
Abstract
Based on mounting clinical and translational evidence demonstrating the impact of exogenously administered inflammatory stimuli on the brain and behavior, increased endogenous inflammation has received attention as one pathophysiologic process contributing to psychiatric illnesses and particularly depression. Increased endogenous inflammation is observed in a significant proportion of depressed patients and has been associated with reduced responsiveness to standard antidepressant therapies. This chapter presents recent evidence that inflammation affects neurotransmitters and neurocircuits to contribute to specific depressive symptoms including anhedonia, motor slowing, and anxiety, which may preferentially improve after anti-cytokine therapies in patients with evidence of increased inflammation. Existing and novel pharmacological strategies that target inflammation or its downstream effects on the brain and behavior will be discussed in the context of a need for intelligent trial design in order to meaningfully translate these concepts and develop more precise therapies for depressed patients with increased inflammation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
16.1 Introduction
Based on mounting clinical and translational evidence demonstrating the impact of exogenously administered inflammatory stimuli on the brain and behavior, increased endogenous inflammation has received attention as a pathophysiologic process that may contribute to psychiatric illnesses and particularly depression. A rich literature describes increased inflammation in patients with depression and other psychiatric disorders, as evidenced by elevated peripheral and central inflammatory cytokines and acute phase proteins. This endogenous inflammation may arise from numerous sources including risk factors for psychiatric illness (e.g., stress, obesity or metabolic dysfunction, genetics, and lifestyle factors) and has been associated with reduced responsiveness to standard antidepressant therapies. As both increased inflammation and treatment resistance occur in a significant proportion of patients with major depressive disorder (MDD), new conceptual frameworks are needed to identify relevant targets and develop novel therapies.
This chapter will present recent evidence that inflammation affects neurotransmitters and neurocircuits to contribute to specific depressive symptoms including anhedonia and motor retardation. Converging findings from neuroimaging studies involving the administration of exogenous inflammatory stimuli or characterization of depressed patients with increased endogenous inflammation will be discussed in relation to growing evidence that inhibition of inflammation with anti-cytokine therapies in patient with mood disorder including depression specifically reduces anhedonia in patients with evidence of increased inflammation. This chapter will then focus on potential pharmacological strategies based on the neurobiological mechanisms by which inflammation affects neurotransmitters, circuits, and symptoms. Such strategies include the use of existing compounds, either by employing biomarkers to guide selection of antidepressants for patients with high inflammation or by repurposing of existing compounds indicated for other conditions as novel therapies to target inflammation or its downstream effects on the brain and behavior. The need for novel immune-modulatory or redesigned, next-generation anti-cytokine therapies will also be discussed in light of design considerations for clinical trials that are required to meaningfully translate these concepts and develop more precise therapies for patients with increased inflammation.
16.2 Increased Inflammation in Depression: Sources, Symptoms, and Role in Treatment Resistance
16.2.1 Inflammation in Depression: Causes and Consequences
Numerous studies including meta-analyses have reported increased peripheral and central inflammatory markers like the acute phase reactant, C-reactive protein (CRP) and the inflammatory cytokines interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF), in MDD [1,2,3,4]. Longitudinal studies have also found that increased inflammatory markers predicted subsequent depression symptoms [5,6,7], even above and beyond prior depression severity [8]. Of note, similar increases in inflammatory markers have been described in other psychiatric disorders that share commons symptom domains like anhedonia, motor slowing, and anxiety, including bipolar disorder, schizophrenia, anxiety disorders, and post-traumatic stress disorder (PTSD) [9,10,11,12]. In addition inflammation-related genetic risk [13], gene expression in peripheral blood immune cells of MDD patients have also revealed activation of oxidative stress pathways and inflammatory cytokines as well as canonical inflammatory signaling pathways including toll-like receptors, nuclear factor kappa B, and the NLRP3 inflammasome complex [14,15,16,17,18]. Activation of these pathways reflects innate immune responses to both pathogen-associated molecular patterns and danger-associated molecular patterns that are generated by host cells under stress, including psychological stress [19]. Indeed, in MDD patients who are otherwise medically healthy, genetic predisposition may interact with not only stress and trauma but also a range of environmental and lifestyle factors to activate the innate immune system and contribute to low-grade systemic “sterile” inflammation in the absence of pathogens including disturbed sleep, physical inactivity, obesity and metabolic disturbances, Western diet, aging, and smoking [20]. Many of these causes of inflammation are risk also factors for both psychiatric and major medical illnesses, suggesting shared pathophysiologic processes that may explain notable comorbidity between psychiatric disorders and cardiovascular disease, diabetes, and cancer [21].
While most studies report relationships between increased peripheral inflammatory markers and depression and many sources of inflammation per above are from the body, inflammatory cytokines and activated immune cells can access the CNS to directly influence neurotransmitters and circuits and to activate local inflammatory processes. Increased inflammatory markers have been described in the cerebrospinal fluid (CSF) in MDD [22, 23]. Postmortem studies have identified evidence of increased inflammatory signaling in brain parenchyma, including increased TLR expression, expression of inflammatory cytokines, and evidence of both peripheral immune cell trafficking to the brain and activation of microglia [24,25,26,27]. Positron emission tomography (PET) imaging of increased translocator protein (TSPO), which is thought to reflect activated microglia and macrophage, has also been reported throughout the brain in MDD but did not relate to peripheral inflammatory markers [28]. While TSPO binds activated microglia in response to acute inflammation [29], it is not clear whether this is specific to inflammatory microglia versus those that activated to perform physiologic roles like synaptic pruning, how much of the signal is due to binding to other cells including astrocytes and neurons [30], or whether signal is confounded by uptake in the periphery of patients with increased inflammation [31] in MDD. It should be noted however that recent data show that inflammatory pathways can disrupt the blood brain barrier (BBB) in discrete subcortical brain regions, particularly those that regulate motivation and reward [32] and correspond to the impact of inflammation on specific circuits and symptoms, as detailed below.
16.2.2 Increased Inflammation and Antidepressant Treatment Response
Whereas not every patient with MDD has increased inflammation, higher concentrations of inflammatory markers have been reliably observed in patients with reduced responsiveness to conventional antidepressants [33]. Indeed, approximately 25–40% of MDD patients depending on the sample exhibit CRP >3 mg/L, considered high risk for developing cardiovascular disease per American Heart Association guidelines [3, 4, 34, 35], with even more falling in the moderate risk range of CRP 1–3 mg/L, while <1 mg/L is considered normal. Retrospective analyses of longitudinal studies have shown that patients with CRP >1 mg/L prior to therapy are less responsive to antidepressants, especially not only selective serotonin reuptake inhibitors (SSRIs) but also serotonin norepinephrine reuptake inhibitors (SNRIs), over the course of an adequate trial [35,36,37,38]. Measurement of inflammatory cytokine mRNA expression in peripheral immune cells was even more predictive of this effect than CRP [37, 39]. Similarly, in MDD patients with a history of antidepressant nonresponse, higher levels of inflammatory markers including IL-6, TNF, and its soluble receptor 2 and CRP were associated with increasing number of prior failed trials [40, 41]. Moreover, some studies have found that patients with CRP >1 mg/L are more responsive to drugs that affect dopaminergic and noradrenergic pathways including bupropion and nortriptyline [35, 42]. Additionally, MDD patients with higher levels of inflammatory markers have shown better response to adjuvant or therapies that boost monoamine availability or target glutamate [42,43,44] and also electroconvulsive therapy [45]. Together, these data suggest that (1) inflammatory markers may help guide antidepressant treatment selection and (2) better understanding the mechanisms by which inflammation affects the brain may lead to development of novel therapies targeted to the many MDD patients with higher levels of inflammation. Therefore, it is it is important to prospectively consider the role of inflammation in future antidepressant trials and to design studies examining novel treatment avenues using appropriately selected biomarkers and outcome measures relevant to specific symptoms associated with high inflammation in MDD.
16.2.3 Relationships Between Inflammation and Symptom Domains
Consistent with impact of inflammatory cytokines on specific circuits and symptoms as described below, evidence of increased inflammation in MDD has been associated with specific symptoms that are common to other disorders including anhedonia, motor slowing, and anxiety [46,47,48,49]. For example, our group recently identified clusters of cytokines and their soluble receptors in CSF that were associated with higher levels of plasma CRP in otherwise medically stable patients with MDD [4]. These CSF markers, in turn, associated with symptom severity with the strongest relationships between CSF TNF and reduced motivation per a subscale from the Multidimensional Fatigue Inventory and CSF IL-6 soluble receptor and anhedonia per a subscale from the Inventory of Depressive Symptomatology Self-Report (IDS-SR) that correlates with the both the self and clinician-administered Snaith-Hamilton pleasure scale (SHAPS) [50, 51]. These results were confirmed and extended by a study demonstrating that both T- and non-T-cell cytokines were associated with anhedonia severity per the IDS-SR subscale [52]. Furthermore, longitudinal associations between cytokines and anhedonia have been reported in MDD where higher baseline plasma TNF predicted greater severity of anhedonia both at baseline and at a four-month follow-up [53]. Similar relationships between psychomotor slowing and inflammatory markers have also been observed in MMD [54], and many studies have found associations between increased inflammatory markers in schizophrenia and negative symptoms, which include motivational deficits, blunted affect, and social withdrawal among others [55]. In regard to anxiety, a growing literature reports correlations between increased CRP and inflammatory cytokines and symptoms of anxiety [46, 56], including in a longitudinal study [57] and in patients with MDD [58]. Together, these studies provide a clinical framework for the potential role of inflammation in symptoms of anhedonia, motor slowing and anxiety in MDD, and other psychiatric disorders and support the need for mechanistic studies to better understand the impact of inflammation on the brain.
16.2.4 Inhibition of Inflammation in Depression and Symptom Specificity
Numerous studies treating psychiatric patients with rather nonspecific anti-inflammatory agents having multiple off-target effects, e.g., nonsteroidal anti-inflammatory drugs and minocycline [59,60,61], were not targeted to patients with increased inflammation and yield mixed results. Although having limited viability as antidepressants for a myriad of reasons [62, 63], more specific anti-cytokine therapies have shown efficacy for reducing specific depressive symptoms in depressed or medically ill patients with high inflammation. For example, treatment of patients with autoimmune or inflammatory disorders with anti-cytokine therapies reduces depression symptom severity [64]. The TNF antagonist infliximab reduced depression severity with respect to placebo in treatment-resistant MDD patients with higher concentrations of plasma CRP, and anhedonia (work and activities) was the most improved symptom followed by motor slowing (retardation) and anxiety (psychic anxiety) [3]. Moreover, similar results have been seen in two recent studies reporting that anti-TNF or IL-6 therapies in unipolar or bipolar depressed patients with evidence of increased inflammation were primarily effective in reducing anhedonia assessed by SHAPS [3, 65, 66]. These data reinforce specificity for the effects of inflammation on neurobiological pathways that contribute to anhedonia, as well as motor slowing and anxiety, as discussed below.
16.3 Inflammation Effects on the Brain and Behavior
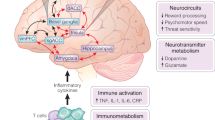
Neuroimaging studies have consistently found that administration of a variety of peripheral inflammatory stimuli, including cytokines and cytokine inducers (e.g., vaccination and subfebrile doses of endotoxin), impact corticostriatal reward and motor circuits to drive reduced motivation and motor slowing as well as anxiety-related brain regions including amygdala, insula, and anterior cingulate cortex (ACC), which may result from cytokine effects on monoamines and glutamate (Fig. 16.1) [67]. Causal evidence for the effects of inflammation on neural circuits and neurotransmitters were initially revealed in patients administered chronic inflammatory cytokines, such as the antiviral and antiproliferative cytokine interferon (IFN)-α, which caused clinical depression in up to half and depressive symptoms in nearly all patients over weeks to months of treatment for infectious diseases or cancer [68, 69]. Like IFN-α, endotoxin and vaccination induce release of classic inflammatory cytokines IL-6, IL-1, and TNF in association with transient increases in depressive symptoms and are commonly used in lab settings to understand their acute effects on the brain [70], as reviewed below. Recent work has also translated findings from these studies investigating causal effects of exogenously administered inflammatory stimuli to study relationships between endogenous inflammation, neurotransmitters, and circuits in MDD patients.

Mechanisms of inflammation effects on the brain and behavior and targets for intervention in depression. Inflammation is increased in otherwise medically stable patients with major depressive disorder (MDD) due to environmental exposures, genetics, psychosocial stressors, diet, and other lifestyle factors. Innate immune cell activation and the release of inflammatory cytokines cause both increased CRP production from the liver and effects on brain neurotransmitters and circuits to drive relevant behavioral changes. Evidence indicates that inflammation and cytokines may preferentially affects dopamine and glutamate systems to disrupt circuits involved in reward and motor activity, as well as those involved anxiety and emotional regulation. In terms of potential novel therapies that may target inflammation or its effects on the brain, there is current interest in (1) compounds that increase dopamine or decrease glutamate signaling, (2) therapies that directly target the immune system to decrease inflammation, and (3) alternative strategies via lifestyle changes or treatments that modify the sources of inflammation. CRP C-reactive protein, dACC dorsal anterior cingulate cortex, DMF dimethyl fumarate, FMT fecal microbiota transplant, IDO indoleamine 2,3 dioxygenase, L-DOPA levodopa, NAC N-acetyl cysteine, NMDA n-methyl-d-aspartate, P2X7 purinergic ATP receptor 7, SAMe S-adenosylmethionine, vmPFC ventromedial prefrontal cortex
16.3.1 Impact of Inflammation on Reward and Motor Regions and Circuits
Early positron emission tomography (PET) studies investigating broad effects of chronic inflammatory cytokines on the brain found that resting glucose metabolism was increased in basal ganglia and decreased in frontal cortex [71, 72], whereby increased metabolism in the left putamen and left nucleus accumbens correlated with IFN-α-induced anergia and fatigue [71]. This pattern of increased glucose metabolism in basal ganglia nuclei is similar to that seen in patients with Parkinson’s disease (PD) [73], which thought to indicate increased oscillatory burst activity secondary to loss of inhibitory dopamine input [74]. Complementary PET using radio-labeled dopamine precursor, [18F]fluorodopa, in IFN-α-treated patients also showed both increased uptake and decreased turnover of FDOPA, reflecting decreased availability of dopamine/precursor and impaired packaging or release of newly synthesized dopamine, in the caudate, putamen, and VS [68]. Magnetic resonance spectroscopy (MRS) further showed increased glutamate in left basal ganglia in patients treated with IFN-α that correlated with reduced motivation [75]. Complementary to these chronic studies, acute challenge with IFN-α caused rapid (4 hours) changes in striatal microstructure that predicted subsequent development of fatigue [76, 77].
Functional impact of the effects of peripheral inflammation on brain regions relevant to reduced motivation and motor activity and involving dopamine and glutamate have also been revealed by functional MRI (fMRI). Indeed, decreased ventral striatal (VS) neural activation to win versus loss was seen in a gambling task after chronic IFN-α, which correlated with self-reported reduced motivation [68]. Studies in healthy controls using vaccination and subfebrile doses of endotoxin have also assessed acute effects of inflammation on reward processing. Reduced activation of VS to reward-predicting cues during a monetary incentive delay task (MIDT) were associated with increased self-reported depressed mood [78] and with cytokine responses in women but not men hours after endotoxin [79]. In a probabilistic instrumental learning task combined with fMRI, typhoid vaccine compared with saline control reduced behavioral attractiveness of rewards while making punishments more aversive, in association with opposing change in VS responses that were decreased to positive feedback but increased to negative feedback [80]. This corresponds with a study showing that greater inflammatory responses to laboratory stress correlated with decreased VS sensitivity to positive feedback [81]. Additionally, typhoid vaccination affected task-based activity in the substantia nigra that correlated with both psychomotor slowing and increased peripheral blood concentrations of IL-6 [82, 83]. Finally, acute administration of IFN-α or typhoid vaccination has been shown to acutely decrease functional connectivity (FC) within motivation-relevant brain regions including VS and the ventromedial prefrontal cortex (vmPFC) [84, 85].
16.3.2 Impact of Inflammation on Regions and Circuits for Fear, Anxiety, and Emotional Processing
Similar to reports of increased reactivity in MDD as well as anxiety disorders and PTSD, exogenous administration of peripheral inflammatory stimuli has been shown to increase neural activity in amygdala, dorsal ACC, and insula [86]. For example, acute IFN-α (4 hour) administration enhanced right amygdala responses to sad versus neutral faces, which correlated with subsequent IFN-α-induced depression severity [87]. Increased IL-6 and TNF after administration of endotoxin to healthy subjects was also shown to increase amygdala activity in response to socially threatening images, which correlated with feelings of social disconnection [88]. Greater dorsal ACC activation was also seen in IFN-α -treated patients that highly correlated with task-related errors [89], and this may be due to increased glutamate in dorsal ACC as measured by MRS in patients administered IFN-a that correlated with depressive symptom severity [90]. In participants administered endotoxin prior to a neuroimaging session in which they were socially excluded during a virtual ball-tossing game, increases in IL-6 were associated with increases in social pain-related neural activity in both dorsal ACC and anterior insula in females but not males [91]. Another study administering typhoid vaccination also reported increased activation of amygdala and dorsal ACC as well as insula, during presentation of congruent and incongruent stimuli [92]. Given the role of the insula in interoception, it is not surprising that this brain region also had increased resting glucose metabolism as measured by PET after endotoxin [93]. These findings suggest that increased inflammatory cytokines in the periphery may contribute to altered neural activity in circuits involving amygdala, dorsal ACC, and insula to disrupt emotional processing in MDD and anxiety-related disorders.
16.3.3 Endogenous Inflammation and Circuit Dysfunction in Patients with Depression
In light of converging evidence of the impact of exogenously induced inflammation on circuits and symptoms relevant to reduced motivation, motor slowing, and anxiety (as described above), recent studies have examined a potential role for increased endogenous inflammation in relevant circuit deficits that are frequently observed in patients with MDD and other psychiatric disorders [94,95,96]. For example, in medically stable and unmedicated MDD patients, endogenous inflammation as measured by plasma CRP and inflammatory cytokines was associated with lower left VS to vmPFC FC, which in turn positively correlated with anhedonia per the IDS-SR subscale [51]. These targeted findings were corroborated by parcellation-based network analysis in MDD revealing vmPFC and VS (a region parcellated as anterior ventral caudate) as the two most significant hubs, respectively, in a widely distributed network of low FC within 63 features in relation to CRP, subsets of which were highly predictive of anhedonia as measured by the IDS-SR subscale and SHAPS [97]. Of note, relationships between increased inflammation and low FC among several regions, dorsal striatal regions, the vmPFC and pre-supplementary motor area, and key components of corticostriatal circuitry involved in linking motivation to motor output [98, 99], were correlated with objective measures of psychomotor slowing in these studies [51, 97]. Furthermore, FC was shown to mediate relationships between CRP and anhedonia and psychomotor symptom severity [51].
Similar relationships between increased inflammation and low FC in primarily left VS to vmPFC reward circuitry were also observed in treatment-resistant MDD patients [100] and in trauma-exposed women in relation to an anhedonia subscale via Beck Depression Inventory [101]. Further evidence of associations between increased endogenous inflammation and functional changes in reward circuits include reduced striatal activation during reward anticipation in MDD patients with higher CRP and inflammatory cytokines [102, 103]. While the above findings generally indicate a role for reduced dopamine signaling, high inflammation in MDD was associated with increased glutamate concentrations in left basal ganglia that correlated with anhedonia [104]. Patients with combined elevations in CRP and glutamate displayed both high anhedonia and low regional homogeneity in left basal ganglia, indicating disrupted local coherence of activity that may be driven by increased glutamate [105].
Regarding threat and anxiety-related circuitry, inflammatory markers have been associated with similar deficits in amygdala-vmPFC circuitry as those reported to characterize individuals with high anxiety, MDD, and/or PTSD [106,107,108]. For example, we previously found that higher concentrations of plasma CRP and inflammatory cytokines correlated with lower right amygdala-vmPFC FC in patients with a primary diagnosis of MDD in association with anxiety symptoms, particularly in patients with comorbid anxiety-related disorders including PTSD [109]. Recent studies in adolescents have also found relationships between endogenous inflammation and altered FC in circuits relevant to threat, anxiety, and emotional processing [110, 111]. Interestingly, acute blockade of TNF in inflammatory arthritis patients with infliximab decreased right amygdala reactivity to emotional (sad, happy, and neutral) faces in association with reduced depressive symptoms at 24 hours [87], suggesting emotional reactivity as a potential target for efficacy of anti-inflammatory therapies.
16.4 Treatment Targets for Depressed Patients with Increased Inflammation
Given the mounting evidence of differential response to antidepressants in MDD patients with higher versus lower levels of inflammation and the reproducible effects of inflammation on behavior, there is a need to consider inflammation in studies examining antidepressant outcomes and for development of novel therapies that block inflammation or its consequences on the brain (Fig. 16.1).
Numerous clinical trials have addressed this concern by the use of agents with putative anti-inflammatory activity as adjuvant or therapy in patients with psychiatric disorders, primarily in depression or schizophrenia [112,113,114]. Meta-analyses of the use of such drugs including cyclooxygenase (COX)-2 inhibitors, anti-cytokine therapies, minocycline, statins, pioglitazone, glucocorticoids, and omega-3 fatty acids suggest modest efficacy [60, 113,114,115,116,117] despite small sample sizes, heterogeneity across studies, and numerous design issues. Most studies used therapies as adjuvant to conventional antidepressants and did compare with placebo, patients were rarely selected to have high inflammation, only a few studies measured inflammatory markers to stratify patients or establish anti-inflammatory effect, and importantly, many of the chosen therapies convey only mild anti-inflammatory activity in the context of numerous “off target” effects that can confound data interpretation [63, 118]. In the largest randomized controlled trial to date using the COX-2 inhibitor celecoxib and minocycline (an antibiotic thought to stabilize microglia but also disrupt microbiota [119]), both failed to separate from placebo in reducing depressive symptoms in depressed bipolar patients [61]. Two studies did, however, consider inflammation levels in treatment-resistant or bipolar depression and found that higher CRP (>3 mg/L) or IL-6 concentrations prior to treatment were predictive of response to minocycline [120, 121], with reduced serum IL-6 after treatment seen only in bipolar depressed responders [121]. While existing cytokine antagonists may not be viable antidepressants [62, 63], they have demonstrated efficacy in depressed patients with high inflammation with specificity for symptoms consistent with the known effects of inflammation on the brain [3, 65, 66], thus providing a foundation for enrolment and outcomes strategies for testing novel therapies.
Given the above-described inconsistencies and challenges in studying anti-inflammatory therapies for depression, existing or novel compounds that target the neurotransmitters impacted by inflammation, like dopamine and glutamate, may serve as more proximal approaches for translating these concepts into patients (Fig. 16.1(1)). Discussed below are the multiple pharmacological interventions that can be used to block inflammation or its downstream effects on the brain, starting with the potential for informed selection of available antidepressants and reuse or repurposing of existing compounds that affect neurotransmitters. Because the above-described use of COX-2 inhibitors, anti-cytokine therapies, minocycline, fatty acids, and the like have been reviewed extensively elsewhere, discussion on immune targets will focus on novel agents or redesign of existing therapies (Fig. 16.1(2)), along with mention of alternative treatments that may exert efficacy via effects on neural and physiologic processes that modulate inflammation (Fig. 16.1(3)). Biomarker and study design considerations for patient selection and target engagement of the brain and behavior will also be discussed.
16.4.1 Compounds That Increase Dopamine Synthesis, Synaptic Availability, and Receptor Signaling
Dopamine reuptake
Decreased response to conventional antidepressant therapies like SSRIs in patients with high inflammation may be due to decreased monoamine synthesis and availability, or to facilitatory effects of cytokines on serotonin transporters, which may circumvent or interfere with their action [122, 123]. Alternatively, reduced response may be due to a preferential access and effects of peripheral inflammation on reward and motor-related brain regions that receive primarily dopamine input [67, 124], consistent with evidence of increased responsiveness in these patients to antidepressants with catecholamine activity and particularly bupropion [42]. Bupropion, an FDA approved and effective medication for MDD [125] that functions primarily by inhibiting dopamine and norepinephrine reuptake, has been shown to increase high effort activity in rats [126]. Some trials also suggest preferential response of anhedonia to bupropion [127, 128]. Together, these findings warranted further investigation of potential efficacy in MDD with high inflammation, such as a recent trial prospectively examining the ability of bupropion versus escitalopram to increase FC in reward circuitry and improve motivation in patients with high CRP (NCT04352101). It should be noted that although classical psychostimulant medications with potent effects on dopamine reuptake or release increase motivation in rodent models and acutely in healthy humans [126, 129, 130], they have demonstrated only limited efficacy in chronically treating fatigue and other dopamine-related symptoms in trials for patients with cancer and other medical illnesses that are associated with inflammation [131,132,133,134,135,136,137,138,139,140], or as augmentation therapy for depression [141,142,143,144,145].
Dopamine synthesis
While compounds that inhibit dopamine reuptake may exert efficacy in high inflammation patients, the primary mechanisms of inflammation effects are likely through the inhibition of key components of dopamine synthesis like tetrahydrobiopterin (BH4) [146, 147], a pivotal cofactor for the enzymes that synthesize dopamine and other monoamines. Indeed, inflammation reduces BH4 availability through oxidation and excessive conversion to BH2 during generation of nitric oxide by nitric oxide synthase [148]. Therefore, depressed patients with high inflammation may benefit from therapies that increase BH4 stability or activity including sapropterin and folic acid, L-methylfolate, and S-adenosylmethionine (SAMe). Low serum folate has been associated with increased risk of depression and non-response to antidepressant treatment and an increased likelihood of depression relapse [149], yet clinical trials using L-methylfolate (marketed as Deplin and Zervalx) and SAMe have shown mixed results [150, 151]. Post hoc analysis of two parallel-sequential adjuvant trials of L-methylfolate in patients with MDD [150] did however reveal that a combination of increased concentrations of leptin, CRP, and inflammatory cytokines or high BMI was associated with greater symptom improvement [44], supporting potential value of targeting such therapies to MDD patients with high inflammation.
Another strategy to address impaired dopamine synthesis is the administration of its precursor, levodopa (L-DOPA). Indeed, in monkeys experiencing similar behavioral responses including reduced effort-based sucrose consumption after chronic IFN-α exposure [152], decreases in striatal dopamine release were reversed by L-DOPA administered via reverse in vivo microdialysis [153]. Replacement of dopamine with L-DOPA improves motor function and was also shown to increase motivation in patients with PD [154]. Whether L-DOPA (in combination with carbidopa) versus placebo improves FC in reward circuitry in association with improved motivation and anhedonia in MDD patients with higher levels of CRP is currently being studied (NCT04723147). Open-label L-DOPA-carbidopa administration to aged depressed patients with motor slowing, a group likely to exhibit increased inflammation, also showed a positive antidepressant response [155], and a similar ongoing study in this population aims to better understand mechanisms of these findings including the potential role of inflammation (NCT04469959).
Dopamine agonists
Dopamine receptor agonists have received attention as efficacious augmentation strategies for depression. For example, antiparkinsonian agents like pramipexole have demonstrated efficacy to reduce depressive symptoms in patients with treatment-resistant depression [156,157,158,159]. Although it is unknown whether this effect is specific to high inflammation in depressed patients [160], it has been shown to block endotoxin-induced degeneration of nigrostriatal dopamine cells in rodents [161].
A growing body of evidence also supports the use of atypical antipsychotics as an augmentation strategy in MDD, including meta-analyses suggesting these agents are more effective than placebo for both response and remission [162, 163]. In addition to more serotonergic activity, newer generation antipsychotics like aripiprazole and amisulpride appear to act as D2/3 partial agonists by facilitate dopamine signaling at lower doses or in states of low endogenous ligand [164], such as with increased inflammation, while preventing overstimulation when endogenous dopamine levels are high.
16.4.2 Therapies That Target Glutamate Transmission
Modulation of the kynurenine pathway
Immune-mediated activation of indoleamine 2,3 dioxygenase (IDO) catabolizes tryptophan (TRP), the primary amino-acid precursor of serotonin, to kynurenine (KYN), downstream metabolites of which affect glutamate transmission in the brain [165, 166]. Peripheral blood KYN/TRP ratio in combination with TNF defines a population of MDD patients with increased anhedonia and treatment resistance [167]. With regard to preventing activation of the KYN pathway, the IDO antagonist, 1-methyl tryptophan (1-MT) has been shown to abrogate depressive-like behavior in animal models of inflammatory challenge or infection [168, 169]. Given the importance of serotonin in T-cell activation, there has been interest in developing IDO inhibitors as a pharmacologic strategy to enhance T-cell function against cancer [170], but compounds like 1-MT have not yet been translated outside of oncology. As leucine competes with KYN for the large amino acid transporter, it can inhibit transport of KYN into the brain and reduce production of neuroactive metabolites like quinolinic acid (QUIN) from KYN in microglia [171]. Thus, high dose leucine (8 mg/d for 2 weeks) is currently being tested in MMD patients, although inflammatory markers that have been associated with KP metabolites in the periphery and CNS will only be examined in post hoc analyses (NCT03079297).
Glutamate receptor modulators
Inflammation can promote excitotoxic glutamate transmission through several mechanisms including decreased buffering by astrocytic expression of excitatory amino acid transporters (EEATs), increase release of glutamate from astrocytes and activated microglia [165, 172,173,174], and increased QUIN, as described above, which directly activates the n-methyl-d-aspartate (NMDA) receptor [175, 176]. Therefore, glutamate receptor antagonists may be useful in preventing excitotoxicity and oxidative stress and may reverse or prevent inflammation-related behavioral change. Indeed, in rodents, the NMDA antagonist ketamine reversed endotoxin-induced depressive-like behavior including anhedonic behavior, while having no effect on inflammation or activation of IDO in the brain [177]. Moreover, blockade of AMPA receptors was able to reverse ketamine’s effects on endotoxin-induced depressive-like behavior, indicating that the effects of ketamine were specific to its impact on glutamate signaling. Moreover, in an animal model of treatment-resistant depression, ketamine responsiveness was predicted by baseline peripheral blood levels of CRP and TNF [178].
In humans, one study in treatment-resistant depression found that patients who were most responsive to ketamine were those with the highest concentrations of serum IL-6 [43]. However, another study found that although treatment-resistant depressed patients exhibited increased IL-6 compared with controls, IL-6 and other inflammatory cytokines were not associated with response to ketamine [179]. Given the restrictions to ketamine and esketamine use (i.e., administration route, post-dose monitoring), alternative agents with equal efficacy and favorable tolerability and safety profile are being actively investigated. One example is AXS-05, a combination oral pill containing the NMDA receptor antagonist/sigma-1 receptor agonist dextromethorphan given with bupropion (to boost dextromethorphan blood levels through CYP2D6 inhibition). As phase 2 results appeared promising, with significant improvements in response and remission at 6 weeks compared with bupropion alone [180], this therapy might be particularly well-suited for treatment of depressed patients with high inflammation.
Glutamate stabilizers
As downstream effects of inflammation both stimulate glutamate receptors and disrupt balance of intracellular and extracellular glutamate, strategies targeting reuptake mechanisms via EAATs may be beneficial in patients with high inflammation. Riluzole is one agent that may support glutamate by facilitating EAAT activity to protect against excitotoxicity [181] and has some evidence of benefit in MDD. One small open-label study found benefit in treatment-resistant depressed patients over the course of 6 weeks with riluzole monotherapy [182], while another small trial showed benefit of riluzole augmentation [183], thus providing support for future studies in MDD with high inflammation.
16.4.3 Therapies That Affect the Immune System
Anti-inflammatory drugs
Results from many trials in psychiatry using anti-inflammatory therapies are mixed at best [59], and only a handful of studies have enriched for patients with evidence of increased inflammation and/or used drugs with known anti-inflammatory activity and little off-target effects [184]. Studies using cytokine antagonists that have potent anti-inflammatory effects have reported encouraging results for improved symptoms of anhedonia in depressed patients with increased inflammation [3, 65, 66]. Translation of these therapies is unfortunately limited, for example, by increased risk for infection, and blockade of potentially beneficial effects of innate immune signaling on other neurobiological pathways such a myelination [62, 63]. Fortunately, immunotherapies are evolving with even more specificity for inflammatory signaling pathways [185]. For example, XPro1595, a novel, first in class selective “dominant-negative” mutant variant of the human TNF protein [186], rapidly binds to and inhibits “inflammatory” signaling driven by the soluble form of TNF through soluble TNF receptors, while having no effect on the immunologic and neuro“protective” signaling driven by the transmembrane form of TNF [186,187,188]. XPro1595 has demonstrated preclinical efficacy in multiple laboratory animal models of depression [189,190,191], including a treatment-resistance model where XPro1595 neutralized TNF of the rat and decreased peripheral blood CRP concentrations [192]. Of note, laboratory animal studies have exemplified that crossing the BBB is not required for the antidepressant effects of traditional cytokine antagonists [193], which are relatively large molecules, as reducing peripheral inflammation in MDD is the primary target [3]. However, the novel TNF antagonist XPro1595 has significant brain penetrance [194], suggesting potential human benefit for diseases like depression above and beyond the reduced risk of infection or demyelination as compared with traditional anti-cytokine therapies.
Drugs targeting inflammatory signaling pathways, such as baricitinib, an oral Janus Kinase (JAK 1/2) inhibitor FDA-approved for the treatment of rheumatoid arthritis with recent additional FDA-approval for emergency use authorization for the treatment of patients hospitalized with COVID-19, are being studied [195,196,197,198,199]. Baricitinib significantly reduces plasma IL-6, TNF, and CRP within days after administration in humans across disease states [196,197,198,199,200,201,202] and might exert benefit in MDD with high inflammation. Other similar drugs in development include those that inhibit other intracellular immune pathways, toll-like receptor signaling, cell adhesion molecules, or chemokine receptors. Additionally, inhibitors of inflammasome activation via the purinergic P2X7 receptor are also being explored, including current testing in depressed patients with incomplete response to monoaminergic antidepressants and CRP ≥1 mg/L (NCT04116606).
Despite considerable interest in the role of the immune system in MDD and other psychiatric disorders and its therapeutic implications, little information exists regarding the specific immunologic mechanisms required to design therapies engaging specific immune cell types. While a monocyte phenotype has traditionally been thought to represent high inflammation in MDD, a recent study clustering patients into inflammatory subgroups suggested distinct populations of high inflammation patients represented either by myeloid cells in one case or lymphoid populations in the other [203]. Accordingly, vast array of other anti-cytokine therapies that selectively target T cell cytokines include anti-IL-17 and anti-IL-12/23, which are FDA-approved; maraviroc which inhibits CCR5 for prevention of HIV; and plerixafor, a CXCR4 antagonist that mobilizes stem cells [204].
Immunometabolic modulation
Evidence of the metabolic and energetic reprograming used by immune cells to sustain inflammatory activation has been observed in medically stable MDD patients who had both high CRP and significant anhedonia [205]. Furthermore, immunometabolic pathways in specific cell types are being targeted for new therapies in autoimmune and inflammatory disorders [206] and align with recent data that rapamycin, an inhibitor of mTORC1 signaling involved in such processes, prolonged the antidepressant benefit of ketamine therapy [207]. Additionally, first-line treatment with dimethyl fumarate (DMF) in patients with multiple sclerosis has recently been shown to inhibit Warburg-like metabolism in immune cells via inhibiting the glyceraldehyde 3-phosphate dehydrogenase, a key enzyme of the glycolytic pathway [208]. Consistent with the role of aerobic glycolysis in activation of specific immune cell subsets, DMF inhibited inflammatory cytokines and lactate production from activated macrophages, Th1 and Th17 cells, while sparing the function of resting macrophages and regulatory T cells. Additionally, N-acetyl cysteine (NAC), a precursor to the antioxidant glutathione that reduces oxidative stress/reactive oxygen species bioproducts of intracellular immunometabolic shifts and inflammation, may both improve mitochondrial function and reduce inflammation [208,209,211]. NAC may also exert antidepressant activity via effects on glutamate signaling through AMPA receptors and the astroglial glutamate exchanger xCT [212, 213]. Given evidence of antidepressant efficacy of NAC, there are ongoing clinical trials including one with inclusion criterion of a CRP >0.85 mg/L (NCT02972398).
Alternative therapies: lifestyle factors, stress reduction, neuroimmunomodulation, and microbiome
The efficacy of modifying environmental exposures and lifestyle factors that contribute to inflammation in depression have also been investigated including exercise, weight reduction, yoga, massage, tai chi, and cognitive behavioral therapy and meditation. Many of these interventions have been shown to induce a variety of immune changes including reduced inflammation [214]. For example, mindfulness meditation over 4 months increased FC between the posterior cingulate cortex and the left dorsolateral PFC, which in turn was associated with decreases in IL-6 [215]. In addition, both cognitive behavioral therapy and tai chi were associated with reduced CRP, monocyte production of inflammatory cytokines, and inflammatory gene expression in elderly patients with insomnia [216]. Both diet and exercise programs have been shown to have antidepressant and anti-anxiety effects [217, 218] and to reduce a variety of inflammatory markers in longitudinal studies [219, 220]. A 3-month hatha yoga program also reduced endotoxin-induced peripheral blood mononuclear cell production of TNF, IL-6, and IL-1beta as well as fatigue in breast cancer survivors [221]. These studies have, however, failed to determine whether changes in inflammation or immune function are required for the efficacy of these interventions [63], or if they are generalizable to MDD patients with high inflammation and anhedonia.
An elegant body of work has described a direct mechanism for neural regulation of the immune response mechanisms by stimulation of efferent vagal fibers to provide acetylcholine-mediated inhibition of the release of TNF and other cytokines from immune cells such as macrophages [222]. This anti-inflammatory cholinergic reflex can be activated by electrically stimulating the vagus and is now being capitalized on via a novel bioelectric platform that recently received designation as an FDA breakthrough device for the treatment of rheumatoid arthritis in patients intolerant of or exhibiting incomplete response to biologic drugs [223]. There is also growing interest in the role of the microbiome in a variety of neuropsychiatric disorders including MDD and its potential as a therapeutic target [224]. While the precise mechanisms by which the microbiome influences behavior are unknown, evidence suggests effects on inflammation as a plausible pathophysiologic pathway [225, 226]. These relationships have been illuminated by translational work showing induction of depressive-like behavior in mice after fecal microbiota transplant (FMT) from donors with major depression [227] and improvements in depressive and anxiety symptoms after FMT in patients with inflammatory or functional bowel disease [228, 229]. Trials examining probiotic supplementation show small but significant improvement of depressive and anxiety symptoms [230] and research on more directed therapies like FMT is warranted.
16.5 Summary and Translational Conclusions
In this chapter, extensive clinical and translational evidence is presented supporting increased inflammation and its effects on the brain as one pathophysiologic pathway to symptoms of anhedonia, reduced motivation and motor slowing as well as anxiety in MDD and other psychiatric disorders. Decreased dopamine availability and excessive glutamate may serve as mechanisms of inflammation’s impact on the brain and potential therapeutic targets for symptom reduction in psychiatric patients with elevated biomarkers of inflammation using existing or novel antidepressant strategies. Moreover, studies employing anti-cytokine therapies in depression have consistently found anhedonia to be the symptom most improved, with some support for motor retardation and anxiety. While translation potential of previously tested anti-inflammatory agents may be limited, there are a myriad of new immune-targeted therapies with promise for therapeutic benefit in depression including redesigned, next-generation cytokine antagonists with improved specificity for inflammatory signaling, intracellular signal transduction inhibitors FDA approved for inflammatory illness, and alternative therapies that modulate immune function via manipulation of the vagal nerve or microbiome to name a few.
Despite consistent causal findings of associations between inflammation and alterations in neurotransmitters and neurocircuits relevant to MDD and other psychiatric illnesses, several challenges and considerations for translation of these concepts exist. Crucial in this regard is the need for informed trial design. For one, given strong evidence of relationships between inflammation and treatment resistance, inflammatory markers should be considered in studies examining predictors of response or selection of patients for existing antidepressant therapies in a prospective rather than post hoc fashion. For treatment development, biomarker-driven approaches should target specific therapies to patients with evidence of high inflammation (i.e., using CRP) and/or relevant symptoms like anhedonia, motor slowing, and/or anxiety and assess not only response and remission but also target engagement of relevant circuits and symptoms. Given that FC in reward and motor circuits has been identified to mediate relationships between endogenous inflammatory markers like CRP and anhedonia and psychomotor speed, functional neuroimaging biomarkers that associate with symptoms of anhedonia, motor slowing, or anxiety can serve as relevant brain biomarkers. Finally, while substantial work has established such relationships in depression, future work is needed to better understand the role of inflammation in the brain and specific behaviors and how it relates to treatment response in other disorders like schizophrenia.
Another challenge for translation is that the FDA and other regulatory bodies do not currently recognize individual symptom domains as appropriate criteria for drug development, despite recent appreciation in the field of symptoms subdomains that cut across disorders and have a similar, well-defined pathophysiological basis [231]. Future efforts will clearly require advocacy in this area, particularly as it relates to treatment resistance and residual symptoms, and considering that the FDA has recognized drugs for cancer based on surrogate markers of specific genes and proteins involved in the growth and survival of cancer cells irrespective of clinical outcome [232]. A similar agnostic approach to treatment based on an emerging understanding of biomarkers such as genes, proteins, neurotransmitters, and circuits that underlie the biological bases of behaviors may be needed to facilitate development and approval of new drugs for treatment of psychiatric disorders.
In sum, an emerging understanding of the mechanisms by which peripheral inflammation can affect neurotransmitters and relevant circuits to impact behavior and contribute to symptoms of MDD and other psychiatric disorders has provided a framework for development of novel therapies. Further identification of a platform of neuroimaging, behavioral, and peripheral biomarkers that can be used to test these therapies lends potential for future personalization of treatments targeted to biologically based subgroups of patients with transdiagnostic presentation of symptoms like anhedonia, motor slowing, and anxiety.
References
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446–57.
Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71(2):171–86.
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiat. 2013;70(1):31–41.
Felger JC, Haroon E, Patel TA, Goldsmith DR, Wommack EC, Woolwine BJ, et al. What does plasma CRP tell us about peripheral and central inflammation in depression? Mol Psychiatry. 2020;25(6):1301–11.
Wium-Andersen MK, Orsted DD, Nielsen SF, Nordestgaard BG. Elevated C-reactive protein levels, psychological distress, and depression in 73, 131 individuals. JAMA Psychiat. 2013;70(2):176–84.
Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A, et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med. 2009;39(3):413–23.
Au B, Smith KJ, Gariepy G, Schmitz N. The longitudinal associations between C-reactive protein and depressive symptoms: evidence from the English Longitudinal Study of Ageing (ELSA). Int J Geriatr Psychiatry. 2015;30(9):976–84.
Bondy E, Norton SA, Voss MD, Marks RB, Bourdreaux MJ, Treadway MT, et al. Inflammation is associated with future depressive symptoms among older adults. Brain Behav Immun Health. 2021;13:100226.
Costello H, Gould RL, Abrol E, Howard R. Systematic review and meta-analysis of the association between peripheral inflammatory cytokines and generalised anxiety disorder. BMJ Open. 2019;9(7):e027925.
Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2015;2(11):1002–12.
Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry. 2016;21(12):1696–709.
Munkholm K, Vinberg M, Vedel KL. Cytokines in bipolar disorder: a systematic review and meta-analysis. J Affect Disord. 2013;144(1–2):16–27.
Barnes J, Mondelli V, Pariante CM. Genetic contributions of inflammation to depression. Neuropsychopharmacology. 2017;42(1):81–98.
Hung YY, Kang HY, Huang KW, Huang TL. Association between toll-like receptors expression and major depressive disorder. Psychiatry Res. 2014;220(1–2):283–6.
Hung YY, Lin CC, Kang HY, Huang TL. TNFAIP3, a negative regulator of the TLR signaling pathway, is a potential predictive biomarker of response to antidepressant treatment in major depressive disorder. Brain Behav Immun. 2017;59:265–72.
Chen RA, Huang TL, Huang KW, Hung YY. TNFAIP3 mRNA level is associated with psychological anxiety in major depressive disorder. Neuroimmunomodulation. 2017;24(4–5):271–5.
Hajebrahimi B, Bagheri M, Hassanshahi G, Nazari M, Bidaki R, Khodadadi H, et al. The adapter proteins of TLRs, TRIF and MYD88, are upregulated in depressed individuals. Int J Psychiatry Clin Pract. 2014;18(1):41–4.
Keri S, Szabo C, Kelemen O. Expression of Toll-Like Receptors in peripheral blood mononuclear cells and response to cognitive-behavioral therapy in major depressive disorder. Brain Behav Immun. 2014;40:235–43.
Fleshner M, Crane CR. Exosomes, DAMPs and miRNA: features of stress physiology and immune homeostasis. Trends Immunol. 2017;38(10):768–76.
Berk M, Williams LJ, Jacka FN, O’Neil A, Pasco JA, Moylan S, et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013;11:200.
Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822–32.
Franzen AD, Lam TT, Williams KR, Nairn AC, Duman RS, Sathyanesan M, et al. Cerebrospinal fluid proteome evaluation in major depressive disorder by mass spectrometry. BMC Psychiatry. 2020;20(1):481.
Felger JC, Haroon E, Patel TA, Goldsmith DR, Wommack EC, Woolwine BJ, et al. What does plasma CRP tell us about peripheral and central inflammation in depression? Mol Psychiatry. 2018;25:1301–11.
Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun. 2014;42:50–9.
Torres-Platas SG, Comeau S, Rachalski A, Bo GD, Cruceanu C, Turecki G, et al. Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflammation. 2014;11:12.
Pandey GN, Rizavi HS, Ren X, Bhaumik R, Dwivedi Y. Toll-like receptors in the depressed and suicide brain. J Psychiatr Res. 2014;53:62–8.
Pandey GN. Inflammatory and innate immune markers of neuroprogression in depressed and teenage suicide brain. Mod Trends Pharmacopsychiatry. 2017;31:79–95.
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiat. 2015;72(3):268–75.
Hannestad J, Gallezot JD, Schafbauer T, Lim K, Kloczynski T, Morris ED, et al. Endotoxin-induced systemic inflammation activates microglia: [(1)(1)C]PBR28 positron emission tomography in nonhuman primates. NeuroImage. 2012;63(1):232–9.
Notter T, Schalbetter SM, Clifton NE, Mattei D, Richetto J, Thomas K, et al. Neuronal activity increases translocator protein (TSPO) levels. Mol Psychiatry. 2021;26(6):2025–37.
Nettis MA, Veronese M, Nikkheslat N, Mariani N, Lombardo G, Sforzini L, et al. PET imaging shows no changes in TSPO brain density after IFN-alpha immune challenge in healthy human volunteers. Transl Psychiatry. 2020;10(1):89.
Dudek KA, Dion-Albert L, Lebel M, LeClair K, Labrecque S, Tuck E, et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc Natl Acad Sci U S A. 2020;117(6):3326–36.
Arteaga-Henríquez G, Simon MS, Burger B, Weidinger E, Wijkhuijs A, Arolt V, et al. Low-grade inflammation as a predictor of antidepressant and anti-inflammatory therapy response in MDD patients: a systematic review of the literature in combination with an analysis of experimental data collected in the EU-MOODINFLAME Consortium. Front Psych. 2019;10:458.
Rapaport MH, Nierenberg AA, Schettler PJ, Kinkead B, Cardoos A, Walker R, et al. Inflammation as a predictive biomarker for response to omega-3 fatty acids in major depressive disorder: a proof-of-concept study. Mol Psychiatry. 2016;21(1):71–9.
Uher R, Tansey KE, Dew T, Maier W, Mors O, Hauser J, et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am J Psychiatry. 2014;171(12):1278–86.
Jha MK, Minhajuddin A, Chin-Fatt C, Greer TL, Carmody TJ, Trivedi MH. Sex differences in the association of baseline c-reactive protein (CRP) and acute-phase treatment outcomes in major depressive disorder: findings from the EMBARC study. J Psychiatr Res. 2019;113:165–71.
Cattaneo A, Gennarelli M, Uher R, Breen G, Farmer A, Aitchison KJ, et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology. 2013;38(3):377–85.
Zhang J, Yue Y, Thapa A, Fang J, Zhao S, Shi W, et al. Baseline serum C-reactive protein levels may predict antidepressant treatment responses in patients with major depressive disorder. J Affect Disord. 2019;250:432–8.
Cattaneo A, Ferrari C, Uher R, Bocchio-Chiavetto L, Riva MA, Consortium MRCI, et al. Absolute measurements of macrophage migration inhibitory factor and interleukin-1-beta mRNA levels accurately predict treatment response in depressed patients. Int J Neuropsychopharmacol. 2016;19(10):pyw045.
Haroon E, Daguanno AW, Woolwine BJ, Goldsmith DR, Baer WM, Wommack EC, et al. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology. 2018;95:43–9.
Chamberlain SR, Cavanagh J, de Boer P, Mondelli V, Jones DNC, Drevets WC, et al. Treatment-resistant depression and peripheral C-reactive protein. Br J Psychiatry. 2018;214:11–9.
Jha MK, Minhajuddin A, Gadad BS, Greer T, Grannemann B, Soyombo A, et al. Can C-reactive protein inform antidepressant medication selection in depressed outpatients? Findings from the CO-MED trial. Psychoneuroendocrinology. 2017;78:105–13.
Yang JJ, Wang N, Yang C, Shi JY, Yu HY, Hashimoto K. Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biol Psychiatry. 2015;77(3):e19–20.
Shelton RC, Pencina MJ, Barrentine LW, Ruiz JA, Fava M, Zajecka JM, et al. Association of obesity and inflammatory marker levels on treatment outcome: results from a double-blind, randomized study of adjunctive L-methylfolate calcium in patients with MDD who are inadequate responders to SSRIs. J Clin Psychiatry. 2015;76(12):1635–41.
Kruse JL, Congdon E, Olmstead R, Njau S, Breen EC, Narr KL, et al. Inflammation and improvement of depression following electroconvulsive therapy in treatment-resistant depression. J Clin Psychiatry. 2018;79(2):17m11597.
Milaneschi Y, Kappelmann N, Ye Z, Lamers F, Moser S, Jones PB, et al. Association of inflammation with depression and anxiety: evidence for symptom-specificity and potential causality from UK Biobank and NESDA cohorts. Mol Psychiatry. 2021;26(12):7393–402.
Swardfager W, Rosenblat JD, Benlamri M, McIntyre RS. Mapping inflammation onto mood: inflammatory mediators of anhedonia. Neurosci Biobehav Rev. 2016;64:148–66.
Lucido MJ, Bekhbat M, Goldsmith DR, Treadway MT, Haroon E, Felger JC, et al. Aiding and abetting anhedonia: impact of inflammation on the brain and pharmacological implications. Pharmacol Rev. 2021;73(3):1084–117.
Michopoulos V, Powers A, Gillespie CF, Ressler KJ, Jovanovic T. Inflammation in fear- and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology. 2017;42(1):254–70.
Ameli R, Luckenbaugh DA, Gould NF, Holmes MK, Lally N, Ballard ED, et al. SHAPS-C: the Snaith-Hamilton pleasure scale modified for clinician administration. PeerJ. 2014;2:e429.
Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. 2016;21(10):1358–65.
Jha MK, Miller AH, Minhajuddin A, Trivedi MH. Association of T and non-T cell cytokines with anhedonia: role of gender differences. Psychoneuroendocrinology. 2018;95:1–7.
Rengasamy M, Marsland A, McClain L, Kovats T, Walko T, Pan L, et al. Longitudinal relationships of cytokines, depression and anhedonia in depressed adolescents. Brain Behav Immun. 2021;91:74–80.
Bekhbat M, Goldsmith DR, Woolwine BJ, Haroon E, Miller AH, Felger JC. Transcriptomic signatures of psychomotor slowing in peripheral blood of depressed patients: evidence for immunometabolic reprogramming. Mol Psychiatry. 2021;26(12):7384–92.
Goldsmith DR, Rapaport MH. Inflammation and negative symptoms of schizophrenia: implications for reward processing and motivational deficits. Front Psych. 2020;11:46.
Ye Z, Kappelmann N, Moser S, Davey Smith G, Burgess S, Jones PB, et al. Role of inflammation in depression and anxiety: tests for disorder specificity, linearity and potential causality of association in the UK Biobank. EClinicalMedicine. 2021;38:100992.
van Eeden WA, El Filali E, van Hemert AM, Carlier IVE, Penninx B, Lamers F, et al. Basal and LPS-stimulated inflammatory markers and the course of anxiety symptoms. Brain Behav Immun. 2021;98:378–87.
Gaspersz R, Lamers F, Wittenberg G, Beekman ATF, van Hemert AM, Schoevers RA, et al. The role of anxious distress in immune dysregulation in patients with major depressive disorder. Transl Psychiatry. 2017;7(12):1268.
Eyre HA, Air T, Proctor S, Rositano S, Baune BT. A critical review of the efficacy of non-steroidal anti-inflammatory drugs in depression. Prog Neuro-Psychopharmacol Biol Psychiatry. 2015;57:11–6.
Rosenblat JD, McIntyre RS. Efficacy and tolerability of minocycline for depression: a systematic review and meta-analysis of clinical trials. J Affect Disord. 2018;227:219–25.
Husain MI, Chaudhry IB, Khoso AB, Husain MO, Hodsoll J, Ansari MA, et al. Minocycline and celecoxib as adjunctive treatments for bipolar depression: a multicentre, factorial design randomised controlled trial. Lancet Psychiatry. 2020;7(6):515–27.
Dreyer L, Magyari M, Laursen B, Cordtz R, Sellebjerg F, Locht H. Risk of multiple sclerosis during tumour necrosis factor inhibitor treatment for arthritis: a population-based study from DANBIO and the Danish Multiple Sclerosis Registry. Ann Rheum Dis. 2016;75(4):785–6.
Miller AH, Haroon E, Felger JC. Therapeutic implications of brain-immune interactions: treatment in translation. Neuropsychopharmacology. 2017;42(1):334–59.
Kappelmann N, Lewis G, Dantzer R, Jones PB, Khandaker GM. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry. 2018;23(2):335–43.
Salvadore G, Nash A, Bleys C, Hsu B, Saad Z, Gause A, et al. A double-blind, placebo-controlled, multicenter study of Sirukumab as adjunctive treatment to a monoaminergic antidepressant in adults with major depressive disorder, in ACNP 57th annual meeting: poster session II, Hollywood, FL. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2018;43(1):228–382.
Lee Y, Mansur RB, Brietzke E, Carmona NE, Subramaniapillai M, Pan Z, et al. Efficacy of adjunctive infliximab vs. placebo in the treatment of anhedonia in bipolar I/II depression. Brain Behav Immun. 2020;88:631–9.
Felger JC, Treadway MT. Inflammation effects on motivation and motor activity: role of dopamine. Neuropsychopharmacology. 2017;42(1):216–41.
Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry. 2012;69(10):1044–53.
Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, et al. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002;26(5):643–52.
Lasselin J, Lekander M, Benson S, Schedlowski M, Engler H. Sick for science: experimental endotoxemia as a translational tool to develop and test new therapies for inflammation-associated depression. Mol Psychiatry. 2021;26(8):3672–83.
Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology. 2007;32(11):2384–92.
Juengling FD, Ebert D, Gut O, Engelbrecht MA, Rasenack J, Nitzsche EU, et al. Prefrontal cortical hypometabolism during low-dose interferon alpha treatment. Psychopharmacology. 2000;152(4):383–9.
Mentis MJ, McIntosh AR, Perrine K, Dhawan V, Berlin B, Feigin A, et al. Relationships among the metabolic patterns that correlate with mnemonic, visuospatial, and mood symptoms in Parkinson’s disease. Am J Psychiatry. 2002;159(5):746–54.
Wichmann T, DeLong MR. Functional neuroanatomy of the basal ganglia in Parkinson’s disease. Adv Neurol. 2003;91:9–18.
Haroon E, Felger JC, Woolwine BJ, Chen X, Parekh S, Spivey JR, et al. Age-related increases in basal ganglia glutamate are associated with TNF, reduced motivation and decreased psychomotor speed during IFN-alpha treatment: preliminary findings. Brain Behav Immun. 2015;46:17–22.
Dowell NG, Bouyagoub S, Tibble J, Voon V, Cercignani M, Harrison NA. Interferon-alpha-induced changes in NODDI predispose to the development of fatigue. Neuroscience. 2019;403:111–7.
Dowell NG, Cooper EA, Tibble J, Voon V, Critchley HD, Cercignani M, et al. Acute changes in striatal microstructure predict the development of interferon-alpha induced fatigue. Biol Psychiatry. 2016;79(4):320–8.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748–54.
Moieni M, Irwin MR, Jevtic I, Olmstead R, Breen EC, Eisenberger NI. Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology. 2015;40(7):1709–16.
Harrison NA, Voon V, Cercignani M, Cooper EA, Pessiglione M, Critchley HD. A neurocomputational account of how inflammation enhances sensitivity to punishments versus rewards. Biol Psychiatry. 2016;80(1):73–81.
Treadway MT, Admon R, Arulpragasam AR, Mehta M, Douglas S, Vitaliano G, et al. Association between interleukin-6 and striatal prediction-error signals following acute stress in healthy female participants. Biol Psychiatry. 2017;82(8):570–7.
Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry. 2008;63(11):1022–9.
Harrison NA, Cercignani M, Voon V, Critchley HD. Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology. 2015;40(4):831–8.
Dipasquale O, Cooper EA, Tibble J, Voon V, Baglio F, Baselli G, et al. Interferon-alpha acutely impairs whole-brain functional connectivity network architecture - a preliminary study. Brain Behav Immun. 2016;58:31–9.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry. 2009;66(5):407–14.
Felger JC. Imaging the role of inflammation in mood and anxiety-related disorders. Curr Neuropharmacol. 2018;16(5):533–58.
Davies KA, Cooper E, Voon V, Tibble J, Cercignani M, Harrison NA. Interferon and anti-TNF therapies differentially modulate amygdala reactivity which predicts associated bidirectional changes in depressive symptoms. Mol Psychiatry. 2021;26(9):5150–60.
Inagaki TK, Muscatell KA, Irwin MR, Cole SW, Eisenberger NI. Inflammation selectively enhances amygdala activity to socially threatening images. NeuroImage. 2012;59(4):3222–6.
Capuron L, Pagnoni G, Demetrashvili M, Woolwine BJ, Nemeroff CB, Berns GS, et al. Anterior cingulate activation and error processing during interferon-alpha treatment. Biol Psychiatry. 2005;58(3):190–6.
Haroon E, Woolwine B, Chen X, Pace T, Parekh S, Spivey J, et al. IFN-alpha-induced cortical and subcortical glutamate changes assessed by magnetic resonance spectroscopy. Neuropsychopharmacology. 2014;39(7):1777–85.
Eisenberger NI, Lieberman MD, Satpute AB. Personality from a controlled processing perspective: an fMRI study of neuroticism, extraversion, and self-consciousness. Cogn Affect Behav Neurosci. 2005;5(2):169–81.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ, et al. Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry. 2009;66(5):415–22.
Hannestad J, Subramanyam K, Dellagioia N, Planeta-Wilson B, Weinzimmer D, Pittman B, et al. Glucose metabolism in the insula and cingulate is affected by systemic inflammation in humans. J Nucl Med. 2012;53(4):601–7.
Whitton AE, Treadway MT, Pizzagalli DA. Reward processing dysfunction in major depression, bipolar disorder and schizophrenia. Curr Opin Psychiatry. 2015;28(1):7–12.
Kaiser RH, Andrews-Hanna JR, Wager TD, Pizzagalli DA. Large-scale network dysfunction in major depressive disorder: a meta-analysis of resting-state functional connectivity. JAMA Psychiat. 2015;72(6):603–11.
Cullen KR, Westlund MK, Klimes-Dougan B, Mueller BA, Houri A, Eberly LE, et al. Abnormal amygdala resting-state functional connectivity in adolescent depression. JAMA Psychiat. 2014;71(10):1138–47.
Yin L, Xu X, Chen G, Mehta ND, Haroon E, Miller AH, et al. Inflammation and decreased functional connectivity in a widely-distributed network in depression: centralized effects in the ventral medial prefrontal cortex. Brain Behav Immun. 2019;80:657–66.
Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2010;35(1):4–26.
Samanez-Larkin GR, Knutson B. Decision making in the ageing brain: changes in affective and motivational circuits. Nat Rev Neurosci. 2015;16(5):278–89.
Rengasamy M, Brundin L, Griffo A, Panny B, Capan C, Forton C, et al. Cytokine and reward circuitry relationships in treatment-resistant depression. Biol Psych Global Open Sci. 2022;2(1):45–53.
Mehta ND, Stevens JS, Li Z, Gillespie CF, Fani N, Michopoulos V, et al. Inflammation, reward circuitry and symptoms of anhedonia and PTSD in trauma-exposed women. Soc Cogn Affect Neurosci. 2020;15(10):1046–55.
Burrows K, Stewart JL, Kuplicki R, Figueroa-Hall L, Spechler PA, Zheng H, et al. Elevated peripheral inflammation is associated with attenuated striatal reward anticipation in major depressive disorder. Brain Behav Immun. 2021;93:214–25.
Costi S, Morris LS, Collins A, Fernandez NF, Patel M, Xie H, et al. Peripheral immune cell reactivity and neural response to reward in patients with depression and anhedonia. Transl Psychiatry. 2021;11(1):565.
Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T, et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry. 2016;21(10):1351–7.
Haroon E, Chen X, Li Z, Patel T, Woolwine BJ, Hu XP, et al. Increased inflammation and brain glutamate define a subtype of depression with decreased regional homogeneity, impaired network integrity, and anhedonia. Transl Psychiatry. 2018;8(1):189.
Tang Y, Kong L, Wu F, Womer F, Jiang W, Cao Y, et al. Decreased functional connectivity between the amygdala and the left ventral prefrontal cortex in treatment-naive patients with major depressive disorder: a resting-state functional magnetic resonance imaging study. Psychol Med. 2013;43(9):1921–7.
Stevens JS, Jovanovic T, Fani N, Ely TD, Glover EM, Bradley B, et al. Disrupted amygdala-prefrontal functional connectivity in civilian women with posttraumatic stress disorder. J Psychiatr Res. 2013;47(10):1469–78.
Kim MJ, Gee DG, Loucks RA, Davis FC, Whalen PJ. Anxiety dissociates dorsal and ventral medial prefrontal cortex functional connectivity with the amygdala at rest. Cereb Cortex. 2011;21(7):1667–73.
Mehta ND, Haroon E, Xu X, Woolwine BJ, Li Z, Felger JC. Inflammation negatively correlates with amygdala-ventromedial prefrontal functional connectivity in association with anxiety in patients with depression: preliminary results. Brain Behav Immun. 2018;73:725–30.
Swartz JR, Carranza AF, Tully LM, Knodt AR, Jiang J, Irwin MR, et al. Associations between peripheral inflammation and resting state functional connectivity in adolescents. Brain Behav Immun. 2021;95:96–105.
Nusslock R, Brody GH, Armstrong CC, Carroll AL, Sweet LH, Yu T, et al. Higher peripheral inflammatory Signaling associated with lower resting-state functional brain connectivity in emotion regulation and central executive networks. Biol Psychiatry. 2019;86(2):153–62.
Deakin B, Suckling J, Barnes TRE, Byrne K, Chaudhry IB, Dazzan P, et al. The benefit of minocycline on negative symptoms of schizophrenia in patients with recent-onset psychosis (BeneMin): a randomised, double-blind, placebo-controlled trial. Lancet Psychiatry. 2018;5(11):885–94.
Zheng W, Cai DB, Yang XH, Ungvari GS, Ng CH, Muller N, et al. Adjunctive celecoxib for schizophrenia: a meta-analysis of randomized, double-blind, placebo-controlled trials. J Psychiatr Res. 2017;92:139–46.
Xiang YQ, Zheng W, Wang SB, Yang XH, Cai DB, Ng CH, et al. Adjunctive minocycline for schizophrenia: a meta-analysis of randomized controlled trials. Eur Neuropsychopharmacol. 2017;27(1):8–18.
Cai DB, Zheng W, Zhang QE, Ng CH, Ungvari GS, Huang X, et al. Minocycline for depressive symptoms: a meta-analysis of randomized, double-blinded, placebo-controlled trials. Psychiatry Q. 2020;91(2):451–61.
Liao Y, Xie B, Zhang H, He Q, Guo L, Subramaniapillai M, et al. Efficacy of omega-3 PUFAs in depression: a meta-analysis. Transl Psychiatry. 2019;9(1):190.
Kohler-Forsberg O, Nicolaisen Lydholm C, Hjorthoj C, Nordentoft M, Mors O, Benros ME. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Acta Psychiatr Scand. 2019;139(5):404–19.
Miller AH, Raison CL. Are anti-inflammatory therapies viable treatments for psychiatric disorders? Where the rubber meets the road. JAMA Psychiatry. 2015;72(6):527–8.
Thompson KG, Rainer BM, Antonescu C, Florea L, Mongodin EF, Kang S, et al. Minocycline and its impact on microbial dysbiosis in the skin and gastrointestinal tract of acne patients. Ann Dermatol. 2020;32(1):21–30.
Nettis MA, Lombardo G, Hastings C, Zajkowska Z, Mariani N, Nikkheslat N, et al. Augmentation therapy with minocycline in treatment-resistant depression patients with low-grade peripheral inflammation: results from a double-blind randomised clinical trial. Neuropsychopharmacology. 2021;46(5):939–48.
Savitz JB, Teague TK, Misaki M, Macaluso M, Wurfel BE, Meyer M, et al. Treatment of bipolar depression with minocycline and/or aspirin: an adaptive, 2x2 double-blind, randomized, placebo-controlled, phase IIA clinical trial. Transl Psychiatry. 2018;8(1):27.
Kitagami T, Yamada K, Miura H, Hashimoto R, Nabeshima T, Ohta T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003;978(1–2):104–14.
Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology. 2010;35(13):2510–20.
Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. 2017;20(12):1752–60.
Clayton AH, Croft HA, Horrigan JP, Wightman DS, Krishen A, Richard NE, et al. Bupropion extended release compared with escitalopram: effects on sexual functioning and antidepressant efficacy in 2 randomized, double-blind, placebo-controlled studies. J Clin Psychiatry. 2006;67(5):736–46.
Randall PA, Lee CA, Podurgiel SJ, Hart E, Yohn SE, Jones M, et al. Bupropion increases selection of high effort activity in rats tested on a progressive ratio/chow feeding choice procedure: implications for treatment of effort-related motivational symptoms. Int J Neuropsychopharmacol. 2015;18(2):pyu017.
Tomarken AJ, Dichter GS, Freid C, Addington S, Shelton RC. Assessing the effects of bupropion SR on mood dimensions of depression. J Affect Disord. 2004;78(3):235–41.
Thase ME, Clayton AH, Haight BR, Thompson AH, Modell JG, Johnston JA. A double-blind comparison between bupropion XL and venlafaxine XR: sexual functioning, antidepressant efficacy, and tolerability. J Clin Psychopharmacol. 2006;26(5):482–8.
Yohn SE, Thompson C, Randall PA, Lee CA, Muller CE, Baqi Y, et al. The VMAT-2 inhibitor tetrabenazine alters effort-related decision making as measured by the T-maze barrier choice task: reversal with the adenosine A2A antagonist MSX-3 and the catecholamine uptake blocker bupropion. Psychopharmacology. 2015;232(7):1313–23.
Soder HE, Cooper JA, Lopez-Gamundi P, Hoots JK, Nunez C, Lawlor VM, et al. Dose-response effects of d-amphetamine on effort-based decision-making and reinforcement learning. Neuropsychopharmacology. 2021;46(6):1078–85.
Mar Fan HG, Clemons M, Xu W, Chemerynsky I, Breunis H, Braganza S, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer. 2008;16(6):577–83.
Moraska AR, Sood A, Dakhil SR, Sloan JA, Barton D, Atherton PJ, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol. 2010;28(23):3673–9.
Butler JM Jr, Case LD, Atkins J, Frizzell B, Sanders G, Griffin P, et al. A phase III, double-blind, placebo-controlled prospective randomized clinical trial of d-threo-methylphenidate HCl in brain tumor patients receiving radiation therapy. Int J Radiat Oncol Biol Phys. 2007;69(5):1496–501.
Sugawara Y, Akechi T, Shima Y, Okuyama T, Akizuki N, Nakano T, et al. Efficacy of methylphenidate for fatigue in advanced cancer patients: a preliminary study. Palliat Med. 2002;16(3):261–3.
Pucci E, Branas P, D’Amico R, Giuliani G, Solari A, Taus C. Amantadine for fatigue in multiple sclerosis. Cochrane Database Syst Rev (Online). 2007;(1):CD002818.
Stankoff B, Waubant E, Confavreux C, Edan G, Debouverie M, Rumbach L, et al. Modafinil for fatigue in MS: a randomized placebo-controlled double-blind study. Neurology. 2005;64(7):1139–43.
Bruera E, Yennurajalingam S, Palmer JL, Perez-Cruz PE, Frisbee-Hume S, Allo JA, et al. Methylphenidate and/or a nursing telephone intervention for fatigue in patients with advanced cancer: a randomized, placebo-controlled, phase II trial. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(19):2421–7.
Ruddy KJ, Barton D, Loprinzi CL. Laying to rest psychostimulants for cancer-related fatigue? J Clin Oncol Off J Am Soc Clin Oncol. 2014;32(18):1865–7.
Escalante CP, Meyers C, Reuben JM, Wang X, Qiao W, Manzullo E, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J. 2014;20(1):8–14.
Gong S, Sheng P, Jin H, He H, Qi E, Chen W, et al. Effect of methylphenidate in patients with cancer-related fatigue: a systematic review and meta-analysis. PLoS One. 2014;9(1):e84391.
Patkar AA, Masand PS, Pae C-U, Peindl K, Hooper-Wood C, Mannelli P, et al. A randomized, double-blind, placebo-controlled trial of augmentation with an extended release formulation of methylphenidate in outpatients with treatment-resistant depression. J Clin Psychopharmacol. 2006;26(6):653–6.
Ravindran AV, Kennedy SH, O’Donovan MC, Fallu A, Camacho F, Binder CE. Osmotic-release oral system methylphenidate augmentation of antidepressant monotherapy in major depressive disorder: results of a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2008;69(1):87.
Michelson D, Adler LA, Amsterdam JD, Dunner DL, Nierenberg AA, Reimherr FW, et al. Addition of atomoxetine for depression incompletely responsive to sertraline: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(4):582–7.
Pary R, Scarff JR, Jijakli A, Tobias C, Lippmann S. A review of psychostimulants for adults with depression. Fed Pract. 2015;32(Suppl 3):30S–7S.
Dunlop BW, Crits-Christoph P, Evans DL, Hirschowitz J, Solvason HB, Rickels K, et al. Coadministration of modafinil and a selective serotonin reuptake inhibitor from the initiation of treatment of major depressive disorder with fatigue and sleepiness: a double-blind, placebo-controlled study. J Clin Psychopharmacol. 2007;27(6):614–9.
Felger JC, Li L, Marvar PJ, Woolwine BJ, Harrison DG, Raison CL, et al. Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain Behav Immun. 2013;31:153–60.
Zoller H, Schloegl A, Schroecksnadel S, Vogel W, Fuchs D. Interferon-alpha therapy in patients with hepatitis C virus infection increases plasma phenylalanine and the phenylalanine to tyrosine ratio. J Interferon Cytokine Res. 2012;32(5):216–20.
Cunnington C, Channon KM. Tetrahydrobiopterin: pleiotropic roles in cardiovascular pathophysiology. Heart. 2010;96(23):1872–7.
Papakostas GI, Petersen T, Mischoulon D, Ryan JL, Nierenberg AA, Bottiglieri T, et al. Serum folate, vitamin B12, and homocysteine in major depressive disorder, part 1: predictors of clinical response in fluoxetine-resistant depression. J Clin Psychiatry. 2004;65(8):1090–5.
Papakostas GI, Shelton RC, Zajecka JM, Etemad B, Rickels K, Clain A, et al. L-methylfolate as adjunctive therapy for SSRI-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267–74.
Sarris J, Price LH, Carpenter LL, Tyrka AR, Ng CH, Papakostas GI, et al. Is S-adenosyl methionine (SAMe) for depression only effective in males? A re-analysis of data from a randomized clinical trial. Pharmacopsychiatry. 2015;48(4–5):141–4.
Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR, et al. Chronic interferon-alpha decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology: official publication of the American college of. Neuropsychopharmacology. 2013;38(11):2179–87.
Felger JC, Hernandez CR, Miller AH. Levodopa reverses cytokine-induced reductions in striatal dopamine release. Int J Neuropsychopharmacol. 2015;18(4):1–5.
Czernecki V, Pillon B, Houeto JL, Pochon JB, Levy R, Dubois B. Motivation, reward, and Parkinson’s disease: influence of dopatherapy. Neuropsychologia. 2002;40(13):2257–67.
Rutherford BR, Slifstein M, Chen C, Abi-Dargham A, Brown PJ, Wall MW, et al. Effects of L-DOPA monotherapy on psychomotor speed and [(11)C]Raclopride binding in high-risk older adults with depression. Biol Psychiatry. 2019;86(3):221–9.
Franco-Chaves JA, Mateus CF, Luckenbaugh DA, Martinez PE, Mallinger AG, Zarate CA Jr. Combining a dopamine agonist and selective serotonin reuptake inhibitor for the treatment of depression: a double-blind, randomized pilot study. J Affect Disord. 2013;149(1–3):319–25.
Cusin C, Iovieno N, Iosifescu DV, Nierenberg AA, Fava M, Rush AJ, et al. A randomized, double-blind, placebo-controlled trial of pramipexole augmentation in treatment-resistant major depressive disorder. J Clin Psychiatry. 2013;74(7):636–41.
Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety. 2000;11(2):58–65.
Cassano P, Lattanzi L, Fava M, Navari S, Battistini G, Abelli M, et al. Ropinirole in treatment-resistant depression: a 16-week pilot study. Can J Psychiatr. 2005;50(6):357–60.
Escalona R, Fawcett J. Pramipexole in treatment resistant-depression, possible role of inflammatory cytokines. Neuropsychopharmacology. 2017;42(1):363.
Iravani MM, Sadeghian M, Leung CC, Tel BC, Rose S, Schapira AH, et al. Continuous subcutaneous infusion of pramipexole protects against lipopolysaccharide-induced dopaminergic cell death without affecting the inflammatory response. Exp Neurol. 2008;212(2):522–31.
Nelson JC, Papakostas GI. Atypical antipsychotic augmentation in major depressive disorder: a meta-analysis of placebo-controlled randomized trials. Am J Psychiatr. 2009;166(9):980–91.
Zhou X, Keitner GI, Qin B, Ravindran AV, Bauer M, Del Giovane C, et al. Atypical antipsychotic augmentation for treatment-resistant depression: a systematic review and network meta-analysis. Int J Neuropsychopharmacol. 2015;18(11):pyv060.
Admon R, Kaiser RH, Dillon DG, Beltzer M, Goer F, Olson DP, et al. Dopaminergic enhancement of striatal response to reward in major depression. Am J Psychiatry. 2017;174(4):378–86.
Dantzer R, Walker AK. Is there a role for glutamate-mediated excitotoxicity in inflammation-induced depression? J Neural Transm. 2014;
Dantzer R, O’Connor JC, Lawson MA, Kelley KW. Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinology. 2011;36(3):426–36.
Haroon E, Welle JR, Woolwine BJ, Goldsmith DR, Baer W, Patel T, et al. Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology. 2020;45(6):998–1007.
O’Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J, et al. Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. J Immunol. 2009;182(5):3202–12.
O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2008;
Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2008;222:206–21.
Walker AK, Wing EE, Banks WA, Dantzer R. Leucine competes with kynurenine for blood-to-brain transport and prevents lipopolysaccharide-induced depression-like behavior in mice. Mol Psychiatry. 2019;24(10):1523–32.
Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85(10):2059–70.
Ida T, Hara M, Nakamura Y, Kozaki S, Tsunoda S, Ihara H. Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci Lett. 2008;432(3):232–6.
Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflammation. 2012;9:275.
Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303(1):1–10.
Tavares RG, Schmidt AP, Abud J, Tasca CI, Souza DO. In vivo quinolinic acid increases synaptosomal glutamate release in rats: reversal by guanosine. Neurochem Res. 2005;30(4):439–44.
Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. 2013;38(9):1609–16.
Walker AJ, Foley BM, Sutor SL, McGillivray JA, Frye MA, Tye SJ. Peripheral proinflammatory markers associated with ketamine response in a preclinical model of antidepressant-resistance. Behav Brain Res. 2015;293:198–202.
Kiraly DD, Horn SR, Van Dam NT, Costi S, Schwartz J, Kim-Schulze S, et al. Altered peripheral immune profiles in treatment-resistant depression: response to ketamine and prediction of treatment outcome. Transl Psychiatry. 2017;7(3):e1065.
Anderson A, Iosifescu DV, Jacobson M, Jones A, Kennon K, O’Gorman C, et al. in ASCP Annual Meeting 28–31 (2019).
Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578(2–3):171–6.
Zarate CA Jr, Payne JL, Quiroz JA, Sporn J, Denicoff KK, Luckenbaugh DA, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatr. 2004;161(1):171–4.
Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, et al. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry. 2007;61(6):822–5.
Manabe T, Togashi H, Uchida N, Suzuki SC, Hayakawa Y, Yamamoto M, et al. Loss of cadherin-11 adhesion receptor enhances plastic changes in hippocampal synapses and modifies behavioral responses. Mol Cell Neurosci. 2000;15(6):534–46.
Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116(5):1218–22.
Steed PM, Tansey MG, Zalevsky J, Zhukovsky EA, Desjarlais JR, Szymkowski DE, et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science. 2003;301(5641):1895–8.
Zalevsky J, Secher T, Ezhevsky SA, Janot L, Steed PM, O’Brien C, et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J Immunol. 2007;179(3):1872–83.
Alexopoulou L, Kranidioti K, Xanthoulea S, Denis M, Kotanidou A, Douni E, et al. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur J Immunol. 2006;36(10):2768–80.
Eidson LN, deSousa Rodrigues ME, Johnson MA, Barnum CJ, Duke BJ, Yang Y, et al. Chronic psychological stress during adolescence induces sex-dependent adulthood inflammation, increased adiposity, and abnormal behaviors that are ameliorated by selective inhibition of soluble tumor necrosis factor with XPro1595. Brain Behav Immun. 2019;81:305–16.
Bedrosian TA, Weil ZM, Nelson RJ. Chronic dim light at night provokes reversible depression-like phenotype: possible role for TNF. Mol Psychiatry. 2013;18(8):930–6.
Aguilar-Valles A, Haji N, De Gregorio D, Matta-Camacho E, Eslamizade MJ, Popic J, et al. Translational control of depression-like behavior via phosphorylation of eukaryotic translation initiation factor 4E. Nat Commun. 2018;9(1):2459.
Price J, Maltais D, Butters K, McGee S, Tye SJ. Antidepressant and anxiolytic effects of tumor necrosis factor inhibition with XPro1595 in a rodent model of antidepressant-resistance. Soc Neurosci. 2018;15:650833.
Karson A, Demirtas T, Bayramgurler D, Balci F, Utkan T. Chronic administration of infliximab (TNF-alpha inhibitor) decreases depression and anxiety-like behaviour in rat model of chronic mild stress. Basic Clin Pharmacol Toxicol. 2013;112(5):335–40.
Karamita M, Barnum C, Möbius W, Tansey MG, Szymkowski DE, Lassmann H, et al. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight. 2017;2(8):e87455.
Gavegnano C, Haile WB, Hurwitz S, Tao S, Jiang Y, Schinazi RF, et al. Baricitinib reverses HIV-associated neurocognitive disorders in a SCID mouse model and reservoir seeding in vitro. J Neuroinflammation. 2019;16(1):182.
Hoang TN, Pino M, Boddapati AK, Viox EG, Starke CE, Upadhyay AA, et al. Baricitinib treatment resolves lower-airway macrophage inflammation and neutrophil recruitment in SARS-CoV-2-infected rhesus macaques. Cell. 2021;184(2):460–75 e21.
Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med. 2020;384:795–807.
Marconi VC, Moser C, Gavegnano C, Deeks SG, Lederman MM, Overton ET, et al. Randomized trial of ruxolitinib in antiretroviral-treated adults with HIV. Clin Infect Dis. 2022;74(1):95–104.
Titanji BK, Farley MM, Mehta A, Connor-Schuler R, Moanna A, Cribbs SK, et al. Use of baricitinib in patients with moderate to severe coronavirus disease 2019. Clin Infect Dis. 2021;72(7):1247–50.
Urits I, Israel J, Hakobyan H, Yusin G, Lassiter G, Fackler N, et al. Baricitinib for the treatment of rheumatoid arthritis. Reumatologia. 2020;58(6):407–15.
Gavegnano C, Detorio M, Montero C, Bosque A, Planelles V, Schinazi RF. Ruxolitinib and tofacitinib are potent and selective inhibitors of HIV-1 replication and virus reactivation in vitro. Antimicrob Agents Chemother. 2014;58(4):1977–86.
Haile WB, Gavegnano C, Tao S, Jiang Y, Schinazi RF, Tyor WR. The Janus kinase inhibitor ruxolitinib reduces HIV replication in human macrophages and ameliorates HIV encephalitis in a murine model. Neurobiol Dis. 2016;92(Pt B):137–43.
Lynall ME, Turner L, Bhatti J, Cavanagh J, de Boer P, Mondelli V, et al. Peripheral blood cell-stratified subgroups of inflamed depression. Biol Psychiatry. 2020;88(2):185–96.
Szollosi DE, Manzoor MK, Aquilato A, Jackson P, Ghoneim OM, Edafiogho IO. Current and novel anti-inflammatory drug targets for inhibition of cytokines and leucocyte recruitment in rheumatic diseases. J Pharm Pharmacol. 2018;70:18–26.
Bekhbat M, Treadway MT, Goldsmith DR, Woolwine BJ, Haroon E, Miller AH, et al. Gene signatures in peripheral blood immune cells related to insulin resistance and low tyrosine metabolism define a sub-type of depression with high CRP and anhedonia. Brain Behav Immun. 2020;88:161–5.
O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65.
Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology. 2020;45(6):990–7.
Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 2018;360(6387):449–53.
Mocelin R, Marcon M, D’Ambros S, Mattos J, Sachett A, Siebel AM, et al. N-acetylcysteine reverses anxiety and oxidative damage induced by unpredictable chronic stress in zebrafish. Mol Neurobiol. 2019;56(2):1188–95.
Wright DJ, Renoir T, Smith ZM, Frazier AE, Francis PS, Thorburn DR, et al. N-acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl Psychiatry. 2015;5:e492.
Zhang Q, Ju Y, Ma Y, Wang T. N-acetylcysteine improves oxidative stress and inflammatory response in patients with community acquired pneumonia: a randomized controlled trial. Medicine. 2018;97(45).
Nasca C, Bigio B, Zelli D, de Angelis P, Lau T, Okamoto M, et al. Role of the astroglial glutamate exchanger xCT in ventral hippocampus in resilience to stress. Neuron. 2017;96(2):402–13 e5.
Linck VM, Costa-Campos L, Pilz LK, Garcia CR, Elisabetsky E. AMPA glutamate receptors mediate the antidepressant-like effects of N-acetylcysteine in the mouse tail suspension test. Behav Pharmacol. 2012;23(2):171–7.
Bower JE, Irwin MR. Mind-body therapies and control of inflammatory biology: a descriptive review. Brain Behav Immun. 2016;51:1–11.
Creswell JD, Taren AA, Lindsay EK, Greco CM, Gianaros PJ, Fairgrieve A, et al. Alterations in resting-state functional connectivity link mindfulness meditation with reduced Interleukin-6: a randomized controlled trial. Biol Psychiatry. 2016;80(1):53–61.
Irwin MR, Olmstead R, Breen EC, Witarama T, Carrillo C, Sadeghi N, et al. Cognitive behavioral therapy and tai chi reverse cellular and genomic markers of inflammation in late-life insomnia: a randomized controlled trial. Biol Psychiatry. 2015;78(10):721–9.
Schuch FB, Vancampfort D, Richards J, Rosenbaum S, Ward PB, Stubbs B. Exercise as a treatment for depression: a meta-analysis adjusting for publication bias. J Psychiatr Res. 2016;77:42–51.
Fabricatore AN, Wadden TA, Higginbotham AJ, Faulconbridge LF, Nguyen AM, Heymsfield SB, et al. Intentional weight loss and changes in symptoms of depression: a systematic review and meta-analysis. Int J Obes (Lond). 2011;35(11):1363–76.
Forsythe LK, Wallace JM, Livingstone MB. Obesity and inflammation: the effects of weight loss. Nutr Res Rev. 2008;21(2):117–33.
Woods JA, Vieira VJ, Keylock KT. Exercise, inflammation, and innate immunity. Immunol Allergy Clin N Am. 2009;29(2):381–93.
Kiecolt-Glaser JK, Bennett JM, Andridge R, Peng J, Shapiro CL, Malarkey WB, et al. Yoga’s impact on inflammation, mood, and fatigue in breast cancer survivors: a randomized controlled trial. J Clin Oncol Off J Am Soc Clin Oncol. 2014;32(10):1040–9.
Tracey KJ. The inflammatory reflex. Nature. 2002;420(6917):853–9.
Pavlov VA, Chavan SS, Tracey KJ. Bioelectronic medicine: from preclinical studies on the inflammatory reflex to new approaches in disease diagnosis and treatment. Cold Spring Harb Perspect Med. 2020;10(3):a034140.
Bastiaanssen TFS, Cussotto S, Claesson MJ, Clarke G, Dinan TG, Cryan JF. Gutted! Unraveling the role of the microbiome in major depressive disorder. Harv Rev Psychiatry. 2020;28(1):26–39.
Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. 2016;16(6):341–52.
Foster JA, Baker GB, Dursun SM. The relationship between the gut microbiome-immune system-brain axis and major depressive disorder. Front Neurol. 2021;12(1660):1–9.
Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatry. 2016;21(6):786–96.
Kilinçarslan S, Evrensel A. The effect of fecal microbiota transplantation on psychiatric symptoms among patients with inflammatory bowel disease: an experimental study. Actas Esp Psiquiatr. 2020;48(1):1–7.
Kurokawa S, Kishimoto T, Mizuno S, Masaoka T, Naganuma M, Liang KC, et al. The effect of fecal microbiota transplantation on psychiatric symptoms among patients with irritable bowel syndrome, functional diarrhea and functional constipation: an open-label observational study. J Affect Disord. 2018;235:506–12.
Liu RT, Walsh RFL, Sheehan AE. Prebiotics and probiotics for depression and anxiety: a systematic review and meta-analysis of controlled clinical trials. Neurosci Biobehav Rev. 2019;102:13–23.
Cuthbert BN. Research domain criteria: toward future psychiatric nosologies. Dialogues Clin Neurosci. 2015;17(1):89–97.
Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol. 2015;12(1):3–20.
Financial Disclosure
Dr. Felger previously consulted for Otsuka on a topic unrelated to this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Felger, J.C. (2023). Increased Inflammation and Treatment of Depression: From Resistance to Reuse, Repurposing, and Redesign. In: Macaluso, M., Preskorn, S.H., Shelton, R.C. (eds) Drug Development in Psychiatry. Advances in Neurobiology, vol 30. Springer, Cham. https://doi.org/10.1007/978-3-031-21054-9_16
Download citation
DOI: https://doi.org/10.1007/978-3-031-21054-9_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-21053-2
Online ISBN: 978-3-031-21054-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)