
Abstract
Ageing is a multifactorial process that affects almost every living organism. The fact that we have already uncovered many of the molecular pathways that determine the ageing phenotype in humans has allowed us to start studying strategies to slow down or prevent the deleterious effects of time in tissue physiology. Several ongoing lines of research are aimed at defining new therapies that could be used in regenerative medicine, based on the knowledge accumulated on the molecular physiology of ageing. Among those, the most promising focus on preventing the accumulation of old cells, also known as senescent cells. These have been shown to disrupt tissue physiology and to be responsible, at least in part, for many of the symptoms associated with ageing. Consistent with this, clearance of senescent cells results in prolonged lifespan and health span in mammals and thus could be a potent anti-ageing therapy. In this chapter, we will discuss these and other novel approaches for regenerative medicine being developed in the context of the current understanding of cellular ageing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Understanding Ageing
Finding a way to stop the damaging effects that ageing has on the human body has been a long-standing ambition for mankind. Numerous legends tell of searches for the fountains of youth and other mythical sources of immortality, but until the raise of modern biology there has been no real opportunity to interfere with the inevitable degradation that time imposes onto organisms. Over the past decades, we have acquired substantial information about the molecular physiology of ageing, but current interventions are limited at those of cosmetic nature. More research has to be conducted before the first true drug that modulates ageing reaches the market.
Despite the fact that we still lack a proper therapy that has a biological effect on the mechanisms involved in ageing, millions of dollars are currently spent annually on chemicals sold as anti-ageing drugs. This is mainly due to the fact that current laws in many countries allow compounds to be labelled as “supplements” or “cosmetics” instead of “medicines”, which would force them to undergo more severe assessments of their efficacy. This loophole has allowed this market to bloom and underscores the immense interest on these products at consumer level.
The key to designing interventions that would indeed slow down or revert the effects of ageing lies on our ability to characterize the molecular pathways involved in the changes that can be observed at the cellular level. Thus, carefully studying cell ageing (also known as senescence) is likely to provide the insights necessary to design the first strategies aimed at modifying organismal ageing. The combination of new genetic techniques and the recent advances in biochemistry are bringing us closer to understanding how the processes that contribute to the ageing phenotype are determined. The first consequence of these advances is that modulation of ageing in the lab is already possible. Fly, worms, mice and other animals that age faster than usual or that survive for more than the normal amount of time can be generated through chemical treatments and genetic manipulation. This is a proof of principle that ageing is not an irreversible and uncontrollable mechanism, as once thought, and that, like every biological process, it can be subjected to manipulation once it has been properly characterized.
Our knowledge of the ageing process in humans is still far from complete. Nevertheless, it has already been hypothesized that our lifespan and, more importantly, health span could be extended by a chemical intervention that interrupted the signalling pathways that determine senescence, as we will discuss later. The limits of such interventions and the impact that they may have in society are still being debated.
Ageing Is a Result of the Cellular Responses to Damage
Different theories have been proposed over the years to explain the molecular basis of cell ageing. These include the accumulation of toxic residues inside and outside the cells, the shortening of telomeres or the accumulation of damage in the mitochondria that limits the amount of energy they can produce [1]. All of them relate in one way or another to the chronic induction of damage signalling pathways that push the cell towards the process known as senescence. This, which could also enhance the progressive loss of potency of the adult stem cell niche involved in regenerative processes, could explain the impact of time on tissue physiology. Thus, ageing could be seen as an excessive accumulation of damaged cells that adopt a senescent (or “old”) phenotype. It has also been proposed that the side effects of certain processes necessary for organismal survival can accelerate the processes that trigger ageing. For instance, the mechanisms that protect cells against cancer can induce senescence as well and contribute to this accumulation [2].
Within the framework of the “ageing as a result of damage” hypothesis, it is important to consider that the oxygen needed to sustain life causes important disruption to several of the components of cells, which then contributes to the progressive deterioration that will eventually lead to cell senescence. This is due to the fact that the breakdown products of oxygen, known as reactive oxygen species (ROS), produce small but measurable damage to the DNA and other macromolecules [3]. The steady accumulation of these lesions has indeed been shown to trigger cellular ageing [4]. This forms the central core of the classic oxidative theory of ageing, which is now part of a wider framework that aims to explain all the changes involved in the phenotypical changes observed in ageing [1]. Consistent with oxidation not being the sole cause of cellular senescence, it has been observed that antioxidants have only limited effects on the ageing of organisms, while they can actually increase other pathologies [5].
In the following pages, we will summarize our understanding of the main factors currently known to be involved in the molecular physiology of cellular ageing, and based on this, we will explore the interventions that could be part of the regenerative medicine tools in the future, mostly by preventing a build-up of senescent cells.
The Physiological Importance of Senescence
Ageing Cells as a Way to Prevent Cancer
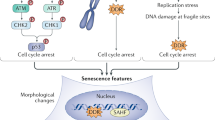
Senescence is a well-known cellular mechanism with a critical role not only in ageing but also in cancer, as a tumour suppressor mechanism [6]. Senescence is usually defined as a permanent cell cycle arrest in which cells remain metabolically active and adopt characteristic phenotypic changes [7]. Senescent cells appear multinucleated, large and extended, and exhibit spindle and vacuolization features [8]. The onset of this phenotype is believed to be triggered by different types of damage, either as a result of telomere shortening after a number of cell divisions (replicative senescence, related to the actual age of the cell, as determined by the length of the telomeres) or as a response to a range of stress stimuli (stress-induced premature senescence, SIPS) [8, 9]. Expression of oncogenes, such as Ras, cyclin E, E2F3 and Raf can also trigger senescence in vitro, which underscores its tumour suppressing properties [10,11,12]. Indeed, the presence of senescent cells in vivo is often observed in the premalignant stages of a tumour, after which they gradually disappear.
In view of this, senescence has been considered one of the two main processes that prevent the emergence of transformed cells, together with apoptosis [13]. Since senescence stops the progression of cancer in vivo [7] and it is known to be increased in response to many therapies [6], the presence of senescent cells in tumours could be considered an indication of a controlled or less advanced disease. Thus, the percentage of senescent cells in tumours could have a utility as a prognostic tool in cancer [14].
Other Functions of Senescence
Although the antineoplastic effects of senescence are the most well-known and studied, recently it has also been reported that it contributes to wound healing, fibrosis and embryonic development [15, 16]. Senescent fibroblasts appear and aggregate as part of normal wound healing and tissue repair [17]. Senescent myofibroblasts in mice liver are able to control fibrosis formation, while mice without senescence effectors (such as p53 and p16) suffered from extreme fibrosis and delay in wound healing [15]. Accumulation of myofibroblasts after liver injury leads to excessive extracellular matrix (ECM) secretion, liver fibrosis and, finally, cirrhosis [15]. All these observations can be explained by the ability of senescent cells to secrete proteins that degrade ECM and thus prevent fibrosis and enhance wound healing.
The involvement of senescence in normal tissue development is just beginning to emerge. For instance, it has been found that megakaryocytes undergo senescence as part of the maturation process that leads to the production of platelets [18]. In addition, senescence also was observed during the normal maturation of syncytiotrophoblasts [19]. Finally, senescent cells are found throughout the embryo, including the apical ectodermal ridge and the neural roof plate, two known signalling centres in embryonic patterning, suggesting that senescence is a mechanism essential for development [16].
The Impact of Senescence on Organismal Ageing
It has been observed that the percentage of senescent cells in tissues in vivo increases over time [20, 21]. All data obtained in rodents and primates suggest that the augment in cell senescence must play a role in age-dependent organismal changes [22,23,24]. Indeed, accumulation of senescent cells has actually been shown to contribute to the functional impairment of different organs [25]. This has led to the hypothesis that senescence is an antagonistically pleiotropic process, with beneficial effects in the early decades of life, mostly as a tumour suppressor, but detrimental to fitness and survival in later stages as senescent cells become more prevalent, due to its contribution to the tissue disruption that leads to age-related pathologies [26].
Because of this, senescent cells are currently thought to be at the core of the physiological changes observed in an organism during the process of ageing. Being able to prevent senescent cell accumulation, or perhaps finding a way to clear them from tissues once they become present, could be an effective strategy to regenerate tissues and maintain their functionality. Such interventions are already being considered, but they would first require a proper understanding of the molecular mechanisms that define the senescent phenotype.
The Molecular Mechanisms of Cellular Ageing
Despite the considerable knowledge accumulated in the 50 years since Leonard Hayflick first described the phenomenon of cell senescence [27], the pathways involved in this process have not been yet fully characterized [28]. One of the well-known features of both replicative senescence and SIPS is the participation of the p53-p21 and/or p16-Rb axis in triggering and maintaining the phenotype. Although in vivo suppression of p53 and/or its upstream regulator ARF is enough to prevent senescence in some models [29], other cell types rely primarily on p16 for its induction [30]. p21, a p53 target gene, has often been considered critical for establishing senescence, whereas p16 could be more involved in the maintenance of the phenotype [31]. This effect would be reinforced by an increase in intracellular ROS [32, 33], thus linking senescence with the classic hypothesis of oxidative stress and ageing. Although p21 is the main cell cycle inhibitor of the p53 pathway, it can also be activated in a p53-independent manner, for example, in response to retinoic acid, IFN and TGFβ [34].
Replicative Versus Stress-Induced Senescence
The two main routes of inducing senescence (replicative or stress-induced) have many common features but diverge in the mechanisms involved in triggering the response. The main difference is that the former features a shortening of the telomeres, while the latter happens in the presence of telomeres of normal length [35,36,37].
Telomeres are structures located at the end of each chromosome, composed of a repeat of the TTAGGG sequence and the proteins that associate with them [38]. Consistent proliferative propagation of cells leads to shortening of telomeres [39], which causes a proliferative arrest mediated by the induction of senescence [8]. Reduction in the length of telomeres is a hallmark of tissue ageing [1]. Once telomere length reaches a limit, this triggers a DNA damage response that leads to the activation of the p53-p21 and p16-Rb pathways, similar to what is observed in SIPS [40].
Telomerase is an enzyme that adds TTAGG repeats to these sites, thus maintaining telomere length and allowing cells to continue dividing [41]. Telomerase is not expressed in most normal cells, but limited to stem cells that need to maintain their proliferative capacity. Telomerase expression can bypass senescence and this is a mechanism that many cancer cells use to avoid a permanent growth arrest [41].
The p53-p21 Pathway in Senescence
The main role of the tumour suppressor p53 is to mediate cellular responses to DNA damage [42]. p53 is a transcription factor that, among other functions, prevents the transformation of cells by triggering protective mechanisms such as cell cycle arrest, senescence or apoptosis [43, 44]. p53 is mainly regulated posttranslationally through many different modifications, including phosphorylation, methylation and acetylation [44,45,46,47,48]. Specifically, its N-terminal region has an important role in its stability because the E3 ligase MDM2 binds to it and ubiquitinates p53, which is then targeted for proteasomal degradation [49]. Different stresses lead to phosphorylation of residues of the N-terminal region by damage-dependent kinases such as ATM and ATR, including serine 15, which disrupts the MDM2-p53 interaction and thus increases the half-life of p53 [50, 51].
Although p53 can trigger the onset of either apoptosis [51, 52] or arrest/senescence [27, 34], the mechanisms involved in the decision between these cellular responses are not well understood. Cell type, presence of growth factors or oncogenes, the intensity of the stress signal and the cellular level of p53 have been cited as important factors in determining a specific p53-induced response [7, 12, 52, 53]. Posttranslational modifications of p53 also have been reported to influence the response observed. For example, p53 phosphorylation by different kinases in response to stress can select for arrest or apoptosis, suggesting the involvement of upstream modifiers in cell fate decisions [29]. Moreover, p53 mutants that can induce growth arrest but not apoptosis, or vice versa, have been identified [12, 49, 54], consistent with the concept that certain p53 mutations may cause selective loss of the ability to transactivate certain p53-responsive promoters [35].
Several p53 target genes have been reported to be specifically involved in apoptosis. These include KILLER/DR5 [55], Bax [39], IGF-BP3 [6], PIG3 [45], PAG608 [24], PERP [1], Noxa [43], PIDD [33], p53AIP1 [44], APAF-1 [46], FDXR [23] and PUMA [41, 56]. Some of these genes, like PIG3 and FDXR, are involved in ROS-related pathways [45]. In fact, apoptosis triggered by p53 has been reported to be dependent on an increase of ROS and the release of apoptotic factors resulting from mitochondrial damage [25]. Despite all the data accumulated in relation to the pro-apoptotic functions of p53, the p53 target genes involved in senescence have not been properly characterized, although it is believed that p21 is its main effector.
p21 is a necessary mediator of p53-induced cell cycle arrest, as indicated by the fact that p53 cannot induce arrest after DNA damage in p21-null mice [53]. p21 is a member of a family of cell cycle inhibitors that includes p27 and p57, and it is capable of inhibiting cyclin-dependent kinases (CDKs) [57], key regulators of the cell cycle. It also acts to block DNA replication by binding to proliferating cell nuclear antigen (PCNA) [58]. p21 expression has been observed in cultured human fibroblasts after prolonged passage, during which such cells undergo senescence [55]. Moreover, p21 has been shown to be capable of inducing permanent growth arrest/senescence in a p53-independent manner [33, 56].
The p16-Rb Pathway in Senescence
Rb is a tumour suppressor protein that regulates the transition phase between G1 and S phases and can thus induce an arrest phenotype that can eventually evolve into senescence [59]. The main role of Rb is to inhibit the E2F family of transcription factors, which is crucial for DNA replication and cell cycle progression [60]. Rb can be inactivated by oncogenes that are encoded by viruses, such as SV40 and E1A, resulting in the release of E2F and senescence bypass [60]. Overexpression of cyclin-dependant kinases (CDKs), which is common in many cancer cells, can also repress Rb and suppress senescence [60]. The CDK inhibitor p16 can maintain Rbin an active state by decreasing CDK4/6 activity [28]. The p16-Rb pathway can be induced by DNA damage signals, which leads to senescence induction in association with the p53-p21 axis [60].
Other Modulators of the Senescence
There are many regulators that directly or indirectly affect the induction of senescence, mainly through their effects on the p53 and Rb pathways. For instance, PML has an essential role in tumour suppression through modulation of the activity of both p53 and Rb, by sequestering inhibitory proteins to the nuclear bodies [54]. As a result, cells that lack PML exhibit impairment in senescence induction by the p53-dependent pathway [61]. On the other hand, PML upregulates histone deacetylases that increase Rb functions [62]. PPP1CA is another effector of senescence that responds to oncogene activation. In the absence of PPP1CA, Ras is unable to induce senescence [63]. SMURF2 is an E3 ubiquitin ligase that, when activated, can induce senescence in fibroblasts independently of p21 [64]. During replicative senescence, the expression of SMURF2 is high and correlates to telomere attrition and p16 upregulation.
BTK is a non-receptor tyrosine kinase that is mutated in the inherited immunodeficiency disease X-linked agammaglobulinaemia [65]. It is expressed in myeloid and lymphoid cells but not in T cells and it is a member of the highly conserved Tec family of kinases, which play an important role in B cell receptor (BCR) signalling [66, 67]. In B cells, BTK is activated after an antigen binds to the BCR, which leads to its phosphorylation at tyrosine 551 by SRC family kinases and its autophosphorylation at tyrosine 223 [68]. Although BTK is mainly located at the cell membrane, it can also be found in the nucleus [69]. A pathological BTK upregulation has been shown in different B cell malignancies, such as chronic lymphocytic leukaemia, mantle cell lymphoma and multiple myeloma [70,71,72]. Because of this, several small molecule inhibitors of BTK have been developed to treat these diseases [73]. BTK was found to be induced in senescent cells and shown to be involved in the p53 pathway as a novel modulator of p53 activity through its phosphorylation [74]. In the absence of BTK, p53-induced senescence was abrogated, showing the importance of BTK in this pathway.
The Importance of Oxidation in Senescence
As we have discussed, increases in intracellular levels of ROS have been implicated at many levels in the pathways of cellular senescence [10]. Senescent cells have higher levels of ROS than normal cells [20], and oncogenic Ras, p21 and p53 induce senescence in association with increased intracellular ROS [30, 32, 36, 75, 76]. It has also been reported that oxidative stress caused by sublethal doses of H2O2 [11] or hyperoxia [58] can force human fibroblasts to arrest in a senescent-like fashion [9]. Moreover, cells can be subjected to oxidative stress due to the effects of many cancer therapeutics, which could increase the presence of senescent cells in tissues [3, 77, 78].
ROS are generated by normal oxidative processes related to cell metabolism [79,80,81]. They are produced initially by the reduction of singlet O2 to superoxide anion and then H2O2 that, if not eliminated, generates highly reactive hydroxyl free radical that causes DNA damage [3, 82]. Increased levels of ROS can be induced by inflammatory responses, certain pathological processes and exposure to agents such as ionizing radiation [83, 84]. Depending on the level of oxidative stress and the extent of the induced DNA damage, cell fate can vary from temporary arrest to death [84, 85]. For instance, exposure to H2O2 has been shown to induce apoptosis or necrosis depending on concentrations and cellular context [85,86,87,88], whereas low concentrations of oxidants can force normal human fibroblasts to permanently arrest in a senescent-like state [4, 86, 89,90,91,92,93].
When proliferating cells are subjected to oxidative stress, the cell cycle temporarily pauses either at the G1, S or G2 phases. Arrest at these checkpoints prevents DNA replication and mitosis in the presence of DNA damage and presumably allows time for DNA repair to occur. The proportion of cells that arrest in each phase after oxidative damage depends on cell type, growth conditions, type of damage and the checkpoints operative in the cells. The G1 checkpoint depends on activation of the tumour suppressor p53, which through p21 induction inhibits cyclin-CDK complexes [94, 95]. Since p53 functions are lost in most neoplasias [96, 97], cancer cells often have a defective G1 checkpoint response to oxidants. Arrest at the G2 checkpoint results primarily from activation of the Chk1 protein kinase, which maintains mitotic cyclin B/Cdc2 complexes in an inactive state [98, 99]. Consistent with this, peroxides such as H2O2 or tert-butyl hydroperoxide (tBH) have been shown to induce both a p53-dependent G1 checkpoint arrest, which can be attenuated by using antioxidants [100, 101], and a G2 checkpoint response [101, 102].
The biochemical responses of normal cells to oxidative stress have been investigated in detail with respect to p53 functions. Oxidants have been shown to promote phosphorylation of p53 at serine 15, which can be blocked by antioxidants [103], and to induce an increase in p53 levels accompanied by elevation of p21 [89]. Although the activation of the p53 pathway in response to oxidative damage contributes importantly to the resulting arrest or cell death responses observed [84], there have been several studies on responses to oxidative stress in cells lacking intact p53 functions [104]. It has been proposed that genotoxic stresses can induce senescence in p53-null as well as wild type p53-containing cancer cells [105] and that this response plays a role in the suppression of tumour growth by chemo- and radiotherapy. However, other studies have indicated that cancer cell lines without functional p53 pathways do not undergo senescence in response to a variety of chemotherapeutic agents [106,107,108].
The fact that oxidative stress triggers a p53 response through DNA damage signals could be a common trigger of senescence and may play an important role in ageing. p53 overexpression has also been shown to cause the accumulation of ROS, presumably mediated by p53 transcriptional influence on pro-oxidant genes [32, 109]. Conversely, overexpression of antioxidant genes like superoxide dismutase or catalase causes extension of lifespan in Drosophila [79]. This can also be observed in cell cultures maintained in low oxygen environments [110]. All of these findings point to a strong relationship between oxidative damage, senescence and ageing.
The Senescence-Associated Secretory Phenotype
Cellular senescence results in the secretion of growth factors, chemokines and cytokines, collectively known as the senescence-associated secretory phenotype (SASP). It has been found that SASP may have a positive effect on cell proliferation and angiogenesis, as well as a role in promoting ageing and tumourigenesis [111, 112]. It can also promote migration of leukocytes and tumour cells, which in turn may induce tumour metastasis [113]. Thus, the presence of SASP could explain many of the negative consequences of senescent cell accumulation, including the pro-ageing effects, and could be a target for regenerative therapies, as we will discuss in more detail later.
Common Markers of Senescent Cells
In order to prevent or stop the accumulation of senescent cells, a limiting factor is the ability to selectively detect them in vivo. Several features have been proposed as being shared by most senescent cells, although none of the currently available markers are sufficient on their own for conclusively identifying senescent cells in vivo or in vitro. This underscores the need for better characterization tools [114].
During cell cycle arrest, many genes that are involved in cell division are supressed, for example, PCNA, E2F or cyclins, and this could be used as an indication of senescence, although it is not specific. Similarly, increased expression of intracellular and/or secreted proteins, such as p21, p16, macroH2A, IL-6, phosphorylated p38 MAPK, PPP1A, SMURF2 or PGM, has been used as surrogate markers of senescence [29, 63, 64, 114,115,116].
Senescent cells display different modifications in the organization of chromatin that can help identify them as well. In normal cells, DNA staining reveals completely uniform colour outlines, whereas senescent cells usually show dot-like patterns, known as senescence-associated heterochromatic foci (SAHF), that appear due to intensive remodelling in the chromatin and a lower susceptibility for digestion by nucleases [117, 118]. SAHF development is not necessary for the establishment of senescence and its presence depends on cell type and the triggering stimuli [119].
Apart from these factors, the most distinctive measurable feature of senescent cells is the presence of a specific β-galactosidase enzymatic activity at pH 6.0, different from the normally observed at pH 4.0 within lysosomes [120]. This has been named senescence-associated β-galactosidase (SA-β-Gal), and it is thought to be a consequence of the enlargement in the structures of lysosome in senescent cells, and it does not have a known role in the establishment or maintenance of the phenotype [121]. Although it is currently the standard for detecting senescent cells in the laboratory, several conditions, such as high cell confluence or treatment with hydrogen peroxide, can also independently stimulate SA-β-Gal activity, leading to many false positives [122].
Recently, a series of membrane markers highly expressed in senescent cells have been identified [14]. This knowledge could contribute to define the interactions of aged cells with the microenvironment and help explain how the mechanisms of senescent cell clearance work normally and stop working with time [123, 124]. Also, specific cell membrane proteins with extracellular epitopes could be useful to rapidly detect senescent cells in vitro and in vivo [125].
Some of these membrane markers, like EBP50 and STX4, are preferentially induced by the p53-p21 pathway, while others, such as DEP1, NTAL and ARMCX3, are dependent on p16-Rb [14]. Thus, they could be used to distinguish between different triggers of senescence. Many of the new markers (such as DEP1, NTAL, ARMCX3, LANCL1, B2MG, PLD3 and VPS26A) have extracellular epitopes, which could be useful in the future to design strategies that could specifically deliver a toxic payload into senescent cells, thus providing a mechanism for clearing them. Of note, many of these proteins play a role in vesicle trafficking (including STX4, VAMP3, VPS26A and PLD3) [126,127,128,129,130,131], which underscores the importance of protein secretion in the senescent phenotype.
The Role of Cell Senescence in Age-Associated Symptoms and Illnesses
It is widely accepted that senescent cells accumulate in vivo in different tissues with time [132]. In addition, there are many age-associated diseases in which it has been shown that accumulation of senescent cells contribute to the onset or maintenance of the symptoms, such as lung and liver fibrosis, neurodegenerative diseases or arthritis [133]. In atherosclerosis, for instance, there is evidence of a link to increased senescence of endothelial and vascular smooth muscle cells [134]. Cell senescence is also induced in myocardial ischemia and hypoxia [135]. In this context, senescent fibroblasts could be a source of fibrosis and collagen accumulation after myocardial infarction. Accumulation of senescent cells has been associated with ocular disorders as well, such as glaucoma and cataracts [123, 136].
In addition, senescence has been shown to be involved in type 2 diabetes, through a p53-dependent increase in insulin resistance in adipose tissue [137]. SA-β-gal activity and p53 and p21 levels are higher in visceral fat from diabetic patients compared to non-diabetic individuals [137]. Similarly, ageing muscle stem cells become senescent with age and the ability to delay senescence increases the potential of their regeneration [138]. In kidney transplantation, the presence of cell senescence in grafted organs associates with poor prognosis [139]. Finally, senescent chondrocytes accumulate in the articular cartilage of people with osteoarthritis [140]. These data together suggest that amelioration of all these diseases could be achieved by preventing the increase of senescent cells in tissues.
How the SASP May Define the Biological Effects of Ageing
The mechanisms by which senescent cells contribute to the symptoms related to ageing are not fully understood. A likely explanation is that impairment of organ function is due to the fact that senescent cells cannot perform their normal roles [123, 136]. However, it has recently been proposed that the paracrine impact of SASP on surrounding cells may be even more relevant for the negative effects of senescent cells, due to its ability to trigger a chronic inflammatory response and facilitate neoplastic transformation [141, 142].
Several of the changes in gene expression observed in senescence are associated with growth factors, chemokines and cytokines that, when secreted, are collectively known as SASP [111, 112]. The SASP likely evolved to create an immune response against senescent cells aimed at their clearance from tissues by phagocytosis. However, this seems to be impaired with time, for reasons that are not known. The SASP from precancerous senescent hepatocytes attract CD4+ cells and are cleared by specific Th1, showing that senescence surveillance is mediated by an adaptive immune clearance [124].
Although the SASP was first described in replicative senescent fibroblasts, it is now known that different cell types have different secretomes [143]. Secretion of inflammatory cytokines triggers proliferation and can also promote migration of leukocytes and tumour cells, which in turn may induce tumour metastasis [113]. Inhibition of the SASP could be an effective way of reducing the impact of senescent cells on tissue physiology [144, 145].
Regenerative Medicine Strategies Aimed at Preventing Ageing
A series of essential hallmarks of ageing have recently been proposed [1]. It is implied that the elimination of each of them should lead to the amelioration of the symptoms associated with ageing. Within this context, the induction of cellular senescence is the endpoint of many of the stimuli associated with ageing. As we have mentioned, the genes involved in triggering senescence belong to tumour suppressor pathways, which suggests that ageing could be, at least in part, a consequence of the natural antineoplastic defences of an organism. Thus, inactivation of these genes can result in increased risk of death from cancer at early ages. Since interfering with the induction of senescence in vivo may prove problematic, a safer approach for regenerative medicine could be to eliminate the senescent cells after they are being formed.
Identifying and Clearing Senescent Cells
The first in vivo proof that accumulation of senescent cells contributes to the deleterious effects of ageing was provided recently using two mouse models in which senescent cells were driven to apoptosis as they started expressing p16 [123, 136]. The absence of senescent cells in tissues importantly delayed the onset of age-associated changes, thus increasing lifespan and health span. These results were reproduced in both fast-ageing and normal mice and confirm that senescent cells could be a target for anti-ageing and regenerative therapies in humans. Moreover, this supports the hypothesis that senescent cell targeting could ameliorate age-related diseases such as cataracts, diabetes and atherosclerosis.
However, performing senescent cell clearance in humans is challenging. The use of anti-senescence drugs, also called senolytics (such as rapamycin, quercetin, dasatinib or navitoclax), could delay senescent cell accumulation in human tissue, but it might contribute to malignant transformation [114]. An alternative would be to use methods to selectively deliver apoptotic drugs to senescent cells using some of the previously described markers of senescence [14]. One possible way would be to use antibody-drug conjugates (ADCs), which have been previously proven to be effective in targeting cancer cells [146, 147]. ADCs are specific monoclonal antibodies bound to a toxic payload by a linker. Once the antibody recognizes an epitope, for instance, in the extracellular domain of a plasma membrane protein, it binds to it and is internalized. The toxin is then released inside the cell by cleavage of the linker. ADCs against markers of senescence are an alternative for designing a regenerative therapy that is currently being investigated.
Other Potential Approaches
Impairment in protein homeostasis (or proteostasis) has also been associated with ageing disorders, especially conditions such as Alzheimer’s and Parkinson’s diseases or muscle atrophy [148]. These are usually the result from impairment in the protein folding mechanisms and regulators of proteostasis normally act through repairing or eliminating misfolded proteins. They could potentially be used as drugs to prevent the protein damage that can contribute to the induction of senescence.
Reducing caloric intake (up to as much as 60%) has been associated with the induction of longevity and healthy life in different animal models, including non-human primates [149]. In fact, it is currently one of the most effective ways to slow down ageing in an experimental context. However, it is difficult to design a trial to assess its relevance in human unless proper markers of tissue ageing are established first. The delay in ageing phenotypes through caloric restriction is thought to be mediated by nutrient signalling mechanisms such as the growth hormone, insulin receptor, IGF-1 and mTOR pathways, and decreases in these factors have been shown to increase in lifespan in vivo [150]. Interestingly, the level of IGF-1 and growth hormone is low in old age and premature ageing syndromes [151].
Consistent with this, pharmacological inhibition of mTOR by rapamycin, a drug produced by Streptomyces hygroscopicus, can delay ageing in mice models [152]. However, it is also a strong immunosuppressant [153], which makes it an unlikely choice for an anti-ageing drug. Resveratrol, a compound found in grapes and other fruits, has been proposed as an alternative. Its effects on ageing seem more complex than was initially anticipated and its mechanism of action is still being discussed, although it seems to be based on the activation of the sirtuin family of deacetylases [154]. Resveratrol may not increase the lifespan of healthy lab animals [155], although it has an important effect on mice being fed a high-fat diet [156].
Finally, the other promising anti-ageing drug being studied intensively is metformin, currently being used to control mild diabetes. Due to its effects on metabolism, metformin has already demonstrated protection against age-related diseases in humans and has been shown to ameliorate ageing in diabetic populations [157]. Its effects on healthy individuals are still being characterized.
Conclusions
Ageing could be seen as a series of symptoms caused by tissues that have stop working properly. Finding a way to restore their function could not only prevent a considerable number of diseases but even prolong lifespan. There are several approaches that could achieve the regeneration of tissues needed to delay ageing and extend quality of life. Here, we have focused on potential strategies to prevent the accumulation of senescent cells, which is thought to be one of the main triggers of organismal ageing.
Anti-ageing drugs need to be highly specific while having virtually no side effects, since they would need to be taken chronically by a population of largely healthy individuals. Current clinical trials with metformin, the first putative anti-ageing drug to reach this stage, will be highly informative and will set the template for future avenues to be tested [157].
Our knowledge of the molecular and cellular physiology of ageing has allowed us for the first time to propose strategies that may have a biological effect on lifespan and health span. It is possible that we will see one or more succeed in the near future, and then regenerative medicine approaches based on chemically mediated lifespan and health span extension will finally become a reality.
References
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Campisi J. From cells to organisms: can we learn about aging from cells in culture? Exp Gerontol. 2001;36(4–6):607–18.
Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65(1):27–33.
Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A. 1995;92(10):4337–41.
Liu D, Xu Y. p53, oxidative stress, and aging. Antioxid Redox Signal. 2011;15(6):1669–78.
Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14(8):547–58.
Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10(1):51–7.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24(22):2463–79.
Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–40.
Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21(4):379–84.
Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9(5):493–505.
Majumder PK, Grisanzio C, O'Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14(2):146–55.
Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432(7015):307–15.
Althubiti M, Lezina L, Carrera S, Jukes-Jones R, Giblett SM, Antonov A, et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014;5:e1528.
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–67.
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155(5):1119–30.
Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12(7):676–85.
Besancenot R, Chaligne R, Tonetti C, Pasquier F, Marty C, Lecluse Y, et al. A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol. 2010;8(9)
Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, et al. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013;27(21):2356–66.
Campisi J. The role of cellular senescence in skin aging. J Investig Dermatol Symp Proc. 1998;3(1):1–5.
Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–22.
Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311(5765):1257.
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8(3):311–23.
Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007;128(1):36–44.
Drummond-Barbosa D. Stem cells, their niches and the systemic environment: an aging network. Genetics. 2008;180(4):1787–97.
Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98(21):12072–7.
Hayflick L, Moorehead P. The serial cultivation of human diploid strains. Exp Cell Res. 1961;25:585–621.
Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28(2):99–114.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602.
Jarrard DF, Sarkar S, Shi Y, Yeager TR, Magrane G, Kinoshita H, et al. p16/pRb pathway alterations are required for bypassing senescence in human prostate epithelial cells. Cancer Res. 1999;59
Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19(3):2109–17.
Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaronson SA. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol. 2003;23(23):8576–85.
Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002;21(9):2180–8.
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–14.
Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A. 1992;89(21):10114–8.
Campisi J, Kim S, Lim CS, Rubio M. Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol. 2001;36(10):1619–37.
Smith JR, Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science. 1996;273(5271):63–7.
Hemann MT, Greider CW. G-strand overhangs on telomeres in telomerase-deficient mouse cells. Nucleic Acids Res. 1999;27(20):3964–9.
Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–60.
d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–8.
Low KC, Tergaonkar V. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem Sci. 2013;38(9):426–34.
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–6.
Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–83.
Marouco D, Garabadgiu AV, Melino G, Barlev NA. Lysine-specific modifications of p53: a matter of life and death? Oncotarget. 2013;4(10):1556–71.
Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16(11):528–36.
Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8(6):1243–54.
Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432(7015):353–60.
Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F, et al. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol. 2007;27(19):6756–69.
Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13(2):83–96.
Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13(2):225–31.
Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8(1):37–45.
Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9(9):702–12.
Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377(6549):552–7.
Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108(2):165–70.
Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211(1):90–8.
Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene. 1999;18(18):2789–97.
Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9(10):1149–63.
Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369(6481):574–8.
Shay JW, Pereirasmith OM, Wright WE. A role for both Rb and P53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196(1):33–9.
Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–12.
de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell. 2004;13(4):523–35.
Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14(16):2015–27.
Castro ME, Ferrer I, Cascon A, Guijarro MV, Lleonart M, Ramon Y, Cajal S, et al. PPP1CA contributes to the senescence program induced by oncogenic Ras. Carcinogenesis. 2008;29(3):491–9.
Zhang H, Cohen SN. Smurf2 up-regulation activates telomere-dependent senescence. Genes Dev. 2004;18(24):3028–40.
Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361(6409):226–33.
de Weers M, Verschuren MC, Kraakman ME, Mensink RG, Schuurman RK, van Dongen JJ, et al. The Bruton's tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol. 1993;23(12):3109–14.
Bradshaw JM. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 2010;22(8):1175–84.
Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science. 1996;271(5250):822–5.
Gustafsson MO, Hussain A, Mohammad DK, Mohamed AJ, Nguyen V, Metalnikov P, et al. Regulation of nucleocytoplasmic shuttling of Bruton's tyrosine kinase (Btk) through a novel SH3-dependent interaction with ankyrin repeat domain 54 (ANKRD54). Mol Cell Biol. 2012;32(13):2440–53.
Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–96.
Chang BY, Francesco M, De Rooij MF, Magadala P, Steggerda SM, Huang MM, et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood. 2013;122(14):2412–24.
Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122(10):3456–63.
Aalipour A, Advani RH. Bruton's tyrosine kinase inhibitors and their clinical potential in the treatment of B-cell malignancies: focus on ibrutinib. Therap Adv Hematol. 2014;5(4):121–33.
Althubiti M, Rada M, Samuel J, Escorsa JM, Najeeb H, Lee KG, et al. BTK modulates p53 activity to enhance apoptotic and senescent responses. Cancer Res. 2016;76(18):5405–14.
Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274(12):7936–40.
Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275(5306):1649–52.
Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–41.
Renschler MF. The emerging role of reactive oxygen species in cancer therapy. Eur J Cancer. 2004;40(13):1934–40.
Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263(5150):1128–30.
Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280(5363):590–2.
Shiloh Y, Kastan MB. ATM: genome stability, neuronal development, and cancer cross paths. Adv Cancer Res. 2001;83:209–54.
Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–47.
Bai J, Cederbaum AI. Catalase protects HepG2 cells from apoptosis induced by DNA-damaging agents by accelerating the degradation of p53. J Biol Chem. 2003;278(7):4660–7.
Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA Repair. 2004;3(8–9):1109–15.
Davies KJ. The broad spectrum of responses to oxidants in proliferating cells: a new paradigm for oxidative stress. IUBMB Life. 1999;48(1):41–7.
Caldini R, Chevanne M, Mocali A, Tombaccini D, Paoletti F. Premature induction of aging in sublethally H2O2-treated young MRC5 fibroblasts correlates with increased glutathione peroxidase levels and resistance to DNA breakage. Mech Ageing Dev. 1998;105(1–2):137–50.
Hampton MB, Orrenius S. Redox regulation of apoptotic cell death in the immune system. Toxicol Lett. 1998;102-103:355–8.
Sun X, Majumder P, Shioya H, Wu F, Kumar S, Weichselbaum R, et al. Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J Biol Chem. 2000;275(23):17237–40.
Chen QM, Bartholomew JC, Campisi J, Acosta M, Reagan JD, Ames BN. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem J. 1998;332(Pt 1):43–50.
von Zglinicki T, Saretzki G, Docke W, Lotze C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res. 1995;220(1):186–93.
Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A. 1994;91(10):4130–4.
Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, Mazarati J, et al. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med. 2000;28(3):361–73.
de Magalhaes JP, Chainiaux F, de Longueville F, Mainfroid V, Migeot V, Marcq L, et al. Gene expression and regulation in H2O2-induced premature senescence of human foreskin fibroblasts expressing or not telomerase. Exp Gerontol. 2004;39(9):1379–89.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–25.
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51(23 Pt 1):6304–11.
Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54(18):4855–78.
Vogelstein B. Cancer. A deadly inheritance [news; comment]. Nature. 1990;348(6303):681–2.
Kaufmann WK, Levedakou EN, Grady HL, Paules RS, Stein GH. Attenuation of G2 checkpoint function precedes human cell immortalization. Cancer Res. 1995;55(1):7–11.
Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408(6811):433–9.
Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8(21):2540–51.
Clopton DA, Saltman P. Low-level oxidative stress causes cell-cycle specific arrest in cultured cells. Biochem Biophys Res Commun. 1995;210(1):189–96.
Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS. The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J Biol Chem. 2001;276(24):21951–9.
Hammond EM, Dorie MJ, Giaccia AJ. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem. 2003;278(14):12207–13.
Hwang ES. Replicative senescence and senescence-like state induced in cancer- derived cells. Mech Ageing Dev. 2002;123(12):1681–94.
Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63(11):2705–15.
te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62(6):1876–83.
Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65(7):2795–803.
Chiu CC, Li CH, Ung MW, Fuh TS, Chen WL, Fang K. Etoposide (VP-16) elicits apoptosis following prolonged G2-M cell arrest in p53-mutated human non-small cell lung cancer cells. Cancer Lett. 2005;223(2):249–58.
Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis [see comments]. Nature. 1997;389(6648):300–5.
Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5(8):741–7.
Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–18.
Krtolica A, Parrinello S, Lockett S, Desprez P-Y, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. P Natl Acad Sci U S A. 2001;98:12072–7.
Mantovani A. Chemokines in neoplastic progression. Semin Cancer Biol. 2004;14(3):147–8.
van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439–46.
Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65(1):177–85.
Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22(10):3389–403.
Narita M, Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, et al. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126(3):503–14.
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703–16.
Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle. 2011;10(3):457–68.
Dimri GP, Lee XH, Basile G, Acosta M, Scott C, Roskelley C, et al. A biomarker that identifies senescent human-cells in culture and in aging skin in-vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–7.
Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–95.
Yang NC, Hu ML. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp Gerontol. 2005;40(10):813–9.
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–6.
Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–51.
Althubiti M, Macip S. Detection of senescent cells by extracellular markers using a flow Cytometry-based approach. Methods Mol Biol. 2017;1534:147–53.
Kean MJ, Williams KC, Skalski M, Myers D, Burtnik A, Foster D, et al. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J Cell Sci. 2009;122(Pt 22):4089–98.
Chen YA, Scheller RH. SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol. 2001;2(2):98–106.
Polgar J, Chung SH, Reed GL. Vesicle-associated membrane protein 3 (VAMP-3) and VAMP-8 are present in human platelets and are required for granule secretion. Blood. 2002;100(3):1081–3.
Olson AL, Knight JB, Pessin JE. Syntaxin 4, VAMP2, and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol. 1997;17(5):2425–35.
Bugarcic A, Zhe Y, Kerr MC, Griffin J, Collins BM, Teasdale RD. Vps26A and Vps26B subunits define distinct retromer complexes. Traffic. 2011;12(12):1759–73.
Osisami M, Ali W, Frohman MA. A role for phospholipase D3 in myotube formation. PLoS One. 2012;7(3):e33341.
Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454(7208):1065–71.
Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–96.
Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111(2):245–59.
Zhu F, Li Y, Zhang J, Piao C, Liu T, Li HH, et al. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS One. 2013;8(9):e74535.
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9.
Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15(9):1082–7.
Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506(7488):316–21.
Naesens M. Replicative senescence in kidney aging, renal disease, and renal transplantation. Discov Med. 2011;11(56):65–75.
Martin JA, Brown TD, Heiner AD, Buckwalter JA. Chondrocyte senescence, joint loading and osteoarthritis. Clin Orthop Relat Res. 2004;427(Suppl):S96–103.
Cahu J. SASP: roadblock for tissue re-organization. Aging. 2013;5(9):641–2.
Herranz N, Gallage S, Gil J. TORn about SASP regulation. Cell Cycle. 2015;14(24):3771–2.
Zhu Y, Armstrong JL, Tchkonia T, Kirkland JL. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr Opin Clin Nutr Metab Care. 2014;17(4):324–8.
Alimbetov D, Davis T, Brook AJ, Cox LS, Faragher RG, Nurgozhin T, et al. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology. 2016;17(2):305–15.
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–72.
de Goeij BE, Lambert JM. New developments for antibody-drug conjugate-based therapeutic approaches. Curr Opin Immunol. 2016;40:14–23.
Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets. 2009;9(8):982–1004.
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–91.
Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–4.
Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489(7415):318–21.
Schumacher B, van der Pluijm I, Moorhouse MJ, Kosteas T, Robinson AR, Suh Y, et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 2008;4(8):e1000161.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–5.
Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol. 2005;56(1):47–60.
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–6.
Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66(2):191–201.
Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016;17(11):679–90.
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab. 2016;23(6):1060–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Macip, S., Althubiti, M. (2019). The Molecular Physiology of Ageing: New Targets for Regenerative Medicine. In: Pinto, H., Fontdevila, J. (eds) Regenerative Medicine Procedures for Aesthetic Physicians. Springer, Cham. https://doi.org/10.1007/978-3-030-15458-5_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-15458-5_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-15457-8
Online ISBN: 978-3-030-15458-5
eBook Packages: MedicineMedicine (R0)