age-related diseases across multiple tissues. For example, shortened telomere length has been linked to both metabolic and cardiac dysfunction (Benetos et al. 2013; D'Mello et al. 2015), while levels of key SASP factors within the blood are known to correlate with chronic disease in advanced age (Fabbri et al. 2015). Increased levels of senescent markers such as p16 have also been identified in heart failure (Chimenti et al. 2003; Schafer et al. 2017), osteoarthritis (Loeser 2009; Jeon et al. 2017) and diabetes (Minamino et al. 2009), and recent studies have even linked senescence within the central nervous system to Alzheimer’s disease (Bhat et al. 2012; Zhang et al. 2019). Tau-containing neurons have demonstrated a senescence-like expression profile in mice, while senescent markers including SA-β-Gal, DNA damage and p16 have been found in retinopathies (Oubaha et al. 2016).
Studies of heart health in ageing have reported elevated cell death and hypertrophy of heart tissue with advanced age, as well as accumulation of p16INK4a-positive cells (Chimenti et al. 2003). In a mouse model prone to atherosclerotic plaque formation, plaques exhibit higher levels of p16 and SA-β-Gal activity. Conversely, plaque formation has been effectively reduced in both p16-3MR and INK-ATTAC mice by removing p16-positive cells, which also subsequently suppressed the SASP (Childs et al. 2016). Inflammatory macrophages have also been found to express high levels of p16 in human atherosclerotic plaques (Holdt et al. 2011). Interestingly, some studies have found that senescent cell accumulation may both promote and prevent aspects of atherosclerosis. Increased expression of p16INK4a, ARF and p15INK4b has been associated with lower incidence of atherosclerotic vascular disease (Liu et al. 2009). However, while the anti-proliferative role of senescent cells is favourable for preventing disease development, the pro-inflammatory aspect promotes atherogenesis which is relieved by targeted clearance of p16INK4a-expressing cells (Childs et al. 2016).
In osteoarthritis models, greater IL-6 secretion (Livshits et al. 2009), telomeric dysfunction (Fragkiadaki et al. 2020; Manoy et al. 2020) and p16 expression has been observed (Malaise et al. 2019), while selective clearance of senescent cells with senolytic treatment has shown success in relieving SASP secretion, slowing disease progression, and improving bone health (Peilin et al. 2019). p53 expression and increased oxidative stress have also been linked to insulin resistance in diabetes, and adipose tissue samples from patients with type 2 diabetes exhibit prominent markers of senescence, including SA-β-Gal activity and pro-inflammatory cytokine expression (Minamino et al. 2009).
The SASP has also been proposed as a major regulator in the development of age-related disease, as chronic inflammation is observed in many conditions such as atherosclerosis (Zhou et al. 2006) and cancer (Thangavel et al. 2011). Work by Xu et al. (2018) has demonstrated that young mice given senescent cells by transplantation resulted in chronic physical dysfunction. This dysfunction was then relieved by senolytic treatment. Increased levels of SASP factors such as IL-6 and tumour necrosis factor (TNF) receptor in the blood have also been identified as predictors of chronic disease with old age (Fabbri et al. 2015).
Anti-senescent Interventions
Given the wide-ranging roles of senescent cell accumulation in ageing and age-related diseases, there has been much research aimed at identifying treatments targeted at senescent cells specifically. Studies have demonstrated that clearance of senescent cells can both prolong lifespan in model organisms and alleviate age-related pathologies (Baker et al. 2011; Zhu et al. 2015; Roos et al. 2016). Two broad categories of therapeutic intervention have emerged over the years; one aiming at complete degradation and clearance of senescent cells (senolytics) (Zhu et al. 2015) and one targeting the harmful aspects of the senescent phenotype (primarily SASP) while allowing the cells themselves to remain (senomorphics/senostatics). Senolytics commonly target pathways which promote cell survival and protect against apoptosis. Many studies have investigated the use of senolytics and senostatics to alleviate the senescent burden in vivo, with varying degrees of success.
Pre-clinical Evidence
In avoiding apoptotic cell death, senescent cells upregulate several anti-apoptotic factors from within the BCL-2 family. Thus, numerous studies have attempted to overcome this apoptotic resistance by treating senescent cells with BCL-2 inhibitors. One of the most commonly tested senolytic treatments is the combination of the tyrosine kinase inhibitor dasatinib with PI3K inhibitor quercetin (D + Q).
D + Q treatment reduces the expression of anti-apoptotic BCL-xL, allowing clearance of senescent cells. This has resulted in improved health span in multiple mouse models of cellular senescence (Xu et al. 2018). Studies have shown that this treatment is able to improve vasomotor function in hypercholesterolaemia (Roos et al. 2016) and lung function in pulmonary fibrosis (Schafer et al. 2017). Recently, D + Q treatment has also been shown to lower SASP factor secretion and senescent cell burden in a mouse model of age-related intervertebral disc degeneration, improving cell viability and limiting degradation (Novais et al. 2021). In a mouse model of Alzheimer’s disease, researchers have found that D + Q treatment is also able to selectively remove senescent cells from Aβ plaques, along with an associated reduction in neuroinflammation and improved cognitive deficits (Zhang et al. 2019).
Navitoclax is another well-established senolytic which inhibits anti-apoptotic members of the BCL-2 family, promoting the release of pro-apoptotic factors (Zhu et al. 2016). Navitoclax-induced clearance of senescent cells in vivo has been found to improve ageing-related pathologies of haematopoietic and skeletal muscle stems cells in both irradiated and naturally aged mice (Chang et al. 2016). It has also been shown to improve atherosclerosis pathology (Childs et al. 2016). However, its use in humans is limited by side effects (Vogler et al. 2011).
Other notable senolytics include ABT-737, another inhibitor of the anti-apoptotic BCL-2 family members (Oltersdorf et al. 2005, Ritschka et al. 2017), EF24 (Li et al. 2019), azithromycin (Ozsvari et al. 2018) and HDAC inhibitors (Di Micco et al. 2011; Samaraweera et al. 2017). HDAC inhibitors are able to promote senescent cell clearance by stimulating chromatin relaxation. This reduces the formation of SAHF which dampen DDR signalling. With reduced SAHF presence the resulting increase in DDR signalling promotes apoptotic cell death (Di Micco et al. 2011) and has been corroborated by studies of the senolytic HDAC inhibitor Panobinostat (Samaraweera et al. 2017).
Senolytics have now been successfully used to alleviate disease pathology in mouse models of numerous diseases, reviewed in detail by Kirkland and Tchkonia (2020).
Rapamycin has also been identified as an effective senostatic and targets the mechanistic target of rapamycin complex 1 (mTORC1) (Brown et al. 1994; Sabers et al. 1995). This inhibition of mTORC1 interferes with signalling pathways required for cell growth and proliferation (Chung et al. 1992). Studies have demonstrated that rapamycin treatment can extend health span, activate autophagy and improve CI activity (Carames et al. 2012; Miwa et al. 2014). Other reported effects include the reduction of elevated mitochondrial mass, suppression of p21 and pro-tumorigenic SASP factor expression, and reduced prevalence of DNA damage foci (Herranz et al. 2015; Correia-Melo et al. 2016).
In vivo studies have shown that rapamycin treatment reduces IL-1β expression and alleviates cartilage degradation in experimental models of osteoarthritis (Carames et al. 2012). In rat models of type 2 diabetes, rapamycin promotes insulin sensitivity and reduces inflammation (Zhou and Ye 2018). Furthermore, rapamycin has shown success in preventing Alzheimer’s disease pathology by protecting against tau-induced neurodegeneration and neuroinflammation (Siman et al. 2015) and improved cognitive and cerebrovascular function in apolipoprotein E ε4 transgenic mice (Lin et al. 2017).
Antisense oligonucleotides are also a new line of investigation in the field of senostatics, aimed at inhibiting DDR signalling. This has proven useful in mouse models of accelerated ageing, which have shown that telomeric ASOs effectively alleviated a number of senescent markers, improved tissue function and extended mouse lifespan (Aguado et al. 2019).
Metformin is a widely used anti-diabetic drug, but has more recently shown promise in the treatment of other age-related diseases. Treatment with metformin has been reported to extend both lifespan and health span in in vivo studies in mice and worms (Anisimov et al. 2008; De Haes et al. 2014). Studies investigating the repurposing of metformin now cover a wide range of age-related diseases including cancer (Heckman-Stoddard et al. 2017), cardiovascular disease (Han et al. 2019) and neurodegenerative diseases (Kickstein et al. 2010; Mor et al. 2020). Chronic treatment with low dose metformin is able to inhibit age-associated atherosclerotic plaque formation in ApoE−/− mice (Karnewar et al. 2018). In models of myocardial infarction, metformin treatment has been shown to protect against hypertrophic and apoptotic remodelling in vivo, while in vitro it has demonstrated reduced hypertrophic and apoptotic responses to stress (Loi et al. 2019). Notably, studies have reported that metformin treatment can improve neuronal insulin signalling in a model of Alzheimer’s disease, though this was accompanied by an increase in Aβ levels (Zhang et al. 2015). In cancer cell models, metformin inhibits cell proliferation via the ATM-AMPK-p53/p21CIP1 pathway, and promotes apoptotic cell death following irradiation (Storozhuk et al. 2013).
More recently, metformin has shown promise as a senostatic treatment in a wide range of models, with the added benefit of a known safety profile in humans (Anisimov et al. 2008; Algire et al. 2012; Chen et al. 2016; Hu et al. 2020; Jiang et al. 2020; Hansel et al. 2021; Le Pelletier et al. 2021; Fielder et al. 2022).
Clinical Evidence
Evidence of beneficial effects of senolytic treatment is now being reported in humans. In the case of D + Q, clinical trials have shown that patients with idiopathic pulmonary fibrosis experienced improved physical function after 3 weeks of treatment (Justice et al. 2019). In another study of diabetic kidney disease, patients had reduced senescent cell burden and circulating pro-inflammatory cytokines within 11 days, along with decreased p16, p21 and SA-β-Gal activity (Hickson et al. 2019).
The use of mTOR inhibitors clinically is limited by known side effects (Pallet and Legendre 2013; Duran et al. 2014), though some trials have found success at low doses (Mannick et al. 2014). For example, mouse model studies have found that while acute treatment with rapamycin improves insulin sensitivity (Tremblay and Marette 2001; Krebs et al. 2007), longer treatment periods worsen hyperglycemia (Fraenkel et al. 2008) and glucose intolerance (Chang et al. 2009) in diabetes. It is also important to note that disruption of mTORC2 can exacerbate insulin resistance (Lamming et al. 2012). Rapamycin treatment has, however, been approved as a method of immunosuppression following the identification of a major role for mTOR in regulating SASP and tumour progression (Herranz et al. 2015; Thapa et al. 2017). However, there is debate as to the risk-benefit value of rapamycin treatment in some circumstances (Knoll et al. 2014; Hahn et al. 2019).
Metformin is a popular avenue of investigation for clinical senostatic use, given its well-known safety profile and current use in humans as a diabetic treatment. Clinical trials of metformin as an ageing-related therapy have found that the expression of DNA repair genes is regulated by metformin in muscle biopsies of older adults (Kulkarni et al. 2018). Trials in cancer therapeutics have also reported that metformin reduces serum markers of breast cancer risk in women who have undergone chemotherapy and radiotherapy treatment courses (Goodwin et al. 2008; Campagnoli et al. 2012). Review studies have also determined that coronary artery disease patients experience reduced cardiovascular mortality, cardiovascular events and all-cause mortality when given metformin (Han et al. 2019).
Future Directions
How to Better Target Senescent Cells?
While a number of senolytic and senostatic treatments have shown promise in clinical trials, and have even been approved for the treatment of some age-related diseases, many are limited by side effect profiles and require much optimisation of dosage and treatment timings. Moreover, it has not yet been confirmed that long-term effects following senolytic treatment do not become toxic or detrimental to the organism. There is also little data on any differing effects of senolytic treatment when given in old age with an assumed greater presence of senescent cells, or potentially toxicity in young age, for example treating particular conditions associated with senescent cells. More study is required to truly appreciate the safety and effectiveness of senolytic and senostatic drug treatments in different cell types, senescent triggers, and treatment regimens.
Nevertheless, novel approaches to senescent-targeted treatments are being reported regularly. The use of senescent-specific fluorescent tracers are becoming an interesting method of tracking senescent cell burden and therapeutic response (Wang et al. 2019), improving the ability to observe and amend the effects of senolytic or senostatic treatment in real time, at least in pre-clinical studies. A number of new studies are targeting specific aspects of the senescent phenotype to prevent or reduce the impact of senescent cell accumulation. For example, Bernardes de Jesus and colleagues have shown that viral delivery of telomerase-encoding gene (Tert) to prevent critical telomere shortening alleviates a number of other senescent markers and promotes lifespan extension in mice (Bernardes de Jesus et al. 2012). Another approach targets the characteristic increase in SA-β-Gal activity of senescence by coating fluorophores or cytotoxic chemicals with galacto-oligosaccharides to direct the delivering nanoparticles to cells with higher SA-β-Gal activity (Gonzalez-Gualda et al. 2020).
Other novel methods of targeting senescent cells concentrate on improving immune-mediated cell clearance. Chimeric antigen receptor T (CAR T) cell therapy is an exciting route of investigation, and studies have recently confirmed that this strategy can relieve a number of senescence-associated disorders (Amor et al. 2020). An important consideration in these studies, however, is the requirement of specific marker recognition which may vary between cell type or senescent stimuli. Another immune-mediated strategy is to target the decoy receptor 2 (DCR2) which is heavily expressed by senescent cells and can be targeted by natural killer (NK) cells via perforin-mediated granule exocytosis. The removal of this receptor subsequently allows the death receptors 4 and 5 (DR4/5) to be targeted by cytotoxic cells for degradation (Sagiv and Krizhanovsky 2013). Furthermore, consistent senolysis such as by immunotherapy-based strategies might become problematic for example, when senescent cells are required for wound repair.
Important Considerations in Senolytics Treatments
A number of key considerations are emerging from the literature on senolytic treatments. Primarily, many senolytic drugs require precise optimisation of dosage, treatment timing and duration in order to avoid toxic side effects. More work is needed in order to identify the minimum effective intervention for therapies which result in senescent cell clearance, particularly when considering the function of the targeted tissue. This is another important consideration to be aware of, as tissue such as muscle, liver or kidney which have major functional roles in human development and function may experience greater detrimental effect following senescent cell clearance if the senescent burden is significant. This should be taken into account when administering senolytics for particular disorders. For example, some mouse studies have demonstrated potential toxicity of senolysis in the liver and perivascular tissue resulting from the removal of senescent endothelial cells, adipocytes and macrophages, impacting dramatically on tissue function (Grosse et al. 2020).
It should also be noted that senolytic efficacy can vary between cell type, particularly if effective treatment relies on the recognition of specific cell markers. Similarly, not all senescent phenotypes are created equal, and there is much heterogeneity between phenotypes induced by different stimuli. In order to obtain a more universally effective treatment, targets would have to be identified that are common to the most forms of senescence. Alternatively, senescent treatment would require stimuli-specific phenotype targets for individual sub-types, such as MiDAS, replicative, OIS, or stress-induced premature senescence (SIPS).
Additionally, a factor to be aware of when deciding between senolytic versus senostatic treatment strategies is the potential benefits of “one time” senolytic treatment compared to continuous administration of senostatic treatment. Though senolytics may indeed require repeated doses, this is likely to be far less than the continuous inhibition of SASP and other senescent markers.
Concluding Remarks
Senescence is emerging as a more dynamic and heterogenous phenotype than first assumed, which must be considered when designing treatments targeted at the clearance or modification of senescent cells. As presented here, there are a number of distinct senescent sub-types with differing stimuli and phenotypic presentations. Though there are many overlapping similarities between these phenotypes, their unique features and regulators should not be discounted. Moreover, it is becoming clear that cellular senescence has cell type-specific roles throughout human development, as well as cell type-specific presentations, and thus their removal may not have universally beneficial effects. It will be important in future work to address these specificities of the senescent phenotype, and consider the many variables that may influence treatment efficacy. Development of methodologies to non-invasively assess senescent cell abundance in vivo will also be necessary. Importantly, senolysis in humans has not yet been proven safe or effective, and long-term effects have not been assessed in in vivo models. Much has now been revealed of the heterogeneous nature of cellular senescence, but there is much still to be discovered. The next stages of investigation may reveal a path to both a more detailed picture of individual senescent phenotypes and a more tailored approach to senolytic and senostatic treatment.
References
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M et al (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15(8):978–990
Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133(6):1006–1018
Aguado J, Sola-Carvajal A, Cancila V, Revechon G, Ong PF, Jones-Weinert CW, Wallen Arzt E, Lattanzi G, Dreesen O, Tripodo C, Rossiello F, Eriksson M, d'Adda di Fagagna F (2019) Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson-Gilford progeria syndrome. Nat Commun 10(1):4990
Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, Le L, Alicea G, Yang TL, Johnson FB, Noma KI, Zhang R (2016) HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol 215(3):325–334
Algire C, Moiseeva O, Deschenes-Simard X, Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G, Pollak MN (2012) Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev Res (Phila) 5(4):536–543
Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, Kulick A, Houlihan S, Peerschke E, Friedman SL, Ponomarev V, Piersigilli A, Sadelain M, Lowe SW (2020) Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583(7814):127–132
Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D et al (2019) Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J 38(5)
Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Yurova MV, Kovalenko IG, Poroshina TE, Semenchenko AV (2008) Metformin slows down aging and extends life span of female SHR mice. Cell Cycle 7(17):2769–2773
Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, Haupt Y, Hannan RD, Pearson RB (2012) AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene 31(15):1949–1962
Bae NS, Baumann P (2007) A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol Cell 26(3):323–334
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530(7589):184–189
Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 36(7):744–749
Baker DJ, Jin F, van Deursen JM (2008a) The yin and yang of the Cdkn2a locus in senescence and aging. Cell Cycle 7(18):2795–2802
Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederlander NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, Eberhardt NL, Terzic A, van Deursen JM (2008b) Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol 10(7):825–836
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479(7372):232–236
Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC et al (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444(7119):633–637
Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS (2006) The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res 66(2):794–802
Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J (2003) Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22(16):4212–4222
Benetos A, Kark JD, Susser E, Kimura M, Sinnreich R, Chen W, Steenstrup T, Christensen K, Herbig U, von Bornemann Hjelmborg J, Srinivasan SR, Berenson GS, Labat C, Aviv A (2013) Tracking and fixed ranking of leukocyte telomere length across the adult life course. Aging Cell 12(4):615–621
Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, Blasco MA (2012) Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med 4(8):691–704
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C (2012) Astrocyte senescence as a component of Alzheimer's disease. PLoS One 7(9):e45069
Birch J, Anderson RK, Correia-Melo C, Jurk D, Hewitt G, Marques FM, Green NJ, Moisey E, Birrell MA, Belvisi MG, Black F, Taylor JJ, Fisher AJ, De Soyza A, Passos JF (2015) DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 309(10):L1124–L1137
Bird TG, Muller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, Lu WY, Jamieson T, Govaere O, Campbell AD et al (2018) TGFbeta inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med 10(454)
Blackburn EH (1991) Structure and function of telomeres. Nature 350(6319):569–573
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279(5349):349–352
Boumendil C, Hari P, Olsen KCF, Acosta JC, Bickmore WA (2019) Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev 33(3–4):144–149
Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369(6483):756–758
Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276(45):42462–42467
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ (2018) Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562(7728):578–582
Campagnoli C, Pasanisi P, Abba C, Ambroggio S, Biglia N, Brucato T, Colombero R, Danese S, Donadio M, Venturelli E, Zito G, Berrino F (2012) Effect of different doses of metformin on serum testosterone and insulin in non-diabetic women with breast cancer: a randomized study. Clin Breast Cancer 12(3):175–182
Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M (2012) Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis 71(4):575–581
Chang GR, Wu YY, Chiu YS, Chen WY, Liao JW, Hsu HM, Chao TH, Hung SW, Mao FC (2009) Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol 105(3):188–198
Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18(8):495–506
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22(1):78–83
Chen D, Xia D, Pan Z, Xu D, Zhou Y, Wu Y, Cai N, Tang Q, Wang C, Yan M, Zhang JJ, Zhou K, Wang Q, Feng Y, Wang X, Xu H, Zhang X, Tian N (2016) Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis 7(10):e2441
Chen K, Shen W, Zhang Z, Xiong F, Ouyang Q, Luo C (2020) Age-dependent decline in stress response capacity revealed by proteins dynamics analysis. Sci Rep 10(1):15211
Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP (2005) Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436(7051):725–730
Chiche A, Le Roux I, von Joest M, Sakai H, Aguin SB, Cazin C, Salam R, Fiette L, Alegria O, Flamant P, Tajbakhsh S, Li H (2017) Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell 20(3):407–414 e4
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW (2011) Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25(20):2125–2136
Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM (2016) Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354(6311):472–477
Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A, Leri A, Maseri A, Anversa P (2003) Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res 93(7):604–613
Chung J, Kuo CJ, Crabtree GR, Blenis J (1992) Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69(7):1227–1236
Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40(2):179–204
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6(12):2853–2868
Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J (2011) Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 286(42):36396–36403
Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A et al (2016) Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35(7):724–742
d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426(6963):194–198
d'Adda di Fagagna F, Teo SH, Jackson SP (2004) Functional links between telomeres and proteins of the DNA-damage response. Genes Dev 18(15):1781–1799
D'Mello MJ, Ross SA, Briel M, Anand SS, Gerstein H, Pare G (2015) Association between shortened leukocyte telomere length and cardiometabolic outcomes: systematic review and meta-analysis. Circ Cardiovasc Genet 8(1):82–90
Da Silva-Alvarez S, Guerra-Varela J, Sobrido-Camean D, Quelle A, Barreiro-Iglesias A, Sanchez L, Collado M (2020) Cell senescence contributes to tissue regeneration in zebrafish. Aging Cell 19(1):e13052
da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, Ishaq A, Saretzki G, Nagaraja-Grellscheid S, Nelson G, von Zglinicki T (2019) The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 18(1):e12848
Dalle Pezze P, Nelson G, Otten EG, Korolchuk VI, Kirkwood TB, von Zglinicki T, Shanley DP (2014) Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLoS Comput Biol 10(8):e1003728
Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J (2013) p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201(4):613–629
De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, Braeckman BP, Schoofs L, Temmerman L (2014) Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci U S A 111(24):E2501–E2509
Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM et al (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7(2):165–176
Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31(6):722–733
Di Leonardo A, Linke SP, Clarkin K, Wahl GM (1994) DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 8(21):2540–2551
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d'Adda di Fagagna F (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444(7119):638–642
Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d'Ario G, Montani E, Mercurio C, Hahn WC, Gorgoulis V, Minucci S, d'Adda di Fagagna F (2011) Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 13(3):292–302
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92(20):9363–9367
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z et al (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550(7676):402–406
Dupre A, Boyer-Chatenet L, Gautier J (2006) Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat Struct Mol Biol 13(5):451–457
Duran I, Goebell PJ, Papazisis K, Ravaud A, Weichhart T, Rodriguez-Portal JA, Budde K (2014) Drug-induced pneumonitis in cancer patients treated with mTOR inhibitors: management and insights into possible mechanisms. Expert Opin Drug Saf 13(3):361–372
Dutto I, Tillhon M, Cazzalini O, Stivala LA, Prosperi E (2015) Biology of the cell cycle inhibitor p21(CDKN1A): molecular mechanisms and relevance in chemical toxicology. Arch Toxicol 89(2):155–178
Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, Heikenwalder M, Wang XW, Zender L, Greten TF (2016) Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30(4):533–547
Eliezer Y, Argaman L, Rhie A, Doherty AJ, Goldberg M (2009) The direct interaction between 53BP1 and MDC1 is required for the recruitment of 53BP1 to sites of damage. J Biol Chem 284(1):426–435
Fabbri E, An Y, Zoli M, Simonsick EM, Guralnik JM, Bandinelli S, Boyd CM, Ferrucci L (2015) Aging and the burden of multimorbidity: associations with inflammatory and anabolic hormonal biomarkers. J Gerontol A Biol Sci Med Sci 70(1):63–70
Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S (2017) Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23(9):1072–1079
Fielder E, Wan T, Alimohammadiha G, Ishaq A, Low E, Weigand BM, Kelly G, Parker C, Griffin B, Jurk D, Korolchuk VI, von Zglinicki T, Miwa S (2022) Short senolytic or senostatic interventions rescue progression of radiation-induced frailty and premature ageing in mice. elife 11. https://doi.org/10.7554/eLife.75492
Fitzner B, Muller S, Walther M, Fischer M, Engelmann R, Muller-Hilke B, Putzer BM, Kreutzer M, Nizze H, Jaster R (2012) Senescence determines the fate of activated rat pancreatic stellate cells. J Cell Mol Med 16(11):2620–2630
Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, Leibowitz G (2008) mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 57(4):945–957
Fragkiadaki P, Nikitovic D, Kalliantasi K, Sarandi E, Thanasoula M, Stivaktakis PD, Nepka C, Spandidos DA, Tosounidis T, Tsatsakis A (2020) Telomere length and telomerase activity in osteoporosis and osteoarthritis. Exp Ther Med 19(3):1626–1632
Freund A, Patil CK, Campisi J (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30(8):1536–1548
Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, Herbig U, Longhese MP, d'Adda di Fagagna F (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14(4):355–365
Fumagalli M, Rossiello F, Mondello C, d'Adda di Fagagna F (2014) Stable cellular senescence is associated with persistent DDR activation. PLoS One 9(10):e110969
Gal H, Lysenko M, Stroganov S, Vadai E, Youssef SA, Tzadikevitch-Geffen K, Rotkopf R, Biron-Shental T, de Bruin A, Neeman M, Krizhanovsky V (2019) Molecular pathways of senescence regulate placental structure and function. EMBO J 38(18):e100849
Galbiati A, Beausejour C, d'Adda di Fagagna F (2017) A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell 16(2):422–427
Georgakilas AG, Martin OA, Bonner WM (2017) p21: a two-faced genome Guardian. Trends Mol Med 23(4):310–319
Gnani D, Crippa S, Della Volpe L, Rossella V, Conti A, Lettera E, Rivis S, Ometti M, Fraschini G, Bernardo ME, Di Micco R (2019) An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell 18(3):e12933
Gonzalez-Gualda E, Paez-Ribes M, Lozano-Torres B, Macias D, Wilson JR 3rd, Gonzalez-Lopez C, Ou HL, Miron-Barroso S, Zhang Z, Lerida-Viso A et al (2020) Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19(4):e13142
Goodwin PJ, Pritchard KI, Ennis M, Clemons M, Graham M, Fantus IG (2008) Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer 8(6):501–505
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G et al (2019) Cellular senescence: defining a path forward. Cell 179(4):813–827
Gray-Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel-Malek ZA, Marais R, Wynford-Thomas D, Bennett DC (2006) Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer 95(4):496–505
Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, Bulavin DV (2020) Defined p16(high) senescent cell types are indispensable for mouse Healthspan. Cell Metab 32(1):87–99 e6
Gustafson B, Nerstedt A, Smith U (2019) Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat Commun 10(1):2757
Hahn D, Hodson EM, Hamiwka LA, Lee VW, Chapman JR, Craig JC, Webster AC (2019) Target of rapamycin inhibitors (TOR-I; sirolimus and everolimus) for primary immunosuppression in kidney transplant recipients. Cochrane Database Syst Rev 12:CD004290
Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319(5868):1352–1355
Hampel B, Malisan F, Niederegger H, Testi R, Jansen-Durr P (2004) Differential regulation of apoptotic cell death in senescent human cells. Exp Gerontol 39(11–12):1713–1721
Han Y, Xie H, Liu Y, Gao P, Yang X, Shen Z (2019) Effect of metformin on all-cause and cardiovascular mortality in patients with coronary artery diseases: a systematic review and an updated meta-analysis. Cardiovasc Diabetol 18(1):96
Hansel C, Barr S, Schemann AV, Lauber K, Hess J, Unger K, Zitzelsberger H, Jendrossek V, Klein D (2021) Metformin protects against radiation-induced acute effects by limiting senescence of bronchial-epithelial cells. Int J Mol Sci 22(13)
Hanzelmann S, Beier F, Gusmao EG, Koch CM, Hummel S, Charapitsa I, Joussen S, Benes V, Brummendorf TH, Reid G, Costa IG, Wagner W (2015) Replicative senescence is associated with nuclear reorganization and with DNA methylation at specific transcription factor binding sites. Clin Epigenetics 7:19
Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345(6274):458–460
Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37:614–636
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621
Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe VV, Ford LG (2017) Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 60(9):1639–1647
Helman A, Klochendler A, Azazmeh N et al (2016) p16Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med 22:412–420. https://doi.org/10.1038/nm.4054
Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A et al (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17(9):1205–1217
Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3:708
Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL et al (2019) Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 47:446–456
Holdt LM, Sass K, Gabel G, Bergert H, Thiery J, Teupser D (2011) Expression of Chr9p21 genes CDKN2B (p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human atherosclerotic plaque. Atherosclerosis 214(2):264–270
Hu Q, Peng J, Jiang L, Li W, Su Q, Zhang J, Li H, Song M, Cheng B, Xia J, Wu T (2020) Metformin as a senostatic drug enhances the anticancer efficacy of CDK4/6 inhibitor in head and neck squamous cell carcinoma. Cell Death Dis 11(10):925
Huang Z, Zhong Z, Zhang L, Wang X, Xu R, Zhu L, Wang Z, Hu S, Zhao X (2015) Down-regulation of HMGB1 expression by shRNA constructs inhibits the bioactivity of urothelial carcinoma cell lines via the NF-kappaB pathway. Sci Rep 5:12807
Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, Wu H, Berger SL, Adams PD (2013) Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202(1):129–143
Jackson JG, Pereira-Smith OM (2006) p53 is preferentially recruited to the promoters of growth arrest genes p21 and GADD45 during replicative senescence of normal human fibroblasts. Cancer Res 66(17):8356–8360
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461(7267):1071–1078
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT (2006) Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443(7110):421–426
Jeck WR, Siebold AP, Sharpless NE (2012) Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell 11(5):727–731
Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, Elisseeff JH (2017) Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23(6):775–781
Jiang X, Ruan XL, Xue YX, Yang S, Shi M, Wang LN (2020) Metformin reduces the senescence of renal tubular epithelial cells in diabetic nephropathy via the MBNL1/miR-130a-3p/STAT3 pathway. Oxidative Med Cell Longev 2020:8708236
Jun JI, Lau LF (2010) Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2(9):627–631
Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron K, von Zglinicki T (2012) Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11(6):996–1004
Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R, Hewitt G, Pender SL, Fullard N, Nelson G, Mann J, van de Sluis B, Mann DA, von Zglinicki T (2014) Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun 2:4172
Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, Kirkland JL (2019) Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40:554–563
Karnewar S, Neeli PK, Panuganti D, Kotagiri S, Mallappa S, Jain N, Jerald MK, Kotamraju S (2018) Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: relevance in age-associated vascular dysfunction. Biochim Biophys Acta Mol basis Dis 1864(4 Pt A):1115–1128
Katlinskaya YV, Katlinski KV, Yu Q, Ortiz A, Beiting DP, Brice A, Davar D, Sanders C, Kirkwood JM, Rui H, Xu X, Koumenis C, Diehl JA, Fuchs SY (2016) Suppression of type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep 15(1):171–180
Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Kohler A, Glossmann H, Schneider R, Sutherland C, Schweiger S (2010) Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A 107(50):21830–21835
Kirkland JL, Tchkonia T (2020) Senolytic drugs: from discovery to translation. J Intern Med 288(5):518–536
Kirschner K, Samarajiwa SA, Cairns JM, Menon S, Perez-Mancera PA, Tomimatsu K, Bermejo-Rodriguez C, Ito Y, Chandra T, Narita M, Lyons SK, Lynch AG, Kimura H, Ohbayashi T, Tavare S, Narita M (2015) Phenotype specific analyses reveal distinct regulatory mechanism for chronically activated p53. PLoS Genet 11(3):e1005053
Knoll GA, Kokolo MB, Mallick R, Beck A, Buenaventura CD, Ducharme R, Barsoum R, Bernasconi C, Blydt-Hansen TD, Ekberg H et al (2014) Effect of sirolimus on malignancy and survival after kidney transplantation: systematic review and meta-analysis of individual patient data. BMJ 349:g6679
Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Furnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M (2007) The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes 56(6):1600–1607
Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443(7110):453–457
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134(4):657–667
Kulkarni AS, Brutsaert EF, Anghel V, Zhang K, Bloomgarden N, Pollak M, Mar JC, Hawkins M, Crandall JP, Barzilai N (2018) Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell 17(2)
Kurz DJ, Decary S, Hong Y, Erusalimsky JD (2000) Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113(Pt 20):3613–3622
Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335(6076):1638–1643
Lau L, Porciuncula A, Yu A, Iwakura Y, David G (2019) Uncoupling the senescence-associated secretory phenotype from cell cycle exit via Interleukin-1 inactivation unveils its Protumorigenic role. Mol Cell Biol 39(12)
Lavin MF, Kozlov S, Gatei M, Kijas AW (2015) ATM-dependent phosphorylation of all three members of the MRN complex: from sensor to adaptor. Biomol Ther 5(4):2877–2902
Lawless C, Wang C, Jurk D, Merz A, Zglinicki T, Passos JF (2010) Quantitative assessment of markers for cell senescence. Exp Gerontol 45(10):772–778
Lazzerini Denchi E, Attwooll C, Pasini D, Helin K (2005) Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol 25(7):2660–2672
Le ON, Rodier F, Fontaine F, Coppe JP, Campisi J, DeGregori J, Laverdiere C, Kokta V, Haddad E, Beausejour CM (2010) Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 9(3):398–409
Le Pelletier L, Mantecon M, Gorwood J, Auclair M, Foresti R, Motterlini R, Laforge M, Atlan M, Feve B, Capeau J, Lagathu C, Bereziat V (2021) Metformin alleviates stress-induced cellular senescence of aging human adipose stromal cells and the ensuing adipocyte dysfunction. elife 10
Li W, He Y, Zhang R, Zheng G, Zhou D (2019) The curcumin analog EF24 is a novel senolytic agent. Aging (Albany NY) 11(2):771–782
Lin AL, Jahrling JB, Zhang W, DeRosa N, Bakshi V, Romero P, Galvan V, Richardson A (2017) Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer's disease. J Cereb Blood Flow Metab 37(1):217–226
Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, Ibrahim JG, Thomas NE, Sharpless NE (2009) INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One 4(4):e5027
Livshits G, Zhai G, Hart DJ, Kato BS, Wang H, Williams FM, Spector TD (2009) Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: the Chingford study. Arthritis Rheum 60(7):2037–2045
Loeser RF (2009) Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr Cartil 17(8):971–979
Loi H, Boal F, Tronchere H, Cinato M, Kramar S, Oleshchuk O, Korda M, Kunduzova O (2019) Metformin protects the heart against hypertrophic and apoptotic remodeling after myocardial infarction. Front Pharmacol 10:154
Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science 288(5470):1425–1429
Malaise O, Tachikart Y, Constantinides M, Mumme M, Ferreira-Lopez R, Noack S, Krettek C, Noel D, Wang J, Jorgensen C, Brondello JM (2019) Mesenchymal stem cell senescence alleviates their intrinsic and Seno-suppressive paracrine properties contributing to osteoarthritis development. Aging (Albany NY) 11(20):9128–9146
Mallette FA, Ferbeyre G (2007) The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle 6(15):1831–1836
Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (2014) mTOR inhibition improves immune function in the elderly. Sci Transl Med 6(268):268ra179
Manoy P, Yuktanandana P, Tanavalee A, Tanpowpong T, Ittipanichpong T, Honsawek S (2020) Telomere shortening is associated with poor physical performance in knee osteoarthritis. Biomed Rep 13(4):27
Mathon NF, Malcolm DS, Harrisingh MC, Cheng L, Lloyd AC (2001) Lack of replicative senescence in normal rodent glia. Science 291(5505):872–875
Matias I, Diniz LP, Damico IV, Araujo APB, Neves LDS, Vargas G, Leite REP, Suemoto CK, Nitrini R, Jacob-Filho W, Grinberg LT, Hol EM, Middeldorp J, Gomes FCA (2022) Loss of Lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 21(1):e13521
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436(7051):720–724
Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I (2009) A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 15(9):1082–1087
Mirzayans R, Andrais B, Hansen G, Murray D (2012) Role of p16(INK4A) in replicative senescence and DNA damage-induced premature senescence in p53-deficient human cells. Biochem Res Int 2012:951574
Mitsui Y, Schneider EL (1976) Increased nuclear sizes in senescent human diploid fibroblast cultures. Exp Cell Res 100(1):147–152
Miwa S, Jow H, Baty K, Johnson A, Czapiewski R, Saretzki G, Treumann A, von Zglinicki T (2014) Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun 5:3837
Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G (2006) DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell 17(4):1583–1592
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443(7110):448–452
Mor DE, Sohrabi S, Kaletsky R, Keyes W, Tartici A, Kalia V, Miller GW, Murphy CT (2020) Metformin rescues Parkinson's disease phenotypes caused by hyperactive mitochondria. Proc Natl Acad Sci U S A 117(42):26438–26447
Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW (1999) Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet 21(1):115–118
Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M (2013) Programmed cell senescence during mammalian embryonic development. Cell 155(5):1104–1118
Nakamura AJ, Rao VA, Pommier Y, Bonner WM (2010) The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle 9(2):389–397
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113(6):703–716
Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T (2018) The senescent bystander effect is caused by ROS-activated NF-kappaB signalling. Mech Ageing Dev 170:30–36
Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T (2012) A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11(2):345–349
Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, Miettinen TP, Vaites LP, Soares LM, Paulo JA, Harper JW, Buratowski S, Manalis S, van Werven FJ, Holt LJ, Amon A (2019) Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell 176(5):1083–1097 e18
Novais EJ, Tran VA, Johnston SN, Darris KR, Roupas AJ, Sessions GA, Shapiro IM, Diekman BO, Risbud MV (2021) Long-term treatment with senolytic drugs Dasatinib and quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat Commun 12(1):5213
Ock S, Lee WS, Ahn J, Kim HM, Kang H, Kim HS, Jo D, Abel ED, Lee TJ, Kim J (2016) Deletion of IGF-1 receptors in Cardiomyocytes attenuates cardiac aging in male mice. Endocrinology 157(1):336–345
Ogrodnik M, Evans SA, Fielder E, Victorelli S, Kruger P, Salmonowicz H, Weigand BM, Patel AD, Pirtskhalava T, Inman CL et al (2021) Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 20(2):e13296
Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, Gorgoulis VG, d'Adda di Fagagna F (2014) Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ 21(6):998–1012
Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B, Hofman P, Rocchi S, Peyron JF, Lacour JP, Ballotti R, Bertolotto C (2011) Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev 25(12):1245–1261
Oltersdorf T, Elmore S, Shoemaker A et al (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435:677–681. https://doi.org/10.1038/nature03579
Ortiz-Montero P, Londono-Vallejo A, Vernot JP (2017) Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal 15(1):17
Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA, Bourdel G, Popovic N, Rezende FA, Kaufman RJ, Mallette FA, Sapieha P (2016) Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med 8(362):362ra144
Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira A, Windheim J, Tsoory M, Schirmbeck R, Amit I, Geiger H, Krizhanovsky V (2018) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9(1):5435
Ozsvari B, Nuttall JR, Sotgia F, Lisanti MP (2018) Azithromycin and Roxithromycin define a new family of "senolytic" drugs that target senescent human fibroblasts. Aging (Albany NY) 10(11):3294–3307
Pallet N, Legendre C (2013) Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf 12(2):177–186
Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5(8):741–747
Patil P, Dong Q, Wang D, Chang J, Wiley C, Demaria M, Lee J, Kang J, Niedernhofer LJ, Robbins PD, Sowa G, Campisi J, Zhou D, Vo N (2019) Systemic clearance of p16(INK4a) -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 18(3):e12927
Peilin W, Songsong T, Chengyu Z, Zhi C, Chunhui M, Yinxian Y, Lei Z, Min M, Zongyi W, Mengkai Y, Jing X, Tao Z, Zhuoying W, Fei Y, Chengqing Y (2019) Directed elimination of senescent cells attenuates development of osteoarthritis by inhibition of c-IAP and XIAP. Biochim Biophys Acta Mol basis Dis 1865(10):2618–2632
Prattichizzo F, De Nigris V, Spiga R, Mancuso E, La Sala L, Antonicelli R, Testa R, Procopio AD, Olivieri F, Ceriello A (2018) Inflammageing and metaflammation: the yin and yang of type 2 diabetes. Ageing Res Rev 41:1–17
Rebbaa A, Zheng X, Chou PM, Mirkin BL (2003) Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene 22(18):2805–2811
Reichert S, Stier A (2017) Does oxidative stress shorten telomeres in vivo? A review. Biol Lett 13(12)
Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM (2017) The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31(2):172–183
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11(8):973–979
Roninson IB (2003) Tumor cell senescence in cancer treatment. Cancer Res 63(11):2705–2715
Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, Miller JD (2016) Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15(5):973–977
Rossiello F, Aguado J, Sepe S, Iannelli F, Nguyen Q, Pitchiaya S, Carninci P, d'Adda di Fagagna F (2017) DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat Commun 8:13980
Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, Chen CC, Ho YJ, Sanchez-Rivera FJ, Feucht J, Baslan T, Tian S, Chen HA, Romesser PB, Poirier JT, Rudin CM, de Stanchina E, Manchado E, Sherr CJ, Lowe SW (2018) NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362(6421):1416–1422
Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT (1995) Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 270(2):815–822
Sagiv A, Krizhanovsky V (2013) Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology 14(6):617–628
Samaraweera L, Adomako A, Rodriguez-Gabin A, McDaid HM (2017) A novel indication for Panobinostat as a Senolytic drug in NSCLC and HNSCC. Sci Rep 7(1):1900
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y et al (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532
Seluanov A, Gorbunova V, Falcovitz A, Sigal A, Milyavsky M, Zurer I, Shohat G, Goldfinger N, Rotter V (2001) Change of the death pathway in senescent human fibroblasts in response to DNA damage is caused by an inability to stabilize p53. Mol Cell Biol 21(5):1552–1564
Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA (1996) Role of the INK4a locus in tumor suppression and cell mortality. Cell 85(1):27–37
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88(5):593–602
Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ (2000) Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res 257(1):162–171
Shao L, Feng W, Li H, Gardner D, Luo Y, Wang Y, Liu L, Meng A, Sharpless NE, Zhou D (2014) Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood 123(20):3105–3115
Siman R, Cocca R, Dong Y (2015) The mTOR inhibitor rapamycin mitigates Perforant pathway neurodegeneration and synapse loss in a mouse model of early-stage Alzheimer-type Tauopathy. PLoS One 10(11):e0142340
Sousa-Victor P, Garcia-Prat L, Serrano AL, Perdiguero E, Munoz-Canoves P (2015) Muscle stem cell aging: regulation and rejuvenation. Trends Endocrinol Metab 26(6):287–296
Stein GH, Dulic V (1995) Origins of G1 arrest in senescent human fibroblasts. BioEssays 17(6):537–543
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155(5):1119–1130
Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G, Tsakiridis T (2013) Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer 108(10):2021–2032
Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, Watanabe S, Kawamoto S, Miyata K, Barber GN, Ohtani N, Hara E (2018) Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9(1):1249
Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13(17):1549–1556
Thangavel C, Dean JL, Ertel A, Knudsen KE, Aldaz CM, Witkiewicz AK, Clarke R, Knudsen ES (2011) Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer 18(3):333–345
Thapa RK, Nguyen HT, Jeong JH, Kim JR, Choi HG, Yong CS, Kim JO (2017) Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles. Sci Rep 7:43299
Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A (2019) Targeted elimination of senescent Beta cells prevents type 1 diabetes. Cell Metab 29(5):1045–1060 e10
Thornell LE, Lindstom M, Renault V, Klein A, Mouly V, Ansved T, Butler-Browne G, Furling D (2009) Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol Appl Neurobiol 35(6):603–613
Tremblay F, Marette A (2001) Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276(41):38052–38060
Turenne GA, Paul P, Laflair L, Price BD (2001) Activation of p53 transcriptional activity requires ATM's kinase domain and multiple N-terminal serine residues of p53. Oncogene 20(37):5100–5110
Victorelli S, Lagnado A, Halim J, Moore W, Talbot D, Barrett K, Chapman J, Birch J, Ogrodnik M, Meves A, Pawlikowski JS, Jurk D, Adams PD, van Heemst D, Beekman M, Slagboom PE, Gunn DA, Passos JF (2019) Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J 38(23):e101982
Vijayachandra K, Lee J, Glick AB (2003) Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res 63(13):3447–3452
Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, Perez-Garcia A, Kiourtis C, Dasgupta N, Lei X, Kruger PJ, Nixon C, Clark W, Jurk D, Bird TG, Passos JF, Berger SL, Dou Z, Adams PD (2020) Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev 34(5–6):428–445
Vogler M, Hamali HA, Sun XM, Bampton ET, Dinsdale D, Snowden RT, Dyer MJ, Goodall AH, Cohen GM (2011) BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 117(26):7145–7154
von Zglinicki T (2002) Oxidative stress shortens telomeres. Trends Biochem Sci 27(7):339–344
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T (2009) DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8(3):311–323
Wang Y, Liu J, Ma X, Cui C, Deenik PR, Henderson PKP, Sigler AL, Cui L (2019) Real-time imaging of senescence in tumors with DNA damage. Sci Rep 9(1):2102
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520(7548):553–557
Wang MJ, Chen F, Li JX, Liu CC, Zhang HB, Xia Y, Yu B, You P, Xiang D, Lu L, Yao H, Borjigin U, Yang GS, Wangensteen KJ, He ZY, Wang X, Hu YP (2014) Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology 60(1):349–361
Weyand CM, Goronzy JJ (2016) Aging of the immune system. Mechanisms and therapeutic targets. Ann Am Thorac Soc 13(Suppl 5):S422–S428
Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23(2):303–314
Woo RA, Poon RY (2004) Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev 18(11):1317–1330
Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T, Curran GL, Ogrodnik M, Jurk D, Johnson KO, Lowe V, Tchkonia T, Westendorf JJ, Kirkland JL (2017) Transplanted senescent cells induce an osteoarthritis-like condition in mice. J Gerontol A Biol Sci Med Sci 72(6):780–785
Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG et al (2018) Senolytics improve physical function and increase lifespan in old age. Nat Med 24(8):1246–1256
Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, Giorgadze N, Jensen MD, LeBrasseur NK, Kirkland JL (2015) JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A 112(46):E6301–E6310
Xu X, Shen X, Wang J, Feng W, Wang M, Miao X, Wu Q, Wu L, Wang X, Ma Y, Wu S, Bao X, Wang W, Wang Y, Huang Z (2021) YAP prevents premature senescence of astrocytes and cognitive decline of Alzheimer's disease through regulating CDK6 signaling. Aging Cell 20(9):e13465
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445(7128):656–660
Yang C, Jiao Y, Wei B, Yang Z, Wu JF, Jensen J, Jean WH, Huang CY, Kuo CH (2018) Aged cells in human skeletal muscle after resistance exercise. Aging (Albany NY) 10(6):1356–1365
Yang NC, Hu ML (2005) The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp Gerontol 40(10):813–819
Yoon KB, Park KR, Kim SY, Han SY (2016) Induction of nuclear enlargement and senescence by Sirtuin inhibitors in glioblastoma cells. Immune Netw 16(3):183–188
Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, Malka F, Jou MJ, Martinou JC, Yoon G (2006) Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol 209(2):468–480
Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, Ben-Porath I, Krizhanovsky V (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7:11190
Yun MH, Davaapil H, Brockes JP (2015) Recurrent turnover of senescent cells during regeneration of a complex structure. elife 4
Zhang H, Cohen SN (2004) Smurf2 up-regulation activates telomere-dependent senescence. Genes Dev 18(24):3028–3040
Zhang L, Zhang S, Maezawa I, Trushin S, Minhas P, Pinto M, Jin LW, Prasain K, Nguyen TD, Yamazaki Y, Kanekiyo T, Bu G, Gateno B, Chang KO, Nath KA, Nemutlu E, Dzeja P, Pang YP, Hua DH, Trushina E (2015) Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer's disease. EBioMedicine 2(4):294–305
Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe M, Mattson MP (2019) Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat Neurosci 22(5):719–728
Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD (2005) Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 8(1):19–30
Zhou W, Ye S (2018) Rapamycin improves insulin resistance and hepatic steatosis in type 2 diabetes rats through activation of autophagy. Cell Biol Int 42(10):1282–1291
Zhou X, Perez F, Han K, Jurivich DA (2006) Clonal senescence alters endothelial ICAM-1 function. Mech Ageing Dev 127(10):779–785
Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, Wren JD, Niedernhofer LJ, Robbins PD, Kirkland JL (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15(3):428–435
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M et al (2015) The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14(4):644–658
Zwetsloot KA, Childs TE, Gilpin LT, Booth FW (2013) Non-passaged muscle precursor cells from 32-month old rat skeletal muscle have delayed proliferation and differentiation. Cell Prolif 46(1):45–57
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Reed, R., Miwa, S. (2023). Cellular Senescence and Ageing. In: Harris, J.R., Korolchuk, V.I. (eds) Biochemistry and Cell Biology of Ageing: Part III Biomedical Science. Subcellular Biochemistry, vol 102. Springer, Cham. https://doi.org/10.1007/978-3-031-21410-3_7
Download citation
DOI: https://doi.org/10.1007/978-3-031-21410-3_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-21409-7
Online ISBN: 978-3-031-21410-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)